
Abstract
Background
Diabetes mellitus (DM) is among the most common risk factors for cardiovascular disease in the world with prevalence of more than 500 million population in 2021. Cardiac fibrosis with its complex process has been hypothesized as one of the mechanisms explaining development of heart failure in diabetic patients. Recently, the biomolecular mechanism of cardiac fibrosis in the hyperglycemia setting has been focusing around transforming growth factor β-1 (TGFβ-1) as a major factor. However, there is interplay role of several factors including microRNAs (miRNAs) which acts as a potential regulator of cardiac fibrosis connected with TGFβ-1. In this review, we explored interplay role of several factors including microRNAs which acts as a potential regulator of cardiac fibrosis connected with TGFβ-1 in diabetes mellitus. This narrative review included articles from the PubMed and Science Direct databases published in the last 10 years (2012–2022).
Main text
In diabetic patients, excessive activation of myofibroblasts occurs and triggers pro-collagen to convert into mature collagen to fill the cardiac interstitial space resulting in a pathological process of extracellular matrix remodeling. The balance between matrix metalloproteinase (MMP) and its inhibitor (tissue inhibitor of metalloproteinase, TIMP) is crucial in degradation of the extracellular matrix. Diabetes-related cardiac fibrosis is modulated by increasing level of TGF-β1 mediated by cellular components, including cardiomyocyte and non-cardiomyocyte cells involving fibroblasts, vascular pericytes smooth muscle cells, endothelial cells, mast cells, macrophages, and dendritic cells. Several miRNAs such as miR-21, miR-9, miR-29, miR-30d, miR-144, miR-34a, miR-150, miR-320, and miR-378 are upregulated in diabetic cardiomyopathy. TGF-β1, together with inflammatory cytokines, oxidative stress, combined sma and the mothers against decapentaplegic (smad) protein, mitogen-activated protein kinase (MAPK), and microRNAs, is interconnectedly involved in extracellular matrix production and fibrotic response. In this review, we explored interplay role of several factors including microRNAs which acts as a potential regulator of cardiac fibrosis connected with TGFβ-1 in diabetes mellitus.
Conclusions
Long-term hyperglycemia activates cardiac fibroblast via complex processes involving TGF-β1, miRNA, inflammatory chemokines, oxidative stress, smad, or MAPK pathways. There is increasing evidence of miRNA’s roles lately in modulating cardiac fibrosis.
Similar content being viewed by others


Background
Diabetes mellitus has become a worldwide health concern due to the rise of sufferers to 537 million in 2021 [1]. According to the World Health Organization (WHO), diabetes was recorded as the ninth leading cause of death, with about 1.5 million deaths in 2019 [2]. The uncontrolled hyperglycemic conditions can lead to fatal cardiovascular complications, such as hypertension [3, 4], stroke [5], atherosclerosis [6, 7], myocardial infarction [8, 9], atrial fibrillation [10], cardiac fibrosis [11,12,13,14], diabetic cardiomyopathy [15,16,17], and sudden cardiac death [18,19,20]. Moreover, the significant increase in diabetes mellitus prevalence raises awareness of cardiovascular system complications [1], in which heart rhythm disturbances [21] and heart failure [22, 23] are the most common cardiovascular complications resulting from prolonged high blood sugar levels.
Diabetes-induced heart failure is a result of the pathological stimulus of prolonged hyperglycemia leading to cardiac remodeling [24]. In general, cardiac remodeling involves changes of the myocardium structure, metabolism, and electrical conduction of the heart as a result of stimulation of endogenous and exogenous factors, which in turn will cause structural changes and cause biological effects on the heart ventricles [25]. Typical features of diabetes-induced cardiac remodeling are endothelial changes, cardiomyocyte hypertrophy and apoptosis, and extracellular matrix accumulation leading to cardiac fibrosis. Initially, the accumulation of extracellular matrix is actually a beneficial protective mechanism for wound healing and tissue regeneration. However, when the extracellular matrix collagen accumulation occurs excessively and continuously, over time, it will cause disturbances in tissue function [24]. The hypothetical mechanism that is thought to underlie the accumulation of extracellular matrix in patients with diabetes mellitus is a decrease in the activity of an enzyme responsible for the degradation of the extracellular matrix, namely matrix metalloproteinase (MMP) and the resultant increase in tissue inhibitor of metalloproteinase (TIMP) [26, 27]. In the case of diabetes, type III collagen is expressed more than type I collagen [28].
In addition to hyperglycemic conditions, the presence of advanced glycation end products (AGEs), reactive oxygen species (ROS), and neurohumoral activation triggers the activation of cardiac fibroblast cells, induces a proliferative response, and triggers the synthesis of matrix phenotypes. Secretion of fibrogenic growth factors and modulation of fibroblast phenotype can also be stimulated by cardiomyocytes and endothelial cells. High plasma concentrations of inflammatory cytokines due to release by adipose cell hypertrophy in diabetes result in systemic inflammation and activation of endothelial cells that will stimulate monocyte infiltration and transformation into macrophages. Activated macrophages will produce transforming growth factor-β (TGF-β) which plays an important role in fibroblast stimulation and initiates the secretion of pro-fibrotic mediators [5].
TGF- β is a prototype factor in the form of a strong profibrotic cytokine and has an active role in the formation of fibrosis, including cardiac fibrosis [29, 30]. In other words, TGF-β is one of the general mediators that play a role in the development of fibrosis formation. For example, in cases of cardiac injury, modulation of profibrotic mediators such as TGF-β, angiotensin II, endothelin-1 will activate cardiac fibroblasts resulting in proliferation and migration of cardiac fibroblasts. This situation will also trigger extracellular matrix protein deposition and myofibroblast differentiation to form cardiac fibrosis. It is therefore believed that inhibiting TGF-β signaling, either by molecular inhibition, antibody, or genetic deletion, will provide a favorable outcome for the treatment of cardiac fibrosis [31].
In addition to the case of heart injury, other cases that trigger the formation of cardiac fibrosis will also increase TGF-β. Studies with animal models have shown that both type-1 diabetes mellitus (T1DM) and type-2 diabetes mellitus (T2DM) still experience an increase in TGF-β [32, 33]. On the other hand, inhibiting TGF-β will reduce the incidence of cardiac fibrosis [14].
Interestingly, cardiac fibrosis has recently been associated with cytokine secretion by cardiac fibroblasts and cardiomyocytes modulated by microRNAs (miRNAs) [34]. miRNA is a small (22–26 nucleotide) single-stranded endogenous non-coding RNA whose function is to control the expression of target messenger RNAs (mRNAs). The expression can be either mRNA degradation or translational repression [35]. The biological role is formed from direct binding to the target 3'UTR mRNA so that it can affect the stability and translation of mRNA [36] by regulating mRNA expression in the transcriptional phase [37]. In its role, miRNA affects the fibrosis process positively and negatively depending on the gene to which it binds. Some miRNAs trigger antifibrotic activity by inhibiting the signaling activity of the TGF-β component in myofibroblasts, but on the other hand, the remaining miRNAs play a pro-fibrotic role by upregulating the expression of TGF-β signaling molecules, one of which is miR-21 [38]. Unfortunately, there is unclear cut explanation about role of diabetes mellitus-induced heart failure involving TGF-β and microRNA stimulation. Taken together, this review proposes a wider comprehensive aspect of molecular mechanism explaining interrelated hypothetical pathways of TGFβ-1, hyperglycemia, and miRNA.
This narrative review included articles from the PubMed and Science Direct databases published in the last 10 years (2012–2022). The search used a combination of medical subject heading (MeSH) terms, i.e., [(cardiac fibrosis) OR (cardiac remodeling) OR (myocardial fibrosis)] AND [(diabetes mellitus) OR (type-2 dm) OR (type-1 dm) OR (hyperglycemia)]. We excluded review articles and book chapters; then, articles met eligibility criteria were further evaluated meticulously by screening each article.
Main text
The advantages and disadvantages of fibrosis for the heart
Cardiac fibrosis is a condition where scar tissue is formed due to the accumulation of collagen deposition, as well as activation and differentiation of cardiac fibroblasts into myofibroblasts [39]. When an injury appears due to cardiac disorders, such as myocardial infarction, hypertensive heart disease, hypertrophic diabetic cardiomyopathy, or idiopathic dilated cardiomyopathy, the heart exhibits a self-repairing response by remodeling or regeneration using the remaining cells [39,40,41]. An extracellular matrix deposition is a protective mechanism for wound repair and tissue regeneration. The reason is that collagen compensates for cells that die due to injury; therefore, at a particular level, collagen's role is highly beneficial because it maintains cardiac structural integrity and protects from cardiac rupture. However, a massive and continuous process can be detrimental and even lead to death. Excessive collagen deposition is found to cause heart damage and to impair cardiac compliance because it leads to increased cardiomyopathy and reduced cardiac elasticity due to the formation of scar tissues [42,43,44]. The formation of cardiac fibrosis as a pathological process due to extracellular matrix remodeling leads to abnormalities in the composition and quality of the extracellular matrix. This condition will affect the cardiac function, i.e., decreasing the ejection fraction due to myocardial matrix stiffness from fibrotic injuries, disruption of the cardiac electrical conduction, and eventually death [41].
Fibrosis mechanism in diabetic patients
In diabetic patients, scar tissue is formed in the cardiac interstitial cells due to long-term hyperglycemic conditions through a pathological process of extracellular matrix remodeling [13, 24, 39, 40, 45].
Cardiac fibroblast activation and myofibroblasts
Fibroblasts are cells that have a significant role in the formation of cardiac fibrosis [46]. Under normal conditions, fibroblasts are in a resting or inactive state; however, they can be activated by the remaining healthy cardiac tissue whenever cardiac cell injury occurs [47].
After the activation, fibroblasts are called myofibroblasts, which are pro-inflammatory, hypersecretory, and hypermigratory. Myofibroblasts have anti-inflammatory and pro-angiogenic phenotypes; thus, they can express cytokines, extracellular matrix, and other paracrine factors used in wound healing due to the injuries [48].
Collagen deposition
Dynamic regulation of the extracellular matrix plays a vital role in maintaining cardiac structural integrity. A disturbance in the regulation of extracellular matrix synthesis, collagen deposition, and extracellular matrix degradation will induce the pathogenesis of cardiac fibrosis. Precursor cells will be triggered to activate myofibroblasts in diabetic patients with long-term uncontrolled hyperglycemia. Excessive activation of myofibroblasts triggers pro-collagen to convert into mature collagen, expressing type I and type III collagen to fill the cardiac interstitial space [10]. The expressed collagen then cross-links with lysyl oxidase to produce fibers that inhibit protein degradation. Type I collagen is related to stiffness, while type III collagen is related to wall tension elasticity. Collagen turnover involves several types of cells, i.e., inflammatory and mediator cells, such as cytokine cells, growth factors, and hormones [49, 50]. These factors help in myofibroblast differentiation and extracellular matrix remodeling [50].
Cardiac fibrosis
Cardiac fibrosis is a pathological condition occurring as an excess accumulation of extracellular matrix due to cardiac remodeling process involving profibrotic cells, growth factors, and expression of inflammatory cytokines. A higher level of profibrotic phenotype in fibroblasts was found in diabetic patients than in non-diabetic patients [10]. Myofibroblast activation is the main effector in cardiac fibrosis; however, monocytes, lymphocytes, mast cells, vascular cells, and cardiomyocytes also produce a fibrotic response by expressing fibrinogen mediators. The related mediators include inflammatory cytokines and chemokines, ROS, mast cell-derived proteases, endothelin-1, renin–angiotensin–aldosterone-system (RAAS), cellular components, and growth factors such as TGF-β [51]. The aspects that cause cardiac fibrosis affect the nature of fibrosis, e.g., the resulting cardiac fibrosis is more localized in myocardial infarction. Meanwhile, in diabetic cardiac fibrosis, the fibrosis is formed more diffusely and is located in the cardiac interstitial part. There are several types of cardiac fibrosis based on the cause. Replacement fibrosis is formed due to myocardial infarction, sarcoidosis, myocarditis, toxic cardiomyopathy, and chronic renal insufficiency. Infiltrative interstitial fibrosis is present in amyloidosis and Anderson–Fabry disease. Meanwhile, reactive interstitial fibrosis is formed in hypertension, diabetes, non-ischemic dilated cardiomyopathy, hypertrophic cardiomyopathy, sarcoidosis, and chronic renal insufficiency [43, 44].
The myocardial interstitial space is a cardiac tissue compartment consisting of stromal cells and an extracellular matrix complex composed of structural and non-structural proteins and bioactive signaling molecules. Structural proteins include collagen and elastin, while non-structural proteins include glycoproteins, proteoglycans, and glycosaminoglycans. The extracellular matrix is structurally complex, dynamic, and organized, thus, maintaining structural integrity, cardiac cycle transmission, communication signals between cells, and repair response after cardiac injury [52].
Histopathology of cardiac fibrosis
The formation of perivascular and interstitial fibrosis in diabetic patients has been studied through histopathological studies, and its process is independent of the pathogenesis of coronary atherosclerosis and hypertension [53]. Histological findings of the left ventricular endomyocardial biopsy revealed increased cardiac fibrosis formation and deposition of advanced glycation end products in heart failure with reduced ejection fraction (HFrEF). Meanwhile, an increase in cardiomyocyte resting tension was discovered in heart failure with preserved ejection fraction (HFpEF) [54] (Fig. 1).

Different types of cardiac fibrosis based on histopathological features of model rat preparations using Sirius-Red staining. A Replacement fibrosis is characterized by the sudden loss of large numbers of cardiomyocytes, mostly caused by myocardial infarction. B Interstitial fibrosis is characterized by increased collagen deposition in the cardiac interstitial space due to significantly reduced cardiomyocyte cells. C Perivascular fibrosis is characterized by expansion of the vascular adventitia matrix. D Illustration of cardiac fibrosis in the cardiac interstitial space influenced by the primary effector cells, i.e., myofibroblasts, and other mediators that play a role, such as macrophages, lymphocytes, mast cells, vascular endothelial cells, and cardiomyocytes. With written permission from Frangogiannis NG [51]
Cardiac remodeling
Diabetes-related cardiac remodeling can cause various manifestations referring to the restrictive or dilated left ventricle. Compared to non-diabetic conditions, there is an increase in cardiac hypertrophy, narrow left ventricular end-diastolic and end-systolic volume indices, higher E/E’ ratio, and an increased prevalence of left ventricular geometry abnormalities, such as concentric left ventricle remodeling, concentric hypertrophy, and eccentric hypertrophy in diabetes [55].
The change in cardiac structure due to diabetic condition shows different features in HFrEF and HFpEF. In HFrEF, there is an increase in cardiac cell death followed by fibrotic scar tissue formation triggered by the stimulation of protein kinase C in cardiac fibroblasts in hyperglycemic situations [56]. The remodeling process in diabetic cardiac fibrosis involves the interaction of profibrotic cells, growth factors, and the expression of inflammatory cytokines [53]. Lipotoxicity occurs in diabetic conditions due to triglyceride or fatty acid accumulation [56]. In addition, deposition of advanced glycation end products that trigger inflammation, immune cell infiltration, and subsequent apoptosis also happens in diabetes condition. Meanwhile, in HFpEF, cardiac cell stiffness and hypertrophy occur due to hyperinsulinemia and endothelial dysfunction, a microvascular complication of diabetes mellitus [57].
Factors influencing cardiac fibrosis
Cellular components
Diabetes-related cardiac fibrosis is mediated by cellular components, including cardiomyocyte and non-cardiomyocyte cells that bind to each other in the interstitial matrix involving fibroblasts, vascular pericytes smooth muscle cells, endothelial cells, mast cells, macrophages, and dendritic cells [58].
Fibroblasts are the primary cells that play a role in forming diabetic-associated cardiac fibrosis [59]. After myocardial infarction or myocardial remodeling, fibroblasts differentiate into myofibroblasts, express contractile proteins such as α-smooth muscle actin (α-SMA), and synthesize extracellular proteins [60].
Monocytes and macrophages are abundant in the myocardium when cardiac fibrosis develops, triggering a fibrogenic phenotype to activate fibroblasts. In type I and type II diabetes, monocytes and macrophages have contributed to fibrotic remodeling through fibrogenic mediation [61]. Lymphocyte cells could also modulate the fibroblast phenotype and trigger fibrosis during myocardial remodeling [62].
Endothelial and pericytes cells are known to contribute to the formation of cardiac fibrosis. Endothelial-to-mesenchymal transition (EndMT) helps increase fibroblast activation. In diabetic conditions, EndMT expands in the cardiac interstitial space. Pericytes cells modulate cardiac fibrosis by increasing myofibroblast conversion and through vascular cells secrete mediators that activate fibroblasts [63]. Mast cells are thought to produce proteases and fibrogenic growth factors in the formation of cardiac fibrosis. In diabetic conditions, delaying mast cell accumulation affects healing [64, 65].
Cardiomyocytes, in diabetic conditions, release toxic substances that are damaging and can cause cell death. Fibrosis in diabetic conditions is in the form of scar tissue that replaces damaged or dead cardiomyocytes due to the released toxic substances. In addition, in hyperglycemic conditions, fibrogenic phenotypes in cardiomyocytes are expressed, such as triggering the synthesis and release of growth factors and cytokines, which stimulate the proliferation and activation of fibroblasts. Cardiomyocytes also produce pro-inflammatory mediators that help activate immune cells and trigger the formation of fibrotic tissue [58].
Mitogen-activated protein kinases
Mitogen-activated protein kinases (MAPKs) play a significant role in biological processes, i.e., proliferation, differentiation, metabolism, motility, cell survival, and cell apoptosis. MAPKs consist of four subfamilies, namely extracellular-signal-regulated kinase 1/2 (ERK 1/2), Jun N-terminal Kinase (JNK), p38, and extracellular-signal-regulated kinase 5 (ERK 5). Currently, MAPKs are one of the targets of pharmacological therapy or genetic manipulation of heart development, heart function, and heart disease [66].
Studies with genetic manipulation of JNK1 deletions have led to increased formation of cardiac fibrosis [67]. Similarly, studies using JNK1 inhibitor therapy in hamster cardiomyopathy models show increased apoptosis and cardiac fibrosis [68]. Another in-vivo study shows that increased JNK activity through loss of β1-integrins is associated with increased MMP-2 activity rather than MMP-9, which lead to reduced cardiac fibrosis formation [69].
The p-38 MAPK also demonstrated a similar thing. During tissue stress and cardiac remodeling, myocardial p-38 activation results in restrictive cardiomyopathy followed by a significant increase in the amount of interstitial fibrosis [70]. MAPK p-38 is also thought to stimulate the release of pro-inflammatory cytokines in myocytes, such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), TGF-β, and vascular cell adhesion molecules-1 (VCAM-1) which affects cardiac remodeling through regulation of proliferation, differentiation, and function of immune cells [71, 72].
Smad protein
Activation of smad-2 and smad-3 as R-smad is primarily significant in fibrotic infiltration of fibroblasts, while smad-1,5,8 is not yet understood. Smad-3 affects the formation of cardiac fibrosis by stimulating myofibroblast phenotype activation, extracellular matrix synthesis, integrin expression, and secretion of proteases and anti-proteases [73]. Smad-3 also stimulates myofibroblast conversion, increases the expression of α-SMA protein, and stimulates the transcription of extracellular matrix structure, including regulating type I collagen, type III collagen, fibronectin, periostin, and tenascin-C, in vitro. In addition, smad-3 reduces MMP expression and stimulates TIMP expression [73,74,75]. Meanwhile, Smad-2 helps modulate fibronectin and periostin. Smad-2 and Smad-3 together affect fibroblast gene transcription, as well as nuclear activation and translocation. Meanwhile, smad 6, as an i-smad, inhibits the response of bone morphogenetic protein (BMP), while smad-7 inhibits both TGF-β and BMP [73].
Smad is a group of proteins that serves as intracellular mediators against the binding formed by TGF-β on the cell surface. Smad activation initiates translocation from the cytoplasm to the nucleus. Along with transcription factors, it plays an important role in transcriptional regulation to regulate target gene expression. Smad combines sma protein and the mothers against decapentaplegic (mad) gene [76, 77]. Based on its function, smad is classified into three groups, i.e., receptors activated smad (R-smads), common smad (Co-smad), and inhibitory smad (i-smad). R-smad, responsible for stimulating TGF-β signaling, consists of smad-1, smad-2, smad-3, smad-5, and smad-8. Co-smad, which binds to R-smad and helps form a signaling complex, consists only of smad-4. Meanwhile, i-smad, a negative R-smad regulator, consists of smad-6 and smad-7 [78].
Transforming growth factor-β 1
TGF-β, expressed from activated macrophages, is a cytokine that plays a role in forming fibrosis in various tissues, including cardiac tissue [79]. TGF-β plays a role in the activation of myofibroblast infiltration and is responsible for extracellular matrix production and fibrotic response. This process increases left ventricular diastolic stiffness leading to HFpEF [5].
TGF-β1 is the dominant isoform in the cardiovascular system, which is more abundant than other TGF-β isoforms. In cases of cardiac fibrosis, it is only limited to TGF-β1. In a healthy heart condition, TGF-β1 is expressed in a latent form and is not associated with its receptors; however, when a heart injury occurs, conversion of TGF-β1 occurs from the latent to the active form. Only a small amount of activated TGF-β1 is required to induce the cellular response system. Various mediators have been appointed as TGF-β1 activators in cardiac fibrosis, one of which is MMP-2. In contrast with MMP, TIMP inhibits MMP performance [51].
In vitro administration of empagliflozin, a sodium glucose co-transporter 2 inhibitor (SGLT2i) as an oral antidiabetic drug, was able to reduce TGF-β expression, fibroblast activation and extracellular matrix remodeling, and the formation of profibrotic markers, such as collagen type I alpha I (COL1A1), actin alpha 2 (ACTA2), αSMA, connective tissue growth factor (CTGF), fibronectin-1 (FN1), and MMP2 [80]. Empagliflozin exerts an antifibrotic effect by inhibiting the TGFβ-Smad pathway. A low dose of canagliflozin, another variant of SGLT2i, also exerted antifibrotic effects by reducing fibronectin, collagen, and TGF-1 in db/db mice [81, 82]. TGF-β is predicted to modulate miR-21 by targeting Jagged 1 and Smad7. TGF-β accelerates the division of pri-miR-21 into pre-miR-21, thereby increasing mature miR-21 [83].
Matrix metalloproteinase
Matrix metalloproteinase (MMP) regulates the degradation of the extracellular matrix. The human body expresses 23 types of MMPs classified according to their constituent substrates. The collagenase group includes MMP-1, MMP-13, and MMP-8; the gelatinase group includes MMP-2 and MMP-9; stromelysins or matrilysins groups include MMP-3 and MMP-7; the membrane group includes MMP-14 [84]. MMP-2 is a specific marker for the fibroblast population [85].
In diabetic patients, the MMP2 activity, a regulator of enzyme activity that plays a role in extra-cellular-matrix (ECM) degradation, changes [52]. In its application, MMP-2 is used as a diagnostic model for left ventricular hypertrophy development and cases of developing heart failure (HF) [86, 87]. A study using an animal model discovered that genetic deletion of MMP-2 resulted in a reduction in the accumulation rate of myocardial collagen fibrils and improved the left ventricular diastolic function [88]. MMP-2 expression is thought to affect ECM stability, induce ECM structural changes, and improve profibrotic signaling pathways [84].
It has been reported that diabetic-associated hypertrophy is blocked by inhibiting the expression of TGF-β1 receptor signaling or inhibiting MMP2 and MMP9 in vitro. Preventing activation of MMP2 or MMP9 inhibits cell hypertrophy induced by high sugar levels [89].
Tissue inhibitor of metalloproteinase
In contrast with MMP, TIMP is an MMP inhibitor that plays a significant role in heart fibrosis and HF processes [90]. An imbalance between MMP and TIMP leads to cardiac fibrosis and heart failure [49]. There are four groups of TIMP with high affinity for MMP binding, namely TIMP-1, TIMP-2, TIMP-3, and TIMP-4. Initially, it was thought that the TIMPs had similar functional characteristics; however, each of the TIMPs had specific functions [84]. TIMP-1 is predicted to influence the collagen degradation process associated with interstitial myocardial fibrosis [52]. An increased TIMP-1 affects MMP expression and reduces ECM degradation [91, 92].
Advanced glycation end products and its receptors
Advanced glycation end products (AGEs) are accumulated under prolonged exposure to hyperglycemia. This condition induces collagen cross-linking that can increase the formation of cardiac fibrosis, followed by increased cardiac rigidity and impaired cardiac relaxation [93]. AGEs increase the production of ROS and receptors for advanced glycation end products (RAGE) [94]. A prolonged hyperglycemic condition triggers the neutrophil derivative of calcium-binding protein S100 A8/A9 (S100A8/A9) in its interaction with Kupffer cells and triggers the release of IL-6. SGLT2-i helps inhibit the bond formed between S100A8/A9 and RAGE [95]. A low dose of SGLT2-i (Empagliflozin 10 mg/kg/day) did not significantly affect the reduction in AGE and ROS or the formation of RAGE. However, a threefold dose of empagliflozin showed reduced expression of AGE, RAGE, and ROS [94].
Inflammation
Long-term hyperglycemia increases plasma concentrations of inflammatory cytokines and activates intracellular cardiomyocyte cytokines. The systemic inflammation and activation of these endothelial cells stimulate monocyte infiltration to transform into macrophages, which also play a role in ECM overproduction [5].
Chemokines are chemotactic cytokines that affect leukocytes and are involved in the fibrotic process. The chemokine C–C ligand 2 (CCL2) or monocyte chemotactic protein-1 (MCP-1) is primarily associated with heart disease, including ischemic heart and cardiac fibrosis. It was discovered that MCP-1 could reduce cardiac fibrosis and improve diastolic dysfunction without affecting blood pressure or systolic function but did not reduce cardiomyocyte hypertrophy [96].
Oxidative stress
Diabetic patients tend to experience long-term exposure to oxidative stress, which plays a role in various complications, including cardiovascular-related [97]. In long-term hyperglycemia, mitochondria of the cardiac cells are unable to cope with the conditions, increasing the ROS and apoptotic cascade [98].
ROS directly regulates the quantity and quality of the interstitial extracellular matrix through modulation of matrix protein expression and metabolism. Increased oxidative stress can activate MMP and decrease fibrillar collagen synthesis in cardiac fibroblasts. ROS also increases extracellular matrix deposition in the cardiac interstitial via TGF-β activation. As a mediator, ROS plays a role in the induction of cytokines and the effect of angiotensin II on fibroblasts. ROS is also involved in the activation of transcription factors, such as activator protein-1 (AP-1), E26 transformation specific (ETS) transcription factor, and nuclear factor kappa-light chain enhancer of activated B cells (NF-kB), which helps MMP transcription [51]. The induction of oxidative stress in cardiac tissue is triggered due to increased cardiomyocyte intracellular inflammatory cytokines. Increased oxidative stress also increases the expression of inducible nitric oxide synthase (iNOS), triggers cell death, and increases the accumulation of ECM proteins, thereby increasing protein rigidity in cardiac muscle cells [5].
Renin–angiotensin aldosterone system
Exposure to hyperglycemia also increases the RAAS activation, causing overproduction of angiotensin II and aldosterone, which triggers cardiac hypertrophy, cardiac fibrosis, and impaired cardiac relaxation. In adult mice, it was discovered that angiotensin II modulates collagen production by fibroblasts via ROS generation. In addition, mitochondrial oxidative stress is understood to mediate fibroticity through angiotensin induction [51, 93, 99].
Micro-ribonucleic acid (micro-RNA, miRNA)
Expression of miRNA almost constantly changes in every step of the biological process. miRNAs are small single-stranded non-coding RNAs that regulate gene expression at the post-transcriptional level [100]. miRNAs are broadly expressed in the nucleus and cytoplasm. However, miRNAs can still be found in blood circulation [101]. As miRNAs are expressed stably in blood, they are likely to be used as potential biomarkers for diagnostic, prognostic, and novel therapeutic strategies related to cardiovascular disease [102]. It has been reported that in diabetic conditions, miRNAs expression is dysregulated in serum/plasma [100]. A number of miRNAs were recorded in plasma and whole blood in diabetic conditions; those are miR-21, miR-320, miR-15a, miR24, miR29b, miR-20b miR-126, miR150, miR197, miR-223, miR191, miR486, miR-28-3p [93], miR-9, miR-29a, miR-30d, miR34a, miR124a, miR-146a, miR375, miR-144, and miR192 [103, 104].
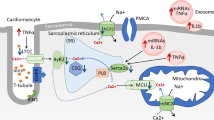
Nine recorded miRNAs were upregulated related to diabetic cardiomyopathy, namely miR-21, miR-9, miR-29, miR-30d, miR-144, miR-34a, miR-150, miR-320, and miR-378. Interestingly, miR-21 was acted via a pro-fibrosis pathophysiological mechanism [102]. miR-21 suppresses the expression of pro-viral integration sites for Moloney murine leukemia virus-1 (Pim1) and B-cell lymphoma-2 (Bcl-2), which are anti-apoptotic and cardioprotective proteins. It increases profibrotic markers, such as TGF-β, collagen, and fibronectin[82]. Overexpression of MiR-21 in heart failure modulates the activation of extracellular signal-regulated kinase mitogen-activated protein kinase (ERK-MAPK) by inhibiting sprouty protein homolog 1 (SPRY1). In addition, the increase in miR-21 leads to changes in cardiofibroblast (CF) viability and increases the accumulation of factors that induce hypertrophy, thereby modulating cardiac remodeling. Increased miR-21 modulates the activation of fibrotic remodeling by inhibiting protein phosphatase and tensin homolog (PTEN) in cardiac fibrosis [83] (Fig. 2).

Interrelated biological mechanism schematic pathways of cardiac fibrosis in hyperglycemia setting lead to heart failure. Long-term hyperglycemia affects cardiac fibroblast activation via inflammatory chemokines, ROS, miRNA, Smad, MAPK, or TGF-β pathways. Cardiac fibrosis is resulted from the increased collagen deposition in the extracellular matrix leading to heart failure
Implication in future cardiac fibrosis therapy
To date, various therapeutic regimens have been used in cardiac fibrosis [41, 105]. For over decades, angiotensin-converting enzyme inhibitors (ACEi) [106], angiotensin receptor blocker (ARB) [107], and mineralocorticoid receptor antagonist (MRA) [108] have been used as heart failure treatment by targeting the RAAS as anti-remodeling drugs [106,107,108]. As a combination, the angiotensin receptor blocker neprilysin inhibitor (ARNI) is considered to reduce interstitial fibrosis and cardiomyocyte hypertrophy in diabetes [109, 110].
The use of SGLT2-i is one of the many promising regimens for cardiac fibrosis despite its original function as an oral antidiabetic drug. As of 2021, heart failure guideline by European Society of cardiology (ESC), followed by American Heart Association guideline in 2022, has included SGLT2-i as one of 4 compulsory drug classes for HFrEF patients to reduce hospitalization and cardiovascular mortality independence of type 2 diabetes presence [111, 112].
However, further study of the biomolecular mechanisms in SGLT2-i is necessary, considering that the SGLT2 receptor is not expressed in the heart [41, 105]. Besides, another promising therapeutic target for cardiac fibrosis has been studied in mice by adding oligonucleotide to bind with certain miRNA (anti-miRNA) [49, 113]. Therefore, new directions for targeted therapy in cardiac fibrosis are needed.
Conclusions
The currently reported biological mechanism of diabetes-associated cardiac fibrosis remains unclear, i.e., reported through the fragments of overlapping pathways. This review provides a hypothesis of the overall pathway and the association between each pathway. The pathway includes the critical roles of TGFβ-1, microRNA, MAPK, ROS, smad protein, and related inflammatory pathways. The hypothesis of the complete pathway is expected to generate further studies, such as potential effective therapies for cardiac fibrosis patients.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during this literature review.
Abbreviations
- ACEi:
-
Angiotensin-converting enzyme inhibitors
- ARB:
-
Angiotensin receptor blocker
- ARNI:
-
Angiotensin receptor blocker neprilysin inhibitor
- ACTA2:
-
Actin alpha 2
- AGEs:
-
Advanced glycation end products
- BMP:
-
Bone morphogenetic protein
- CCL-2:
-
Chemokine C–C ligand-2
- COL1A1:
-
Collagen type i alpha I
- CTGF:
-
Connective tissue growth factor
- DM:
-
Diabetes mellitus
- ECM:
-
Extracellular matrix
- EndMT:
-
Endothelial-to-mesenchymal transition
- ERK:
-
Extracellular signal-regulated kinase
- ERK/MAPK:
-
Extracellular signal-regulated kinase/mitogen-activated protein kinase
- FN1:
-
Fibronectin-1
- HF:
-
Heart failure
- HFrEF:
-
Heart failure with reduced ejection fraction
- HFpEF:
-
Heart failure with preserved ejection fraction
- iNOS:
-
Inducible nitric oxide synthase
- JNK:
-
C-jun n-terminal kinase
- MAPK:
-
Mitogen-activated protein kinase
- MCP-1:
-
Monocyte chemotactic protein-1
- mRNAs:
-
Messenger RNA
- miRNA:
-
Micro-RNA
- miR-21:
-
Micro-RNA-21
- MMP:
-
Matrix metalloproteinase
- MRA:
-
Mineralocorticoid receptor antagonist
- PTEN:
-
Protein phosphatase and tensin homolog
- RAAS:
-
Renin angiotensin aldosterone system
- RAGE:
-
Receptor advanced glycation end products
- ROS:
-
Reactive oxygen species
- SCD:
-
Sudden cardiac death
- SGLT:
-
Sodium glucose co-transporter
- SGLT2-i:
-
Sodium glucose co-transporter 2 inhibitor
- α-SMA:
-
α-Smooth muscle actin
- Spry1:
-
Sprouty homolog-1
- TIMP-1:
-
Tissue inhibitor of metalloproteinase type 1
- TNF-α:
-
Tumor necrosis factor-α
- TGF-β1:
-
Transforming growth factor-β1
- TIMP-1:
-
Tissue inhibitor of metalloprotein-1
- VCAM-1:
-
Vascular cell adhesion molecules-1
- WHO:
-
World Health Organization
References
Boyko EJ, Karuranga S, Magliano DJ, Saedi P, Sun H (2021) International diabetes federation (IDF) diabetes atlas 10th edition [internet]. International Diabetes Federation. www.diabetesatlas.org
World Health Organization (2021) Diabetes [Internet]. https://www.who.int/news-room/fact-sheets/detail/diabetes
Alam S, Hasan MK, Neaz S, Hussain N, Hossain MF, Rahman T (2021) Diabetes mellitus: insights from epidemiology, biochemistry, risk factors, diagnosis, complications and comprehensive management. Diabetology. 2:36–50
Turkbey EB, Backlund JYC, Genuth S, Jain A, Miao C, Cleary PA et al (2011) Myocardial structure, function, and scar in patients with type 1 diabetes mellitus. Circulation 124:1737–1746
Kashiwagi A, Araki S, Maegawa H (2021) Sodium–glucose cotransporter 2 inhibitors represent a paradigm shift in the prevention of heart failure in type 2 diabetes patients. J Diabetes Investig [Internet] 12:6–20. https://doi.org/10.1111/jdi.13329
Pancholia AK (2018) Sodium-glucose cotransporter-2 inhibition for the reduction of cardiovascular events in high-risk patients with diabetes mellitus. Indian Heart J 70:915–921
Emdin CA, Anderson SG, Callender T, Conrad N, Salimi-Khorshidi G, Mohseni H et al (2015) Usual blood pressure, peripheral arterial disease, and vascular risk: cohort study of 4.2 million adults. BMJ [Internet]. https://doi.org/10.1136/bmj.h4865
Bujak M, Frangogiannis NG (2007) The role of TGF-β signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res 74:184–195
Yuan J, Chen H, Ge D, Xu Y, Xu H, Yang Y et al (2017) Mir-21 promotes cardiac fibrosis after myocardial infarction via targeting Smad7. Cell Physiol Biochem 42:2207–2219
Fauchier L, Boriani G, de Groot JR, Kreutz R, Rossing P, Camm AJ (2021) Medical therapies for prevention of cardiovascular and renal events in patients with atrial fibrillation and diabetes mellitus. EP Europ 23:1–19
Lee HC, Shiou YL, Jhuo SJ, Chang CY, Liu PL, Jhuang WJ et al (2019) The sodium-glucose co-transporter 2 inhibitor empagliflozin attenuates cardiac fibrosis and improves ventricular hemodynamics in hypertensive heart failure rats. Cardiovasc Diabetol BioMed Central 18:1–13
Biernacka A, Cavalera M, Wang J, Russo I, Shinde A, Kong P et al (2015) Smad3 signaling promotes fibrosis while preserving cardiac and aortic geometry in obese diabetic mice. Circ Heart Fail 8:788–798
González A, Schelbert EB, Díez J, Butler J (2018) Myocardial interstitial fibrosis in heart failure: biological and translational perspectives. J Am Coll Cardiol 71:1696–1706
Zhang F, Dang Y, Li Y, Hao Q, Li R, Qi X (2016) Cardiac contractility modulation attenuate myocardial fibrosis by inhibiting TGF-β1/Smad3 signaling pathway in a rabbit model of chronic heart failure. Cell Physiol Biochem 39:294–302
Oral EA (2016) Closing the knowledge gap on cardiovascular disease in type 2 diabetes: the EMPA-REG OUTCOME trial and beyond. Drugs Context 5:1–10
Verma S, Klug E, Mareev VY, Kobalava ZD, Connelly KA, Arici M et al (2020) Sodium-glucose cotransporter 2 inhibitors at the intersection of cardiovascular, renal and metabolic care: an integrated and multidisciplinary approach to patient-centered care. Curr Opin Cardiol 35:589–601
Wong TC, Piehler KM, Kang IA, Kadakkal A, Kellman P, Schwartzman DS et al (2014) Myocardial extracellular volume fraction quantified by cardiovascular magnetic resonance is increased in diabetes and associated with mortality and incident heart failure admission. Eur Heart J 35:657–664
Almdal T, Scharling H, Jensen JS, Vestergaard H (2004) The independent effect of type 2 diabetes mellitus on ischemic heart disease, stroke, and death: a population-based study of 13,000 men and women with 20 years of follow-up. Arch Intern Med [Internet] 164:1422–1426
Bertoia ML, Allison MA, Manson JE, Freiberg MS, Kuller LH, Solomon AJ et al (2012) Risk factors for sudden cardiac death in post-menopausal women. J Am Coll Cardiol 60:2674–2682
Münch AJ, Avanesov M, Bannas P, Säring D, Krämer E, Mearini G et al (2016) Serum matrix metalloproteinases as quantitative biomarkers for myocardial fibrosis and sudden cardiac death risk stratification in patients with hypertrophic cardiomyopathy. J Card Fail 22:845–850
Grisanti LA (2018) Diabetes and arrhythmias: pathophysiology, mechanisms and therapeutic outcomes. Front Physiol 9:1–15
Ceriello A, Catrinoiu D, Chandramouli C, Cosentino F, Dombrowsky AC, Itzhak B et al (2021) Heart failure in type 2 diabetes: current perspectives on screening, diagnosis and management. Cardiovasc Diabetol BioMed Central 20:1–19
Dunlay SM, Givertz MM, Aguilar D, Allen LA, Chan M, Desai AS et al (2019) Type 2 diabetes mellitus and heart failure a scientific statement from the American Heart Association and the Heart Failure Society of America. Circulation 140:e294–e324
Murtha LA, Schuliga MJ, Mabotuwana NS, Hardy SA, Waters DW, Burgess JK et al (2017) The processes and mechanisms of cardiac and pulmonary fibrosis. Front Physiol 8:1–15
Kalosakas G, Ngai KL, Flach S (2005) Breather-induced anomalous charge diffusion. Phys Rev E [Internet] 71:061901. https://doi.org/10.1103/PhysRevE.71.061901
Westermann D, Rutschow S, Jäger S, Linderer A, Anker S, Riad A et al (2007) Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: the role of angiotensin type 1 receptor antagonism. Diabetes 56:641–646
van Linthout S, Seeland U, Riad A, Eckhardt O, Hohl M, Dhayat N et al (2008) Reduced MMP-2 activity contributes to cardiac fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol 103:319–327
Shimizu M, Umeda K, Sugihara N, Yoshio H, Ino H, Takeda R et al (1993) Collagen remodelling in myocardia of patients with diabetes. J Clin Pathol 46:32–36
Gressner AM, Weiskirchen R (2006) Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-β β as major players and therapeutic targets. J Cell Mol Med 10:76–99
Engebretsen KVT, Skårdal K, Bjørnstad S, Marstein HS, Skrbic B, Sjaastad I et al (2014) Attenuated development of cardiac fibrosis in left ventricular pressure overload by SM16, an orally active inhibitor of ALK5. Curr Ther Res Clin Exp 76:148–157
Parichatikanond W, Luangmonkong T, Mangmool S, Kurose H (2020) Therapeutic targets for the treatment of cardiac fibrosis and cancer: focusing on tgf-β Signaling. Front Cardiovasc Med 7:1–19
Abed HS, Samuel CS, Lau DH, Kelly DJ, Royce SG, Alasady M et al (2013) Obesity results in progressive atrial structural and electrical remodeling: implications for atrial fibrillation. Heart Rhythm 10:90–100
Gao Y, Kang L, Li C, Wang X, Sun C, Li Q et al (2016) Resveratrol ameliorates diabetes-induced cardiac dysfunction through AT1R-ERK/p38 MAPK signaling pathway. Cardiovasc Toxicol 16:130–137
Li N, Zhou H, Tang Q (2018) miR-133: a suppressor of cardiac remodeling? Front Pharmacol 9:903
Dietrich C, Singh M, Kumar N, Singh SR (2018) The emerging roles of microRNAs in stem cell aging. In: Mettinger KL, Rameshwar P, Kumar V (eds) Exosomes, stem cells and MicroRNA: aging, cancer and age related disorders. Springer, Cham, pp 11–26
Xiao J, Tang X, Li Y, Fang Z, Ma D, He Y et al (2011) Identification of microRNA precursors based on random forest with network-level representation method of stem-loop structure. BMC Bioinform 12:1–8
Calin GA, Croce CM (2006) MicroRNA signatures in human cancers. Nat Rev Cancer [Internet] 6:857–866
Chen C, Ponnusamy M, Liu C, Gao J, Wang K, Li P (2017) MicroRNA as a therapeutic target in cardiac remodeling. Biomed Res Int [Internet] 2017:1–25
Disertori M, Masè M, Ravelli F (2017) Myocardial fibrosis predicts ventricular tachyarrhythmias. Trends Cardiovasc Med 27:363–372
Jellis C, Martin J, Narula J, Marwick TH (2010) Assessment of nonischemic myocardial fibrosis. J Am Coll Cardiol 56:89–97
Hinderer S, Schenke-Layland K (2019) Cardiac fibrosis: a short review of causes and therapeutic strategies. Adv Drug Deliv Rev [Internet] 146:77–82
Kurose H (2021) Cardiac fibrosis and fibroblasts. Cells 10:1716
Karamitsos TD, Arvanitaki A, Karvounis H, Neubauer S, Ferreira VM (2020) Myocardial tissue characterization and fibrosis by imaging. JACC Cardiovasc Imaging 13:1221–1234
Bing R, Dweck MR (2019) Myocardial fibrosis: why image, how to image and clinical implications. Heart 105:1832–1840
De Jong S, Van Veen TAB, Van Rijen HVM, De Bakker JMT (2011) Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol 57:630–638
Umbarkar P, Ejantkar S, Tousif S, Lal H (2021) Mechanisms of fibroblast activation and myocardial fibrosis: lessons learned from FB-specific conditional mouse models. Cells 10:2412
Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD et al (2018) Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Investig 128:2127–2143
Ma Y, Iyer RP, Jung M, Czubryt MP, Lindsey ML (2017) Cardiac fibroblast activation post-myocardial infarction: current knowledge gaps. Physiol Behav 38:448–458
Liu T, Song D, Dong J, Zhu P, Liu J, Liu W et al (2017) Current understanding of the pathophysiology of myocardial fibrosis and its quantitative assessment in heart failure. Front Physiol 8:1–13
Horn MA, Trafford AW (2016) Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J Mol Cell Cardiol 93:175–185
Kong P, Christia P, Frangogiannis NG (2014) The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 71:549–574
Díez J, González A, Kovacic JC (2020) Myocardial interstitial fibrosis in nonischemic heart disease, part 3/4: JACC focus seminar. J Am Coll Cardiol 75:2204–2218
Li L, Zhao Q, Kong W (2018) Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol 68–69:490–506
Van Heerebeek L, Hamdani N, Handoko ML, Falcao-Pires I, Musters RJ, Kupreishvili K et al (2008) Diastolic stiffness of the failing diabetic heart: Importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 117:43–51
Yap J, Tay WT, Teng THK, Anand I, Richards AM, Ling LH et al (2019) Association of diabetes mellitus on cardiac remodeling, quality of life, and clinical outcomes in heart failure with reduced and preserved ejection fraction. J Am Heart Assoc 8:e013114
Paulus WJ, Dal Canto E (2018) Distinct myocardial targets for diabetes therapy in heart failure with preserved or reduced ejection fraction. JACC Heart Fail 6:1–7
Basta G, Schmidt AM, De Caterina R (2004) Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res 63:582–592
Russo I, Frangogiannis NG (2016) Molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol 90:84–93
Shinde AV, Frangogiannis NG (2014) Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol 70:74–82
van Putten S, Shafieyan Y, Hinz B (2016) Mechanical control of cardiac myofibroblasts. J Mol Cell Cardiol 93:133–142
Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T et al (2005) CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res 96:881–889
Frieler RA, Mortensen RM (2015) Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation 131:1019–1030
Humphreys BD (2012) Targeting pericyte differentiation as a strategy to modulate kidney fibrosis in diabetic nephropathy. Semin Nephrol 32:463–470
Nishikori Y, Shiota N, Okunishi H (2014) The role of mast cells in cutaneous wound healing in streptozotocin-induced diabetic mice. Arch Dermatol Res 306:823–835
Zhang W, Chancey AL, Tzeng H-P, Zhou Z, Lavine KJ, Gao F et al (2011) The development of myocardial fibrosis in transgenic mice with targeted overexpression of tumor necrosis factor requires mast cell-fibroblast interactions. Circulation 124:2106–2116
Rose BA, Force T, Wang Y (2010) Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev 90:1507–1546
Tachibana H, Perrino C, Takaoka H, Davis RJ, Naga Prasad SV, Rockman HA (2006) JNK1 is required to preserve cardiac function in the early response to pressure overload. Biochem Biophys Res Commun 343:1060–1066
Kyoi S, Otani H, Matsuhisa S, Akita Y, Tatsumi K, Enoki C et al (2006) Opposing effect of p38 MAP kinase and JNK inhibitors on the development of heart failure in the cardiomyopathic hamster. Cardiovasc Res 69:888–898
Krishnamurthy P, Subramanian V, Singh M, Singh K (2007) Β1 integrins modulate B-adrenergic receptor-stimulated cardiac myocyte apoptosis and myocardial remodeling. Hypertension 49:865–872
Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H et al (2001) The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A 98:12283–12288
Rincón M, Davis RJ (2009) Regulation of the immune response by stress-activated protein kinases. Immunol Rev 228:212–224
Kyriakis JM, Avruch J (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81:807–869
Hanna A, Humeres C, Frangogiannis NG (2021) The role of Smad signaling cascades in cardiac fibrosis. Cell Signal 77:109826
Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G et al (2007) Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 116:2127–2138
Russo I, Cavalera M, Huang S, Su Y, Hanna A, Chen B et al (2019) Protective effects of activated myofibroblasts in the pressure-overloaded myocardium are mediated through smad-dependent activation of a matrix-preserving program. Circ Res 124:1214–1227
Raftery LA, Twombly V, Wharton K, Gelbart WM (1995) Genetic screens to identify elements of the decapentaplegic signaling pathway in Drosophila. Genetics 139:241–254
Kretzschmar M, Massagué J (1998) SMADs: mediators and regulators of TGF-β signaling. Curr Opin Genet Dev 8:103–111
Hata A, Chen Y-G (2016) TGF-β signaling from receptors to smads. Cold Spring Harb Perspect Biol 8:a022061
Chen C, Ponnusamy M, Liu C, Gao J, Wang K, Li P (2017) MicroRNA as a therapeutic target in cardiac remodeling. Biomed Res Int 2017:1278436
Li N, Zhou H (2020) Sglt2 inhibitors: a novel player in the treatment and prevention of diabetic cardiomyopathy. Drug Des Devel Ther 14:4775–4788
Wakisaka M, Kamouchi M, Kitazono T (2019) Lessons from the trials for the desirable effects of sodium glucose co-transporter 2 inhibitors on diabetic cardiovascular events and renal dysfunction. Int J Mol Sci 20:1–15
Borghetti G, Von LD, Eaton DM, Sourij H, Houser SR, Wallner M (2018) Diabetic cardiomyopathy: current and future therapies. Beyond Glycemic Control 9:1–15
Dai B, Wang F, Nie X, Du H, Zhao Y, Yin Z et al (2020) The cell type-specific functions of miR-21 in cardiovascular diseases. Front Genet [Internet] 11:1–16. https://doi.org/10.3389/fgene.2020.563166/full
Spinale FG, Janicki JS, Zile MR, Spinale FG, Janicki JS, Zile MR (2013) Membrane-associated matrix proteolysis and heart failure. Circ Res 112:195–208
del Monte-Nieto G, Fischer JW, Gorski DJ, Harvey RP, Kovacic JC (2020) Basic biology of extracellular matrix in the cardiovascular system, part 1/4: JACC focus seminar. J Am Coll Cardiol 75:2169–2188
Zile MR, DeSantis SM, Baicu CF, Stroud RE, Thompson SB, McClure CD et al (2011) Plasma biomarkers that reflect determinants of matrix composition identify the presence of left ventricular hypertrophy and diastolic heart failure. Circ Heart Fail [Internet] 4:246–256. https://doi.org/10.1161/CIRCHEARTFAILURE.110.958199
Marchesi C, Dentali F, Nicolini E, Maresca AM, Tayebjee MH, Franz M et al (2012) Plasma levels of matrix metalloproteinases and their inhibitors in hypertension: a systematic review and meta-analysis. J Hypertens 30:3–16
Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K et al (2006) Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension 47:711–717
Wu L, Derynck R (2009) Essential role of TGF-β signaling in glucose-induced cell hypertrophy. Dev Cell 17:35–48
Bonnans C, Chou J, Werb Z (2014) Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol [Internet] 15:786–801
Martos R, Baugh J, Ledwidge M, O’Loughlin C, Conlon C, Patle A et al (2007) Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation 115:888–895
López B, González A, Querejeta R, Larman M, Díez J (2006) Alterations in the pattern of collagen deposition may contribute to the deterioration of systolic function in hypertensive patients with heart failure. J Am Coll Cardiol 48:89–96
Zelniker TA, Braunwald E (2020) Mechanisms of cardiorenal effects of sodium-glucose cotransporter 2 inhibitors: JACC state-of-the-art review. J Am Coll Cardiol 75:422–434
Habibi J, Aroor AR, Sowers JR, Jia G, Hayden MR, Garro M et al (2017) Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc Diabetol BioMed Central 16:1–15
Kraakman MJ, Lee MKS, Al-Sharea A, Dragoljevic D, Barrett TJ, Montenont E et al (2017) Neutrophil-derived S100 calcium-binding proteins A8/A9 promote reticulated thrombocytosis and atherogenesis in diabetes. J Clin Investig 127:2133–2147
Kuwahara F, Kai H, Tokuda K, Takeya M, Takeshita A, Egashira K et al (2004) Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension 43:739–745
Arow M, Waldman M, Yadin D, Nudelman V, Shainberg A, Abraham NG et al (2020) Sodium-glucose cotransporter 2 inhibitor Dapagliflozin attenuates diabetic cardiomyopathy. Cardiovasc Diabetol BioMed Central 19:1–12
Dadson K, Kovacevic V, Sweeney G (2015) Mechanisms of cardiac fibrosis and heart failure. Cardiac fibrosis and heart failure: cause or effect? Springer, Cham, pp 279–297
Lijnen P, Papparella I, Petrov V, Semplicini A, Fagard R (2006) Angiotensin II-stimulated collagen production in cardiac fibroblasts is mediated by reactive oxygen species. J Hypertens 24:757–766
Kim M, Zhang X (2019) The profiling and role of miRNAs in diabetes mellitus. J Diabetes Clin Res 1:5–23
Zhao Y, Du D, Chen S, Chen Z, Zhao J (2022) New insights into the functions of MicroRNAs in cardiac fibrosis: from mechanisms to therapeutic strategies. Genes (Basel) [Internet] 13:1390–1406
Guo R, Nair S (2017) Role of microRNA in diabetic cardiomyopathy: from mechanism to intervention. Biochim Biophys Acta Mol Basis Dis 1863:2070–2077
Kong L, Zhu J, Han W, Jiang X, Xu M, Zhao Y et al (2011) Significance of serum microRNAs in pre-diabetes and newly diagnosed type 2 diabetes: a clinical study. Acta Diabetol 48:61–69
Karolina DS, Armugam A, Tavintharan S, Wong MTK, Lim SC, Sum CF et al (2011) MicroRNA 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS ONE [Internet] 6:e22839. https://doi.org/10.1371/journal.pone.0022839
Sweeney M, Corden B, Cook SA (2020) Targeting cardiac fibrosis in heart failure with preserved ejection fraction: mirage or miracle? EMBO Mol Med 12:1–26
Brilla CG, Funck RC, Rupp H (2000) Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation 102:1388–1393
Berezin A (2002) Angiotensin-II receptor antagonist losartan dose-dependently improves the left ventricular remodelling in patients with congestive heart failure. J Clin Basic Cardiol [Internet] 5:83–86
McDiarmid AK, Swoboda PP, Erhayiem B, Bounford KA, Bijsterveld P, Tyndall K et al (2020) Myocardial effects of aldosterone antagonism in heart failure with preserved ejection fraction. J Am Heart Assoc 9:1–10
McMurray JJV, Packer M, Desai AS, Gong J (2017) Angiotensin–neprilysin inhibition versus enalapril in heart failure. Rev Argent Med 5:132–133
Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, Maggioni AP et al (2019) Angiotensin–neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med 381:1609–1620
McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M et al (2021) 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 42:3599–3726
Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM et al (2022) 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on clinical practice guidelines. Circulation 145:e895-1032
Rog-Zielinska EA, Norris RA, Kohl P, Markwald R (2016) The living scar—cardiac fibroblasts and the injured heart. Trends Mol Med 22:99–114
Acknowledgements
Not applicable.
Funding
This paper is supported by grant from Indonesian ministry of Education, Culture, Research and Technology to HD (Contract No: 145/E5/PG.02.00.PT/2022).
Author information
Authors and Affiliations
Contributions
MR wholly conducted this literature review. HD was responsible for conception, review, and funding. MS initiated ideas, drafting, and revision. RL edited and reviewed manuscript as well as assisted funding acquisition. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval is not required.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ridwan, M., Dimiati, H., Syukri, M. et al. Potential molecular mechanism underlying cardiac fibrosis in diabetes mellitus: a narrative review. Egypt Heart J 75, 46 (2023). https://doi.org/10.1186/s43044-023-00376-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43044-023-00376-z