
Abstract
Background
Methanotrophs play an important role in biotechnological applications, with their ability to utilize single carbon (C1) feedstock such as methane and methanol to produce a range of high-value compounds. A newly isolated obligate methanotroph strain, Methylomonas sp. DH-1, became a platform strain for biotechnological applications because it has proven capable of producing chemicals, fuels, and secondary metabolites from methane and methanol. In this study, transcriptome analysis with RNA-seq was used to investigate the transcriptional change of Methylomonas sp. DH-1 on methane and methanol. This was done to improve knowledge about C1 assimilation and secondary metabolite pathways in this promising, but under-characterized, methane-bioconversion strain.
Results
We integrated genomic and transcriptomic analysis of the newly isolated Methylomonas sp. DH-1 grown on methane and methanol. Detailed transcriptomic analysis indicated that (i) Methylomonas sp. DH-1 possesses the ribulose monophosphate (RuMP) cycle and the Embden–Meyerhof–Parnas (EMP) pathway, which can serve as main pathways for C1 assimilation, (ii) the existence and the expression of a complete serine cycle and a complete tricarboxylic acid (TCA) cycle might contribute to methane conversion and energy production, and (iii) the highly active endogenous plasmid pDH1 may code for essential metabolic processes. Comparative transcriptomic analysis on methane and methanol as a sole carbon source revealed different transcriptional responses of Methylomonas sp. DH-1, especially in C1 assimilation, secondary metabolite pathways, and oxidative stress. Especially, these results suggest a shift of central metabolism when substrate changed from methane to methanol in which formaldehyde oxidation pathway and serine cycle carried more flux to produce acetyl-coA and NADH. Meanwhile, downregulation of TCA cycle when grown on methanol may suggest a shift of its main function is to provide de novo biosynthesis, but not produce NADH.
Conclusions
This study provides insights into the transcriptomic profile of Methylomonas sp. DH-1 grown on major carbon sources for C1 assimilation, providing in-depth knowledge on the metabolic pathways of this strain. These observations and analyses can contribute to future metabolic engineering with the newly isolated, yet under-characterized, Methylomonas sp. DH-1 to enhance its biochemical application in relevant industries.
Similar content being viewed by others


Background
Single-carbon (C1) feedstock such as methane and methanol has great potential for a number of applications and has shown flexibility when used with biocatalysts and in bioconversion processes to produce various products [1, 2]. Methane is the main component of natural (and shale) gas and biogas. Recently, expansion of the global methane market has improved accessibility, resulting in decreasing gas prices. This has made methane an attractive next-generation feedstock [3]. Generally, chemical conversions of methane to other chemicals require a high input of energy because of the high activation energy of the C-H bond [4]. Thus, biological conversions of methane to higher value products using methanotrophs has grown more attractive recently. As intermediates in the aerobic methane oxidation process, methanotrophs are capable of utilizing methanol as a sole carbon source and utilize a similar pathway to that for methane assimilation [2]. Methanol is also an attractive carbon substrate because of its high production volume and low market price [5]. A wide range of methylotrophic bacteria has been used to produce multiple chemically valuable compounds, including single-cell proteins, biopolymers, lipids, methanol, ectoine, and vitamin B12 [6]. In addition, genetically engineered methanotrophs have served as promising and powerful production platforms to over-produce several non-native compounds such as carotenoids, lactic acid, succinic acid, 1,4-butanediol and 2,3-butanediol [7,8,9]. In an effort to expand on the collection of bacterial bioconversion platforms, a novel obligate methanotroph strain, Methylomonas sp. DH-1, was recently isolated from the sludge of a brewery. However, the strain had yet to be characterized for its bioconversion abilities with molecular-level experiments [10]. Methylomonas sp. DH-1 showed highly efficient bioconversion of methane to methanol, with a titer of 1.34 g per liter. This is more efficient than the conversion rate of previously reported biocatalysts [10]. Furthermore, the strain’s ability to tolerate high concentrations of methanol (up to 7% [v/v]) offers advantages for high-titer methanol production [10]. Recently, Methylomonas sp. DH-1 also proved to be a novel and competent biocatalyst for the conversion of propane to acetone, with a titer of 16.62 mM under mild reaction conditions [11]. Furthermore, Methylomonas sp. DH-1 may become a biotechnologically important biocatalyst for its ability to produce several carotenoids (unpublished report).
Development of next-generation sequencing technology enabled the sequencing of genomes of several methanotrophs, and the genomes of those methanotrophs provided essential information for the reconstruction of methane metabolism in methanotrophs [12, 13]. In addition, multi-omics studies (which integrate transcriptomics, metabolomics, proteomics, and genomics) have provided insights to assess various metabolic engineering targets for methanotrophs [8]. Indeed, several previous studies using a multi-omics approach to analyze multiple model methanotrophs have been reported [14,15,16,17,18]. Recently, in our previous work, a complete genome sequence for Methylomonas sp. DH-1 was determined [19]. The strain contains one 4.86 Mb chromosome and one 278 kb plasmid, pDH1 [19]. The availability of the complete genome sequence of Methylomonas sp. DH-1 provided an essential background for revisiting a genome-based reconstruction of methane metabolism. But to date, a comprehensive transcriptome analysis of Methylomonas sp. DH-1 is still elusive, and mechanisms responsible for the methanol tolerance of Methylomonas sp. DH-1 have yet to be investigated. An RNA-sequencing approach has been used to investigate the transcriptome and has provided insights into the methane metabolism of type I [16,17,18] and type II [14, 15] methanotrophs. In this study, we first detail the genome-wide transcriptional responses of methane metabolism and secondary metabolite production in Methylomonas sp. DH-1 during growth on methane. We then offer a comparative transcriptomic analysis conducted with cells grown on methane and methanol. This analysis revealed differences in the transcriptional response of multiple metabolic pathways which are relevant to C1 assimilation, secondary metabolite production, and oxidative stress.
Methods
Bacterial growth conditions
Methylomonas sp. DH-1 was isolated from the activated sludge of a brewery plant based on NMS medium using enrichment culture with methane as sole carbon source as described in our previous work [10]. Liquid pre-cultures were grown in 180 ml baffled-flask with 10 ml NMS with a supplement of 30% methane (v/v) as a sole carbon source at 30 °C and 230 rpm, sealed with a screw cap. The precultures were then inoculated in a 500 ml baffled flask sealed with a screw cap containing 50 ml of NMS and methane also was supplied to a final concentration of 30% (v/v) by gas substitution using a gas-tight syringe, and the headspace was refreshed every day. The methanol-cultured Methylomonas sp. DH-1 was cultured in the same medium containing 1% (v/v) methanol without added methane. All cultures were grown in triplicate for subsequent RNA extraction and sequencing.
Total RNA isolation and sequencing
For sequencing library preparation, 5 ml of microbial culture broths containing either methane- or methanol-grown cultures in mid-exponential phase were harvested, and total RNA was stabilized using the bacterial reagent RNAprotect (Qiagen, Hilden, Germany). Cells were lysed and total RNA was extracted using a GeneJET RNA Purification Kit (Thermo Fisher Scientific, USA), following the manufacturer’s protocol. Total RNA quality and quantity were measured using an RNA 6000 Nano kit with the Agilent Bioanalyzer (Agilent Technologies, CA, USA). Next, rRNA was removed using the Ribo-Zero rRNA removal kit for gram-negative bacteria (Epicentre, Madison, WI, USA), and the remaining RNA was used for creating the sequencing library using the TruSeq Stranded Total RNA Sample Prep Kit (Illumina, USA) according to the manufacturer’s instructions. Transcriptome sequencing was conducted using the Illumina/Hiseq-2000 RNA sequencing platform (Macrogen, Korea).
Quantification of differentially expressed genes
After evaluating the quality of the raw sequence data with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), further quantitative analyses were performed. Illumina sequencing (of triplicates) was aligned against the genome sequence of Methylomonas sp. DH-1 (NZ_CP014360 for the chromosome and NZ_CP014361 for the plasmid). The Bowtie tool was used with a maximum insert size of 1000 bp and with two maximum mismatches after trimming 3 bp at the 3′ ends under the default options. The sequence alignment/map (SAM) files were post-processed using SAMTools (http://samtools.sourceforge.net/), by first converting to binary alignment/map (BAM) files and then sorting and indexing those BAM files. Indexed and sorted BAM files generated from SAMTools were analyzed by Cufflinks and Cuffdiff to calculate values for fragments per kilobase of exon per million fragments (FPKM) and differential expression, respectively (with default options and a library type of dUTP RNA-seq). Genes from the Cuffdiff output showing differential expression with a log2 fold change ≥1.0 and a false discovery rate (FDR) value ≤0.01 were considered as differentially expressed genes in our study.
Clusters of orthologous groups (COGs) functional assignment and ortholog calculation
All of the CDS regions were assigned to different functional categories based on the Clusters of orthologous genes (COGs) designation [20]. The ortholog calculation was performed using InParanoid software [21].
Results
Genome-wide transcriptome profiling
Gene expression analysis was carried out on Methylomonas sp. DH-1 grown in NMS medium with supplementation of methane or methanol as a sole carbon and energy source. All of the experiments were performed using biological triplicates. RNA-seq was performed as described in the Materials and Methods section. On average, 30 million reads were generated per sample, with a Q30 value higher than 96%, which is considered to be large enough for differential expression analysis in bacteria [22]. The Bowtie algorithm was used for sequence read alignment onto the Methylomonas sp. DH-1 reference sequence (NZ_CP014360 and NZ_CP014361). On average, 98% of reads were mapped onto the Methylomonas sp. DH-1 reference genome. The Bowtie was run with the following options: a maximum insert size of 1000 bp and 2 maximum mismatches after trimming 3 bp at the 3′ ends, with default parameters for the other options. The relative expression level, generated as FPKM values, was calculated to compare gene expression levels within and across biological replicates. A total of 4318 CDS regions in the chromosome were analyzed, while 129 CDS regions without sufficient alignments were removed from further analysis. For the plasmid, 7 among 242 CDS regions were excluded because the number of mapped reads was low. Using the calculated relative expression levels, genes were grouped into 6 expression categories (omitting rRNA genes) following the method by Vorobev et al. [15]: very high (500 FPKM), high (500 to 200 FPKM), intermediate (200 to 50 FPKM), low (50 to 10 FPKM), very low (10 to 2 FPKM), and not expressed (below 2 FPKM) (Table 1). The majority of expression levels fell into the intermediate/low category, covering 72.64% of genes from methanol-grown culture and 71.83% of genes from methane-grown culture. A small proportion of genes showed very high/high expression, covering 6.83 and 5.57% of genes in methane and methanol, respectively (Table 1). Interestingly, most of the genes in the endogenous plasmid (90% in methane- and 87.6% in methanol-grown cultures) showed very high expression (Table 1). Differential expression analysis of Methylomonas sp. DH-1 grown on methanol and methane indicated that 261 and 723 genes were upregulated and downregulated, respectively, with a fold-change ≥2 and P ≤ 0.05. In the ten most highly expressed genes from cultures grown in methane and methanol, there were two genes for non-coding RNA (ncRNA), one gene encoding transfer-messenger RNA (tmRNA), 3 genes encoding the particulate methane monooxygenase (pmo) operon, and four genes encoding hypothetical proteins (Additional file 1: Table S1). Genome analysis indicated that Methylomonas sp. DH-1 harbors RNase P, three ncRNAs (RNA component class A [rnpB], 6S RNA [ssrS], and signal recognition particle sRNA [small type, ffs]), and one tmRNA (ssrA). Among the three ncRNAs, rnpB, which is an essential and ubiquitous ribozyme responsible for the maturation of tRNA [23], was the most highly expressed in Methylomonas sp. DH-1, followed by ssrS (with the third highest expression in this strain). ssrA encodes a unique tmRNA that showed the second highest expression in Methylomonas sp. DH-1. Furthermore, expression levels of ssrS, which typically interacts with the primary holoenzyme form of RNA polymerase and functions as a global regulator that downregulates transcription for modulating stress and optimizing survival during nutrient limitation [24], were strongly downregulated under methanol growth, suggesting that methanol can be a stress factor affecting the growth of Methylomonas sp. DH-1. It was speculated that ncRNA (ssrS and rnpB) and tmRNA might serve important roles in the gene regulation of Methylomonas sp. DH-1. Additionally, from transcriptional profiling analysis, 1482 genes coding for hypothetical proteins were expressed. Among these genes, 85 showed very high expression levels. These findings suggest that unknown, functional proteins might be important in the metabolism of Methylomonas sp. DH-1, and that the functional annotation of these hypothetical proteins needs to be performed.
Expression of genes involved methane oxidation of Methylomonas sp. DH-1
An overview of the methane metabolism of Methylomonas sp. DH-1 grown on methane is summarized in Fig. 1. The relative expressions of genes (FPKM values) are shown in the additional files for growth on methane or methanol. Since Methylomonas sp. DH-1 has pathways for C1 assimilation, it was postulated that the genes involved in C1 assimilation would be highly expressed when grown on methane or methanol. As postulated, genes in the pathways for methane or methanol oxidation were highly expressed or very highly expressed. Compared to the C1 metabolic genes in typical obligate methanotrophs, the Methylomonas DH-1 genome harbors one copy of the particulate methane monooxygenase (pmo) gene cluster and does not contain genes encoding soluble methane monooxygenase (smo). The pmo gene cluster was the most highly expressed when grown on methane. Among the three genes in the pmoCAB gene cluster, which was also very highly expressed, expression of pmoC was approximately 2.5-fold higher than the other genes in the same operon (Fig. 3a and Additional file 2: Table S2). Despite pmo genes being part of a canonical operon, the transcript abundance of pmoC was higher than that of pmoA and pmoB. This is consistent with previous findings in the alpha-proteobacterial methanotrophs Methylosinus trichosporium OB3b [14] and Methylocystis sp. strain SB2 [15], and with previous findings in the gamma-proteobacterial methanotrophs Methylomicrobium alcaliphilum 20Z [16] and Methylomicrobium buryatense 5GB1 [17]. In addition, a sequence-divergent particulate monooxygenase that grouped with the non-canonical form pmxABC was found in the Methylomonas sp. DH-1 genome, similar to findings in other species from the genera Methylomonas, Methylobacter, and Methylomicrobium [25]. In contrast to the pmo operon, the expression of pmxABC was very low (Additional file 2: Table S2). When grown on methanol, the expression level of the pmoCAB operon was dramatically downregulated, with 2.87, 5.46, and 2.74-fold changes observed respectively for each gene (Fig. 2, Fig. 3a). However, the expression level of these genes on methanol remained much higher than pmxABC. The expression level of the first two genes in the pmxABC operon, pmxA and pmxB, did not change significantly, while the expression of pmxC was downregulated on methanol (Fig. 3b). In summary, these results clearly indicated that the pmo genes play an important role in methane metabolism, and that methane may be a key enhancer for the expression of the pmo operon. The existence of a non-canonical form of ammonia/methane monooxygenase, pmxABC, was found in the genome of Methylomonas sp. DH-1. However, expression of pmx was very low, which suggested that the protein products of this operon may not be actively involved in the catalytic processes of Methylomonas sp. DH-1 when grown on methane or methanol.

Overview of the central metabolic pathways in Methylomonas sp. DH-1 predicted from the genomic annotation and the transcriptomic data mapping. The colors indicate the relative gene expression level. Ru5P: ribulose 5-phosphate, H6P: hexulose 6-phosphate, F6P: fructose 6-phosphate, KDPG: 2-keto-3-deoxy 6-phosphogluconate, F1,6BP: fructose 1,6-bisphosphate, DHAP: dihydroxyacetone phosphate, G3P: glyceraldehyde 3-phosphate, PEP: phosphoenolpyruvate, OAA: oxaloacetic acid

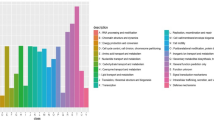
The central metabolism and the secondary metabolites of Methylomonas sp. DH-1 grown on methane or methanol as a sole carbon/energy source. Genes highlighted in red and blue were significantly upregulated and downregulated (respectively) on methanol

Differential expression of genes involved in C1 metabolism: a methane oxidation, b non-canonical form pxmABC, c methanol oxidation, d formaldehyde oxidation in Methylomonas sp. DH-1 grown on methane (black) and methanol (gray). * Significantly different expression of genes between methane- and methanol-grown cultures (P < 0.05)
Pyrroloquinoline quinone (PQQ)-dependent methanol dehydrogenase (mxaFJGI) and the PQQ biosynthesis gene cluster (pqqABCDE) were identified in the Methylomonas sp. DH-1 genome. The mxaFJGI operon for methanol oxidation (encoding two subunits of methanol dehydrogenase, cytochrome, and a protein of unknown function [mxaJ]) showed relatively high expression levels. While the expression of mxaFJGI actually decreased on methanol, the fold change was less than 2 (Fig. 2, Fig. 3c, Additional file 2: Table S2). The genes downstream of the mxaFJGI operon (which are chaperones, regulators, or have Ca2+ insertion functions) showed an intermediate or low level of expression that was 14- to 55-fold lower in comparison to the first four genes in the methanol dehydrogenase (MDH) operon (Additional file 2: Table S2). The PQQ biosynthesis gene cluster (pqqABCDE) encodes an essential system that provides cofactors for methanol oxidation. The expression level of this gene cluster was high (Additional file 2: Table S2). A homolog of mxaF, known as xoxF, was found in Methylomonas sp. DH-1. This gene showed a high expression level (Fig. 3c and Additional file 2: Table S2) when grown on methane, but was much lower than the expression of mxaF. Similar to the pmo operon, the expression of xoxF was significantly downregulated (3.65-fold change) on methanol (Fig. 3c). The homolog xoxF was firstly reported as part of a regulatory complex essential for the expression of methanol dehydrogenase in Methylobacterium extorquens AM1 [26] and currently has been identified as a predominant methanol dehydrogenase in M. buryatense 5GB1 [27]. Thus, it is possible that a high expression level of xoxF may contribute to methanol oxidation in Methylomonas sp. DH-1. Furthermore, direct coupling mode, in which methanol oxidation supplies electrons for methane oxidation, is most suitable mode of methane oxidation in gamma-proteobacterial methanotrophic bacteria [17]. Surprisingly, xoxF was highly downregulated during growth on methanol, similar to the pattern seen for pmo. Thus, we can postulate that xoxF may also play a role in methane oxidation in Methylomonas sp. DH-1 during growth on methane, and the exact contribution of xoxF in methane and methanol oxidation should be experimentally explored.
From the genomic analysis a group of genes in conventional type II methanotrophic metabolic pathways (such as the tetrahydromethanopterin (H4MPT) pathway and the tetrahydrofolate [H4F] pathway) were also identified in the genome of Methylomonas sp. DH-1. Previous studies have indicated that there are three possible pathways for formaldehyde oxidation, including H4MPT, H4F, and direct oxidation by formaldehyde dehydrogenase [28]. Genes for the direct oxidation of formaldehyde by formaldehyde dehydrogenase were not found in the Methylomonas sp. DH-1 genome. However, a broad-specificity aldehyde dehydrogenase (ald) was predicted from the genomic analysis, and the transcriptome analysis showed an intermediate expression level for this aldehyde dehydrogenase (Additional file 2: Table S2). Recently, the H4F pathway has drawn attention for its assimilatory function to convert formate to methylene-H4F. This pathway also contributes to formaldehyde oxidation in M. extorquens AM1 and M. trichosporium OB3b [14, 29]. All of the genes involved in the H4F pathway in Methylomonas sp. DH-1 were expressed at an intermediate level (Additional file 2: Table S2). During growth on methanol, the expression levels of genes in the H4F pathway were slightly increased compared to growth on methane (Fig. 3d). In particular, methenyltetrahydromethanopterin cyclohydrolase (fch) showed a fold change of 2.18 (Additional file 2: Table S2). Among the three different formaldehyde oxidation processes, the H4MPT pathway serves as the key pathway in the type II model methanotroph, M. trichosporium OB3b [14]. Formaldehyde-activating enzyme (fae), which condenses formaldehyde with tetrahydromethanopterin (H4MPT) to produce methylene-H4MPT, is the first enzyme of the formaldehyde detoxification pathway through H4MPT. Interestingly, Methylomonas sp. DH-1 has three homologs of fae at different genomic locations. The orthologs fae1 and fae3 appear to be expressed at very low and at high levels on methane-grown cultures, respectively (Additional file 2: Table S2). However, the ortholog fae2 was expressed at a very high level (10-fold higher than fae3) (Fig. 1 and Additional file 2: Table S2). The rest of the genes encoding key enzymes in the H4MPT pathway were also expressed at a high level on methane (Fig. 1 and Additional file 2: Table S2). Thus, our transcriptomic data analysis revealed that the genes in the H4MPT pathway showed high expression at the transcriptional level, indicating this pathway was likely key for formaldehyde oxidation in Methylomonas sp. DH-1. Interestingly, the expression of fae2 was upregulated on methanol compared to methane (Fig. 2, Fig. 3d), meaning formaldehyde oxidation via H4MPT was more active during growth on methanol. Most methanotrophs use a NAD-dependent formate dehydrogenase to oxidize formate to CO2 [30]. It has been reported that most of the reducing power for methane metabolism was provided by formate oxidation to CO2 [31]. Methylomonas sp. DH-1 has the formate dehydrogenase (fdsABGCD) gene cluster, encoding a NAD-dependent formate dehydrogenase and an additional single copy of the alpha subunit (fdhA). Most of the genes in the operon fdsABGCD were expressed at a high level, and no significantly different expression of fdsABGCD was observed at the transcription level between methane and methanol (Fig. 3d and Additional file 2: Table S2). Overall, the transcriptomic analysis illustrate that the H4MPT pathway may serve as the key pathway for formaldehyde oxidation in Methylomonas sp. DH-1, as the pathway genes were highly expressed. Similarly, activation of H4F and the H4MPT pathway in M. buryatense strain 5GB1 and M. alcaliphilum 20ZR under methane growth were also observed [17, 18].
A complete set of functional genes for formaldehyde fixation through the ribulose monophosphate (RuMP) cycle, Embden-Meyerhof Parnas (EMP) pathway, the Entner-Doudoroff (ED) pathway, and the pentose phosphate (PP) pathway was also identified in the Methylomonas sp. DH-1 genome. More interestingly, the complete gene set implementing the serine cycle without the interconnected ethylmalonyl-CoA (EMC) cycle and TCA cycle existed in the genome of Methylomonas sp. DH-1. A transcriptional analysis of these key formaldehyde assimilation pathways is described below.
Gene expression of C1 assimilation pathway under methane growth and its response to substrate shifts
The RuMP pathway has been assumed to be the main pathway for C1 assimilation in the type I methanotrophs [30]. All genes for a complete RuMP cycle were identified in Methylomonas sp. DH-1, but those genes were transcribed at different levels (Fig. 1, Fig. 4a and Additional file 3: Table S3). Two key enzymes of the RuMP cycle are hexulose phosphate synthase (hps) and phophohexulo isomerase (phi). As expected, they were expressed at a very high level. In addition, another copy of hexulose phosphate synthase (AYM39_RS02745) was found in Methylomonas sp. DH-1, which was also expressed at a very high level. The expression of hps and phi did not change significantly between growth on methane and methanol (Fig. 2, Fig. 4a and Additional file 3: Table S3). This could be because those enzymes are regulated by concentration of formaldehyde [32]. The transcriptional expression of enzymes involved in the downstream portion of the RuMP cycle (from fructose-6-phosphate) was at an intermediate level (18- to 49-fold lower than hpi and hps). Interestingly, the abundance of transcripts encoding the Embden–Meyerhof–Parnas (EMP) pathway was 3 to 5-fold higher than that for Entner–Doudoroff (EDD) pathway enzymes (Fig. 1 and Fig. 4b, and Additional file 3: Table S3). Furthermore, a putative pyruvate kinase (pk) showed a very high expression level. It seemed that significant carbon flux could occur via the EMP pathway. Recently, 13C-labeling analysis have shown that the dominant pathway for generating pyruvate is the EMP pathway, which replenishes up to 75% of pyruvate in Methylomicrobium alcaliphilum 20Z and Methylomonas sp. LW13 [16]. In our previous study, comparative genomic and phylogenetic analysis was performed for 17 Methylomonas strains including Methylomonas sp. DH-1 and Methylomonas sp. LW13, and we found that Methylomonas sp. DH-1 and Methylomonas sp. LW13 showed high average nucleotide identity [19]. Along with the similar of gene expression profile as M. alcaliphilum 20Z and Methylomonas sp. LW13, a similar carbon isotopic distribution in pyruvate may exist and the EMP pathway likely serves as the main pathway for C1 assimilation in Methylomonas sp. DH-1.

Differential expression of genes involved in C1 metabolism: a RuMP cycle, b ED and EMP pathways, c serine cycle, d TCA cycle in Methylomonas sp. DH-1 grown on methane (black) and methanol (gray). * Significantly different expression of genes between methane- and methanol-grown cultures (P < 0.05)
In a comparison between the transcriptome profiles of methane- and methanol-grown cultures, the expression levels of most genes in the RuMP cycle did not change significantly, with these exceptions: glucose-6-phosphate dehydrogenase (gpd1), 6-phosphogluconolactonase (pgls), and ribose 5-phosphate isomerase (rpi). Upregulation of some genes in the EMP pathway, including fructose-bisphosphatase (fbp), triose-phosphate isomerase (tpi), and phosphoglycerate mutase (gpml), was observed. Transcription of pyruvate kinase was also upregulated on methanol compared to methane (Fig. 4a, b, and Additional file 3: Table S3).
Targeted transcriptomic analysis of serine and TCA cycle suggested flux shifts under methanol-grown cultures
The genomic analysis suggested that Methylomonas sp. DH-1 possesses a complete serine cycle [19]. This is a typical C1 assimilation pathway of the type II methanotrophs, and is not a common feature in type I methanotrophs. All of the genes involved in the serine cycle were identified in the genome of Methylomonas sp. DH-1. Interestingly, but not surprisingly, those genes were expressed when grown on methane or methanol (Fig. 1, Fig. 4c, and Additional file 4: Table S4). Among them, D-glycerate dehydrogenase (dgd) and glycerate kinase (gck) were expressed at rather low expression levels; in contrast, phosphopyruvate hydratase (eno) and malate dehydrogenase (mdh) were highly expressed. It was reported that a partial serine cycle without phosphoenolpyruvate carboxylase (ppc) in M. buryatense 5G was predicted to have a minor flux during growth on methane by steady state 13C metabolic flux analysis [33] and in silico simulation [17]. Conversely, the existence and expression in Methylomonas sp. DH-1 of phosphoenolpyruvate carboxylase (ppc), a key enzyme in the serine cycle that serves as a major entry point for CO2 in alpha-proteobacterial methanotrophs [34], may provide the capability for CO2 fixation and malate production. Notably, the expression of ppc was significantly downregulated on methanol compared to methane. There are two types of ppc involved in a serine cycle: a “non-regulated” type where enzymatic activity is not controlled by intermediates of the TCA cycle or the glycolysis pathway, and a “regulated” type where the enzymatic activity of ppc is subject to control by various metabolic effectors [30, 35]. An ortholog analysis suggests that ppc of Methylomonas sp. DH-1 belongs to the regulated group (Additional file 5: Figure S1). Transcription of ppc decreased dramatically (9.6-fold) on methanol compared to methane, possibly because ppc was subject to metabolic effectors that are better produced on methane. As noted above, the H4F pathway can function as a part of an assimilatory pathway (via serine) that converts formate to methylene H4F. In accordance with this, upregulation of the H4F pathway was observed in the transcriptome data for growth on methanol, and this in turn may have affected expression of the serine cycle (Fig. 2 and Additional file 4: Table S4). Replenishment of glyoxylate is an essential function of the serine cycle [30, 34, 36]. However, no homolog of isocitrate lyase or malate synthase in the glyoxylate shunt was found in Methylomonas sp. DH-1, similar to other obligate methanotrophs [16, 17]. In addition, an ethylmalonyl-CoA (EMC) cycle was not identified. Thus, Methylomonas sp. DH-1 appears to have a complete serine cycle for carbon conversion to acetyl-CoA and for CO2 fixation. Among the genes in the serine cycle, the key genes such as serine-glyoxylate aminotransferase (sga) and mdh was significantly upregulated, with a 2.1-fold and 1,7-fold change, respectivelly (Fig. 2). Especially, the malyl-coA lyase (mclA), which directly produce acetyl-CoA via serine cycle, has been upregulated with a 2,78-fold change on methanol-grown cells. That suggested an increase flux towards the serine cycle to contribute acetyl-CoA production during growth on methanol.
Typically, type I and type X methanotrophs differ from type II methanotrophs because the former groups possess an incomplete tricarboxylic acid (TCA) cycle and do not have 2-oxoglutarate dehydrogenase enzyme activity [33, 37, 38]. It has been suggested that the main function of the TCA cycle in methanotrophs is to provide precursors for de novo biomass synthesis, as opposed to providing reducing power to the system [30]. However, a recent study using 13C tracer analysis demonstrated that a complete oxidative TCA cycle operates in M. buryatense [33]. Similar to M. buryatense, our genome analysis indicate that Methylomonas sp. DH-1 encodes all of the essential genes for the TCA cycle, and these genes are expressed on both methane and methanol (Fig. 1, Fig. 4d, Additional file 4: Table S4). Most genes in the TCA cycle were expressed at a high or intermediate level under methane growth, except for fumarate hydratase (fh), which was expressed at a low level. The 2-oxoglutarate dehydrogenase complex, which plays a key role in the TCA cycle, also was expressed at a high level. In order to confirm whether Methylomonas sp. DH-1 operates a complete TCA cycle, the ability to convert 2-oxoglutarate to succinyl-CoA or succinate needed to be tested. A succinate dehydrogenase mutant was generated to confirm any 2-oxoglutarate dehydrogenase activity. Interestingly, the mutant strain showed no difference in its growth rate compared to the wild-type strain, and succinate was accumulated in the media (data not shown). This observation supports the hypothesis that Methylomonas sp. DH-1 possesses a complete oxidative TCA cycle. This finding might be useful for future metabolic engineering of the TCA cycle in Methylomonas sp. DH-1 to produce relevant products. During growth on methanol, the expression of key genes in the TCA cycle was significantly downregulated such as succinate-coA ligase (sucCD), succinate dehydrogenase (sdh) and isocitrate dehydrogenase (idh1) (Fig. 2, Additional file 4: Table S4) which suggested the decreasing flux towards TCA cycle under methanol growth.
Upregulation of carotenoid and hopanoid biosynthesis pathways under methanol growth
The ability to produce various carotenoids demonstrates another potential value for Methylomonas sp. DH-1 in industrial use. Methylomonas sp. DH-1 carries a complete MEP pathway for carotenoid production, with two copies of 1-deoxy-D-xylulose-5-phosphate synthase (AYM39_RS06125 and AYM39_RS06125) [19]. Transcriptional profiling of Methylomonas sp. DH-1 grown on methane indicated that most of the genes in the MEP pathway were expressed at an intermediate or low level, with the exception of 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (ispH), 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase (ispE), and 1-deoxy-D-xylulose-5-phosphate reductoisomerase (dxr), which were highly expressed (Fig. 5a and Additional file 6: Table S5). Among the genes in the MEP pathway, ispE showed the highest expression, and two homologs of dxs showed the lowest expression. Interestingly, a transcriptional comparison between the two carbon and energy sources indicated that many genes in the MEP pathway were significantly upregulated on methanol (Fig. 2). Among the upregulated genes in the MEP pathway, dxs showed particularly high upregulation on methanol: 2.5 and 3.1-fold changes were observed for dxs1 and dxs2, respectively. Another important gene in the carotenoid synthesis pathway, the squalene/phytoene synthase gene (sqs), showed a low expression level on methane. Surprisingly, this gene was strongly upregulated on methanol, with a 5.7-fold change. On the other hand, the association of squalene, carotenoid and hopanoid biosynthesis response to stress in bacteria were reported [39, 40]. Thus, we can postulate that methanol might serve as a stress factor that induces the expression of genes in the MEP pathway. Consistent with the transcriptome data, a carotenoid profiling analysis showed that carotenoid production increased on methanol in Methylomonas sp. DH-1 (data not shown).

Differential expression of genes involved in secondary metabolites: a carotenoid and MEP pathways, b hopanoid biosynthesis pathway in Methylomonas sp. DH-1 grown on methane (black) and methanol (gray). * Significantly different expression of genes between methane- and methanol-grown cultures (P < 0.05)
The carotenoid biosynthetic pathways share an upstream pathway of hopanoid biosynthesis [41]. Hopanoids are a group of natural pentacyclic compounds derived from the basic chemical structure of hopane. Studies in a range of bacteria suggest that hopanoids play a role in regulating membrane properties through an interaction with the outer leaflet of the outer membrane. From our genomic analysis, a complete hopanoid biosynthesis pathway is predicted in Methylomonas sp. DH-1. We predict the strain should therefore convert squalene to various hopanoids (including adenosylhopane, ribosylhopane, formylhopane, bacteriohopanetetrol), aminobacteriohopanetriol, and 3-methyl ABH) by hopanoid biosynthesis, using the associated radical S-adenosylmethionine (SAM) protein (hpnH), phosphorylase (hpnG), aminotransferase (hpnO), and hopanoid C-3 methylase (hpnR). Moreover, the existence of hopanoid biosynthesis-associated transporter (hpnN) and hopanoid biosynthesis protein (hpnM) in Methylomonas sp. DH-1 might enable the trafficking of hopanoids [42, 43]. Under methane growth, most of the genes in the hopanoid biosynthesis pathway were expressed at an intermediate level, except for hpnH and hpnR, which were expressed at a high level (Fig. 5b and Additional file 7: Table S6). Interestingly, when a carbon substrate was shifted from methane to methanol, the expression of hopanoid pathway genes increased significantly between 3.4-fold and 6.4-fold (Fig. 2, Fig. 5b, and Additional file 7: Table S6). Among them, hphH showed the highest upregulation, with a 6.4-fold change. It is tempting to speculate that such changes in gene expression in hopanoid biosynthesis pathway related to the ability to tolerate a high methanol concentration in DH-1 via modification of membrane properties.
Transcriptional responses to oxidative stress during growth on methanol
As described above, methanol changed transcriptional levels for the carotenoid and hopanoid biosynthesis pathways, which both provide antioxidants [44]. In addition, a pigmented carotenoid in Methylomonas exhibited high antioxidative activities [45]. Based on the changes in the carotenoid and hopanoid profiles, we hypothesized that methanol might induce oxidative stress in Methylomonas sp. DH-1. Thus, we further attempted to determine transcriptional responses to oxidative stress during growth on methanol. Since CH4 biocatalysis is oxygen dependent, using oxygen-enriched air is a potential strategy for getting high-density growth of aerobic methanotrophs, for maximizing volumetric production of bacterial biomass, and for recombinant protein production. Thus, determining the effect of oxidative stress on the physiology and growth of methanotrophs is necessary. First we examined expression of antioxidant defense systems using scavenging enzymes, such as superoxide dismutase (sod), peroxiredoxin (prdx), and catalase (cat) (Fig. 6a and Additional file 8: Table S7). Both manganese superoxide dismutase and iron superoxide dismutase, which catalyze superoxide radicals into hydrogen peroxide and oxygen, were identified. Cu-Zn superoxide dismutase was not identified. A very high expression level of the gene encoding superoxide dismutase was observed in both of the treatments, but slightly upregulated in methanol. Catalases (cat) that decompose hydrogen peroxide to water and oxygen were also present in the Methylomonas sp. DH-1 genome. Expression of catalase in methane was intermediate, and it was slightly decreased in methanol. Three copies of prdx were identified, and all of them were expressed (but not significantly different) under the two conditions. There were six copies of glutathione S-transferase, which has an antioxidant role [46], and one of them (AYM39_RS19665) was strongly upregulated (2.2-fold change) in methanol (Fig. 6a, Additional file 8: Table S7). Under stressful conditions, however, these enzymes may be insufficient to protect cells from reactive oxygen species (ROS). Two other regulatory defense systems in gram-negative bacteria are induced under oxidative stress conditions: the oxyR system [47] responds to hydrogen peroxide, and the soxR and soxS systems respond to redox-active compounds [48]. Recently, a systems biology approach to decode the oxyR, soxR, and soxS regulatory networks under oxidative stress was reported in E. coli K12 MG1655 [49]. Since the regulators of oxidative stress in methanotrophs remain unclear, we performed an ortholog analysis between E. coli K12 MG1655 and Methylomonas sp. DH-1 using InParanoid [21] to compare the expression changes of regulators and their regulons during culture in methane and methanol. Because the genome annotation of Methylomonas sp. DH-1 still contains many gaps, an ortholog comparison of the DH-1 proteome and an accurate annotation of model strain E. coli K12 MG1655 was deemed a suitable approach for finding corresponding genes between the two strains. Based on the ortholog calculations, oxyR and soxR, but not SoxS, were identified in DH-1; these regulators were expressed at an intermediate level (Fig. 6b and Additional file 8: Table S7). Another copy of oxyR was identified, and it was expressed at a low level. In the case of E. coli K12 MG1655, expression levels of oxyR and soxR were upregulated under the oxidative stress treatment [49]. The expression levels of these regulators in DH-1 were not significantly changed in the methanol cultures. Thus, the regulatory defense system against oxidative stress might be different in methanotrophs compared to E. coli. A total of 68 genes in 51 transcription units (TUs) belong to the oxyR, soxS, and soxR regulons, which have been characterized [49]. Based on those results and our ortholog calculations, we further analyzed the expression of the oxyR and soxRS regulons in Methylomonas sp. DH-1. Thirty genes belonging to the oxyR and soxRS regulons exist in the DH-1 genome. Among them, 16 genes showed expression changes in methanol-grown cultures (Additional file 8: Table S7). Next, we analyzed the functions of those regulons. Among the 16 genes belonging to the oxyR, soxR, and soxS regulons, expression of glucose 6-phosphate dehydrogenase (zwf) was increased 2.1-fold in methanol. It has been reported that oxidative stress induces metabolic responses, such as the activation of zwf by SoxS, to increase NADPH pools and promote antioxidant defense by mediating the reduction of thioredoxins and glutaredoxins [50, 51]. Overexpression of amino acid biosynthesis genes as a means to overcome oxidative stress also has been reported [49]. The expression of 2-dehydro-3-deoxyphosphoheptonate aldolase (aroF), which promotes the synthesis of aromatic amino acids in Methylomonas sp. DH-1, increased possibly to overcome the shortage of essential amino acids. Other genes characterizing the cellular response to oxidative stress and damage repair, such as those involved in iron-sulfur (FeS) clusters, were highly overexpressed in methanol. Methanol likely induced oxidative stress in Methylomonas sp. DH-1 by activating a series of key enzymes in the damage repair and protection pathway, allowing cells to activate robust defenses against oxidative stress.

Differential expression of genes involved in the response to oxidative stress: a primary defense systems with scavenging enzymes, b oxyR, oxyR2, soxR, and their regulon genes in methane (black) and methanol (gray). * Significantly different expression of genes between methane- and methanol-grown cultures (P < 0.05)
High expression of endogenous plasmid genes plays a role in recombination
Endogenous plasmids are present in several methanotroph genomes [13, 19, 52]. The loss of native plasmid in M. buryatense 5G allowed the variant strain to receive small incompatibility group P (IncP) plasmids, which are broad host range vectors, via conjugation [52]. The native plasmid in M. buryatense 5G likely carries no genes essential for growth, since curing of that plasmid did not affect the growth phenotype [52]. Methylomonas sp. DH-1 contains an endogenous plasmid (pDH1) of 277 kb. In classifying the plasmid genes based on the clusters of orthologous groups (COGs) protein database, we found that replication, recombination, repair (category L), and transcription (category K) were overrepresented. Attempts to cure strains of this plasmid have failed (unpublished report) because it likely plays a significant role in the metabolism of DH-1. Conjugation to introduce IncP-based broad host range vectors (for expressing foreign genes) was not successful. This may have been due to the natural restriction-methylation barrier system that cleaves transforming plasmids before they can replicate in the cell. It is equally possible that incompatibility between foreign plasmid and endogenous plasmid caused stability and maintenance issues. The transcriptomic profile of Methylomonas sp. DH-1 showed that most of the genes in the pDH1 endogenous plasmid had very high or high expression levels (Table 1, Additional file 9: Table S8). Expression of the origin of replication in the plasmid (DnaC) was very high, in fact 10-fold higher than expression of the origin of replication in the main chromosome (DnaA). Although the exact copy number of the endogenous plasmid in DH-1 has not yet been quantified, the high expression of its origin of replication protein might reflect a high plasmid copy number. Thus, integrating foreign DNA cassettes into the endogenous plasmid may be an effective way to express heterologous genes in this strain. From a total of 242 protein-coding genes, 105 had significant expression changes. Among them, 46 genes and 59 genes were downregulated and upregulated in methanol-grown cultures, respectively (Additional file 9: Table S8).
Discussion
In order to provide knowledge for methanotroph-based refineries, multi-omics can be used to define which metabolic pathways are active in certain conditions, and how cells response and adapt to new environments. In our previous work, the complete genome sequence of the newly isolated methanotroph Methylomonas sp. DH-1 was reported [19]. In the present study, a comprehensive characterization of the complete transcriptome of Methylomonas sp. DH-1 was provided and analyzed for the first time by an RNA-seq approach. This study provides in-depth knowledge about the metabolic pathways of this strain and reveals key differences in the transcriptional responses for certain metabolic pathways during growth in methane and methanol.
In the well-characterized methanotrophs, pmo is expressed at the highest level for cultures grown on methane [14,15,16,17]. A previous study has determined that transcripts of pmoA are very stable, with a half-life in the range of hours to days [53] which supported the hypothesis that the higher expression levels of pmo compared to other enzymes in the C1 oxidation pathway led to the first step of oxidizing methane is relatively slower compared to subsequent steps. During growth on methanol, the pmo operon was dramatically downregulated, likely because pmo genes are not involved in oxidizing methane. This is consistent with our previous study in which MMO activity has been dropped more than 3-fold when DH-1 growth on methanol [11]. Methane therefore may be a key regulator for the expression of the pmo operon. Similar to that of M. trichosporium OB3b, a type II model methanotroph, the expression of pmo and smo strongly affected by selection of different substrates [54]. The expression level of pmo and smo and their activity extremely dropped when the growth was shifted from methane to methanol [54].
xoxF, Ln3+ − dependent methanol dehydrogenase, has an important enzyme in methylotrophy, providing a new outlook on the distribution of methylotrophy in the bacterial community [55]. Interestingly, xoxF showed highly expression level without presence of Ln3+ and the similar expression pattern of xoxF and pmo supported the assumption that xoxF could contribute to the methane oxidation process in Methylomonas sp. DH-1. In agreement with our hypothesis, in most recent, the structure and function of xoxF in M. buryatense 5GB1C has been reported by investigating the possibility of interaction between pMMO and XoxF [56]. The results indicated a XoxF monomer may bind to pMMO and suggested an alternative structure of MDH-pMMO association. On the other hand, M. trichosporium OB3b showed very low expression level of xoxF1 and xoxF2 in methane and methanol [54]. Furthermore, the expression level of xoxF1, xoxF2 as well as mxaF in M. trichosporium OB3b were decreased when grown on methanol with the presence of 10 μM copper, highlighting the differences in gene expression regulation in response to the type of carbon sources available between Methylomonas sp. DH-1 and M. trichosporium OB3b. It should be noted that while M. trichosporium OB3b exhibited the “copper-switch” to control the expression of alternative forms of methane monooxygenase, the “copper-switch” was not exist in Methylomonas sp. DH-1.
The discovery of typical type II methanotrophs metabolic pathways, such as the H4MPT pathway, H4F pathway, and complete serine cycle, in Methylomonas sp. DH-1 raised questions about the roles of these pathways in the central metabolism of this strain. From a previously published genome-scale model of M. buryatense 5GB1, a minor carbon flux is predicted via the H4MPT and H4F pathways [17, 18, 57]. However, these pathways were more active during growth on methanol, suggesting the improvement of carbon flux towards these pathway. This observation supports our hypothesis that the H4MPT and H4F pathways are mainly responsible for formaldehyde oxidation and contribute to carbon conversion via the serine cycle when grown on methanol.
A partial serine cycle without ppc has been determined in various type I methanotrophs such as M. buryatense 5GB1 and M. alcaliphilum 20ZR which contributed a minor flux during growth in methane [17, 18]. Likewise, the complete gene set implementing the serine cycle in Methylomonas sp. DH-1 should allow the minor carbon flux needed to produce acetyl-coA. In the type II methanotroph M. trichosporium OB3b, which typically uses the serine cycle as a main pathway for C1 assimilation, there are two kinds of ppc gene: ppc1 belongs to the non-regulated group and ppc2 belongs to the regulated group [14]. The existence of two functionally identical but different regulation systems in M. trichosporium OB3b allows control of flux through phosphoenolpyruvate-oxaloacetate in response to the serine cycle, and this flux is never blocked completely [14]. The presence of only regulated ppc in Methylomonas sp. DH-1 indicates that carbon flux through the serine cycle can be blocked in the absence of effectors. During culture on methanol, expression of ppc was strongly downregulated, possibly because metabolite effectors which activate ppc expression were absent. The growth rate of Methylomonas sp. DH-1 in methanol was significantly decreased, perhaps because carbon flux via the serine cycle may have been blocked under methanol growth. However, most of the genes in the serine cycle were upregulated in methanol, suggesting significant shifts occur in C1 assimilation pathways, from RuMP to serine cycle. Along with RuMP cycle, the serine cycle also could take the role of producing acetyl-coA. EMP is main variant of RuMP pathway which play major role for C1 assimilation to produce NADH and ATP in type I methanotrophs [16,17,18]. The shifts decrease flux towards EMP pathway which subsequently decrease ATP production. Instead, the available electrons from methanol oxidation, which not used for methane oxidation under methanol growth, are transferred to the electron transport chain follow by producing ATP via oxidative phosphorylation. In order to determine the detailed rearrangement of metabolic network involved methanol-grown, 13C tracer analysis and constraint-based analysis of genome-scale metabolic network studies are needed. Thus, even the exist of the complete serine cycle in Methylomonas sp. DH-1 could not be main pathway for C1 assimilation, it could contribute to the control of carbon flux when shifting carbon substrates.
One unsolved question surrounding the central metabolism of type I methanotrophs is whether the oxidative TCA cycle is complete. In the recent time, a complete oxidative TCA cycle has been demonstrated to operate in M. buryatense 5GB1, and it has showed three separate pathways for converting 2-oxoglutarate to succinyl-CoA [33]. In another study, highly branched TCA cycle at the 2-oxoglutarate node also has been reported in M. alcaliphilum 20ZR [18]. In this study, we also suggested Methylomonas sp. DH-1 possesses an complete oxidative TCA cycle. However, genomic analysis indicated at 2-oxoglutarate node, Methylomonas sp. DH-1 possesses 2-oxoglutarate dehydrogenase complex only but not 2-oxoglutarate ferredoxin oxidoreductase, succinate semialdehyde dehydrogenase or 2-oxoglutarate decarboxylase. Thus, the presence of highly branched TCA cycle in DH-1 remains to be elucidate. In addition, it seems that carbon flux though TCA cycle was reduced on methanol growth and the critical function of TCA under methanol growth has changed. In methanol-grown cells, TCA cycle mostly provide precursors for de novo synthesis but not reducing power such as NADH. Instead, it appears that the activation of formaldehyde oxidation in methanol growth could produce NADH.
In our previous study, the carotenoid biosynthesis pathways which derived from MEP pathway has been proposed [19]. The dxs is the first and one of the most important rate-limiting step in the MEP pathway, and overexpression of dxs could improve the production of several downstream secondary metabolites such as isoprenoid and carotenoid [58,59,60,61]. The flux shift occurred to MEP pathway via the strong upregulation of two dxs homologs (dxs1 and dxs2) led to the accumulation of carotenoids in methanol-grown cultures. Meanwhile, the extremely upregulation of hopanoid biosynthesis pathway might related to membrane modifications under methanol growth (Fig. 2). The function of hopanoids has been characterized in several organisms, including methylotrophic bacteria [62, 63]. A lack of hopanoid biosynthesis increases sensitivity against toxins and osmotic stress. During growth on single-carbon compounds, methanol is generally converted to formaldehyde in the periplasm, and the formaldehyde is then transported and utilized in the cytoplasm. Given the toxic intermediates in this process, elevated maintenance of the inner and outer membranes is necessary. The role of hopanoids in maintaining membrane robustness and membrane barrier function is likely conserved across bacterial lineages. This function is possibly mediated through an interaction with lipid A in the outer membrane of Methylobacterium extorquens DM4 [63]. In addition, membrane function in the hopanoid-free Methylobacterium extorquens PA1 was lower [62]. Further investigation on the function of hopanoid biosynthesis pathway in property membranes of Methylomonas sp. DH-1 is needed to solve the question if hopanoid biosynthesis pathway could enable resistance to high methanol concentrations in Methylomonas sp. DH-1. Under methanol growth, the upregulation of carotenoid biosynthesis pathway, which produced pigmented carotenoid as antioxidant, and many regulatory defense systems against oxidative stress via damage repair and protection systems have been observed. It is speculated that such changes of these gene expression were induced by methanol which might induces ROS in Methylomonas sp. DH-1. A high expression of MEP pathway genes and an accumulation of carotenoids under stress conditions also describe previously reported in Haematococcus pluvialis [64]. That such speculation must be more rigorously confirmed by apply a system biology approach to reconstruct genome-wide of OxyR, SoxR, and SoxS regulatory networks under oxidative stress condition in methanotrophs.
Conclusions
In conclusion, we have presented genomic and transcriptomic analyses of an industrially promising obligate methanotroph, Methylomonas sp. DH-1. The strain was grown on methane and methanol to analyze the shift of metabolism affecting by selection of substrates (Figs. 1, 2). While some metabolic functions had been reported in previous studies, several novel functions were identified and characterized in this strain. Methylomonas sp. DH-1 possesses the active EMP pathway which main route for C1 assimilation in this strain. In addition, Methylomonas sp. DH-1 also operates a complete oxidative TCA cycle. Along with the existence complete serine cycle, these pathways may function in C1 assimilation and energy production. We also identified a flux shift of metabolism towards formaldehyde oxidation pathway, serine and TCA cycle in Methylomonas sp. DH-1 when substrate was changed from methane and methanol. Furthermore, a significant upregulation of carotenoid and hopanoid biosynthesis pathways under methanol growth might explain the resistance to high methanol concentrations observed in Methylomonas sp. DH-1. It appears that methanotrophs are very dynamic to respond to change of environmental parameters.
Abbreviations
- BAM:
-
Binary alignment/map
- BHT:
-
Bacteriohopanetetrol
- C1:
-
Single-carbon
- CDS:
-
Coding DNA sequence
- COG:
-
Clusters of orthologous genes
- DMAPP:
-
Dimethylallyl diphosphate
- EDD:
-
Entner–Doudoroff
- EMC:
-
Ethylmalonyl-CoA
- EMP:
-
Embden–Meyerhof–Parnas
- FDR:
-
False discovery rate
- FeS:
-
Iron-sulfur
- FPKM:
-
Fragments per kilobase of exon per million fragments
- H4F:
-
Tetrahydrofolate
- H4MPT:
-
Tetrahydromethanopterin
- IPP:
-
Isopentenyl diphosphate
- MDH:
-
Methanol dehydrogenase
- MEP:
-
2-C-methyl-D-erythritol 4-phosphate
- MVA:
-
Mevalonatic acid
- PP:
-
Pentose phosphate
- PQQ:
-
Pyrroloquinoline quinone
- ROS:
-
Reactive oxygen species
- RuMP:
-
Ribulose monophosphate
- SAM:
-
S-adenosylmethionine
- SAM:
-
Sequence alignment/map
- TAC:
-
Tricarboxylic acid
- TU:
-
Transcription unit
References
Dürre P, Eikmanns BJ. C1-carbon sources for chemical and fuel production by microbial gas fermentation. Curr Opin Biotechnol. 2015;35:63–72.
Clomburg JM, Crumbley AM, Gonzalez R. Industrial biomanufacturing: the future of chemical production. Science. 2017;355:aag0804.
Hwang IY, Hur DH, Lee JH, Park CH, Chang IS, Lee JW, Lee EY. Batch conversion of methane to methanol using Methylosinus trichosporium OB3b as biocatalyst. J Microbiol Biotechnol. 2015;25:375–80.
Haynes CA, Gonzalez R. Rethinking biological activation of methane and conversion to liquid fuels. Nat Chem Biol. 2014;10:331–9.
Schrader J, Schilling M, Holtmann D, Sell D, Villela Filho M, Marx A, et al. Methanol-based industrial biotechnology: current status and future perspectives of methylotrophic bacteria. Trends Biotechnol. 2009;27:107–15.
Khmelenina VN, Rozova ON, But SY, Mustakhimov II, Reshetnikov AS, Beschastnyi AP, et al. Biosynthesis of secondary metabolites in methanotrophs: biochemical and genetic aspects. Appl Biochem Microbiol. 2015;51:150.
Hwang IY, Nguyen AD, Nguyen TT, Nguyen LT, Lee OK, Lee EY. Biological conversion of methane to chemicals and fuels: technical challenges and issues. Appl Microbiol Biotechnol. 2018;102(7):3071–80.
Lee OK, Hur DH, Nguyen DT, Lee EY. Metabolic engineering of methanotrophs and its application to production of chemicals and biofuels from methane. Biofuels Bioprod Biorefin. 2016;10:848–63.
Nguyen AD, Hwang IY, Lee OK, Kim D, Kalyuzhnaya MG, Mariyana R, Hadiyati S, Kim MS, Lee EY. Systematic metabolic engineering of Methylomicrobium alcaliphilum 20Z for 2, 3-butanediol production from methane. Metab Eng. 2018;47:323–33.
Hur DH, Na JG, Lee EY. Highly efficient bioconversion of methane to methanol using a novel type I Methylomonas sp. DH-1 newly isolated from brewery waste sludge. J Chem Technol Biotechnol. 2016;92:311–8.
Hur DH, Nguyen TT, Kim D, Lee EY. Selective bio-oxidation of propane to acetone using methane-oxidizing Methylomonas sp. DH-1. J Ind Microbiol Biotechnol. 2017;44:1097–105.
Boden R, Cunliffe M, Scanlan J, Moussard H, Kits KD, Klotz MG, et al. Complete genome sequence of the aerobic marine methanotroph Methylomonas methanica MC09. J Bacteriol. 2011;193:7001–2.
Vuilleumier S, Khmelenina VN, Bringel F, Reshetnikov AS, Lajus A, Mangenot S, et al. Genome sequence of the haloalkaliphilic methanotrophic bacterium Methylomicrobium alcaliphilum 20Z. J Bacteriol. 2012;194:551–2.
Matsen JB, Yang S, Stein LY, Beck D, Kalyuzhnaya MG. Global molecular analyses of methane metabolism in methanotrophic alphaproteobacterium, Methylosinus trichosporium OB3b. Part I: transcriptomic study. Front Microbiol. 2013;4:40.
Vorobev A, Jagadevan S, Jain S, Anantharaman K, Dick GJ, Vuilleumier S, et al. Genomic and transcriptomic analyses of the facultative methanotroph Methylocystis sp. strain SB2 grown on methane or ethanol. Appl Environ Microbiol. 2014;80:3044–52.
Kalyuzhnaya MG, Yang S, Rozova ON, Smalley NE, Clubb J, Lamb A, et al. Highly efficient methane biocatalysis revealed in a methanotrophic bacterium. Nat Commun. 2013;4:2785.
Torre A, Metivier A, Chu F, Laurens LM, Beck DA, Pienkos PT, et al. Genome-scale metabolic reconstructions and theoretical investigation of methane conversion in Methylomicrobium buryatense strain 5G (B1). Microb Cell Factories. 2015;14:188.
Akberdin IR, Thompson M, Hamilton R, Desai N, Alexander D, Henard CA, Guarnieri MT, Kalyuzhnaya MG. Methane utilization in Methylomicrobium alcaliphilum 20Z R: a systems approach. Sci Rep. 2018;8(1):2512.
Nguyen AD, Hwang IY, Lee OK, Hur DH, Jeon YC, Hadiyati S, Kim M, Yoon SH, Jeong H, Lee EY. Functional analysis of Methylomonas sp. DH-1 genome as a promising biocatalyst for bioconversion of methane to valuable chemicals. Catalysts. 2018;8(3):117.
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, et al. The COG database: an updated version includes eukaryotes. BMC Bioinform. 2003;4(1):41.
Sonnhammer EL, Östlund G. InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 2015;43:D234–9.
Haas BJ, Chin M, Nusbaum C, Birren BW, Livny J. How deep is deep enough for RNA-Seq profiling of bacterial transcriptomes? BMC Genomics. 2012;13:734.
Hartmann E, Hartmann RK. The enigma of ribonuclease P evolution. Trends Genet. 2003;19:561–9.
Cavanagh AT, Wassarman KM. 6S RNA, a global regulator of transcription in Escherichia coli, Bacillus subtilis, and beyond. Annu Rev Microbiol. 2014;68:45–60.
Tavormina PL, Orphan VJ, Kalyuzhnaya MG, Jetten MSM, Klotz MG. A novel family of functional operons encoding methane/ammoni monooxygenase-related proteins in gammaproteobacterial methanotrophs. Environ Microbiol Rep. 2011;3:91–100.
Skovran E, Palmer AD, Rountree AM, Good NM, Lidstrom ME. XoxF is required for expression of methanol dehydrogenase in Methylobacterium extorquens AM1. J Bacteriol. 2011;193:6032–8.
Chu F, Lidstrom ME. XoxF acts as the predominant methanol dehydrogenase in the type I methanotroph Methylomicrobium buryatense. J Bacteriol. 2016;198:1317–25.
Vorholt JA, Marx CJ, Lidstrom ME, Thauer RK. Novel formaldehyde-activating enzyme in Methylobacterium extorquens AM1 required for growth on methanol. J Bacteriol. 2000;182:6645–50.
Crowther GJ, Kosály G, Lidstrom ME. Formate as the main branch point for methylotrophic metabolism in Methylobacterium extorquens AM1. J Bacteriol. 2008;190:5057–62.
Anthony C. The biochemistry of methylotrophs. London: Academic press. 1982;17:254.
Hanson RS, Hanson TE. Methanotrophic bacteria. Microbiol Rev. 1996;60(2):439–71.
Eshinimaev BT, Khmelenina VN, Sakharovskii VG, Suzina NE, Trotsenko YA. Physiological, biochemical, and cytological characteristics of a haloalkalitolerant methanotroph grown on methanol. Microbiology. 2002;71:512–8.
Fu Y, Li Y, Lidstrom M. The oxidative TCA cycle operates during methanotrophic growth of the type I methanotroph Methylomicrobium buryatense 5GB1. Metab Eng. 2017;42:43–51.
Shishkina VN, Trotsenko YA. Multiple enzymic lesions in obligate methanotrophic bacteria. FEMS Microbiol Lett. 1982;13:237–42.
Kai Y, Matsumura H, Izui K. Phosphoenolpyruvate carboxylase: three-dimensional structure and molecular mechanisms. Arch Biochem Biophys. 2003;414:170–9.
Peyraud R, Kiefer P, Christen P, Massou S, Portais JC, Vorholt JA. Demonstration of the ethylmalonyl-CoA pathway by using 13C metabolomics. Proc Natl Acad Sci. 2009;106:4846–51.
Kaluzhnaya M, Khmelenina V, Eshinimaev B, Suzina N, Nikitin D, Solonin A, et al. Taxonomic characterization of new alkaliphilic and alkalitolerant methanotrophs from soda lakes of the southeastern Transbaikal region and description of Methylomicrobium buryatense sp. nov. Syst Appl Microbiol. 2001;24:166–76.
Davey JF, Whittenbury R, Wilkinson JF. The distribution in the methylobacteria of some key enzymes concerned with intermediary metabolism. Arch Mikrobiol. 1972;87:359–66.
Bosak T, Losick RM, Pearson A. A polycyclic terpenoid that alleviates oxidative stress. Proc Natl Acad Sci. 2008;105(18):6725–9.
Welander PV, Hunter RC, Zhang L, Sessions AL, Summons RE, Newman DK. Hopanoids play a role in membrane integrity and pH homeostasis in Rhodopseudomonas palustris TIE-1. J Bacteriol. 2009;191(19):6145–56.
Sohlenkamp C, Geiger O. Bacterial membrane lipids: diversity in structures and pathways. FEMS Microbiol Rev. 2016;40:133–59.
Doughty DM, Coleman ML, Hunter RC, Sessions AL, Summons RE, Newman DK. The RND-family transporter, HpnN, is required for hopanoid localization to the outer membrane of Rhodopseudomonas palustris TIE-1. Proc Natl Acad Sci. 2011;108:E1045–51.
Schmerk CL, Welander PV, Hamad MA, Bain KL, Bernards MA, Summons RE, et al. Elucidation of the Burkholderia cenocepacia hopanoid biosynthesis pathway uncovers functions for conserved proteins in hopanoid-producing bacteria. Environ Microbiol. 2015;17:735–50.
Fiedor J, Burda K. Potential role of carotenoids as antioxidants in human health and disease. Nutrients. 2014;6:466–88.
Guo W, Li D, He R, Wu M, Chen W, Gao F, Zhang Z, Yao Y, Yu L, Chen S. Synthesizing value-added products from methane by a new Methylomonas. J Appl Microbiol. 2017;123(5):1214–27.
Veal EA, Toone WM, Jones N, Morgan BA. Distinct roles for glutathione S-transferases in the oxidative stress response in Schizosaccharomyces pombe. J Biol Chem. 2002;277:35523–31.
Zheng M, Åslund F, Storz G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science. 1998;279:1718–22.
Nunoshiba T, Hidalgo E, Cuevas CA, Demple B. Two-stage control of an oxidative stress regulon: the Escherichia coli SoxR protein triggers redox-inducible expression of the soxS regulatory gene. J Bacteriol. 1992;174:6054–60.
Seo SW, Kim D, Szubin R, Palsson BO. Genome-wide reconstruction of OxyR and SoxRS transcriptional regulatory networks under oxidative stress in Escherichia coli K-12 MG1655. Cell Rep. 2015;12:1289–99.
Henard CA, Bourret TJ, Song M, Vázquez-Torres A. Control of redox balance by the stringent response regulatory protein promotes antioxidant defenses of salmonella. J Biol Chem. 2010;285:36785–9.
Prinz WA, Åslund F, Holmgren A, Beckwith J. The role of the Thioredoxin and Glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J Biol Chem. 1997;272:15661–7.
Puri AW, Owen S, Chu F, Chavkin T, Beck DA, Kalyuzhnaya MG, et al. Genetic tools for the industrially promising methanotroph Methylomicrobium buryatense. Appl Environ Microbiol. 2015;81:1775–81.
Wendeberg A, Zielinski FU, Borowski C, Dubilier N. Expression patterns of mRNAs for methanotrophy and thiotrophy in symbionts of the hydrothermal vent mussel Bathymodiolus puteoserpentis. ISME J. 2012;6:104–12.
Haque MFU, Gu W, Baral BS, DiSpirito AA, Semrau JD. Carbon source regulation of gene expression in Methylosinus trichosporium OB3b. Appl Microbiol Biotechnol. 2017;101(9):3871–9.
Chistoserdova L, Kalyuzhnaya MG. Current trends in Methylotrophy. Trends Microbiol. 2018.
Deng YW, Ro SY, Rosenzweig AC. Structure and function of the lanthanide-dependent methanol dehydrogenase XoxF from the methanotroph Methylomicrobium buryatense 5GB1C. J Biol Inorg Chem. 2018:1–11.
Demidenko A, Akberdin IR, Allemann M, Allen EE, Kalyuzhnaya MG. Fatty acid biosynthesis pathways in Methylomicrobium buryatense 5G (B1). Front Microbiol. 2016;7.
Yang J, Guo L. Biosynthesis of β-carotene in engineered E. Coli using the MEP and MVA pathways. Microb Cell Factories. 2014;13:160.
Estévez JM, Cantero A, Reindl A, Reichler S, León P. 1-deoxy-D-xylulose-5-phosphate synthase, a limiting enzyme for plastidic isoprenoid biosynthesis in plants. J Biol Chem. 2001;276:22901–9.
Kim SJ, Kim MD, Choi JH, Kim SY, Ryu YW, Seo JH. Amplification of 1-deoxy-D-xyluose 5-phosphate (DXP) synthase level increases coenzyme Q10 production in recombinant Escherichia coli. Appl Microbiol Biotechnol. 2006;72:982–5.
Zhao Y, Yang J, Qin B, Li Y, Sun Y, Su S, et al. Biosynthesis of isoprene in Escherichia coli via methylerythritol phosphate (MEP) pathway. Appl Microbiol Biotechnol. 2011;90:1915.
Sáenz JP, Grosser D, Bradley AS, Lagny TJ, Lavrynenko O, Broda M, et al. Hopanoids as functional analogues of cholesterol in bacterial membranes. Proc Natl Acad Sci. 2015;112:11971–6.
Bradley AS, Swanson PK, Muller EE, Bringel F, Caroll SM, Pearson A, et al. Hopanoid-free Methylobacterium extorquens DM4 overproduces carotenoids and has widespread growth impairment. PLoS One. 2017;12:e0173323.
Liang C, Zhang W, Zhang X, Fan X, Xu D, Ye N, et al. Isolation and expression analyses of methyl-D-erythritol 4-phosphate (MEP) pathway genes from Haematococcus pluvialis. J Appl Phycol. 2016;28:209–18.
Acknowledgements
We appreciate Dr. Ok Kyung Lee, In Yeub Hwang, Dong Hoon Hur and Young Chan Jeon for their invaluable discussion.
Funding
This research was supported by the C1 Gas Refinery Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT (2015M3D3A1A01064882). This work was also supported by the 2018 Research Fund (1.180074.01) of UNIST (Ulsan National Institute of Science and Technology). The funding bodies themselves did not participate in the design of the study or the collection, analysis, and interpretation of data, or in writing the manuscript.
Availability of data and materials
The data generated and analyzed in this study are included in this article, its supplementary information files and on the GEO repository with accession number of GSE101494.
Author information
Authors and Affiliations
Contributions
ADN carried out the experimental procedures, performed a complete analysis, and prepared a draft of the manuscript. DK performed the read mapping (from the RNA-seq data to the reference sequences) and the differential expression analysis, and revised the manuscript. EYL coordinated the study and finalized the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Ten genes most highly expressed in Methylomonas sp. DH-1 in methane. (XLSX 12 kb)
Additional file 2:
Table S2. Differential expression of genes involved in methane, methanol and formaldehyde oxidation in methane and methanol. (XLSX 14 kb)
Additional file 3:
Table S3. Differential expression of genes involved in RuMP cycle, EMP and EDD pathway in methane and methanol. (XLSX 14 kb)
Additional file 4:
Table S4. Differential expression of genes involved in serine and TCA cycle in methane and methanol. (XLSX 13 kb)
Additional file 5:
Figure S1. Phylogenetic tree of phosphoenolpyruvate carboxylases. Sequence identifiers follow by species label. Node in red indicated “non-regulated” type and node in green indicated “regulated” type of ppc. Sequences were aligned and tree was created with ClustalX2 2.1 and rendered with iTOL. (DOCX 146 kb)
Additional file 6:
Table S5. Differential expression of genes involved in MEP pathway and carotenoid biosynthesis pathway in methane and methanol. (XLSX 12 kb)
Additional file 7:
Table S6. Differential expression of genes involved in hopanoid biosynthesis pathway in methane and methanol. (XLSX 11 kb)
Additional file 8:
Table S7. Differential expression of genes involved in responding to the oxidative stress in methane and methanol. (XLSX 17 kb)
Additional file 9:
Table S8. Differential expression of genes in pDH1 plasmid in methane and methanol. (XLSX 30 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nguyen, A.D., Kim, D. & Lee, E.Y. A comparative transcriptome analysis of the novel obligate methanotroph Methylomonas sp. DH-1 reveals key differences in transcriptional responses in C1 and secondary metabolite pathways during growth on methane and methanol. BMC Genomics 20, 130 (2019). https://doi.org/10.1186/s12864-019-5487-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5487-6