
Abstract
In recent years, the incidence of diabetes has been increasing rapidly, posing a serious threat to human health. Diabetic cardiomyopathy (DCM) is characterized by cardiomyocyte hypertrophy, myocardial fibrosis, apoptosis, ventricular remodeling, and cardiac dysfunction in individuals with diabetes, ultimately leading to heart failure and mortality. However, the underlying mechanisms contributing to DCM remain incompletely understood. With advancements in molecular biology technology, accumulating evidence has shown that numerous non-coding RNAs (ncRNAs) crucial roles in the development and progression of DCM. This review aims to summarize recent studies on the involvement of three types of ncRNAs (micro RNA, long ncRNA and circular RNA) in the pathophysiology of DCM, with the goal of providing innovative strategies for the prevention and treatment of DCM.
Similar content being viewed by others

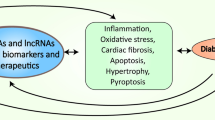
Introduction
Diabetic cardiomyopathy (DCM) is a type of diabetic heart disease with abnormal myocardial structure and function in diabetic patients without other cardiovascular diseases (such as coronary heart disease, hypertension, severe valvular disease, and congenital heart disease) [1, 2]. Individuals with type 2 diabetes mellitus (T2DM) are estimated to face a 75% higher risk of cardiovascular mortality or hospitalization for heart failure compared with patients without diabetes [3]. The underlying mechanisms of DCM, which encompass altered metabolism, mitochondrial dysfunction, oxidative stress, inflammation, cardiac fibrosis, cell death, and extracellular matrix remodeling, have not been fully elucidated and remain subject to debate [4]. Disruption in energy substrate utilization [5,6,7], calcium and sodium homeostasis disorder [8,9,10], insulin resistance [11, 12], potential involvement of epicardial fat [13,14,15,16], and endothelial dysfunction [17,18,19] are believed to contribute to the onset and progression of DCM.
Non-coding RNAs (ncRNAs) are highly functional and dynamic nucleic acids that do not encode proteins. They include RNAs with specific functions, such as rRNAs, tRNAs, snRNAs, snoRNAs, and microRNAs (miRNAs), as well as RNAs with unknown functions. Long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs) are the novel members of non-coding RNA family, but their functions and regulatory mechanisms are still not fully understood. Accumulating evidence indicates that ncRNAs play a crucial role in the regulation of endothelial cells, vascular and smooth muscle cells, cardiac metabolism, ischemia and inflammation. This indicates that ncRNAs hold significant potential in the diagnosis, evaluation, and treatment of DCM.
MicroRNAs
MicroRNAs (miRNAs) are highly conserved, single-stranded ncRNAs typically consisting of 20–22 nucleotides. The typical functionality of miRNAs is to negatively regulate gene expression by binding to target mRNA, leading to either mRNA degradation or inhibition of translation [20]. MiRNAs play diverse roles encompassing cardiac hypertrophy, cardiomyocyte apoptosis, autophagy and pyroptosis, myocardial fibrosis, oxidative stress, and other pathophysiological processes, Figs. 1 and 2 [21,22,23,24]. The expression pattern of miRNAs during DCM were revealed in 2011. Since then, there have been continuous studies on the role of miRNAs in the development and progression of DCM.

Functional miRNAs promote or inhibit cardiac hypertrophy, fibrosis, ROS, mitochondria dysfunction and cell death in the pathology of diabetic cardiomyopathy

MiRNA regulate fibrosis through TGF-β, PI3K/AKT, Notch, and MAPK signaling pathways, and apoptosis through MAPK and p53. P53, AMPK and TGF-β also mediate the role of miRNAs in cardiac hypertrophy. While Nrf2 signaling pathways is the key in ROS
Cardiac hypertrophy
Multiple miRNAs have been identified to modulate cardiac hypertrophy and fibrosis in DCM. Anti-hypertrophic miRNAs, such as miR-1 [25], miR-133a [26], miR-373 [27], miR-181a [28], miR-150 [22], miR-30c [29], miR-378a [30], miR-29a [31] and miR-200c [32]. Pro-hypertrophic miRNAs include miR-208a [33], miR-451 [34], miR-214, miR-212 [35], miR-221 [36], miR-195, miR-125b [37] and miR-199a [38]. For instance, miR-1, a muscle-specific miRNA, attenuates cardiomyocyte hypertrophy by negatively regulating calcium signaling components calmodulin, Gata4 and Mef2a [39]. Overexpression of miR-133a has been shown to prevent hypertrophic changes in DCM by downregulating the serum and glucocorticoid-regulated kinase 1 (SGK1), IGFR1 and myocyte-specific enhancer factor 2C (MEF2C) [26]. Additionally, miR-373 influences MEF2C signaling, a key transcription factor for myocardial hypertrophy and mediates cardiac fibrosis through activation of the p300 gene [27]. MiR-181a and miR-30c could synergistically regulate the p53–p21 pathway in diabetes-induced cardiac hypertrophy [28]. MiR-208a promotes cardiac hypertrophy by inhibiting myostatin, GATA4, and β-myosin heavy chain (MHC) expression [33]. Kuwabara et al. [34] demonstrated that miR-451 could suppress the LKB1/AMPK pathway in cardiac hypertrophy induced by diabetes. Biao et al. [40] reported that miR-195 accelerates cardiomyocyte hypertrophy in vitro induced by high-glucose through downregulating the expression of Smad7 and modulating TGF-β/Smad pathways. Moreover, the endothelial-to-meschenymal transition (EndMT) is a key driver of cardiac fibrosis and plays an important role in the pathogenesis of DCM. Ding et al. [41] reported that silencing miR-195-5p inhibits the TGF-β1-smads-snail pathway by targeting Smad7, thereby attenuating EndMT and reducing myocardial fibrosis in DCM.
Myocardial fibrosis
Myocardial fibrosis stands out as a prominent pathological characteristic of DCM, and its regulation involves various miRNAs such as miR-34a, miR-150-5p, miR-18a-5p, miR-30, miR-199 [38], miR-135b [42], miR-133a [23], miR-125b, miR-200b [43], miR-320 [44], miR-15a/b [45], miR-21 [46], miR-29 [47] and so on. For instance, research by Bernardo et al. [48] highlighted the involvement of miR-34a in cardiac fibroblasts exposed to high glucose, showing its ability to enhance collagen synthesis through decreasing the level of sirtuin 1 (SIRT1) [49, 50]. Che et al. [51] indicated that inhibiting miR-150-5p could ameliorate NF-κB-related inflammation and TGF-β1/Smad-induced cardiac fibrosis through targeting Smad7. Li et al. [52] showed that the miR-21 inhibition decreased cardiac perivascular fibrosis by suppressing EndMT and upregulating Smad7 while activating p-Smad2 and p-Smad3. Additionally, inhibition of miR-21 could reduce fibrosis via blocking the activation of the p38 signaling pathway [46, 53]. MiR-18a-5p was found to downregulate Notch2 expression, thereby suppressing EndMT in human aortic valvular endothelial cells exposed to high glucose [54]. Zhu et al. [55] demonstrated that miR-142-3p could attenuate high glucose-induced EndMT in primary human aortic endothelial cells(HAECs), possibly through blocking the TGF-β1/Smad signaling pathway. In addition, Yang et al. [56] showed that the miR-203 may function as a cardioprotective regulator in DCM, as its up-regulation could reduce myocardial hypertrophy, myocardial fibrosis, myocardial apoptosis by targeting PIK3CA via inactivation of PI3K/Akt signaling pathway.
Mitochondrial damage and oxidative stress
Mitochondrial damage and the accumulation of excessive reactive oxygen species (ROS), including reduced oxygen (O2) metabolites, superoxide anion (O2−), hydroxyl radicals (·OH) and hydrogen peroxide (H2O2), are recognized as significant molecular and cellular mechanisms contributing to cardiac dysfunction and cardiomyopathy in diabetic individuals. Various miRNAs such as miR-340-5p, miR-92a-2-5p, miR-1 [39], miR-22, miR-144, miR-195 [57], miR-200c, miR-221 [36], miR-146a, miR-34a [58], miR-210, miR-19b, miR-125b, miR-155, miR-27a and miR-503 [35] have been implicated in the regulation of hyperglycaemia-induced oxidative stress. For instance, Zhu et al. indicated that overexpression of miR-340-5p [59] in cardiomyocytes led to increased mitochondrial functional loss, oxidative stress, and cardiomyocyte apoptosis in diabetic mice by targeting myeloid cell leukemia 1(Mcl-1). Yu et al. [60] observed that decreased miR-92a-2-5p expression was also detected in high glucose-induced cardiomyocytes. Overexpression of miR-92a-2-5p ameliorated cardiomyocyte oxidative stress injury, by inhibiting MKNK2 expression and leading to decreased phosphorylation of p38-mitogen-activated protein kinase(MAPK) signaling. Overexpression of miR-22 was shown to attenuate oxidative stress by upregulating Sirt 1 in DCM [61]. Furthermore, Yu et al. found that downregulation of miR-144 protected against diabetes-induced cardiac oxidative damage by directly targeting nuclear factor-erythroid 2-related factor 2 (Nrf2) [62]. Members of miR-200 family such as the miR-200a, and miR-200c play a crucial role in oxidative stress in cardiovascular complications of diabetes. MiR-200c was shown to enhance COX-2 expression in endothelial cells by suppressing ZEB1 expression, promoting prostaglandin E2 production, and thereby reducing endothelium-dependent relaxation [63]. Additionally, Miao et al. demonstrated that upregulated expression of miR-503 in DCM was associated with the protective effects of Phase II Enzyme Inducer CPDT via the nuclear factor erythroid 2-related factor 2/anti-oxidant response elements (Nrf2/ARE) signaling pathway, a key anti-oxidant signaling pathway [64, 65].
Cell death
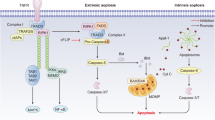
Apoptosis, autophagy, necrosis and pyroptosis are four pathways resulting in cell death, playing important roles in the pathological progression of DCM. Several miRNAs, including miR-1, miR-30, miR-483-3p, miR-144, miR-21 [66, 67], miR-210, miR-212 [68], miR-200b-3p, miR-195, miR-320b, miR-133, miR-221 [36], miR-320 [69], miR-378 [70], miR-34a [71], miR-29 [47], miR-181a [28], have been associated with cell death. For example, the miR-30 family is one of the most abundant miRNAs in the heart, comprising miR-30a, miR-30b, miR-30c, miR-30d and miR-30e, participates in DCM through a variety of mechanisms, including autophagy, apoptosis, oxidative stress, and inflammation [72, 73]. Enforced expression of miR-30a or miR-30b can inhibit apoptosis induced by hydrogen peroxide, by influencing p53 translation [74]. Conversely, upregulation of miR-30d in DCM has been linked to promoting cardiomyocyte pyroptosis, leading to enhanced proinflammatory cytokines IL-1β and IL-18, as well as caspase-1. In addition, microRNA-30d could regulate cardiomyocyte pyroptosis by directly targeting foxo3a in DCM [24]. Qiao et al. found that miR-483-3p [75] was upregulated in streptozotocin-induced diabetic mice, promoting myocardial cell apoptosis by transcriptionally repressing insulin growth factor 1 (IGF1). Furthermore, repression of miR-144 decreased the protein levels of Bax. It phosphorylated c-Jun amino-terminal kinase (p-JNK) promoted cell proliferation and reduced apoptosis of cardiomyocytes treated with high glucose through targeting the CTRP3/JNK signaling pathway [76]. Moreover, Lin et al. demonstrated that miR-210 [77] repression facilitates advanced glycation end-product (AGE)-induced cardiac mitochondrial dysfunction and apoptosis through JNK activation. On the other hand, low expression of miR-200b-3p in DCM was associated with increased cardiocyte apoptosis, and its overexpression could reduce apoptosis by targeting the CD36/PPAR-γ signaling pathway [78]. Upregulation of miR-195 was reported to lead to cardiomyocyte apoptosis. Zheng et al. revealed that the knockdown of miR-195 could inhibit myocardial hypertrophy in diabetes by preventing cardiomyocyte apoptosis in cardiac endothelial cells in response to non-esterified fatty acid (NEFA) such as palmitate [57]. Additionally, Tang et al. reported that enforced expression of miR-22 could attenuate oxidative injury by upregulating Sirt 1 in diabetic cardiomyopathy [61]. These findings underscore the intricate regulatory roles of miRNAs in modulating cell death pathways and their implications for the pathogenesis of DCM.
LncRNAs
lncRNAs were the heterogeneous RNA transcripts, which are longer than 200 nucleotides, and have many epigenetic regulation forms, including DNA methylation, histone modification and regulation of miRNA [79, 80]. lncRNAs play essential roles in multiple biological processes, such as chromatin structural changes, transcriptional regulation, post-transcriptional processing, intracellular trafficking, and regulation of enzyme activity [81, 82]. Recently, growing evidence has suggested that lncRNAs can actively participate in the pathogenesis of diverse cardiovascular diseases, including DCM, Fig. 3.

Involvement of lncRNA in the pathogenesis of diabetic cardiomyopathy
Cardiac hypertrophy and myocardial fibrosis
Myocardial fibrosis is a critical pathological change observed in DCM. Feng et al. [83] reported increased lncRNA DCRF expression and induced autophagy in cardiomyocytes in high glucose-induced rats. Knockdown of DCRF was found to reduce cardiomyocyte autophagy, attenuate myocardial fibrosis and improve cardiac function in diabetic rats by targeting miR-551b-5p. In another study, Liu et al. [84] indicated that lncRNA NORAD was upregulated in diabetic and DCM mice. Silencing NORAD expression could reduce inflammatory responses, and improve cardiac function and fibrosis in DCM mice via the ceRNA network of NORAD/miR-125a-3p/Fyn. Moreover, Qi et al. [85] demonstrated that high glucose-induced lncRNA MIAT upregulation was responsible for Interleukin-17 (IL-17) production in cardiomyocytes, which was a pro-inflammatory cytokine and a key regulator of host inflammation. Knockdown of MIAT could significantly attenuate IL-17 expression, ameliorate cardiac fibrosis and improve cardiac contractility. Recent research has also highlighted the involvement of lncRNA Airn in the progression of cardiac fibroblasts in DCM, demonstrating its ability to alleviate diabetic cardiac fibrosis via a m6A-IMP2-p53 axis [86]. EndMT was induced by high glucose and drove to cardiac fibrosis. LncRNA DANCR could markedly attenuate high glucose-mediated EndMT in vitro by inhibiting the activation of FoxO1 and increasing the expression of DDAH1 [87]. Moreover, Wang et al. revealed that lncRNA TUG1 was upregulated in diabetic mice exposed to high glucose, TUG1 overexpression promoted myocardial fibrosis by suppressing the expression of microRNA-145a-5p [88]. These studies underscore the intricate regulatory roles of various lncRNAs in modulating myocardial fibrosis and cardiac function in the context of DCM.
Mitochondrial damage and oxidative stress
Mitochondrial damage and oxidative stress have a significant involvement in the progression of DCM. Recent research has highlighted the upregulation of lncRNA DACH1 in DCM hearts and high glucose-treated cardiomyocytes. DACH1 aggravates DCM by promoting mitochondrial oxidative stress, cell apoptosis, cardiac fibrosis and hypertrophy via increasing ubiquitination-mediated SIRT3 degradation in mice’s hearts [89]. In a study by Gao et al. [50], it was found that lncRNA HOTAIR expression was significantly decreased in diabetic mice hearts. Knockdown of HOTAIR in high glucose-induced H9c2 cells resulted in increased oxidative injury. HOTAIR could protect against DCM via activating of the Sirtuin 1(SIRT1) expression by sponging miR-34a [90]. Additionally, lncRNA MALAT1 was significantly upregulated in the myocardium of diabetic mice and high glucose-induced cardiomyocytes, mediated oxidative stress, mitochondrial damage and apoptosis through activating the RhoA/ROCK pathway via sponging miR-185-5p. LncRNA H19 is a key lncRNA in DCM, which produces a 2.3-kb non-coding mRNA and is conserved via matriarchal evolution [91]. Wang et al. demonstrated that H19 repressed oxidative stress, endoplasmic reticulum stress (ERS) and apoptosis in vitro, furthermore, it reduced cardiomyocytes apoptosis and improved fibrosis in vivo through PI3K/AKT/mTOR signaling pathway [92].
Cell death
Some lncRNAs have been identified to be correlated with cardiomyocyte apoptosis, pyroptosis, ferroptosis and autophagy during the process of DCM. For instance, lncRNAs MALAT1 not only has been implicated in mitochondrial injury, but also participated in cardiomyocyte apoptosis. Zhang et al. [93] reported that Down-regulation of lncRNA MALAT1 could reduce cardiomyocyte apoptosis and improve left ventricular function in diabetic rats. Furthermore, Wang Chong and colleagues [94] found that MALAT1 recruited the histone methyltransferase EZH2 to the promoter region of miR-22, thereby inhibing its expression. EZH2, in turn, upregulated the expression of ATP-binding cassette transporter A1 (ABCA1), a known target gene of miR-22. Knockdown of EZH2 was found to enhance cardiac function and prevent cardiomyocyte apoptosis in db/db mice and mouse cardiomyocytes cultured inhigh glucose conditions in the presence of MALAT1. MALAT1 was involved in the processes of cardiac function and cardiomyocyte apoptosis via the EZH2/miR-22/ABCA1 signaling cascade. lncRNA TINCR participated in pyroptosis in DCM progression, which positively regulated NLRP3 by increasing its mRNA stability, downregulating TINCR could suppress pyroptosis and DCM [95]. Recently, Xie et al. indicated that lncRNA ZNF593-AS directly interacted with the functional domain of interferon regulatory factor 3 (IRF3), thereby inhibiting the fatty acid-induced phosphorylation and activation of IRF3. This interaction ultimately let to mitigation of cardiac cell death and inflammation in DCM [96]. Moreover, lncRNA MIAT was demonstrated to be involve in the progression of cell death in DCM. Xiao et al. [97] reported that MIAT played a vital role in regulating of pyroptosis in DCM via targeting miR-214-3p. Zhou et al. [98] indicated that MIAT knockdown could reduce DAPK2 expression by increasing miR-22-3p, and inhibit apoptosis in cardiomyocytes exposed to high glucose. Ferroptosis is an iron-dependent regulated necrosis associated with a new form of regulatory cell death [99]. Ni et al. [100] showed that inhibition of lncRNA ZFAS1 could alleviate the development of DCM by reducing ferroptosis via stabilizing miR-150-5p to activate CCND2. Interactions of ncRNA are also involved in cardiomyocyte apoptosis. The interaction among NORAD, miRNA-150-5p and ZEB1 has been clarified to influencing the proliferation and apoptosis in HG-induced AC16 cells [101].
Circular RNAs
CircRNAs are produced from precursor mRNAs by the back-splicing of exons in eukaryotes and are widely expressed in a tissue-specific and developmental stage-specific pattern. However, knowledge of these species has remained limited due to their difficult study through traditional methods of RNA analysis [21, 102]. CircRNAs differ from linear RNAs in that they are circular molecules with covalently closed loop structures, which are involved in a wide range of biological processes, the expression disorder of circRNAs might lead to abnormal cellular functions and disease. CircRNAs may inhibit the translation of mRNAs, altering gene expression by regulating splicing or transcription and by interacting with RNA-binding proteins [103]. However, the regulation of circRNAs in cardiovascular diseases remains largely unexplored Fig. 4.

The role of circRNA in the pathogenesis of diabetic cardiomyopathy
Compared with miRNAs and lncRNAs, the understanding of circRNAs in the molecular mechanisms of DCM is still needs to be improved. Yuan et al. revealed that circRNA DICAR was downregulated in diabetic mice hearts and was associated with cardiac dysfunction, cardiac cell hypertrophy, and cardiac fibrosis [104]. Yang et al. showed the involvement of another circRNA in the regulation of diabetic myocardial fibrosis. They found that circRNA-0076631 was increased both in high glucose-induced cardiomyocytes and in the serum of diabetic patients, modulated miR-214-3p and its target gene caspase-1 and mediated fibrosis-associated protein resection [105]. Zhou et al. reported that circRNA-010567 sponged miR-141 and upregulated target gene TGF-β1, mediated fibrosis-associated protein resection in the diabetic mice myocardial fibrosis model. Silencing the expression of circRNA-010567 could suppress fibrosis-associated protein resection, including Col I, Col III and α-SMA in the regulation of diabetic myocardial fibrosis [106]. CircRNA homeodomain interacting protein kinase 3 (circHIPK3) is a particularly abundant circRNA involved in metabolic dysregulation and tumorigenesis [107,108,109]. Wang et al. found that circHIPK3 was upregulated in a DCM model of streptozotocin (STZ)-induced diabetic mice [110]. CircHIPK3 increased the expression of fibrosis-associated genes, such as COL1A2, COL3A1 and α-SMA, via sponging miR-29b-3p in AngII-induced mouse myocardium [111]. Knockdown of circHIPK3 could ameliorate myocardial fibrosis and improve cardiac function in vivo, while decreasing the proliferation of CFs treated with Ang II via miR-29b-3p/Col1a1-Col3a1 in vitro. circRNA circular cerebellar degeneration-related protein 1 antisense (circCDR1as) is degraded by sponging miR-671 via protein Argonaute 2 [112]. CircCDR1as was upregulated in DCM hearts of STZ-induced diabetic mice, which promoted cardiomyocyte apoptosis through activating the MST1-Hippo pathway in vivo and in HG-treated primary cardiomyocytes. Knocking down CDR1as inhibited cardiomyocyte apoptosis in DCM [113]. Recently, a novel circRNA mitogen-activated protein kinase kinase kinase 5 (circMAP3K5) was found to regulate apoptosis of cardiomyocytes in DCM. Shen et al. indicated that circMAP3K5 upregulated in in high glucose-induced H9c2 cardiomyocytes, accelerated cardiomyocytes apoptosis through the miR-22-3p/death-associated protein kinase 2 (DAPK2) axis [114]. Fu et al. found that circ-0071269 was significantly overexpressed in H9c2 cells upon treatment with high glucose. Circ_0071269 could promote the development of DCM through the miR-145/GSDMA axis. Knockdown of circ_0071269 promoted cell viability and inhibited the inflammatory response, cytotoxicity, and pyroptosis of H9c2 cells in vitro [115].
Clinical application
NcRNAs are involved in the development and progression of DCM, presenting in the blood are extremely stable and can be potentially used as diagnostic and prognostic biomarkers for cardiovascular diseases, consequently allowing early intervention. Furthermore, ncRNAs are modulating various biological pathways, suggesting that these molecules may be harnessed as a novel therapeutic strategy in treating DCM.
Vildagliptin is an oral hypoglycemic drug that reduces hyperglycemia in T2DM. Li et al. reported that vildagliptin could enhance cardiac function in type 2 diabetic mice by restoring autophagy and alleviated fibrosis through the miR-21/SPRY1/ERK/mTOR pathway [66]. Melatonin is a hormone produced by the pineal gland, and it has extensive beneficial effects on various tissues and organs. Che et al. showed that melatonin administration significantly ameliorated cardiac dysfunction and reduced collagen production via inhibiting lncRNA MALAT1/miR-141-mediated NLRP3 inflammasome and TGF-β1/Smads signaling pathway, while the expression of TGF-β1, p-Smad2, p-Smad3, NLRP3, ASC, cleaved caspase-1, mature IL-1β, and IL-18 were downregulated in the heart of mice with diabetes mellitus following melatonin treatment [116]. Furthermore, melatonin was reported to alleviate cardiac dysfunction and cardiomyocyte apoptosis in diabetic rats, notably by downregulating lncRNA H19/MAPK and upregulating miR-29c levels [117]. Diallyl trisulfide (DATS) is an anti-oxidant in garlic oil, can inhibit stress-induced cardiac apoptosis and can be used as a cardioprotective agent. Lin et al. found that the DATS could mediate AGE-induced cardiac cell apoptosis attenuation by promoting FoxO3a nuclear transactivation to enhance miR-210 expression and regulate JNK activation [77]. Pomegranate peel extract (PPE) exhibits a cardioprotective effect due to its anti-oxidant and anti-inflammatory properties, which could significantly ameliorate cardiac hypertrophy in diabetic rats and increase the survival rate. The protective effect of PPE on DCM could be due to the inhibition of the NLRP3/caspase-1/IL-1β signaling pathway and downregulation of lncRNA-MALAT1 [118]. Berberine (BBR) is a natural compound extracted from a Chinese herb (Rhizoma coptidis; known as ‘Huang Lian’ in Chinese). It has been traditionally used in Chinese medicine for treating inflammatory disorders and cardiovascular injury induced by diabetes mellitus [119]. Yang et al. indicated that BBR alleviated DCM by inhibiting miR‑18a-3p-mediated gasdermin D (Gsdmd) activation [120]. Citronellal (CT), a monoterpenoid natural product extracted from the grass plant Citronella, has demonstrated anti-thrombotic, anti-hypertensive and anti-diabetic cardiomyopathy properties. Qiu et al. reported that CT significantly reduced vascular plate area and decreased endothelial lipid and cholesterol deposition in the common carotid artery of mice. CT upregulated the expression of activated protein 2α (AP-2α/TFAP2A) and circRNA_102979 in vascular endothelium. This led to an enhanced binding capability of circRNA_102979 to miR-133a, counteracting the inhibitory effect of miR-133a on target genes. Consequently, this mechanism helped alleviate vascular endothelial injury [121]. Ranolazine, a piperazine derivative approved by the US Food and Drug Administration in 2006 for the treatment of stable angina pectoris, has shown effectiveness in treating cardiovascular disease [122]. Ranolazine increased miR-135b expression in cardiac fibroblasts exposed to high glucose. Furthermore, miR-135b directly interacted with caspase-1. Thereby, ranolazine could reduce pyroptosis, inhibit collagen deposition and improve cardiac function in rats by upregulating miR-135b [42]. Activation of cardiac miR-132 leads to adverse remodeling and pathological hypertrophy. CDR132L, a synthetic antisense oligonucleotide that selectively blocks pathologically elevated miR-132, has shown promisingeffects on heart failure (HF) in the early stage following myocardial infarction (MI) in phase I/II trials [123,124,125]. There are many opportunities for further advancement in cardiovascular medicine, particularly in the new therapeutics to target ncRNAs for diabetic DCM, through conducting large-animal studies and phase I/II trials involving humans.
Conclusions
In the present review, we provide an overview of the recent advancements in understanding the role of ncRNAs in the pathogenesis of DCM. Various ncRNAs play crucial roles in regulating cardiomyocyte hypertrophy, myocardial fibrosis, apoptosis and autophagy, oxidative stress and inflammatory response, all of which are key mechanisms associated with DCM, Table 1. With the growing epidemic of diabetes mellitus and its related cardiac complications, the potential of ncRNA as promising attractive biomarkers and therapeutic targets for DCM and heart failure has captured significant attention within the scientific community. The identification and characterizations of ncRNAs and the pathways they influence may pave the way for the development of innovative treatments to manage or combat diabetic cardiomyopathy in the near future.
Availability of data and materials
No datasets were generated or analysed during the current study.
Abbreviations
- ABCA1:
-
ATP binding cassette subfamily A member 1
- ·OH:
-
Hydroxyl radicals
- ABCA1:
-
ATP-binding cassette transporter A1
- AGE:
-
Advanced glycation end-product
- Akt:
-
Akt kinase
- AMPK:
-
AMP-activated protein kinase
- AP-2α:
-
Activated protein 2α
- ARE:
-
Anti-oxidant response elements
- ASC:
-
Apoptosis-associated speck-like protein containing a caspase recruitment domain
- Bax:
-
BCL2 associated X, apoptosis regulator
- BBR:
-
Berberine
- Bcl-2:
-
BCL2 apoptosis regulator
- Cab39:
-
Calcium binding protein 39
- CASP1:
-
Caspase 1
- CCND2:
-
Cyclin D2
- CD36:
-
CD36 molecule
- circCDR1as:
-
CircRNA circular cerebellar degeneration-related protein 1 antisense (circCDR1as)
- circHIPK3:
-
CircRNA homeodomain-interacting protein kinase 3
- circMAP3K5:
-
CircRNA mitogen-activated protein kinase kinase kinase 5
- circRNA:
-
Circular RNA
- Col I:
-
Collagen I
- Col III:
-
Collagen III
- Col1a2:
-
Collagen Type I Alpha 2 Chain
- Col3a1:
-
Collagen Type III Alpha 1 Chain
- COX-2:
-
Cyclooxygenase-2
- CPDT:
-
Phase II Enzyme Inducer
- CT:
-
Citronellal
- CTRP3:
-
C1QTNF3 C1q and TNF related 3
- DAPK2:
-
Death-associated protein kinase 2
- DATS:
-
Diallyl trisulfide
- DCM:
-
Diabetic cardiomyopathy
- DDCH1:
-
Dimethylarginine dimethylaminohydrolase 1
- DUSP8:
-
Dual specificity phosphatase 8
- ELAVL1:
-
ELAV like RNA binding protein 1
- EndMT:
-
Endothelial-to-meschenymal transition
- ERS:
-
Endoplasmic reticulum stress
- EZH2:
-
Enhancer of zeste 2 polycomb repressive complex 2 subunit
- FoxO1:
-
Forkhead box O1
- foxo3a:
-
Forkhead box O3
- Fyn:
-
Fyn proto-oncogene
- Gata4:
-
GATA binding protein 4
- GSDMA:
-
Gasdermin A
- Gsdmd:
-
Gasdermin D
- H2O2:
-
Hydrogen peroxide
- HAECs:
-
Human aortic endothelial cells
- IGF1:
-
Insulin like growth factor 1
- IL-17:
-
Interleukin-17
- IL-18:
-
Interleukin-18
- IL-1β:
-
Interleukin-1β
- IMP2:
-
Insulin-like growth factor 2 mRNA binding protein 2
- IGF1:
-
Insulin growth factor 1
- IRF3:
-
Interferon regulatory factor 3
- LKB1:
-
Liver kinase B1
- lncRNA:
-
Long ncRNA
- lncRNA DACH1:
-
LncRNA dachshund homolog 1
- lncRNA DANCR:
-
LncRNA Differentiation antagonizing nonprotein coding RNA
- lncRNA DCRF:
-
LncRNA DCM-related factor
- lncRNA HOTAIR:
-
LncRNA HOX transcript antisense RNA
- lncRNA MIAT:
-
LncRNA myocardial infarction associated transcript
- lncRNA NORAD:
-
LncRNA non-coding RNA activated by DNA damage
- lncRNA TINCR:
-
LncRNA terminal differentiation-induced non-coding RNA
- lncRNA TUG1:
-
LncRNA taurine-upregulated gene 1
- lncRNA ZFAS1:
-
LncRNA zinc finger NFX1-type containing 1 antisense RNA 1
- lncRNA ZNF593-AS:
-
LncRNA Zinc Finger Protein 593 AS
- LncRNAs MALAT1:
-
LncRNA metastasis associated lung adenocarcinoma transcript 1
- LXRα:
-
Liver X receptor α
- m6A:
-
N6-methyladenosine
- MAPK:
-
Mitogen-activated protein kinase
- Mcl-1:
-
Myeloid cell leukemia 1
- Mef2a:
-
Myocyte enhancer factor 2A
- MEF2C:
-
Myocyte-specific enhancer factor 2C
- MHC:
-
Myosin heavy chain
- miRNA:
-
Micro RNA
- MKNK2:
-
MAPK interacting serine/threonine kinase 2
- MST1:
-
Macrophage stimulating 1
- mTOR:
-
Mammalian target of rapamycin
- ncRNAs:
-
Non-coding RNAs
- NEFA:
-
Non-esterified fatty acid
- NLRP3:
-
NLR family pyrin domain containing 3
- Notch2:
-
Notch receptor 2
- Nrf2:
-
Nuclear factor-erythroid 2-related factor 2
- O2 :
-
Oxygen
- O2 − :
-
Superoxide anion
- p21:
-
Cyclin-dependent kinase inhibitor p21
- p27:
-
P27 protein
- p38:
-
Mitogen-activated protein kinase 14
- p53:
-
Tumor suppressor gene p53
- PCDH17:
-
Protocadherin 17
- PI3K:
-
Phosphatidylinositol 3-kinase
- PIK3CA:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- p-JNK:
-
Phosphorylated c-Jun amino-terminal kinase
- PPAR-γ:
-
Peroxisome proliferator activated receptor gamma
- PPE:
-
Pomegranate peel extract
- p-Smad2:
-
Phosphorylated SMAD family member 2
- p-Smad3:
-
Phosphorylated SMAD family member 3
- RhoA:
-
Ras homolog family member A
- ROCK:
-
Rho associated coiled-coil containing protein kinase
- ROS:
-
Reactive oxygen species
- SAPK:
-
Mitogen-activated protein kinase 9
- SGK1:
-
Serum and glucocorticoid-regulated kinase 1
- SIRT1:
-
Sirtuin 1
- SIRT3:
-
Sirtuin 3
- Smad7:
-
SMAD family member 7
- SPRY1:
-
Sprouty RTK signaling antagonist 1
- STZ:
-
Streptozotocin
- T2DM:
-
Type 2 diabetes mellitus
- TFAP2A:
-
Transcription factor AP-2 alpha
- TGF-β:
-
Transforming growth factor beta
- ZEB1:
-
Zinc finger E-box binding homeobox 1
- α-SMA:
-
α-Smooth muscle actin
References
Lorenzo-Almoros A, Tunon J, Orejas M, Cortes M, Egido J, Lorenzo O. Diagnostic approaches for diabetic cardiomyopathy. Cardiovasc Diabetol. 2017;16(1):28.
Lorenzo-Almoros A, Cepeda-Rodrigo JM, Lorenzo O. Diabetic cardiomyopathy. Rev Clin Esp (Barc). 2022;222(2):100–11.
Kristensen SL, Mogensen UM, Jhund PS, Petrie MC, Preiss D, Win S, Kober L, McKelvie RS, Zile MR, Anand IS, et al. Clinical and echocardiographic characteristics and cardiovascular outcomes according to diabetes status in patients with heart failure and preserved ejection fraction: a report from the I-preserve trial (Irbesartan in Heart failure with preserved ejection fraction). Circulation. 2017;135(8):724–35.
Hu X, Bai T, Xu Z, Liu Q, Zheng Y, Cai L. Pathophysiological Fundamentals of Diabetic Cardiomyopathy. Compr Physiol. 2017;7(2):693–711.
Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA-REG OUTCOME trial: a thrifty substrate hypothesis. Diabetes Care. 2016;39(7):1108–14.
Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol. 2018;15(8):457–70.
Taegtmeyer H. Failing heart and starving brain: ketone bodies to the rescue. Circulation. 2016;134(4):265–6.
Mustroph J, Wagemann O, Lucht CM, Trum M, Hammer KP, Sag CM, Lebek S, Tarnowski D, Reinders J, Perbellini F, et al. Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. 2018;5(4):642–8.
Bertero E, Maack C. Calcium Signaling and reactive oxygen species in Mitochondria. Circ Res. 2018;122(10):1460–78.
Lambert R, Srodulski S, Peng X, Margulies KB, Despa F, Despa S. Intracellular na + concentration ([Na+]i) is elevated in Diabetic hearts due to enhanced Na+-Glucose cotransport. J Am Heart Assoc. 2015;4(9):e002183.
Taegtmeyer H, McNulty P, Young ME. Adaptation and maladaptation of the heart in diabetes: part I: general concepts. Circulation. 2002;105(14):1727–33.
Kruger M, Babicz K, von Frieling-Salewsky M, Linke WA. Insulin signaling regulates cardiac titin properties in heart development and diabetic cardiomyopathy. J Mol Cell Cardiol. 2010;48(5):910–6.
Packer M. Leptin-Aldosterone-Neprilysin Axis: identification of its distinctive role in the pathogenesis of the three phenotypes of heart failure in people with obesity. Circulation. 2018;137(15):1614–31.
Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation. 2017;136(1):6–19.
Gruzdeva OV, Akbasheva OE, Dyleva YA, Antonova LV, Matveeva VG, Uchasova EG, Fanaskova EV, Karetnikova VN, Ivanov SV, Barbarash OL. Adipokine and Cytokine profiles of Epicardial and Subcutaneous Adipose tissue in patients with Coronary Heart Disease. Bull Exp Biol Med. 2017;163(5):608–11.
Udell JA, Cavender MA, Bhatt DL, Chatterjee S, Farkouh ME, Scirica BM. Glucose-lowering drugs or strategies and cardiovascular outcomes in patients with or at risk for type 2 diabetes: a meta-analysis of randomised controlled trials. Lancet Diabetes Endocrinol. 2015;3(5):356–66.
Eriksson L, Nystrom T. Antidiabetic agents and endothelial dysfunction—beyond glucose control. Basic Clin Pharmacol Toxicol. 2015;117(1):15–25.
Dunlay SM, Roger VL, Weston SA, Jiang R, Redfield MM. Longitudinal changes in ejection fraction in heart failure patients with preserved and reduced ejection fraction. Circ Heart Fail. 2012;5(6):720–6.
De Keulenaer GW, Brutsaert DL. Systolic and diastolic heart failure are overlapping phenotypes within the heart failure spectrum. Circulation. 2011;123(18):1996–2004. discussion 2005.
Wang L, Lv Y, Li G, Xiao J. MicroRNAs in heart and circulation during physical exercise. J Sport Health Sci. 2018;7(4):433–41.
Zhang W, Xu W, Feng Y, Zhou X. Non-coding RNA involvement in the pathogenesis of diabetic cardiomyopathy. J Cell Mol Med. 2019;23(9):5859–67.
Duan Y, Zhou B, Su H, Liu Y, Du C. miR-150 regulates high glucose-induced cardiomyocyte hypertrophy by targeting the transcriptional co-activator p300. Exp Cell Res. 2013;319(3):173–84.
Chen S, Puthanveetil P, Feng B, Matkovich SJ, Dorn GW 2nd, Chakrabarti S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J Cell Mol Med. 2014;18(3):415–21.
Li X, Du N, Zhang Q, Li J, Chen X, Liu X, Hu Y, Qin W, Shen N, Xu C, et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. 2014;5(10):e1479.
Ikeda S, He A, Kong SW, Lu J, Bejar R, Bodyak N, Lee KH, Ma Q, Kang PM, Golub TR, et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol. 2009;29(8):2193–204.
Feng B, Chen S, George B, Feng Q, Chakrabarti S. miR133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes Metab Res Rev. 2010;26(1):40–9.
Shen E, Diao X, Wang X, Chen R, Hu B. MicroRNAs involved in the mitogen-activated protein kinase cascades pathway during glucose-induced cardiomyocyte hypertrophy. Am J Pathol. 2011;179(2):639–50.
Raut SK, Singh GB, Rastogi B, Saikia UN, Mittal A, Dogra N, Singh S, Prasad R, Khullar M. miR-30c and miR-181a synergistically modulate p53-p21 pathway in diabetes induced cardiac hypertrophy. Mol Cell Biochem. 2016;417(1–2):191–203.
Raut SK, Kumar A, Singh GB, Nahar U, Sharma V, Mittal A, Sharma R, Khullar M. miR-30c mediates Upregulation of Cdc42 and Pak1 in Diabetic Cardiomyopathy. Cardiovasc Ther. 2015;33(3):89–97.
Florczyk-Soluch U, Polak K, Sabo R, Martyniak A, Stepniewski J, Dulak J. Compromised diabetic heart function is not affected by miR-378a upregulation upon hyperglycemia. Pharmacol Rep. 2023;75(6):1556–70.
Ridwan M, Dimiati H, Syukri M, Lesmana R. Potential molecular mechanism underlying cardiac fibrosis in diabetes mellitus: a narrative review. Egypt Heart J. 2023;75(1):46.
Singh GB, Raut SK, Khanna S, Kumar A, Sharma S, Prasad R, Khullar M. MicroRNA-200c modulates DUSP-1 expression in diabetes-induced cardiac hypertrophy. Mol Cell Biochem. 2017;424(1–2):1–11.
Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. 2009;119(9):2772–86.
Kuwabara Y, Horie T, Baba O, Watanabe S, Nishiga M, Usami S, Izuhara M, Nakao T, Nishino T, Otsu K, et al. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ Res. 2015;116(2):279–88.
Costantino S, Paneni F, Luscher TF, Cosentino F. MicroRNA profiling unveils hyperglycaemic memory in the diabetic heart. Eur Heart J. 2016;37(6):572–6.
Su M, Wang J, Wang C, Wang X, Dong W, Qiu W, Wang Y, Zhao X, Zou Y, Song L, et al. MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. 2015;22(6):986–99.
Xu CR, Fang QJ. Inhibiting glucose metabolism by miR-34a and miR-125b protects against Hyperglycemia-Induced Cardiomyocyte Cell Death. Arq Bras Cardiol. 2021;116(3):415–22.
Song XW, Li Q, Lin L, Wang XC, Li DF, Wang GK, Ren AJ, Wang YR, Qin YW, Yuan WJ, et al. MicroRNAs are dynamically regulated in hypertrophic hearts, and miR-199a is essential for the maintenance of cell size in cardiomyocytes. J Cell Physiol. 2010;225(2):437–43.
Yildirim SS, Akman D, Catalucci D, Turan B. Relationship between downregulation of miRNAs and increase of oxidative stress in the development of diabetic cardiac dysfunction: junctin as a target protein of miR-1. Cell Biochem Biophys. 2013;67(3):1397–408.
Biao K, Dongli S, Tao R, Guohui Z. Inhibition of microRNA195 attenuates high-glucose induced neonatal cardiomyocytes hypertrophy in vitro. Zhonghua Xin Xue Guan Bing Za Zhi. 2015;43(8):712–7.
Ding H, Yao J, Xie H, Wang C, Chen J, Wei K, Ji Y, Liu L. MicroRNA-195-5p downregulation inhibits endothelial mesenchymal transition and myocardial fibrosis in Diabetic Cardiomyopathy by Targeting Smad7 and inhibiting transforming growth factor Beta 1-Smads-snail pathway. Front Physiol. 2021;12:709123.
Ren L, Chen X, Nie B, Qu H, Ju J, Bai Y. Ranolazine inhibits pyroptosis via regulation of miR-135b in the treatment of Diabetic Cardiac Fibrosis. Front Mol Biosci. 2022;9:806966.
Feng B, Cao Y, Chen S, Chu X, Chu Y, Chakrabarti S. miR-200b mediates endothelial-to-mesenchymal transition in Diabetic Cardiomyopathy. Diabetes. 2016;65(3):768–79.
Ghosh N, Fenton S, van Hout I, Jones GT, Coffey S, Williams MJA, Sugunesegran R, Parry D, Davis P, Schwenke DO, et al. Therapeutic knockdown of miR-320 improves deteriorated cardiac function in a pre-clinical model of non-ischemic diabetic heart disease. Mol Ther Nucleic Acids. 2022;29:330–42.
Rawal S, Munasinghe PE, Nagesh PT, Lew JKS, Jones GT, Williams MJA, Davis P, Bunton D, Galvin IF, Manning P, et al. Down-regulation of miR-15a/b accelerates fibrotic remodelling in the type 2 diabetic human and mouse heart. Clin Sci (Lond). 2017;131(9):847–63.
Liu S, Li W, Xu M, Huang H, Wang J, Chen X. Micro-RNA 21Targets dual specific phosphatase 8 to promote collagen synthesis in high glucose-treated primary cardiac fibroblasts. Can J Cardiol. 2014;30(12):1689–99.
van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105(35):13027–32.
Bernardo BC, Yildiz GS, Kiriazis H, Harmawan CA, Tai CMK, Ritchie RH, McMullen JR. In vivo inhibition of miR-34a modestly limits cardiac enlargement and fibrosis in a mouse model with established type 1 diabetes-induced cardiomyopathy, but does not improve diastolic function. Cells. 2022;11(19):3117.
Chen R, Chen H, Yang Z, Zhu L, Bei Y, Chen W, Qiu Y. Danlou tablet inhibits high-glucose-induced cardiomyocyte apoptosis via the miR-34a-SIRT1 axis. Heliyon. 2023;9(3):e14479.
Gao L, Wang X, Guo S, Xiao L, Liang C, Wang Z, Li Y, Liu Y, Yao R, Liu Y, et al. LncRNA HOTAIR functions as a competing endogenous RNA to upregulate SIRT1 by sponging miR-34a in diabetic cardiomyopathy. J Cell Physiol. 2019;234(4):4944–58.
Che H, Wang Y, Li Y, Lv J, Li H, Liu Y, Dong R, Sun Y, Xu X, Zhao J, et al. Inhibition of microRNA-150-5p alleviates cardiac inflammation and fibrosis via targeting Smad7 in high glucose-treated cardiac fibroblasts. J Cell Physiol. 2020;235(11):7769–79.
Li Q, Yao Y, Shi S, Zhou M, Zhou Y, Wang M, Chiu JJ, Huang Z, Zhang W, Liu M, et al. Inhibition of miR-21 alleviated cardiac perivascular fibrosis via repressing EndMT in T1DM. J Cell Mol Med. 2020;24(1):910–20.
Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456(7224):980–4.
Geng H, Guan J. MiR-18a-5p inhibits endothelial-mesenchymal transition and cardiac fibrosis through the Notch2 pathway. Biochem Biophys Res Commun. 2017;491(2):329–36.
Zhu GH, Li R, Zeng Y, Zhou T, Xiong F, Zhu M. MicroRNA-142-3p inhibits high-glucose-induced endothelial-to-mesenchymal transition through targeting TGF-beta1/Smad pathway in primary human aortic endothelial cells. Int J Clin Exp Pathol. 2018;11(3):1208–17.
Yang X, Li X, Lin Q, Xu Q. Up-regulation of microRNA-203 inhibits myocardial fibrosis and oxidative stress in mice with diabetic cardiomyopathy through the inhibition of PI3K/Akt signaling pathway via PIK3CA. Gene. 2019;715:143995.
Zheng D, Ma J, Yu Y, Li M, Ni R, Wang G, Chen R, Li J, Fan GC, Lacefield JC, et al. Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia. 2015;58(8):1949–58.
Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, Kaluza D, Treguer K, Carmona G, Bonauer A, et al. MicroRNA-34a regulates cardiac ageing and function. Nature. 2013;495(7439):107–10.
Zhu Y, Yang X, Zhou J, Chen L, Zuo P, Chen L, Jiang L, Li T, Wang D, Xu Y, et al. miR-340-5p Mediates cardiomyocyte oxidative stress in diabetes-induced cardiac dysfunction by targeting Mcl-1. Oxid Med Cell Longev 2022;2022:3182931.
Yu M, Sun Y, Shan X, Yang F, Chu G, Chen Q, Han L, Guo Z, Wang G. Therapeutic overexpression of miR-92a-2-5p ameliorated cardiomyocyte oxidative stress injury in the development of diabetic cardiomyopathy. Cell Mol Biol Lett. 2022;27(1):85.
Tang Q, Len Q, Liu Z, Wang W. Overexpression of miR-22 attenuates oxidative stress injury in diabetic cardiomyopathy via Sirt 1. Cardiovasc Ther. 2018;36(2):e12318.
Yu M, Liu Y, Zhang B, Shi Y, Cui L, Zhao X. Inhibiting microRNA-144 abates oxidative stress and reduces apoptosis in hearts of streptozotocin-induced diabetic mice. Cardiovasc Pathol. 2015;24(6):375–81.
Zhang H, Liu J, Qu D, Wang L, Luo JY, Lau CW, Liu P, Gao Z, Tipoe GL, Lee HK, et al. Inhibition of miR-200c restores endothelial function in Diabetic mice through suppression of COX-2. Diabetes. 2016;65(5):1196–207.
Miao Y, Wan Q, Liu X, Wang Y, Luo Y, Liu D, Lin N, Zhou H, Zhong J. miR-503 is involved in the Protective effect of phase II enzyme Inducer (CPDT) in Diabetic Cardiomyopathy via Nrf2/ARE signaling pathway. Biomed Res Int. 2017;2017:9167450.
Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284(20):13291–5.
Li X, Meng C, Han F, Yang J, Wang J, Zhu Y, Cui X, Zuo M, Xu J, Chang B. Vildagliptin attenuates myocardial dysfunction and restores Autophagy via miR-21/SPRY1/ERK in Diabetic mice heart. Front Pharmacol. 2021;12:634365.
Surina S, Fontanella RA, Scisciola L, Marfella R, Paolisso G, Barbieri M. miR-21 in human cardiomyopathies. Front Cardiovasc Med. 2021;8:767064.
Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, Dangwal S, Kumarswamy R, Bang C, Holzmann A, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3:1078.
Wang XH, Qian RZ, Zhang W, Chen SF, Jin HM, Hu RM. MicroRNA-320 expression in myocardial microvascular endothelial cells and its relationship with insulin-like growth factor-1 in type 2 diabetic rats. Clin Exp Pharmacol Physiol. 2009;36(2):181–8.
Knezevic I, Patel A, Sundaresan NR, Gupta MP, Solaro RJ, Nagalingam RS, Gupta M. A novel cardiomyocyte-enriched microRNA, miR-378, targets insulin-like growth factor 1 receptor: implications in postnatal cardiac remodeling and cell survival. J Biol Chem. 2012;287(16):12913–26.
Zhao F, Li B, Wei YZ, Zhou B, Wang H, Chen M, Gan XD, Wang ZH, Xiong SX. MicroRNA-34a regulates high glucose-induced apoptosis in H9c2 cardiomyocytes. J Huazhong Univ Sci Technolog Med Sci. 2013;33(6):834–9.
Zhang X, Dong S, Jia Q, Zhang A, Li Y, Zhu Y, Lv S, Zhang J. The microRNA in ventricular remodeling: the miR-30 family. Biosci Rep. 2019;39(8):BSR20190788.
Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, Golub TR, Pieske B, Pu WT. Altered microRNA expression in human heart disease. Physiol Genomics. 2007;31(3):367–73.
Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010;6(1):e1000795.
Qiao Y, Zhao Y, Liu Y, Ma N, Wang C, Zou J, Liu Z, Zhou Z, Han D, He J, et al. Mir-483-3p regulates hyperglycaemia-induced cardiomyocyte apoptosis in transgenic mice. Biochem Biophys Res Commun. 2016;477(4):541–7.
Song Y, Mai H, Lin Y, Wang Y, Wang X, Gu S. MiR-144 affects proliferation and apoptosis of high glucose-induced AC16 cardiomyocytes by regulating CTRP3/JNK signaling. Int J Clin Exp Pathol. 2020;13(2):142–52.
Lin KH, Ng SC, Lu SY, Lin YM, Lin SH, Su TC, Huang CY, Kuo WW. Diallyl trisulfide (DATS) protects cardiac cells against advanced glycation end-product-induced apoptosis by enhancing FoxO3A-dependent upregulation of miRNA-210. J Nutr Biochem. 2024;125:109567.
Xu L, Chen W, Ma M, Chen A, Tang C, Zhang C, Cai L. Microarray profiling analysis identifies the mechanism of miR-200b-3p/mRNA-CD36 affecting diabetic cardiomyopathy via peroxisome proliferator activated receptor-gamma signaling pathway. J Cell Biochem. 2019;120(4):5193–206.
Li D, Kular L, Vij M, Herter EK, Li X, Wang A, Chu T, Toma MA, Zhang L, Liapi E, et al. Human skin long noncoding RNA WAKMAR1 regulates wound healing by enhancing keratinocyte migration. Proc Natl Acad Sci U S A. 2019;116(19):9443–52.
Cheedipudi SM, Matkovich SJ, Coarfa C, Hu X, Robertson MJ, Sweet M, Taylor M, Mestroni L, Cleveland J, Willerson JT, et al. Genomic reorganization of Lamin-Associated domains in Cardiac myocytes is Associated with Differential Gene expression and DNA methylation in human dilated cardiomyopathy. Circ Res. 2019;124(8):1198–213.
Moran VA, Perera RJ, Khalil AM. Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 2012;40(14):6391–400.
Clark MB, Mattick JS. Long noncoding RNAs in cell biology. Semin Cell Dev Biol. 2011;22(4):366–76.
Feng Y, Xu W, Zhang W, Wang W, Liu T, Zhou X. LncRNA DCRF regulates cardiomyocyte autophagy by targeting miR-551b-5p in diabetic cardiomyopathy. Theranostics. 2019;9(15):4558–66.
Liu Y, Zhu Y, Liu S, Liu J, Li X. NORAD lentivirus shRNA mitigates fibrosis and inflammatory responses in diabetic cardiomyopathy via the ceRNA network of NORAD/miR-125a-3p/Fyn. Inflamm Res. 2021;70(10–12):1113–27.
Qi Y, Wu H, Mai C, Lin H, Shen J, Zhang X, Gao Y, Mao Y, Xie X. LncRNA-MIAT-Mediated mir-214-3p silencing is responsible for IL-17 production and Cardiac Fibrosis in Diabetic Cardiomyopathy. Front Cell Dev Biol. 2020;8:243.
Peng T, Liu M, Hu L, Guo D, Wang D, Qi B, Ren G, Hu C, Zhang F, Chun HJ, et al. LncRNA Airn alleviates diabetic cardiac fibrosis by inhibiting activation of cardiac fibroblasts via a m6A-IMP2-p53 axis. Biol Direct. 2022;17(1):32.
Wu M, Li T, Li G, Niu B, Wu T, Yan L, Wang S, He S, Huang C, Tong W, et al. LncRNA DANCR deficiency promotes high glucose-induced endothelial to mesenchymal transition in cardiac microvascular cells via the FoxO1/DDAH1/ADMA signaling pathway. Eur J Pharmacol. 2023;950:175732.
Wang K, Lin Y, Shen H, Yu S, Xu J. LncRNA TUG1 exacerbates myocardial fibrosis in Diabetic Cardiomyopathy by modulating the microRNA-145a-5p/Cfl2 Axis. J Cardiovasc Pharmacol. 2023;81(3):192–202.
Zhang Q, Li D, Dong X, Zhang X, Liu J, Peng L, Meng B, Hua Q, Pei X, Zhao L, et al. LncDACH1 promotes mitochondrial oxidative stress of cardiomyocytes by interacting with sirtuin3 and aggravates diabetic cardiomyopathy. Sci China Life Sci. 2022;65(6):1198–212.
Wang T, Li N, Yuan L, Zhao M, Li G, Chen Y, Zhou H. MALAT1/miR-185-5p mediated high glucose-induced oxidative stress, mitochondrial injury and cardiomyocyte apoptosis via the RhoA/ROCK pathway. J Cell Mol Med. 2023;27(17):2495–506.
Zhuo C, Jiang R, Lin X, Shao M. LncRNA H19 inhibits autophagy by epigenetically silencing of DIRAS3 in diabetic cardiomyopathy. Oncotarget. 2017;8(1):1429–37.
Wang S, Duan J, Liao J, Wang Y, Xiao X, Li L, Liu Y, Gu H, Yang P, Fu D, et al. LncRNA H19 inhibits ER stress induced apoptosis and improves diabetic cardiomyopathy by regulating PI3K/AKT/mTOR axis. Aging. 2022;14(16):6809–28.
Zhang M, Gu H, Xu W, Zhou X. Down-regulation of lncRNA MALAT1 reduces cardiomyocyte apoptosis and improves left ventricular function in diabetic rats. Int J Cardiol. 2016;203:214–6.
Wang C, Liu G, Yang H, Guo S, Wang H, Dong Z, Li X, Bai Y, Cheng Y. MALAT1-mediated recruitment of the histone methyltransferase EZH2 to the microRNA-22 promoter leads to cardiomyocyte apoptosis in diabetic cardiomyopathy. Sci Total Environ. 2021;766:142191.
Meng L, Lin H, Huang X, Weng J, Peng F, Wu S. METTL14 suppresses pyroptosis and diabetic cardiomyopathy by downregulating TINCR lncRNA. Cell Death Dis. 2022;13(1):38.
Xie R, Fan J, Wen J, Jin K, Zhan J, Yuan S, Tang Y, Nie X, Wen Z, Li H, et al. LncRNA ZNF593-AS alleviates diabetic cardiomyopathy via suppressing IRF3 signaling pathway. Mol Ther Nucleic Acids. 2023;32:689–703.
Xiao W, Zheng D, Chen X, Yu B, Deng K, Ma J, Wen X, Hu Y, Hou J. Long non-coding RNA MIAT is involved in the regulation of pyroptosis in diabetic cardiomyopathy via targeting miR-214-3p. iScience. 2021;24(12):103518.
Zhou X, Zhang W, Jin M, Chen J, Xu W, Kong X. lncRNA MIAT functions as a competing endogenous RNA to upregulate DAPK2 by sponging mir-22-3p in diabetic cardiomyopathy. Cell Death Dis. 2017;8(7):e2929.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Ni T, Huang X, Pan S, Lu Z. Inhibition of the long non-coding RNA ZFAS1 attenuates ferroptosis by sponging mir-150-5p and activates CCND2 against diabetic cardiomyopathy. J Cell Mol Med. 2021;25(21):9995–10007.
Han Y, Tian H, Gao X. NORAD regulates proliferation and apoptosis in cardiomyocytes under high-glucose treatment through miRNA-150-5p/ZEB1 axis. Eur Rev Med Pharmacol Sci. 2020;24(21):11259–65.
Chen LL, Yang L. Regulation of circRNA biogenesis. RNA Biol. 2015;12(4):381–8.
Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17(4):205–11.
Yuan Q, Sun Y, Yang F, Yan D, Shen M, Jin Z, Zhan L, Liu G, Yang L, Zhou Q, et al. CircRNA DICAR as a novel endogenous regulator for diabetic cardiomyopathy and diabetic pyroptosis of cardiomyocytes. Signal Transduct Target Ther. 2023;8(1):99.
Yang F, Li A, Qin Y, Che H, Wang Y, Lv J, Li Y, Li H, Yue E, Ding X, et al. A Novel Circular RNA mediates pyroptosis of Diabetic Cardiomyopathy by functioning as a competing endogenous RNA. Mol Ther Nucleic Acids. 2019;17:636–43.
Zhou B, Yu JW. A novel identified circular RNA, circRNA_010567, promotes myocardial fibrosis via suppressing miR-141 by targeting TGF-beta1. Biochem Biophys Res Commun. 2017;487(4):769–75.
Shan K, Liu C, Liu BH, Chen X, Dong R, Liu X, Zhang YY, Liu B, Zhang SJ, Wang JJ, et al. Circular noncoding RNA HIPK3 mediates retinal vascular dysfunction in diabetes Mellitus. Circulation. 2017;136(17):1629–42.
Li Y, Zheng F, Xiao X, Xie F, Tao D, Huang C, Liu D, Wang M, Wang L, Zeng F, et al. CircHIPK3 sponges miR-558 to suppress heparanase expression in bladder cancer cells. Embo Rep. 2022;23(11):e56102.
Chen G, Shi Y, Liu M, Sun J. circHIPK3 regulates cell proliferation and migration by sponging miR-124 and regulating AQP3 expression in hepatocellular carcinoma. Cell Death Dis. 2018;9(2):175.
Wang W, Zhang S, Xu L, Feng Y, Wu X, Zhang M, Yu Z, Zhou X. Involvement of circHIPK3 in the pathogenesis of diabetic cardiomyopathy in mice. Diabetologia. 2021;64(3):681–92.
Ni H, Li W, Zhuge Y, Xu S, Wang Y, Chen Y, Shen G, Wang F. Inhibition of circHIPK3 prevents angiotensin II-induced cardiac fibrosis by sponging miR-29b-3p. Int J Cardiol. 2019;292:188–96.
Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, Kjems J. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011;30(21):4414–22.
Shao Y, Li M, Yu Q, Gong M, Wang Y, Yang X, Liu L, Liu D, Tan Z, Zhang Y, et al. CircRNA CDR1as promotes cardiomyocyte apoptosis through activating hippo signaling pathway in diabetic cardiomyopathy. Eur J Pharmacol. 2022;922:174915.
Shen M, Wu Y, Li L, Zhang L, Liu G, Wang R. CircMAP3K5 promotes cardiomyocyte apoptosis in diabetic cardiomyopathy by regulating miR-22-3p/DAPK2 Axis. J Diabetes. 2024;16(1):e13471.
Fu L, Zhang J, Lin Z, Li Y, Qin G. CircularRNA circ_0071269 knockdown protects against from diabetic cardiomyopathy injury by microRNA-145/gasdermin A axis. Bioengineered. 2022;13(2):2398–411.
Che H, Wang Y, Li H, Li Y, Sahil A, Lv J, Liu Y, Yang Z, Dong R, Xue H, et al. Melatonin alleviates cardiac fibrosis via inhibiting lncRNA MALAT1/miR-141-mediated NLRP3 inflammasome and TGF-beta1/Smads signaling in diabetic cardiomyopathy. FASEB J. 2020;34(4):5282–98.
Tang H, Zhong H, Liu W, Wang Y, Wang Y, Wang L, Tang S, Zhu H. Melatonin Alleviates Hyperglycemia-Induced Cardiomyocyte Apoptosis via Regulation of Long Non-Coding RNA H19/miR-29c/MAPK Axis in Diabetic Cardiomyopathy. Pharmaceuticals (Basel). 2022;15(7):821.
Abo-Saif MA, Ragab AE, Ibrahim AO, Abdelzaher OF, Mehanyd ABM, Saber-Ayad M, El-Feky OA. Pomegranate peel extract protects against the development of diabetic cardiomyopathy in rats by inhibiting pyroptosis and downregulating LncRNA-MALAT1. Front Pharmacol. 2023;14:1166653.
Feng X, Sureda A, Jafari S, Memariani Z, Tewari D, Annunziata G, Barrea L, Hassan STS, Smejkal K, Malanik M, et al. Berberine in Cardiovascular and metabolic diseases: from mechanisms to therapeutics. Theranostics. 2019;9(7):1923–51.
Yang L, Cheng CF, Li ZF, Huang XJ, Cai SQ, Ye SY, Zhao LJ, Xiong Y, Chen DF, Liu HL, et al. Berberine blocks inflammasome activation and alleviates diabetic cardiomyopathy via the miR–18a–3p/Gsdmd pathway. Int J Mol Med. 2023;51(6):1.
Qiu Y, Zhang X, Li SS, Li YL, Mao BY, Fan JX, Shuang G, Yin YL, Li P. Citronellal can alleviate vascular endothelial dysfunction by reducing ectopic miR-133a expression. Life Sci. 2024;339:122382.
Banerjee K, Ghosh RK, Kamatam S, Banerjee A, Gupta A. Role of Ranolazine in cardiovascular disease and diabetes: exploring beyond angina. Int J Cardiol. 2017;227:556–64.
Bauersachs J, Solomon SD, Anker SD, Antorrena-Miranda I, Batkai S, Viereck J, Rump S, Filippatos G, Granzer U, Ponikowski P et al. Efficacy and safety of CDR132L in patients with reduced left ventricular ejection fraction after myocardial infarction: rationale and design of the HF-REVERT trial. Eur J Heart Fail 2024. https://doi.org/10.1002/ejhf.3139. Online ahead of print.
Batkai S, Genschel C, Viereck J, Rump S, Bar C, Borchert T, Traxler D, Riesenhuber M, Spannbauer A, Lukovic D, et al. CDR132L improves systolic and diastolic function in a large animal model of chronic heart failure. Eur Heart J. 2021;42(2):192–201.
Taubel J, Hauke W, Rump S, Viereck J, Batkai S, Poetzsch J, Rode L, Weigt H, Genschel C, Lorch U, et al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: results of a first-in-human phase 1b randomized, double-blind, placebo-controlled study. Eur Heart J. 2021;42(2):178–88.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from Zhejiang Provincial Natural Science Foundation of China (No. LZ22H050001), Zhejiang provincial program for the Cultivation of High-level Innovative Health talents, National Natural Science Foundation of China, NSFC (82270704 and 82370902), National Infrastructures for Translational Medicine (Shanghai) (TMSK-2021-506), and Primary Research and Development Plan of Zhejiang Province (2024C03165).
Author information
Authors and Affiliations
Contributions
JT and WL contributes to the conception and design of the work; XY and XYH have drafted the work; JT, JC and WL substantively revised it. All authors have approved the submitted version.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
Ethical approval was not applicable for this review.
Consent for publication
Not applicable.
Competing interests
Prof Tian is a co-author of this study and an Editorial Board member of the journal. She was not involved in handling this manuscript during the submission and the review processes. The rest of the authors have no conflict of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yao, X., Huang, X., Chen, J. et al. Roles of non-coding RNA in diabetic cardiomyopathy. Cardiovasc Diabetol 23, 227 (2024). https://doi.org/10.1186/s12933-024-02252-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-024-02252-9