
Abstract
Background
Wet meadows, a type of wetland, are impacted by significant climate fluctuation and human activity, impacting soil microorganisms that play an essential role in ecosystem processes. Understanding the underlying ecological mechanisms and processes of wet meadows depends on the fungal communities associated with the plant roots. We used Illumina MiSeq profiling for amplicon sequencing to determine how environmental factors and elevation affect the fungal communities of the rhizosphere and rhizoplane related to three plant species, Cremanthodium ellisii, Cremanthodium lineare, and Caltha scaposa, in alpine wet meadows.
Results
The phyla Ascomycota and Basidiomycota dominated the rhizosphere (54.5% and 20.9%) and rhizoplane (51.6% and 36.4%), while the predominant fungal genera in the rhizosphere and rhizoplane were Unclassified fungi, Unclassified Ascomycota, Pseudeurotium, Tetracladium, Vishniacozyma, Rhodotorula, Cadophora, and Penicillium. Mantel test and network analysis revealed that the soil water content (SWC), soil organic carbon (SOC), and total nitrogen (TN) were the primary drivers of fungal communities. However, the influence of microbial biomass C (MBC), pH, microbial biomass N (MBN), and elevation varied. Stochastic assembly processes were dominant in both rhizosphere and rhizoplane fungal communities. FUNGuild functional prediction revealed site-specific variation in the trophic level and guild of plant-root-associated fungal communities. The rhizosphere contained 58.5% saprotrophs, 11.7% pathotrophs, and 12.6% symbiotrophs. In addition, 60.4% of the observed OTUs were arbuscular mycorrhizae, 13.2% were endophytes, 20.9% were ectomycorrhizae, and 1.09% were orchid mycorrhizae. The rhizoplane comprised 51.3% of OTUs linked with saprotrophs, 13.9% with pathotrophs, and 7.92% with symbiotrophs. Moreover, 36.1% of OTUs represented arbuscular mycorrhizae, 25.0% were endophytes, 30.6% were ectomycorrhizae, and 2.77% were ascribed to orchid mycorrhizae in the rhizoplane. The abundance of saprotrophs and pathotrophs in the rhizosphere was highest in C. ellisii at SI and SIII, while symbiotrophs were highest in C. lineare at SIII. Similar variations among the plant species and sites were observed in the fungal functional groups (guilds).
Conclusions
It was concluded that although root compartments significantly influenced the fungal communities in the rhizosphere and rhizoplane, environmental factors and plant types exhibited distinct effects. This study explains how physicochemical properties, plant species, and sites can alter the overall structure and functional repertoire of fungal communities in alpine wet meadows.
Graphical Abstract

Similar content being viewed by others


Background
The global concern surrounding the loss of biodiversity has escalated due to the accumulating evidence indicating the detrimental effects of these losses on various components of ecosystems. The decline in soil biodiversity can impede various ecosystem characteristics, such as above-ground plant diversity, nutrient cycling, and nutrient retention [1]. Soil microorganisms are distributed throughout various soil profiles, from the surface horizon enriched with organic matter to the deeper horizons that harbor distinct microorganism communities [2]. Researchers have found that the microbial communities in lower soils vastly differ from those on the surface. These differences are essential for soil formation, ecosystem biogeochemistry, and the breakdown of pollutants [3]. Fungi and bacteria are the salient organisms in the soil microbiota and are involved in the flow of energy and the recycling of nutrients in any environment [4].
Wetlands cover less than 9% of the Earth's geographical area [5] and are critical natural resources [6]. The Ramsar Convention broadly defined wetlands, which included all rivers and lakes, subterranean swamps, marshes, aquifers, wet grasslands, oases, peatlands, tidal flats, estuaries and deltas, mangroves and other littoral habitats, coral reefs, and all artificial sites including rice paddies, salt pans, and reservoirs [7]. The natural wetlands on the Qinghai−Tibetan Plateau (QTP) are estimated to cover 13.36 km2 [8]. Approximately half of the alpine wetlands comprise of wet meadows, typically found on the western periphery of the plateau. Conversely, Marshlands constitute approximately 6% of the alpine wetlands and are predominantly located along the eastern edge of the plateau [9]. Wet meadows and marshlands are becoming more accessible to grazing animals because of climate change and human-caused ditch drainage. As a result, overgrazing and livestock trampling are damaging flora and soil structures in this vulnerable area. These disturbances are linked to a decreased soil organic carbon (SOC), resulting in degradation and substantial wetland loss [10, 11].
Fungi, which are filamentous eukaryotic microorganisms, often constitute a significant proportion of the overall microbial biomass found in soil [12]. Furthermore, fungi play a significant role in compound transformation as they can effectively decompose resilient organic substances; thus, providing plants with essential nutrients [13]. The effects of environmental filtering on ecological function are the best understood by studying fungal nutrient acquisition, especially for species in mutualism with plant hosts. Mycorrhizae are a type of symbiotic fungi (sometimes called mutualists) that form relationships with plant roots in return for resources such as N, C, and P obtained from the soil [14]. Mycorrhizae are very important to the environment because they supply nearly half of a tree's organic N and most of the new carbon that goes into the soil [15]. Moreover, ectomycorrhizal fungi and arbuscular mycorrhizal (mycorrhizal groups) vary in the input of N and P to their hosts [16], and in their extent to C, they contribute to the soil [17].
Wetlands are characterized by the presence of macrophytes, which are predominantly vascular plants [18]. The plant-root system enhances the effectiveness of wetlands by creating distinct microhabitats for soil microorganisms [19], which support the cycling of nutrients [20]. The immediate area of soil around a plant’s root system is known as the rhizosphere, and the area where the root surface is in touch with the soil directly and forms the inner boundary of the rhizosphere is known as the rhizoplane [21, 22]. Changes in the oxygen, soil pH, C/N, and water levels of the environment caused by plants change the rhizosphere and rhizoplane ecosystems [23, 24]. The variety and complexity of microorganisms in the rhizosphere and rhizoplane are critical for sustaining equilibrium in the soil ecosystem [23, 25, 26].
The composition of soil fungi is influenced by biotic factors, such as the plant community and abiotic factors, including nutritional substrates, soil microclimate, and physicochemical properties [27,28,29]. Therefore, the presence of different plant groups directly impacts soil fungi, as it alters the quantity and composition of plant litter within the soil, including factors such as the C:N ratio found in both litter and roots [29]. Soil physicochemical properties, such as moisture and pH, are essential drivers of fungal communities in soil [30]. Maestre et al. [31] studied the diversity and abundance of soil fungi, which decline with decreasing soil pH. Furthermore, it has been reported that the quality and amount of soil nutrient substrates change soil fungal groups [27, 32]. Since most soil fungi are saprophytes, Geml et al. [33] and Zimudzi et al. [34] found that the soil fungi were highly related to N content, C: N ratio, and SOC. In the past, studies stated that species of invasive plants altered the SOC and N sequestration [35], the composition of plant litter [36], and soil physicochemical properties [37].
Conventional methodologies merely provide limited insights into the vast array of soil microorganisms, with a significant majority of approximately 80–90% of soil microbiota constituents remaining unidentified [38]. Because of this, a wide range of detection approaches based on the DNA sequencing procedures have been described [39]. Such studies examined the microbial communities in a variety of soil settings, including the root endospheres, rhizosphere, and rhizoplane [40,41,42,43,44,45,46] in wetlands [47], alpine meadows [48, 49] meadow steppe [50], paddy soil [51], and mud flats [52]. In alpine wet meadows, however, not much is known about the diversity and composition of the rhizosphere and rhizoplane community of fungi in relation to plant species, site characteristics, and soil physicochemical properties. The primary objectives of the present study were to address these gaps in knowledge by utilizing high-throughput sequencing (HTS) techniques to (1) assess the fungal diversity of alpine wet meadow plants within the rhizosphere and rhizoplane; (2) investigate the assembly of fungal communities in the rhizosphere and rhizoplane; (3) examine the influence of various physicochemical properties, such as soil water content (SWC), soil organic carbon (SOC), microbial biomass carbon (MBC), total nitrogen (TN), microbial biomass nitrogen (MBN), pH, and elevation, on the composition of fungal communities; and (4) analyze the impact of plant-root compartments (rhizosphere and rhizoplane), plant species (Cremanthodium lineare, Caltha scaposa, and Cremanthodium ellisii), and different sites on fungal functional profiles using FUNGuild analysis.
Methods
Experimental design and soil collection
Three dominant plant species, Cremanthodium ellisii, Caltha scaposa, and Cremanthodium lineare, were collected from wet meadows at three elevations: 3737 m (SI), 3938 m (SII), and 4134 m (SIII), respectively, in Maqin county (34.4773° N, 100.2396° E), Qinghai Province, Tibetan Plateau, China (Additional file 1: Table S1). The annual precipitation averages 580 mm, and the average air temperature is − 0.6 ℃. The area has various soil types, notably dark meadows, swamps, and peat. Three random plots, separated by at least 1000 m between any two, were selected at each elevation. All three species are herbaceous wet meadow plants, with C. ellisii and C. lineare belonging to the family Asteraceae and C. scaposa belonging to the family Ranunculaceae. Three individuals of each plant species were randomly collected in each plot. All samples were treated separately. Plants were carefully cut using a sterilized knife and placed in a sterilized bag. The rhizosphere and rhizoplane were isolated according to the methods proposed by Oh et al. [53] and Kaiser et al. [54]. The rhizosphere of each plant was collected by jerking the roots three times for 60 s to release the soil. The soil was then vortexed, and the supernatant was discarded to collect the rhizosphere sample. The rhizoplane sample was collected by rinsing the plant roots three times in a phosphate buffer saline (PBS, 140 mM NaCl, 3 mM KCl, 8 mM Na2HPO4, 1.5 mM NaH2PO4, pH 7.0) up to a volume of 50 ml. After being washed, the roots were measured, shaken (350 rpm), vortexed, and sonicated (Branson 2800 Ultrasonic Cleaner, Branson Ultrasonics, Richmond, Virginia, USA) every 30 s with alteration for 3 min to detach microbial cells. The suspension was then filtered through a nylon mesh (pore size, 200 µm, Spectrum Europe BV) to separate microbial cells and centrifuged at 3, 150 ×g for 15 min at 4 °C. The cell pellets were washed twice with a PBS buffer and stored at − 20 °C.
Analysis of soil
The soil samples were air-dried and passed through a 2 mm sieve to ensure a homogeneous sample. TN was examined by a flow injection autoanalyzer (FIAstar 5000 Analyzer, Foss Tecator AB, Hillerød, Denmark) after digestion with sulphuric acid [55], and SOC was acquired using colorimetry after oxidation with a combination of potassium dichromate and sulfuric acid [56]. MBN and MBC were obtained through fumigation−extraction and analyzed using the Elementar Vario EL cube elemental analyzer (Langenselbold, Hesse, Germany) and the Skalar Automatic Analytical Analyzer (BV, Breda, Netherlands). SWC was determined by oven-drying at 105 °C [57], and the pH of the soil filtrate (soil: water = 1:5, w/v) was determined through a pH meter (Benchtop acidity meter PHS-3E, Leici, Shanghai, China).
Microbial community analysis
Microbial community DNA was isolated from soil samples using the DNeasy PowerSoil Pro Kit (Qiagen, USA) following the manufacturer’s instructions. The DNA concentration was assessed with a Nanodrop ND-2000 UV–VIS Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). The internal transcribed spacer (ITS) region was amplified using ITS1 (TCCGTAGGTGAACCTGCGG) and ITS2 (GCTGCGTTCTTCATCGATGC) primers with Illumina Miseq [58] through ABI GeneAmp® 9700 PCR thermocycler (ABI, CA, USA). Amplicons were pooled in equimolar concentrations and sequenced on an Illumina MiSeq platform (2 × 300 bp paired-end reads) at MajorBio Biopharm Technology (China). Raw paired-end reads were merged with FLASH software (v1.2.11). Merged reads were filtered for quality and chimera removal using QIIME (version 1.9.1) and Fastp (version 0.19.6). Operational taxonomic units (OTUs) were clustered at 97% similarity with UPARSE (http://www.drive5.com/uparse/). The OTUs were assigned to different taxonomies of fungi at a 70% confidence threshold using the ribosomal database project (RDP) classifier (http://rdp.cme. msu.edu/) against the ITS fungal database (Unite 8.0) [59]. In total, 4,182 rhizosphere and 1488 rhizoplane fungal OTUs were detected across all samples.
Data transformation and bioinformatics analysis
The Shannon, Simpson, Ace, and Sobs α-diversity indices were estimated via Mothur (version v.1.30.2 https://mothur.org/wiki/calculators/). The Ace (Abundance-based Coverage Estimators) and Sobs (observed species) are α-diversity indices used to quantify species richness within a given community [60, 61]. The Ace α-diversity index estimates the total number of species in the community, considering the potential presence of rare or less abundant species that might be missed in small sample sizes. The index calculates the expected richness by extrapolating the observed data to better estimate the total species richness. In contrast, the Sobs provide a straightforward measure of species richness and calculate the number of unique species observed in a sample without considering their abundance or relative frequency [62, 63]. The Shannon and Simpson α-diversity indices are used to measure the diversity of species in a group [64, 65]. The Shannon diversity index analyzes both the number of species present and their relative abundances, providing a more thorough evaluation of diversity. In contrast, Simpson stresses dominance or the concentration of abundant species in the group. Higher Shannon values suggest greater diversity and equal distribution of species abundances, whereas higher Simpson values indicate less diversity and domination by a few species [66]. The differences in the α-diversity indices of fungal communities between the rhizosphere and rhizoplane were assessed by the two-tailed Wilcoxon’s rank sum test using the stats package in R. The variations in α-diversity of fungal communities among the plant species and elevations were checked via Kruskal–Wallis with False Discovery Rate (FDR) multiple correction tests (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001) through stats package in R. The β-diversity was shown by nonmetric multidimensional scaling (NMDS) analysis employing ANOSIM and hierarchical cluster tree using Bray–Curtis dissimilarity matrix in vegan and ggplot package in R. The differences between the rhizosphere and rhizoplane fungal communities at the genus level were identified via the Metastats differential test with Benjamini–Hochberg multiple corrections. Metastats is a statistical method designed for comparing metagenomic samples of two populations based on the count data (i.e., acquired through sequencing) to detect differentially abundant features [67]. The linear mixed model (LLM) was applied to investigate the effects of plant species, elevations, and root compartments on the α- and β-diversity of fungal communities in SPSS 22. Further, the Null-modeling-based approach (iCAMP) was utilized to infer fungal community assembly mechanisms in the rhizosphere and rhizoplane by the “Picante” package in R [68]. The Null model identifies the community assembly mechanism by assessing the standard deviation of observed ecological patterns as compared to the randomly shuffled ecological patterns generated by the null model [69]. The variations in the ecological patterns were investigated by comparing β-diversity metrics, i.e., beta-nearest taxon index (βNTI) and the Raup−Crick metric (RCbray). The analysis was performed based on the methods described by Stegen and coworkers [70] to classify community pairs into underlying drivers of stochastic processes (homogeneous selection and heterogeneous selection) or deterministic processes (undominated, dispersal limitation, and homogenous dispersal). The relative influence of homogeneous and heterogeneous selection was quantified with |βNTI|< − 2 and |βNTI|> 2. At the same time, the homogeneous dispersal, dispersal limitation, and undominated ecological patterns were quantified with the pairwise comparisons of |βNTI|≤ 2, RC < − 0.95, |βNTI| 2 ≤ , RC < 0.95, and |βNTI|≤ 2, RC ≤ 0.95, respectively [71]. The “mantel_test” function in the dplyr R package was utilized to examine the association amongst the fungal phyla and familiae with physicochemical properties using Pearson's correlation coefficient. The co-occurrence network was constructed to visualize relationships among fungal genera and environmental variables by Spearman's correlation (p < 0.05), using the “corr.test” function in the psych R package, while the network analysis was visualized with the interactive platform gephi. The network-level topology was analyzed in terms of nodes, edges, average weight degree, positive and negative correlations, and several modularities. FUNGuild [72] predicted the functions of rhizosphere and rhizoplane fungal communities. Fungal species were categorized into putative functional groupings by assigning OTUs with a confidence level of “highly probable” or “probable” to trophic modes and guilds.
Results
Physicochemical characteristics of the soil
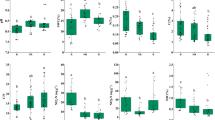
The three plant species, Cremanthodium ellisii, Cremanthodium lineare, and Caltha scaposa, varied in their physicochemical characteristics at each site. As presented in Additional file 1: Table S2 and Fig. 1, the one-way ANOVA exhibited significant differences in SWC (p < 0.01) among plant species, whereas SOC (p < 0.001), TN (p < 0.001), pH (p < 0.001), SWC (p < 0.001), MBC (p < 0.001), and MBN (p < 0.001) contents of the rhizoplane differed among sites. Moreover, the two-way ANOVA (site × plant species) revealed no significant differences (p > 0.05) in the rhizoplane soil (Additional file 1: Table S2 and Fig. 1). The soil parameters of the rhizosphere, including SWC (p < 0.001), SOC (p < 0.001), MBC (p < 0.001), TN (p < 0.001), MBN (p < 0.001), and pH (p < 0.001) varied among sites, whereas TN (p < 0.05) was significant among plant species. Similarly, the two-way ANOVA (sites × plant species) showed no significant differences (p > 0.05) in the rhizosphere compartment (Additional file 1: Table S2 and Fig. 1).

Soil physicochemical properties of rhizosphere and rhizoplane samples associated with Cremanthodium ellisii, Cremanthodium lineare, and Caltha scaposa. Mean (n = 3) with lowercase letters in a column within the rhizoplane and rhizosphere differ at p < 0.05. SI, SIII, and SIII represent sampling sites, ns non-significant, SOC soil organic carbon, TN total nitrogen, SWC soil water content, MBC microbial biomass carbon, MBN microbial biomass nitrogen, *p < 0.05; **p < 0.01 and ***p < 0.001
Fungal community composition
The predominant soil fungal phyla in the rhizosphere of C. ellisii, C. lineare, and C. scaposa were Ascomycota at 54.5% and Basidiomycota at 20.9%, while Rozellomycota, Mortierellomycota, unclassified fungi, and others totaled 24.5% (Additional file 1: Figure S1). Similarly, Ascomycota dominated the rhizoplane region at 51.6% and Basidiomycota at 36.4%, while Rozellomycota, Mortierellomycota, unclassified fungi, and others totaled 11.9% (Additional file 1: Figure S1). Moreover, the rhizosphere and rhizoplane fungal genera also varied among C. ellisii, C. lineare, and C. scaposa at all sites (Fig. 2). The predominant fungal genera in the rhizosphere were Unclassified fungi > Unclassified Ascomycota > Pseudeurotium > Tetracladium > Vishniacozyma, while that of rhizoplane were Rhodotorula > Unclassified fungi > Unclassified Ascomycota > Cadophora > Pencillium (Fig. 2). However, the Unclassified GS11 (p < 0.001), Unclassified Pyronemataceae (p < 0.01), Unclassified Sclerotiniaceae (p < 0.01), Wardomyces (p < 0.05), Unclassified Helotiales (p < 0.05), and Necteria (p < 0.05) were the significant abundant genera among the rhizosphere and rhizoplane fungal communities, respectively (Additional file 1: Table S3).

Distribution of fungal genera in the a rhizosphere and b rhizoplane associated with Cremanthodium ellisii, Cremanthodium lineare, and Caltha scaposa
Variations in fungal communities
The α-diversity indices of the rhizosphere and rhizoplane fungal communities of C. ellisii, C. lineare, and C. scaposa at SI, SII, and SIII are presented in Additional file 1: Tables S4 and S5. The Ace, Sobs, Simpson, and Shannon α-diversity indices significantly differed between the rhizosphere and rhizoplane. As shown in Fig. 3, the Ace (p < 0.001), Sobs (p < 0.001), and Shannon (p < 0.001) indices were significant in the rhizosphere, whereas Simpsons' (p < 0.001) index was substantially different in the rhizoplane. In addition, the α-diversity indices of C. ellisii, C. lineare, and C. scaposa at SI, SII, and SIII varied. For instance, as shown in Additional file 1: Figure S2, the Ace index of the rhizosphere differed between C. ellisii and C. lineare (p < 0.05) at SI and SIII, while the Sobs index differed among C. ellisii and C. scaposa (p < 0.01) at SI, and SIII, respectively. However, the Shannon (p > 0.05) and Simpson (p > 0.05) indices showed no differences among plant species or sites (Additional file 1: Figure S2). In contrast, the Ace, Shannon, Simpson, and Sobs indices of the rhizoplane differed significantly among plants and sites (Additional file 1: Figure S3).

Differences in alpha diversity indices of the rhizosphere and rhizoplane fungal communities based on the Wilcoxon rank sum test; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001
The nonmetric multidimensional scaling (NMDS) analysis among the plant species formed significant clusters of fungal diversity (β-diversity) of the rhizosphere (p < 0.001) and rhizoplane (p < 0.001) in the ordination space (Fig. 4). The NMDS ordination of the rhizosphere revealed that fungal communities of C. ellisii at SI and SII were separate from C. lineare and C. scaposa. In contrast, fungal communities of C. ellisii, C. lineare, and C. scaposa were clustered together at SI, SII, and SIII (Fig. 4). Moreover, the NMDS ordination revealed that the fungal communities in the rhizoplane of C. ellisii, C. lineare, and C. scaposa were separated at SI and SII, while C. scaposa was separated from C. lineare and C. ellisii at SIII. However, C. scaposa and C. lineare at SII and SIII, and C. ellisii at SI and SII were clustered together in the rhizoplane (Fig. 4). The NMDS ordination of fungal communities across different rhizosphere sites was notable; for example, fungal communities of C. ellisii at SI were distinct from those of C. lineare and C. scaposa, which clustered together. Similarly, the fungal communities of C. ellisii differed from those of C. lineare and C. scaposa at SII. The fungal communities of C. ellisii, C. lineare, and C. scaposa at SIII clustered together, indicating that the fungal communities are more comparable between plants (Fig. 4). In comparison, the fungal communities within the rhizoplanes of all three plants formed different clusters at SI, although C. lineare and C. scaposa clustered together at SII, implying similarities. In addition, the fungal communities of C. scaposa appeared to be distinct from those of C. lineare and C. ellisii, which together formed clusters at SIII, as shown in Fig. 4.

Distribution of the fungal communities (β-diversity) in the rhizosphere (a) and rhizoplane (b) associated with Cremanthodium ellisii, Cremanthodium lineare, and Caltha scaposa based on the nonmetric multidimensional scaling (NMDS) analysis using Bray—Curtis dissimilarity matrix and ANOSIM at the number of replacements = 999; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001
The hierarchical cluster tree and clustering heatmap based on the Bray–Curtis distance algorithm also showed that the fungal rhizosphere communities of C. ellisii and C lineare at SII and C. scaposa at all sites were separate, while C. scaposa at SII and SIII and C. lineare at SI, SII, and SIII were clustered together (Additional file 1: Figure S4). The results from the linear mixed model demonstrated that both the rhizosphere and rhizoplane showed a significant effect (p < 0.001) on the α- and β-diversities while plant species and elevations showed varying results on the α- and β-diversity indices (Additional file 1: Table S6). However, the combined effect of all variables (i.e., plants × elevations × compartments) only exhibited a significant effect on Shannon (p < 0.01) and Simpson (p < 0.01) of α-, and bray–Curtis distance (p < 0.01) of β-diversity indices. (Additional file 1: Table S6).
Variations in fungal community assembly
Understanding the ecological forces that govern community formation is a central concern. Communities may form in two ways: niche-based deterministic and neutral-based stochastic. Niche-based deterministic mechanisms include environmental filtering (such as pH, temperature, moisture, and salinity) and various biological interactions (such as competition, facilitation, mutualisms, and predation). In contrast, neutral-based stochastic processes include birth/death, speciation/extinction, and immigration [73]. As shown in Additional file 1: Table S7, variations in rhizosphere fungal communities were strongly influenced by dispersal limitation, i.e., the dispersal limitation of C. ellisii, C. lineare, and C. scaposa was 71.11%, 46.66%, and 64.44%. In contrast, the fungal communities of rhizoplane were shaped by an undominated niche process, i.e., the undominated niche-based selection of C. ellisii, C. lineare, and C. scaposa was 51.11%, 28.88%, and 60.52%, respectively. Overall, the fungal community assembly of rhizosphere and rhizoplane was shaped by stochastic rather than deterministic processes. For example, the stochastic processes within the rhizosphere of C. ellisii, C. lineare, and C. scaposa were 93.33%, 86.66%, and 93.33%, while that of rhizoplane were 93.33%, 28.88%, and 68.42%. In comparison, the deterministic processes within the rhizosphere of C. ellisii, C. lineare, and C. scaposa were 6.66%, 13.33%, and 6.66%, while that of rhizoplane were 6.66%, 71.11%, and 31.57%, respectively (Additional file 1: Table S7 and Figure S5).
Association among fungal communities and physicochemical properties
The association between the fungal communities and the physicochemical properties in alpine wet meadows was analyzed by mantel test and network analysis. The Mantel test investigated the relationship between physicochemical properties and changes in fungal phyla and familiae in the rhizosphere and rhizoplane. The variables SOC, TN, pH, SWC, MBC, MBN, and elevation were shown to be correlated with the fungal phyla in the rhizosphere (r < 0.25; p ≤ 0.05). Although pH (r < 0.25; p = 0.01–0.05), MBN (r = 0.25–0.5; p = 0.01–0.05), and SOC, TN, SWC, MBC, and elevation (r = 0.25–0.5; p < 0.001–0.01) were significantly correlated with the fungal familiae of the rhizosphere (Fig. 5). In comparison, SOC, TN, pH, SWC, MBC, MBN, and elevation substantially correlated (r < 0.25; p ≤ 0.05) with the fungal phyla of the rhizoplane, whereas SOC, TN, pH, MBN, and elevation (r < 0.25; p = 0.01–0.05), and SWC and MBC (r < 0.25; p = 0.001–0.01) correlated with the relative abundance of the fungal familiae within the rhizoplane, respectively (Fig. 5). In addition, the network analysis explained the effect of observed variables on the relative abundances of fungal genera in the rhizosphere and rhizoplane (p < 0.05). The rhizosphere network comprised 118 nodes connected by 433 edges, while the rhizoplane had 63 nodes and 96 edges. The rhizosphere and rhizoplane exhibited more negative than positive links (Fig. 5). Topological features, likewise, network diameter, average degree, modularity, and graph density, described the complex pattern of fungal genera in the rhizosphere and rhizoplane (Additional file 1: Tables S8 and S9). Based on the rhizosphere network, TN, elevation, and SOC were the deterministic observed variables, whereas TN was the most prominent soil variable driving the fungal genera of the rhizoplane (Fig. 5).

Mantels’ test based on the Pearsons’ correlation coefficient among fungal phyla and familiae a rhizosphere, b rhizoplane, and correlation network analysis based on Spearman’s correlation coefficient among the fungal genera with the observed variables of c rhizoplane, d rhizosphere. TN Total nitrogen, SWC soil water content, SOC soil organic carbon, MBC microbial biomass carbon, MBN Microbial biomass nitrogen, EVN elevation, and pH; the size of the nodes = abundance, color = genera or soil properties; the green lines represent positive correlation, while the pink lines represent negative correlations
Functional determination of fungal communities
A total of 1124 (26.9%) of 4181 OTUs in the rhizosphere and 731 (49.1%) of 1488 OTUs in the rhizoplane were assigned to a corresponding fungal trophic mode and functional group (guild) via FUNGuild (Additional file 1: Table S10). We only selected three trophic modes, namely, saprotrophs, symbiotrophs, and pathotrophs, and screened the four guilds, arbuscular mycorrhizae, endophytes, ectomycorrhizae, and orchid mycorrhizae. The percentages of OTUs assigned to saprotrophs, pathotrophs, and symbiotrophs in the rhizosphere with a “highly probable” and “probable” confidence ranking were 58.5%, 11.7%, and 12.6%, whereas that of rhizoplane were 51.3%, 13.9%, and 7.92%, respectively. The percentages of OTUs representing arbuscular mycorrhizae, endophytes, ectomycorrhizae, and orchid mycorrhizae within the rhizosphere were 60.4%, 13.2%, 20.9%, and 1.09%, while that of rhizoplane were 36.1%, 25.0%, 30.6%, and 2.77%, respectively. Moreover, the fungal trophic modes differed among the three plant species and sites. For example, the abundance of saprotrophs and pathotrophs in the rhizosphere was highest in C. ellisii at SI and SIII. In contrast, symbiotrophs were highest in C. lineare at SIII (Fig. 6). Similar variations were observed in the fungal functional groups (guilds) among the plant species and sites (Fig. 6). Interestingly, the fungal taxonomy representing the different functional modes changes with the change in the root compartment. For example, Archaeospora remained the dominant genus of arbuscular mycorrhizae in the rhizoplane, while Dominikia was the dominant arbuscular mycorrhizae in the rhizosphere.

Circle packing characterization of the fungal trophic and guilds: a, b rhizosphere, c, d rhizoplane based on the FUNGuild analysis. The size of each circle represents the abundance of each function detected in the rhizosphere and rhizoplane of C. ellisii, C. lineare, and C. scaposa at SI, SII, and SIII
Discussion
Rhizosphere microbiomes of different plant species often have different structural properties [74], which also applies to microbial rhizoplane communities, albeit not many studies have analyzed the rhizoplane [75,76,77]. Rhizosphere and rhizoplane fungi are vital to plant ecology, evolution, and growth promotion; hence, more studies are needed to understand these microbial communities in wetlands environments, specifically in alpine wet meadows. This study used Illumina-Miseq profiling to determine how environmental factors and elevations affect the fungal communities of the rhizosphere and rhizoplane related to three plant species, Cremanthodium ellisii, Cremanthodium lineare, and Caltha scaposa in Maqin county, Qinghai−Tibetan Plateau, China.
Wetland soil is a complex habitat where soil depth, water level, biocrusts, and physical crust affect soil microbiota. The differences we observed in the SWC of rhizoplane among the plant species and other physicochemical properties among sites were in line with the studies by [78,79,80], indicating that the soil contents of water, N, and C tend to increase along an elevation gradient. The pH, vegetation type, SWC, and elevation can affect soil characteristics. For example, Zhang et al. [81] reported that SOC and TN concentrations increased with altitude, coinciding with a vegetation type change along the altitudinal gradient. The alterations of the C and N profiles along the elevation gradient could be due to the low air temperature, inhibiting microbial processes in mineralizing these compounds. Studies have reported on the reduction in the decomposition of SOM by the microbes with altitude due to low temperature [82, 83]. SWC can stimulate or inhibit the growth of microorganisms, which influences the mineralization of C and N [84]. Moreover, the SWC and water level influence the distribution of microbial species in wetland habitats, which may limit oxygen availability in the soil [6].
Our results agree with Zhang et al. [85], who stated that Basidiomycota and Ascomycota are the predominant phyla in the wetlands. The variation in the abundance of Ascomycota and Basidiomycota in the rhizosphere and rhizoplane can be attributed to disparities in microenvironmental variables and functional distinctions between the soil near the roots and the surface of the rhizoplane [41]. These fungi generally establish connections between processes occurring above and below the ground in wet meadows. However, unclassified fungi could be due to the presence of rich organic matter, that is, SOC, in the wet meadow soils at the three sites [85, 86]. Earlier studies that discussed how SOM and microbial function relate to one another provided evidence for the low abundance of fungi in the wet meadow [86, 87]. For instance, Zhang et al. [85] reported in their study that wetland soil was typically covered with water, causing a drop in oxygen content and limiting the growth of fungi. Likewise, oxygen availability is a limiting factor in alpine wet meadows [87], which most likely inhibits the growth of fungi, resulting in fewer fungi but a more homogenous community. The SWC also limits the available oxygen [88] in soil, which could explain, at least in part, the low diversity of fungal communities within the wetland habitat [89]. In addition, we observed differences in the fungal communities among plant species and sites. Similar results were observed in the fungal communities across plant-root structures [90], plant types [91], and sites [92]. Possible reasons for these differences could be due to the plant–microbe relationship, which is driven by plant exudates that contain signaling compounds (flavonoids, strigolactones) and organic compounds (organic acids, amino acids, proteins, and fatty acids), and by adaptations to different characteristics of their niche, such as temperature, oxygen level, and UV light [93, 94] The abiotic and biotic factors which influence the fungal community differ significantly across different locations, plant-root structures, and plant types [90,91,92]. Comparatively, the rhizoplane fungal community had low species diversity and richness, implying that an inadequate number of species were screened by plant roots [95]. This means that the microbiota in each rhizo-compartment is unique in composition and structure and susceptible to further manipulation by host-controlled processes [41]. Potential variations in plant-fungal communication and mutualistic relationships could exist between the rhizoplane and the rhizosphere. An illustration of this phenomenon can be observed in the symbiotic relationship between mycorrhizal fungi and root cortical cells, wherein the former might potentially influence the colonization of other fungal species [96]. Differential selective pressures may be exerted on the root surface as opposed to the surrounding soil environment due to the interplay between plant immunity and microbial competitiveness. This phenomenon can potentially result in the development of discrete fungal communities suited to the rhizoplane [96,97,98].
The α- and β-diversity showed substantial differences in the rhizosphere and rhizoplane of C. ellisii, C. lineare, and C. scaposa. The findings demonstrated that the plant-root compartments, the plant species, and the sites impacted the diversity of the fungal groups. Changes in the α- and β-diversities of microbial communities were reported with respect to the vegetation and site specificity. These changes were linked to either human activities that disturb plant cover or soil variables that may change with the altitude [99, 100]. According to Bgachelot et al. [29] and Chen et al. [90], differences in the root exudates and litter inputs of various plant species might influence the availability of resources and cause alterations in the alpha diversity of fungal communities in the rhizosphere and rhizoplane. Gradients can also influence fungal alpha diversity in abiotic variables such as moisture, oxygen, pH, and nutrients linked to soil depth, location, or seasonal dynamics [30, 32]. In addition, the capacity of the rhizosphere to select microbes and prevent their penetration into other root compartments, such as the rhizoplane and endosphere, may be responsible for the differences between the rhizosphere and rhizoplane fungal communities [101, 102]. Moreover, the community assembly revealed that neutral-based stochastic processes governed the fungal communities of both rhizosphere and rhizoplane; however, the intra-stochastic processes across the rhizoplane and rhizosphere of C. ellisii, C. lineare, and C. scaposa varied. Overall, the fungal community assembly of rhizosphere and rhizoplane was shaped by stochastic rather than deterministic processes. Our findings are consistent with those of Huang et al. [103], who found that stochastic processes played a more critical role in the formation of fungal communities in wetland soils than deterministic ones, which confirms that stochastic processes play a vital role in the assembly of fungal communities in wetland soil. The different assemblage processes could be linked to the growth strategies of fungi within the succession. Moreover, the undominated process accounts for a large proportion of the fungal assembly [69], which is evident from our results that the undominated neutral-based stochastic process influenced the rhizoplane fungal communities. The differences in the assembly processes could also be linked to the environmental filtration of the given area; for example, the soil N content and pH variations can affect the balance between stochastic and deterministic assembly processes [104]. It has been reported that deterministic processes proliferate in nutrient-poor environments, whereas stochastic processes thrive in nutrient-rich environments [105]. For example, Wang et al. [106] found that fungal communities in the bulk soil were formed by deterministic mechanisms, as opposed to the rhizosphere, where stochastic processes regulated fungal communities. They stated that the bulk soil had inadequate N levels, whereas the rhizosphere soil had appropriate N levels, resulting in a difference in the fungal assembly in the bulk and rhizosphere soil [106]. Thus, it is plausible that the altered fungal assembly processes within the rhizosphere and rhizoplane associated with C. ellisii, C. lineare, and C. scaposa were caused by the alterations in the rhizosphere and rhizoplane soil parameters observed in our study.
The association between the fungal communities and the soil physicochemical properties in alpine wet meadows was analyzed by mantel tests and network analysis. These findings suggested that increasing the positively linked variables promoted fungi growth in the rhizosphere or rhizoplane. The presence of positive relationships between soil physicochemical properties and fungal taxonomic abundance suggests that there are likely direct or indirect influences on fungal growth and activity in the rhizosphere and rhizoplane environments. As an example, the augmentation of moisture and nutrient availability has the potential to directly induce microbial metabolism and proliferation by alleviating limitations imposed by the surrounding environment [84]. As a significant resource, soil organic matter has been found to influence the size of fungal populations positively. Moreover, it is worth noting that fungi have the ability to react indirectly to variations in soil conditions that lead to changes in the patterns of exudation from plant roots. This response is mediated by chemicals such as sugars and organic acids, which serve as significant signals regulating fungal gene expression [94]. Fungal dynamics can be influenced by various intricate biotic interactions, both direct and indirect, which are modulated by soil physicochemical properties. Positive connections may ultimately indicate that some environmental factors facilitate the growth of specific fungal species rather than impede it. However, Pearson’s correlation revealed that the SOC negatively correlated with the fungal phyla of the rhizoplane, suggesting that the SOC may limit the growth of fungal communities within the rhizoplane. The interplay between plant production and soil C is sensitive to the partitioning of several fungal groups. This relation could justify the exclusive and prominent fungal communities in the rhizosphere and rhizoplane of the observed plant species. Most likely, the intricacy of the network in the rhizosphere was caused by frequent changes in the environment of the roots and the soil near them, such as changes in nutrient availability, pH, moisture, oxygen levels, and carbon dioxide levels [107, 108]. These results indicate that the rhizosphere is the most active locus of microbial and plant-microbial interactions. Moreover, it also explains why plant species may not be the primary determinant of the structure of fungal communities, but seasonal changes and soil variables are the main drivers [79, 109]. SWC was correlated with fungal phyla, familiae, and genera in the rhizosphere and rhizoplane, which implies that SWC was a limiting factor for these microorganisms. A change in SWC could lead to alterations in C and N mineralization within the soil, which affects the distribution of microbial species in wetland habitats [6, 84]. Moreover, moisture enables the diffusion of soil nutrients [110], and consequently, a more excellent supply of organic C would be available for the soil microbes, increasing their relative abundance.
FUNGuild analysis revealed differences in the functional traits of fungal communities of rhizosphere and rhizoplane associated with C. ellisii, C. lineare, and C. scaposa. The functional modes and guilds were selected based on the importance of their role in climate change and environmental filtering by forming plant-host associations [111]. Mycorrhizae are symbiotroph fungi that interact with plant hosts and contribute carbon, nitrogen, and phosphorus [15]. It was predicted that plants typically direct 10–20%, but can reach 50%, of their photosynthates to their fungi, with the major mycorrhizal fungi of an ecosystem exerting considerable impact over C cycling and storage [112]. In exchange, mycorrhizal fungi supply up to 80% of the phosphorus (P) requirements of host plants [113], as well as considerable amounts of nitrogen (N) and micronutrients [114]. Therefore, the maximum abundance of fungal trophic modes at SIII could result from more soil C and N matters in the form of SOC, MBC, TN, and MBN. The increased availability of resources, such as SOC and TN, contributes to the provision of additional energy, carbon compounds, and essential nutrients that support the growth and metabolic processes of saprotrophic fungi, which derive their nutrition by decomposing organic matter [29]. The stimulation of saprotroph abundance through bottom-up processes is also intensified by the presence of MBC and MBN pools, which signify a substantial and dynamic population of bacteria and fungi involved in the decomposition process [30, 115]. Conversely, a higher concentration of SOC is also associated with an elevated allocation of carbon below ground by plants. This allocation is believed to facilitate a larger population of symbiotrophic mycorrhizal fungi, which acquire plant-derived photosynthates in exchange for essential mineral nutrients [17]. Several factors, including SOC, MBC, TN, and MBN, influence detrital resources, plant productivity, and soil microbiota. Collectively, these factors provide a conducive environment that supports the growth and activity of free-living decomposer organisms and symbiotic fungi. The difference between the number of OTUs corresponding to the trophic modes and guilds within the rhizosphere and rhizoplane could be due to plant–host specificity, soil chemistry, and the screening effect of the plant-root compartment [116, 117]. Moreover, we also observed changes in the fungal taxonomy with different functional modes, which could be the reason behind the intra- and intercellular fungal association patterns with host plants [118]. FUNGuild has been used extensively to analyze the functions of fungi. However, more in-depth studies focusing on quantifying fungal communities and functional groups are required to strengthen the functional profiles of fungi within the plant-root compartments at different sites.
The present study is subject to several limitations. Firstly, the collection of soil samples was limited to the rhizosphere and rhizoplane for a single year rather than encompassing a longitudinal time series. Secondly, the findings were derived from relative abundance, correlations, modeling, and functional predictions of fungal communities rather than employing quantitative techniques such as qPCR. Thirdly, a hypothetical repeated sampling approach was utilized, which may restrict the generalizability of the results to the specific sampling area of alpine wet meadows rather than wetlands in their entirety. Further research is required in the form of quantitative and time series studies to obtain a more accurate and comprehensive understanding of the fungal communities present in the rhizosphere and rhizoplane, as well as the ecological functions they serve in alpine wet meadows.
Conclusions
The current study provides insights into the spatial distribution of fungal communities in the rhizosphere and rhizoplane of three plant species, namely C. lineare, C. scaposa, and C. ellisii, across three distinct sites, and expands our understanding of fungal ecology in alpine wet meadows. The Unclassified GS11, Unclassified Pyronemataceae, Unclassified Sclerotiniaceae, Wardomyces, Unclassified Helotiales, and Necteria were the significant abundant genera among the rhizosphere and rhizoplane fungal communities. Substantial differences emerged in fungal α-diversity indices in the rhizosphere and rhizoplane, with ACE, Sobs, and Shannon being distinct in the rhizosphere, while Simpson was the highest in the rhizoplane. The intra-fungal community, based on the NMDS analysis, also revealed significant fungal clusters in the rhizosphere and rhizoplane and across different sites. Root compartments significantly influenced the fungal population, although plant species and elevation had varying effects. In addition, the formation of the fungal community showed significant heterogeneity, with stochastic process predominating in both the rhizosphere and the rhizoplane. The fungal communities of rhizosphere and rhizoplane were shaped by total nitrogen (TN), soil water content (SWC), and soil organic carbon (SOC); however, pH, microbial biomass C (MBC), microbial biomass N (MBN), and elevation had a trivial effect. The findings from the FUNGuild analysis provide additional evidence to support the notion that fungal trophic mode and guild variations are influenced by plant-root compartments, plant species, and elevation in alpine wet meadows, thereby influencing the fungal contribution to the ecosystem. However, additional research is necessary to gain a comprehensive understanding of the structural and functional properties of the fungal ecosystem in the rhizosphere and rhizoplane of alpine wet meadows, in conjunction with the metatranscriptome and metaproteome.
Availability of data and materials
The raw reads were deposited in the Sequence Read Archive (SRA) of the National Center for Biotechnology Information under the project accession numbers PRJNA882059 and PRJNA881951 (www.ncbi.nlm.nih.gov/sra).
Abbreviations
- N:
-
Nitrogen
- C:
-
Carbon
- P:
-
Phosphorus
- m:
-
Meter
- mm:
-
Millimeter
- g:
-
Gram
- oC:
-
Celsius
- kg:
-
Kilogram
References
Wagg C, Bender SF, Widmer F, Van Der Heijden MGA (2014) Soil biodiversity and soil community composition determine ecosystem multifunctionality. Proc Natl Acad Sci USA 111:5266–5270. https://doi.org/10.1073/pnas.1320054111
Voyron S, Tonon C, Guglielmone L et al (2022) Diversity and structure of soil fungal communities unveil the building history of a burial mound of ancient Japan (Tobiotsuka Kofun, Okayama Prefecture). J Archaeol Sci 146:105656. https://doi.org/10.1016/j.jas.2022.105656
De Deyn GB, Van Der Putten WH (2005) Linking above-ground and belowground diversity. Trends Ecol Evol 20:625–633. https://doi.org/10.1016/j.tree.2005.08.009
Schulz S, Brankatschk R, Dümig A et al (2013) The role of microorganisms at different stages of ecosystem development for soil formation. Biogeosciences 10:3983–3996. https://doi.org/10.5194/bg-10-3983-2013
Li H, Li T, Sun W et al (2021) Degradation of wetlands on the Qinghai-Tibetan Plateau causing a loss in soil organic carbon in 1966–2016. Plant Soil 467:253–265. https://doi.org/10.1007/s11104-021-05086-6
Iqbal A, Shang Z (2020) Wetlands as a carbon sink: insight into the Himalayan Region. In: Shang Z, Degen AA, Rafiq MK, Squires VR (eds) Carbon management for promoting local livelihood in the Hindu Kush Himalayan (HKH) Region. Springer, Cham, pp 125–144
Fan H, Hu Y, Tian B et al (2023) Long-term ecological changes in China’s Ramsar sites. Ecol Indic 149:110159. https://doi.org/10.1016/j.ecolind.2023.110159
Iqbal A, Shang Z, Rehman MLU et al (2019) Pattern of microbial community composition and functional gene repertoire associated with methane emission from Zoige wetlands, China—a review. Sci Total Environ. https://doi.org/10.1016/j.scitotenv.2019.133675
Wei D, Xu-Ri TT et al (2015) Revisiting the role of CH4 emissions from alpine wetlands on the Tibetan Plateau: evidence from two in situ measurements at 4758 and 4320m above sea level. J Geophys Res Biogeosciences 120:1741–1750. https://doi.org/10.1002/2015JG002974
Zhang Y, Dong S, Gao Q et al (2016) Climate change and human activities altered the diversity and composition of soil microbial community in alpine grasslands of the Qinghai-Tibetan Plateau. Sci Total Environ 562:353–363. https://doi.org/10.1016/j.scitotenv.2016.03.221
Li J, Chen Q, Li Q et al (2021) Influence of plants and environmental variables on the diversity of soil microbial communities in the Yellow River Delta Wetland, China. Chemosphere 274:129967. https://doi.org/10.1016/j.chemosphere.2021.129967
Moore D, Robson GD, Trinci APJ (2020) 21st Century Guidebook to Fungi. Cambridge University Press, Cambridge
Tomer A, Singh R, Singh SK et al (2021) Role of fungi in bioremediation and environmental sustainability. Springer, Cham, pp 187–200
Genre A, Lanfranco L, Perotto S, Bonfante P (2020) Unique and common traits in mycorrhizal symbioses. Nat Rev Microbiol 18:649–660
Zhang Z, Yuan Y, Liu Q, Yin H (2019) Plant nitrogen acquisition from inorganic and organic sources via root and mycelia pathways in ectomycorrhizal alpine forests. Soil Biol Biochem 136:107517. https://doi.org/10.1016/j.soilbio.2019.06.013
Teste FP, Jones MD, Dickie IA (2020) Dual-mycorrhizal plants: their ecology and relevance. New Phytol 225:1835–1851. https://doi.org/10.1111/nph.16190
Averill C, Turner BL, Finzi AC (2014) Mycorrhiza-mediated competition between plants and decomposers drives soil carbon storage. Nature 505:543–545. https://doi.org/10.1038/nature12901
Choung Y, Min BM, Lee KS et al (2021) Categorized wetland preference and life forms of the vascular plants in the Korean Peninsula. J Ecol Environ 45:4–9. https://doi.org/10.1186/s41610-021-00183-0
Hu YH, Zhang XY, Zhang K et al (2021) Tussock microhabitats increase nitrogen uptake by plants in an alpine wetland. Plant Soil 466:569–580. https://doi.org/10.1007/S11104-021-05056-Y/FIGURES/7
Nyer SC, Volkenborn N, Aller RC et al (2022) Nitrogen transformations in constructed wetlands: a closer look at plant-soil interactions using chemical imaging. Sci Total Environ 816:151560. https://doi.org/10.1016/J.SCITOTENV.2021.151560
Sugiyama A (2019) The soybean rhizosphere: metabolites, microbes, and beyond—a review. J Adv Res 19:67–73
Raynaud X, Eickhorst T, Nunan N et al (2017) Spatial colonization of microbial cells on the rhizoplane. NASA/ADS. In: ui.adsabs.harvard.edu. https://ui.adsabs.harvard.edu/abs/2017EGUGA..1918887R/abstract. Accessed 13 Dec 2022
Paterson E, Mwafulirwa L (2021) Root–soil–microbe interactions mediating nutrient fluxes in the rhizosphere. Springer, Berlin
Tosi M, Gaiero J, Linton N et al (2021) Bacterial endophytes: diversity, functional importance, and potential for manipulation. Springer, Berlin, pp 1–49
Oliver IC, Knox OGG, Flavel RJ, Wilson BR (2021) Rhizosphere legacy: plant root interactions with the soil and its biome. Springer, Berlin, pp 129–153. https://doi.org/10.1007/978-981-15-6125-2_6
Pett-Ridge J, Shi S, Estera-Molina K et al (2021) Rhizosphere carbon turnover from cradle to grave: the role of microbe–plant interactions. Springer, Berlin, pp 51–73. https://doi.org/10.1007/978-981-15-6125-2_2
Peay KG, Baraloto C, Fine PVA (2013) Strong coupling of plant and fungal community structure across western Amazonian rainforests. ISME J 7:1852–1861. https://doi.org/10.1038/ismej.2013.66
Prober SM, Leff JW, Bates ST et al (2015) Plant diversity predicts beta but not alpha diversity of soil microbes across grasslands worldwide. Ecol Lett 18:85–95. https://doi.org/10.1111/ele.12381
Bachelot B, Uriarte M, Zimmerman JK et al (2016) Long-lasting effects of land use history on soil fungal communities in second-growth tropical rain forests. Ecol Appl 26:1881–1895. https://doi.org/10.1890/15-1397.1
Leff JW, Jones SE, Prober SM et al (2015) Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proc Natl Acad Sci USA 112:10967–10972. https://doi.org/10.1073/pnas.1508382112
Maestre FT, Delgado-Baquerizo M, Jeffries TC et al (2015) Increasing aridity reduces soil microbial diversity and abundance in global drylands. Proc Natl Acad Sci USA 112:15684–15689. https://doi.org/10.1073/pnas.1516684112
Lauber CL, Strickland MS, Bradford MA, Fierer N (2008) The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biol Biochem 40:2407–2415. https://doi.org/10.1016/j.soilbio.2008.05.021
Geml J, Pastor N, Fernandez L et al (2014) Large-scale fungal diversity assessment in the Andean Yungas forests reveals strong community turnover among forest types along an altitudinal gradient. Mol Ecol 23:2452–2472. https://doi.org/10.1111/mec.12765
Zimudzi J, van der Waals JE, Coutinho TA et al (2018) Temporal shifts of fungal communities in the rhizosphere and on tubers in potato fields. Fungal Biol 122:928–934. https://doi.org/10.1016/j.funbio.2018.05.008
Lee MR, Flory SL, Phillips RP, Wright JP (2018) Site conditions are more important than abundance for explaining plant invasion impacts on soil nitrogen cycling. Ecosphere 9:e02454. https://doi.org/10.1002/ecs2.2454
Liao C, Luo Y, Jiang L et al (2007) Invasion of Spartina alterniflora enhanced ecosystem carbon and nitrogen stocks in the Yangtze Estuary, China. Ecosystems 10:1351–1361. https://doi.org/10.1007/s10021-007-9103-2
Yang W, Jeelani N, Zhu Z et al (2019) Alterations in soil bacterial community in relation to Spartina alterniflora Loisel. Invasion chronosequence in the eastern Chinese coastal wetlands. Appl Soil Ecol 135:38–43. https://doi.org/10.1016/j.apsoil.2018.11.009
Wolfe BE (2018) Using cultivated microbial communities to dissect microbiome assembly: challenges, limitations, and the path ahead. mSystems. https://doi.org/10.1128/msystems.00161-17
Fricker AM, Podlesny D, Fricke WF (2019) What is new and relevant for sequencing-based microbiome research? A mini-review. J Adv Res 19:105–112. https://doi.org/10.1016/j.jare.2019.03.006
Bulgarelli D, Rott M, Schlaeppi K et al (2012) Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488:91–95. https://doi.org/10.1038/nature11336
Gottel NR, Castro HF, Kerley M et al (2011) Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types. Appl Environ Microbiol 77:5934–5944. https://doi.org/10.1128/AEM.05255-11
Inceoǧlu Ö, Salles JF, Van Overbeek L, Van Elsas JD (2010) Effects of plant genotype and growth stage on the betaproteobacterial communities associated with different potato cultivars in two fields. Appl Environ Microbiol 76:3675–3684. https://doi.org/10.1128/AEM.00040-10
Patel JS, Singh A, Singh HB, Sarma BK (2015) Plant genotype, microbial recruitment and nutritional security. Front Plant Sci 6:608. https://doi.org/10.3389/fpls.2015.00608
Peiffer JA, Spor A, Koren O et al (2013) Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci USA 110:6548–6553. https://doi.org/10.1073/pnas.1302837110
Vasco-Palacios AM, Bahram M, Boekhout T, Tedersoo L (2020) Carbon content and pH as important drivers of fungal community structure in three Amazon forests. Plant Soil 450:111–131. https://doi.org/10.1007/s11104-019-04218-3
Zancarini A, Mougel C, Terrat S et al (2013) Combining ecophysiological and microbial ecological approaches to study the relationship between Medicago truncatula genotypes and their associated rhizosphere bacterial communities. Plant Soil 365:183–199. https://doi.org/10.1007/s11104-012-1364-7
Yang W, Zhang D, Cai X et al (2019) Significant alterations in soil fungal communities along a chronosequence of Spartina alterniflora invasion in a Chinese Yellow Sea coastal wetland. Sci Total Environ 693:133548. https://doi.org/10.1016/j.scitotenv.2019.07.354
Li H, Qiu Y, Yao T et al (2021) Nutrients available in the soil regulate the changes of soil microbial community alongside degradation of alpine meadows in the northeast of the Qinghai-Tibet Plateau. Sci Total Environ 792:148363. https://doi.org/10.1016/j.scitotenv.2021.148363
Shi G, Yao B, Liu Y et al (2021) The effects of long-term warming on arbuscular mycorrhizal fungal communities depend on habitat type on the Qinghai-Tibet Plateau. Appl Soil Ecol 167:104030. https://doi.org/10.1016/j.apsoil.2021.104030
Yang X, Zhu K, Loik ME, Sun W (2021) Differential responses of soil bacteria and fungi to altered precipitation in a meadow steppe. Geoderma 384:114812. https://doi.org/10.1016/j.geoderma.2020.114812
Liu C, Ding N, Fu Q et al (2016) The influence of soil properties on the size and structure of bacterial and fungal communities along a paddy soil chronosequence. Eur J Soil Biol 76:9–18. https://doi.org/10.1016/j.ejsobi.2016.06.002
Mohapatra M, Yadav R, Rajput V et al (2021) Metagenomic analysis reveals genetic insights on biogeochemical cycling, xenobiotic degradation, and stress resistance in mudflat microbiome. J Environ Manage 292:112738. https://doi.org/10.1016/j.jenvman.2021.112738
Oh YM, Kim M, Lee-Cruz L et al (2012) Distinctive bacterial communities in the rhizoplane of four tropical tree species. Microb Ecol 64:1018–1027. https://doi.org/10.1007/s00248-012-0082-2
Kaiser O, Pühler A, Selbitschka W (2001) Phylogenetic analysis of microbial diversity in the rhizoplane of oilseed rape (Brassica napus cv. Westar) employing cultivation-dependent and cultivation-independent approaches. Microb Ecol 42:136–149. https://doi.org/10.1007/s002480000121
Ferreira AMR, Lima JLFC, Rangel AOSS (1996) Potentiometric determination of total nitrogen in soils by flow injection analysis with a gas-diffusion unit. Aust J Soil Res 34:503–510. https://doi.org/10.1071/SR9960503
Ruirui C, Xiaoting W (2022) BOOK REVIEW: Analytical methods for soil and agro‐chemistry (in Chinese). Edited by H. Z. Zhu, P. A. He, C. Z. Chen, H. M. Zhou, D. C. Su, J. M. Xu, H. Y. Qin, S. D. Bao, R. K. Lu S. H. Jiang Soil Science Society of China Beijing, China Agricultural Science and Tech. Eur J Soil Sci. https://doi.org/10.1111/ejss.13280
Li Y, Han C, Dong X et al (2022) Soil microbial communities of dryland legume plantations are more complex than non-legumes. Sci Total Environ. https://doi.org/10.1016/j.scitotenv.2022.153560
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. https://doi.org/10.1016/b978-0-12-372180-8.50042-1
Zhou JC, Wang YF, Zhu D, Zhu YG (2023) Deciphering the distribution of microbial communities and potential pathogens in the household dust. Sci Total Environ 872:162250
Chen YP, Tsai CF, Rekha PD et al (2021) Agricultural management practices influence the soil enzyme activity and bacterial community structure in tea plantations. Bot Stud 62:1–13. https://doi.org/10.1186/s40529-021-00314-9
Li Z, Zhou J, Liang H et al (2022) Differences in alpha diversity of gut microbiota in neurological diseases. Front Neurosci 16:879318. https://doi.org/10.3389/FNINS.2022.879318/BIBTEX
Chao A, Jost L (2012) Coverage-based rarefaction and extrapolation: Standardizing samples by completeness rather than size. Ecology 93:2533–2547. https://doi.org/10.1890/11-1952.1
Gotelli NJ, Colwell RK (2001) Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecol Lett 4:379–391
Li Q, Zhang B, Yang X, Ge Q (2018) Deterioration-associated microbiome of stone monuments: structure, variation, and assembly. Appl Environ Microbiol. https://doi.org/10.1128/AEM.02680-17
Xing R, Zhang HC, Gao Q, et al (2022) Bacterial communities associated with mushrooms in the Qinghai-Tibet Plateau are shaped by soil parameters. Int Microbiol 26:231–242. https://doi.org/10.1007/s10123-022-00286-1
Magurran AE (2021) Measuring biological diversity. Curr Biol 31:R1174–R1177. https://doi.org/10.1016/j.cub.2021.07.049
White JR, Nagarajan N, Pop M (2009) Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLOS Comput Biol 5:e1000352. https://doi.org/10.1371/JOURNAL.PCBI.1000352
Kembel SW, Cowan PD, Helmus MR et al (2010) Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26:1463–1464. https://doi.org/10.1093/bioinformatics/btq166
Zhang X, Liu H, Han H et al (2022) Microbial community succession associated with poplar wood discoloration. Plants 11:2420. https://doi.org/10.3390/plants11182420
Stegen JC, Lin X, Fredrickson JK et al (2013) Quantifying community assembly processes and identifying features that impose them. ISME J 7:2069–2079. https://doi.org/10.1038/ismej.2013.93
Hou Y, Jia R, Li B, Zhu J (2022) Apex predators enhance environmental adaptation but reduce community stability of bacterioplankton in crustacean aquaculture ponds. Int J Mol Sci. https://doi.org/10.3390/ijms231810785
Nguyen NH, Song Z, Bates ST et al (2016) FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol 20:241–248. https://doi.org/10.1016/j.funeco.2015.06.006
Bahram M, Kohout P, Anslan S et al (2016) Stochastic distribution of small soil eukaryotes resulting from high dispersal and drift in a local environment. ISME J 10:885–896. https://doi.org/10.1038/ismej.2015.164
Zhao F, Feng X, Guo Y et al (2020) Elevation gradients affect the differences of arbuscular mycorrhizal fungi diversity between root and rhizosphere soil. Agric For Meteorol 284:107894. https://doi.org/10.1016/j.agrformet.2019.107894
Jiang S, Ling N, Ma Z et al (2021) Short-term warming increases root-associated fungal community dissimilarities among host plant species on the Qinghai-Tibetan Plateau. Plant Soil 466:597–611. https://doi.org/10.1007/s11104-021-05073-x
Wang B, Chen C, Xiao Y et al (2023) Temperature thresholds drive the biogeographic pattern of root endophytic fungal diversity in the Qinghai-Tibet Plateau. Sci Total Environ 889:164270. https://doi.org/10.1016/j.scitotenv.2023.164270
Li F, Jin Z, Wang Z et al (2022) Host plant selection imprints structure and assembly of fungal community along the soil-root continuum. mSystems. https://doi.org/10.1128/msystems.00361-22
Xiong X, Lyu M, Deng C et al (2022) Carbon and nitrogen availability drives seasonal variation in soil microbial communities along an elevation gradient. Forests. https://doi.org/10.3390/f13101657
Ma H, Yang X, Guo Q et al (2016) Soil organic carbon pool along different altitudinal level in the Sygera Mountains, Tibetan Plateau. J Mt Sci 13:476–483. https://doi.org/10.1007/S11629-014-3421-6
Xu J, Chen Y, Zhang L et al (2017) Using phylogeny and functional traits for assessing community assembly along environmental gradients: a deterministic process driven by elevation. Ecol Evol 7:5056–5069. https://doi.org/10.1002/ece3.3068
Zhang Y, Ai J, Sun Q et al (2021) Soil organic carbon and total nitrogen stocks as affected by vegetation types and altitude across the mountainous regions in the Yunnan Province, south-western China. CATENA 196:104872. https://doi.org/10.1016/j.catena.2020.104872
He N, Yu Q, Wu L et al (2008) Carbon and nitrogen store and storage potential as affected by land-use in a Leymus chinensis grassland of northern China. Soil Biol Biochem 40:2952–2959. https://doi.org/10.1016/j.soilbio.2008.08.018
Deng L, Liu G, Shangguan Z (2014) Land-use conversion and changing soil carbon stocks in China’s “Grain-for-Green” Program: a synthesis. Glob Chang Biol 20:3544–3556. https://doi.org/10.1111/gcb.12508
Yan N, Marschner P, Cao W et al (2015) Influence of salinity and water content on soil microorganisms. Int Soil Water Conserv Res 3:316–323
Zhang H, Zhang S, Meng X et al (2018) Conversion from natural wetlands to forestland and farmland alters the composition of soil fungal communities in Sanjiang Plain, Northeast China. Biotechnol Biotechnol Equip 32:951–960. https://doi.org/10.1080/13102818.2018.1459208
Cookson WR, Murphy DV, Roper MM (2008) Characterizing the relationships between soil organic matter components and microbial function and composition along a tillage disturbance gradient. Soil Biol Biochem 40:763–777. https://doi.org/10.1016/j.soilbio.2007.10.011
Li Y, Ma J, Gao C et al (2021) Anaerobic ammonium oxidation (anammox) is the main microbial N loss pathway in alpine wetland soils of the Qinghai-Tibet Plateau. Sci Total Environ 787:147714. https://doi.org/10.1016/j.scitotenv.2021.147714
Saedi T, Mosaddeghi MR, Sabzalian MR, Mamedov AI (2021) Effect of Epichloë endophyte-tall fescue symbiosis on rhizosphere aggregate stability and quality indicators under oxygen–limited conditions. Geoderma 381:114624. https://doi.org/10.1016/J.GEODERMA.2020.114624
Zhou H, Zhang D, Jiang Z et al (2019) Changes in the soil microbial communities of alpine steppe at Qinghai-Tibetan Plateau under different degradation levels. Sci Total Environ 651:2281–2291. https://doi.org/10.1016/j.scitotenv.2018.09.336
Chen P, Zhao M, Tang F et al (2020) The effect of plant compartments on the Broussonetia papyrifera-associated fungal and bacterial communities. Appl Microbiol Biotechnol 104:3627–3641. https://doi.org/10.1007/s00253-020-10466-6
Põlme S, Bahram M, Jacquemyn H et al (2018) Host preference and network properties in biotrophic plant–fungal associations. New Phytol 217:1230–1239. https://doi.org/10.1111/nph.14895
Zhang T, Wang N, Yu L (2020) Soil fungal community composition differs significantly among the Antarctic, Arctic, and Tibetan Plateau. Extremophiles 24:821–829. https://doi.org/10.1007/s00792-020-01197-7
Sundin GW, Jacobs JL (1999) Ultraviolet radiation (UVR) sensitivity analysis and UVR survival strategies of a bacterial community from the phyllosphere of field-grown peanut (Arachis hypogeae L.). Microb Ecol 38:27–38. https://doi.org/10.1007/s002489900152
Zhalnina K, Louie KB, Hao Z et al (2018) Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat Microbiol 3:470–480. https://doi.org/10.1038/s41564-018-0129-3
Lee SA, Kim Y, Kim JM et al (2019) A preliminary examination of bacterial, archaeal, and fungal communities inhabiting different rhizocompartments of tomato plants under real-world environments. Sci Rep 9:1–15. https://doi.org/10.1038/s41598-019-45660-8
Parniske M (2008) Arbuscular mycorrhiza: the mother of plant root endosymbioses. Nat Rev Microbiol 6:763–775
Zamioudis C, Hanson J, Pieterse CMJ (2014) ß-Glucosidase BGLU42 is a MYB72-dependent key regulator of rhizobacteria-induced systemic resistance and modulates iron deficiency responses in Arabidopsis roots. New Phytol 204:368–379. https://doi.org/10.1111/nph.12980
Stringlis IA, Yu K, Feussner K et al (2018) MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc Natl Acad Sci USA 115:E5213–E5222. https://doi.org/10.1073/pnas.1722335115
Costello EK, Halloy SRP, Reed SC et al (2009) Fumarole-supported islands of biodiversity within a hyperarid, high-elevation landscape on socompa volcano, puna de atacama, andes. Appl Environ Microbiol 75:735–747. https://doi.org/10.1128/AEM.01469-08
Moon JB, Wardrop DH, Bruns MAV et al (2016) Land-use and land-cover effects on soil microbial community abundance and composition in headwater riparian wetlands. Soil Biol Biochem 97:215–233. https://doi.org/10.1016/j.soilbio.2016.02.021
Sui X, Zhang R, Frey B et al (2019) Land use change effects on diversity of soil bacterial, Acidobacterial and fungal communities in wetlands of the Sanjiang Plain, northeastern China. Sci Rep 9:1–14. https://doi.org/10.1038/s41598-019-55063-4
Lin Y, Zhang Y, Liang X et al (2022) Assessment of rhizosphere bacterial diversity and composition in a metal hyperaccumulator (Boehmeria nivea) and a nonaccumulator (Artemisia annua) in an antimony mine. J Appl Microbiol 132:3432–3443. https://doi.org/10.1111/jam.15486
Huang L, Bai J, Wang J et al (2022) Different stochastic processes regulate bacterial and fungal community assembly in estuarine wetland soils. Soil Biol Biochem 167:108586. https://doi.org/10.1016/j.soilbio.2022.108586
Dini-Andreote F, Stegen JC, Van Elsas JD, Salles JF (2015) Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc Natl Acad Sci USA 112:E1326–E1332. https://doi.org/10.1073/pnas.1414261112
Feng Y, Chen R, Stegen JC et al (2018) Two key features influencing community assembly processes at regional scale: Initial state and degree of change in environmental conditions. Mol Ecol 27:5238–5251. https://doi.org/10.1111/mec.14914
Wang J, Liao L, Wang G et al (2022) N-induced root exudates mediate the rhizosphere fungal assembly and affect species coexistence. Sci Total Environ 804:150148. https://doi.org/10.1016/j.scitotenv.2021.150148
Chaparro JM, Badri DV, Vivanco JM (2014) Rhizosphere microbiome assemblage is affected by plant development. ISME J 8:790–803. https://doi.org/10.1038/ISMEJ.2013.196
Shi S, Nuccio EE, Shi ZJ et al (2016) The interconnected rhizosphere: high network complexity dominates rhizosphere assemblages. Ecol Lett 19:926–936. https://doi.org/10.1111/ELE.12630
Duan Y, Lian J, Wang L et al (2021) Variation in soil microbial communities along an elevational gradient in alpine meadows of the Qilian Mountains, China. Front Microbiol. https://doi.org/10.3389/fmicb.2021.684386
Blagodatsky S, Smith P (2012) Soil physics meets soil biology: towards better mechanistic prediction of greenhouse gas emissions from soil. Soil Biol Biochem 47:78–92
Bui A, Orr D, Lepori-Bui M et al (2020) Soil fungal community composition and functional similarity shift across distinct climatic conditions. FEMS Microbiol Ecol. https://doi.org/10.1093/femsec/fiaa193
Soudzilovskaia NA, van der Heijden MGA, Cornelissen JHC et al (2015) Quantitative assessment of the differential impacts of arbuscular and ectomycorrhiza on soil carbon cycling. New Phytol 208:280–293. https://doi.org/10.1111/nph.13447
Van Der Heijden MGA, Dombrowski N, Schlaeppi K (2017) Continuum of root–fungal symbioses for plant nutrition. Proc Natl Acad Sci USA 114:11574–11576
Smith SE, Smith FA (2011) Roles of arbuscular mycorrhizas in plant nutrition and growth: new paradigms from cellular to ecosystem scales. Annu Rev Plant Biol 62:227–250. https://doi.org/10.1146/annurev-arplant-042110-103846
Iqbal A, Maqsood Ur Rehman M, Sajjad W et al (2023) Patterns of bacterial communities in the rhizosphere and rhizoplane of alpine wet meadows. Environ Res 241:117672. https://doi.org/10.1016/j.envres.2023.117672
Martin MM, Kellogg CA, Hallock P (2019) Microbial associations of four species of algal symbiont-bearing foraminifers from the Florida reef tract, USA. J Foraminifer Res 49:178–190. https://doi.org/10.2113/gsjfr.49.2.178
Veach AM, Chen H, Yang ZK et al (2020) Plant hosts modify belowground microbial community response to extreme drought. mSystems. https://doi.org/10.1128/msystems.00092-20
Field KJ, Pressel S (2018) Unity in diversity: structural and functional insights into the ancient partnerships between plants and fungi. New Phytol 220:996–1011
Acknowledgements
We thank the Central Laboratory of the School of Life Science, Lanzhou University, for providing equipment.
Funding
This work was supported by the Second Tibetan Plateau Expedition (2019QZKK0302), the Natural Science Foundation of China (31961143012, U21A20183, 31870433, 42041005), the Fundamental Research Funds for the Central Universities (lzujbky-2021-ct10), and the ‘111’ Programme 2.0 (BP0719040).
Author information
Authors and Affiliations
Contributions
AW did the laboratory analyses, wrote-original draft, and prepared the figures. ZHS contributed to the conceptualization, funding acquisition, supervision, and allocation of resources. AW, and MMR processed the data. JN contributed during the sample collection. MU, WS, MR, and AAD edited and reviewed the final version of the manuscript. All the authors participated in editing the initial draft and approved the final submission version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Fig. S1. Relative abundance of fungal phyla in the a rhizosphere and b rhizoplane associated with Cremanthodium ellisii, Cremanthodium lineare, and Caltha scaposa. Fig. S2. Differences in the alpha diversity of rhizosphere associated with Cremanthodium lineare, Caltha scaposa, and Cremanthodium ellisii at three sites, SI, SII, and SIII based on the Kruskal−Wallis test with False Discovery Rate (FDR) multiple correction tests; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Fig. S3. Differences in the alpha diversity of rhizoplane associated with Cremanthodium lineare, Caltha scaposa, and Cremanthodium ellisii at three sites, SI, SII, and SIII, based on the Kruskal−Wallis test with False Discovery Rate (FDR) multiple correction tests; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Fig. S4. Hierarchical cluster and clustering heatmap of the rhizosphere (a, b) and rhizoplane (c, d) based on the Bray-Curtis distance algorithm of intra- fungal communities associated with Cremanthodium ellisii, Cremanthodium lineare, and Caltha scaposa at three sites, SI, SII, and SIII. Fig. S5. Fungal community assembly process associated with rhizosphere and rhizoplane, a βNTI results with dashed lines at + 2 and − 2, b RCbray results with dashed lines at + 95 and − 95 confidence intervals, Community assembly process of c rhizosphere, d rhizoplane; HTS heterogenous selection, UD undominated, DL dispersal limitation, HMS homogenous selection, HMD homogenous dispersal, and proportion of stochastic and deterministic processes of e rhizosphere, and f rhizoplane. Table S1. Characteristics of the sampling sites. Table S2. Soil physicochemical properties of the rhizosphere and rhizoplane samples associated with Cremanthodium ellisii, Cremanthodium lineare, and Caltha scaposa. Table S3. Metastats test between the fungal communities of rhizoplane and rhizosphere at the genus level. Table S4. The α-diversity indices of the rhizosphere fungal community associated with Cremanthodium ellisii, Cremanthodium lineare, and Caltha scaposa. Table S5. The α-diversity indices of the rhizoplane fungal community associated with Cremanthodium ellisii, Cremanthodium lineare, and Caltha scaposa. Table S6. Lineare mixed model analysis of fungal α-and β- diversity indices with plants, sites, and root compartments. Table S7. Characteristics of fungal community assembly in the rhizosphere and rhizoplane. Table S8. Topological features of the rhizosphere network analysis. Table S9. Topological features of the rhizoplane network analysis. Table S10. Percent abundance of OTUs representing fungal trophic modes and functional guilds in the rhizosphere and rhizoplane.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Iqbal, A., Maqsood Ur Rehman, M., Usman, M. et al. Unraveling the relationship between plant species and physicochemical properties on rhizosphere and rhizoplane fungal communities in alpine wet meadows. Environ Sci Eur 35, 115 (2023). https://doi.org/10.1186/s12302-023-00823-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12302-023-00823-3