
Abstract
The neuropeptides calcitonin gene-related peptide (CGRP) and pituitary adenylate cyclase-activating polypeptide (PACAP) have emerged as mediators of migraine pathogenesis. Both are vasodilatory peptides that can cause migraine-like attacks when infused into people and migraine-like symptoms when injected into rodents. In this narrative review, we compare the similarities and differences between the peptides in both their clinical and preclinical migraine actions. A notable clinical difference is that PACAP, but not CGRP, causes premonitory-like symptoms in patients. Both peptides are found in distinct, but overlapping areas relevant to migraine, most notably with the prevalence of CGRP in trigeminal ganglia and PACAP in sphenopalatine ganglia. In rodents, the two peptides share activities, including vasodilation, neurogenic inflammation, and nociception. Most strikingly, CGRP and PACAP cause similar migraine-like symptoms in rodents that are manifested as light aversion and tactile allodynia. Yet, the peptides appear to act by independent mechanisms possibly by distinct intracellular signaling pathways. The complexity of these signaling pathways is magnified by the existence of multiple CGRP and PACAP receptors that may contribute to migraine pathogenesis. Based on these differences, we suggest PACAP and its receptors provide a rich set of targets to complement and augment the current CGRP-based migraine therapeutics.
Similar content being viewed by others

Background
Migraine is one of the most disabling neurological disorders in the world [1]. It affects over one billion people with 3:1 prevalence in women. Migraine is a headache lasting 4–72 h with characteristics that often include unilateral pulsating pain of moderate to severe intensity that is aggravated by routine physical activity and is associated with vomiting or nausea and/or photophobia and phonophobia [2]. While the mechanisms of migraine are still poorly understood, insights from clinical and preclinical studies over the past three decades have focused attention on two neuropeptides: calcitonin gene-related peptide (CGRP) and pituitary adenylate cyclase-activating polypeptide (PACAP).
The importance of CGRP in migraine was first realized in reports that CGRP is upregulated during migraine attacks and between attacks in chronic migraine patients [3,4,5], although this is not seen in all studies [6,7,8]. Even more striking was the finding that infusion of CGRP can induce migraine-like attacks in migraine patients, as described below. The importance of CGRP has been fully established over the past 5 years with the efficacy of eight FDA approved CGRP-based therapeutics [9,10,11,12,13]. These drugs include monoclonal antibodies against CGRP or its receptor and small molecule receptor antagonists that are effective for prevention and treatment of migraine. However, in general only about 40–60% of migraine patients are significantly helped by these agents [12, 14, 15], which suggests involvement of other factors beyond CGRP in migraine pathophysiology, such as PACAP [16]. In this context, patients who do not respond well to CGRP-based drugs might respond to drugs that target PACAP and similarly, a combinatorial approach targeting both CGRP and PACAP might improve treatment efficacies.
Like CGRP, PACAP has been linked to migraine pathogenesis [17,18,19]. The PACAP gene encodes two isoforms containing either 27 or 38 amino acids with PACAP-38 being the more prevalent, representing 90% of PACAP forms in mammalian tissues [20, 21]. Unless otherwise indicated, we will refer to both isoforms simply as PACAP. As with CGRP, elevated plasma PACAP levels during migraine have been reported [19, 22], but not consistently observed [23, 24], and infusion of either the PACAP-38 or PACAP-27 isoforms caused migraine in people, as described below [18, 25].
The goal of this narrative review is to briefly compare and contrast the actions of CGRP and PACAP in migraine patients and rodent migraine models. For more extensive reviews on CGRP and PACAP, the reader is referred to a number of excellent reviews [23, 26,27,28,29,30,31,32,33].
Infusion of CGRP and PACAP in patients
A key similarity of CGRP and PACAP is their ability to induce migraine-like headaches when infused into migraine patients (Table 1). Intravenous infusion of CGRP caused a delayed migraine-like headache in about 63% of migraine patients (50–77%), but only a mild immediate headache in control subjects [34,35,36,37,38,39,40]. Similarily, intravenous infusion of PACAP-38 caused a delayed migraine-like headache in about 68% of migraine patients (58–73%), but only rarely in control subjects [17, 18, 38, 41]. This was also seen with the shorter PACAP isoform, PACAP-27 [25]. In addition, both CGRP and PACAP-induced attacks in migraine patients were effectively treated by sumatriptan [35, 42]. However, sumatriptan did not block CGRP-induced headaches in control subjects [43], which emphasizes the importance of doing infusion studies in migraine patients.
The frequencies of CGRP and PACAP induced attacks were comparable to those observed with other migraine triggers (Table 1). These triggers include other members of the CGRP and PACAP families, the nitric oxide donor glyceryl trinitrate (GTN), phosphodiesterase inhibitors that elevate cAMP and cGMP levels, an activator of ATP-sensitive potassium (KATP) channels, and inflammatory agents (histamine, prostaglandins).
With respect to identifying the relevant receptors for CGRP and PACAP involved in migraine, it is informative that other members of the CGRP and PACAP peptide families can induce migraine. Two CGRP-related peptides, adrenomedullin and a synthetic analog of amylin (pramlintide), triggered migraine-like attacks (Table 1) [40, 46]. As discussed below, CGRP, amylin, and adrenomedullin act via a family of related G protein-coupled receptors (GPCRs) and in particular, CGRP binds the amylin 1 (AMY1) receptor with equal affinity as its canonical receptor [26]. In the case of PACAP, the family member vasoactive intestinal peptide (VIP) caused migraine-like headaches comparable to PACAP, but only after prolonged infusion to mimic the longer lasting vascular actions of PACAP [60]. The shared ability of PACAP and VIP is important since while the PACAP1 (PAC1) receptor is preferentially activated by PACAP, the two peptides are equally active at the VIP-PACAP (VPAC) receptors VPAC1 and VPAC2, as discussed below.
In migraine patients, CGRP also induced non-headache symptoms characteristic of migraine, including photophobia, phonophobia, and nausea. In addition to headache, cranial vascular changes were observed with dilation of the middle cerebral artery (MCA) and middle meningeal artery (MMA) [35, 61, 62]. Like CGRP, PACAP also induced photophobia and other non-headache symptoms. As with CGRP, there were cranial vascular changes. PACAP-induced headache was associated with prolonged dilation of the MMA but not the MCA [17, 63]. Both CGRP and PACAP caused side effects likely due to systemic vasodilation (flushing, warm sensation, palpitation, dizziness), although PACAP caused additional effects not seen with CGRP-infusion [37, 38, 41] (Fig. 1).

Clinical symptoms caused by CGRP and PACAP infusions. Both CGRP and PACAP cause migraine-like headache in about 2/3 of migraine patients. PACAP causes more premonitory symptoms and side effects than CGRP. Data are only from studies that included premonitory symptoms [37, 38, 41]. For a comprehensive listing of CGRP and PACAP infusion studies and migraine frequencies, see Table 1. Created with BioRender.com
A difference between CGRP and PACAP was revealed when patients were asked if they developed premonitory symptoms after peptide infusion (Fig. 1). Premonitory symptoms occur prior to the headache in most migraine patients [64, 65]. Premonitory symptoms most commonly observed include fatigue, yawning, neck stiffness, hunger or food cravings, mood swings, poor concentration, and sometimes photophobia and phonophobia, which also occur during the headache phase. After PACAP infusion, a delayed migraine-like headache was reported by 23 of 32 patients (72%) and 11 of those 23 (48%) reported one or more premonitory symptoms prior to the headache [38]. In contrast, after CGRP infusion, while migraine was reported by 25 of 40 patients (63%), only 2 of those 25 (9%) reported premonitory symptoms prior to the headache. This difference in premonitory symptoms between CGRP and PACAP may reflect PACAP's ability, albeit limited, to enter the central nervous system (CNS) [66]. Within the CNS, the hypothalamus has been strongly associated with the premonitory phase by imaging studies [67, 68] and other criteria [69]. Importantly, the hypothalamus has abundant PACAP receptors [70]. However, caution must be exercised in interpreting these results due to several caveats, most notably the lack of placebo and non-migraine control groups [38]. These caveats are particularly important since CGRP and PACAP induced premonitory symptoms to the same extent in patients who did not develop a migraine attack as those who did, which raises the prospect that patients may have been exhibiting peptide responses that were not necessarily premonitory of migraine. Hence, it might be safer to refer to the symptoms as premonitory-like. Nonetheless, it is clear that both CGRP and PACAP can induce a delayed migraine-like headache, and that PACAP can also initiate premonitory-like symptoms.
To understand the mechanism of CGRP and PACAP induced headaches, Ashina and colleagues tested whether CGRP and PACAP share KATP channels as a downstream cellular target. KATP channels are ATP regulated potassium channels located in trigeminovascular neurons and vessels. The rationale of this idea was based on studies showing that the KATP channel opener levcromakalim was a potent inducer of migraine in patients (Table 1) [55, 56], and that both CGRP and PACAP elevate cAMP levels, which in vascular smooth muscle would activate the channels, leading to vasodilation associated with headache [71]. Yet neither CGRP nor PACAP actions were blocked by treatment with an inhibitor of KATP channels, glibenclamide [72, 73]. However, the lack of efficacy of glibenclamide must be tempered by the caveats that the studies were not done in migraine patients and glibenclamide only delayed and did not prevent levcromakalim-induced headaches [74]. Furthermore, preclinical allodynia studies described below showed that glibenclamide inhibits CGRP, but not PACAP actions in mice. Further studies with glibenclamide and other antagonists in migraine patients are needed to help resolve this discrepancy.
Migraine relevant sites of CGRP and PACAP and their receptors
Based on the shared ability of exogenous CGRP and PACAP to cause migraine, a pertinent question is where are endogenous sites of CGRP and PACAP expression and action in the central and peripheral nervous systems? Both peptides and their receptors are found in multiple areas relevant to migraine, ranging from the hypothalamus to the trigeminal ganglia (Fig. 2). These sites largely overlap, but there are differences and few studies have looked at cellular co-expression other than in the trigeminal and sphenopalatine ganglia [75, 76].

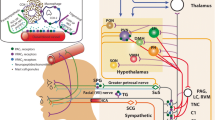
Sites of CGRP, PACAP, and their receptors in the CNS and cranial structures. CGRP, PACAP and their receptors are present in meningeal and vascular cells [77,78,79,80], hypothalamus [75, 76], thalamus [75, 76], amygdala [76, 81,82,83], cerebellum [75, 76], cerebral cortex [75, 76], sphenopalantine ganglia (SPG) [84,85,86,87], bed nucleus of stria terminalis (BST) [81, 83, 88, 89], periaqueductal gray (PAG) [90,91,92], locus coeruleus (LC) [75, 76], trigeminal nucleus caudalis (TNC) [93,94,95], parabrachial nucleus (PBN) [81, 83, 96, 97], trigeminal ganglia (TG) [98,99,100], dorsal root ganglia (DRG) [98, 101,102,103], and spinal cord [91, 93, 104]. Peripheral cranial structures are indicated with a black circle. For peptides, location within a region indicates presence in cell bodies and/or fibers. Relative abundance or cellular resolution of the two peptides or their receptors have generally not been directly compared, with the exception of the TG and SPG, where the relative abundances of CGRP over PACAP in the TG and PACAP over CGRP in the SPG are indicated. For receptors, location in a region is a collective assessment of CGRP receptors (canonical CGRP, AMY1) and PACAP receptors (PAC1, VPAC1, VPAC2, MRGB2/B3/X2). Created with BioRender.com
In the peripheral nervous system, CGRP is predominantly expressed in sensory neurons of the dorsal root and trigeminal ganglia, although it is also found in motor neurons and is abundant within the enteric nervous system [26]. The distribution of CGRP appears to be largely similar across species (reviewed in [26]). In the mouse, rat and human trigeminovascular system, CGRP is primarily found in the perivascular afferents innervating cranial arteries [105, 106]. Within rat and human trigeminal ganglia, PACAP and CGRP are found in neurons, and PACAP receptors are found on both neurons and satellite glia [98, 107,108,109]. While co-localized with CGRP, PACAP is found in far fewer neurons [110]. Although, PACAP receptors PAC1 and VPAC1 are found in rat and human satellite glia [111], their functions are not known. CGRP receptors are also found on subsets of trigeminal ganglia neurons and satellite glia in rats, mice and humans, where they may contribute to peripheral sensitization in migraine [26, 106, 112].
In contrast to CGRP, in the periphery of the cranium, PACAP is mainly expressed in parasympathetic neurons with a much smaller trigeminal distribution than CGRP in rats and humans [99, 110]. The predominant site of PACAP expression in rats and humans is the extracranial parasympathetic sphenopalatine ganglion, which also contains PACAP receptors [84, 85, 113]. Stimulation of the sphenopalatine ganglia likely contributes to autonomic symptoms of migraine since it can increase cerebral blood flow, intracranial and extracranial vasodilation, and dural plasma protein extravasation in humans [114]. Within the sphenopalatine ganglia, CGRP containing fibers from the trigeminal ganglia were found in both rat and human [86, 87]. CGRP was also found in neural cell bodies but only in rats, not humans [86]. Interestingly, PACAP can induce release of CGRP from rat trigeminal neurons [107] and stimulation of the rat superior salivatory nucleus can activate neuronal trigeminovascular actions and cranial autonomic symptoms [115, 116]. These results all suggest the possibility of cross-talk between the sphenopalatine and trigeminal systems.
Within the CNS, both peptides and their receptors are found in migraine-relevant regions across species, especially the hypothalamus (Fig. 2) [26, 70]. CGRP receptors have been identified throughout the CNS and are particularly abundant in the human cerebellum [117]. PACAP is found in the spinal cord and second order neurons of the trigeminal nucleus caudalis (TNC) of rodents and humans [93, 118, 119]. Similar to CGRP, PACAP binds to a variety of sites throughout the CNS, including the hypothalamus, thalamus, various areas throughout the brainstem, and the dorsal horn of the spinal cord across species [120,121,122]. In particular, the PAC1 receptor is expressed throughout the brain, including the neocortex, limbic system, and brainstem [123]. Like CGRP [26], PACAP has been linked to anxiety-like behavior [124, 125]. PACAP and PAC1 knockout mice have decreased anxiety-like behavior. Both knockouts show a variety of neurobehavioral phenotypes including increased hyperactivity, decreased depression-like behavior, and aberrant social interaction [124, 125]. Studies have identified a genetic association with PACAP and the PAC1 receptor with post-traumatic stress disorder in humans and shown that alterations in the PACAP/PAC1 pathway are involved in stress responses in rodents [126]. In addition, chronic stress increased PACAP expression within the rat bed nucleus of the stria terminalis [127]. These findings document a role for PACAP in stress and anxiety, which are both associated with migraine [128]. Hence, the locations of CGRP and PACAP peptides and their receptors are overlapping and well-positioned to contribute to peripheral and central actions in migraine.
CGRP and PACAP migraine-like functions
CGRP and PACAP roles in vasodilation, neurogenic inflammation, and nociception
Both CGRP and PACAP are multifunctional peptides with many roles in the nervous, cardiovascular, respiratory, gastrointestinal, and reproductive systems [26, 123, 129]. We will briefly focus on three processes that are associated with migraine: vasodilation, neurogenic inflammation, and nociception. While the role of vasodilation and neurogenic inflammation in migraine remains a debated topic, and neurogenic inflammation has not led to a successful therapeutic, it does seem likely that the vasculature and local inflammatory signals contribute to peripheral sensitization and hence to migraine [26, 130].
Both CGRP and PACAP are well-characterized vasodilatory peptides [131], and as mentioned above both can act on cranial vessels. It is intriguing that the two peptides, along with another commonly used migraine trigger, GTN (a nitric oxide donor), are all vasodilators [132]. In addition to their contributions via vasodilation in neurogenic inflammation in rats [133, 134], CGRP and PACAP cause mast cell degranulation and release of inflammatory compounds. These CGRP actions are well-documented in the rat dura [80]. PACAP-38 was reported to induce dural mast cell degranulation in rats and was significantly more potent than VIP and PACAP-27 [135, 136]. Like CGRP, PACAP is upregulated following inflammation in sensory neurons [137]. However, the complexity of PACAP actions is highlighted by the fact that in contrast to the dura, PACAP inhibits neurogenic inflammation in rodent skin [138,139,140,141]. Nonetheless, within the meninges, it seems likely both PACAP and CGRP can contribute to neurogenic inflammation.
With respect to nociception, the story is even more complex. While CGRP is recognized as a nociceptive peptide [26], PACAP appears to have both antinociceptive and nociceptive functions. In the periphery, PACAP was reported to be antinociceptive [138,139,140,141]. In contrast, PACAP in the CNS appears to be nociceptive based on studies with PACAP knockout mice suggesting a possible role in central sensitization [141]. Similarly, injection of PACAP into the hypothalamic paraventricular nucleus increased TNC activity in rats, which could be inhibited by a PAC1 receptor antagonist [142] and intrathecal injection of PACAP has been shown to induce hyperalgesia in mice [122]. PACAP also causes a delayed activation and sensitization of central trigeminovascular neurons. Specifically, the central PAC1 receptors have been implicated in pro-nociceptive transmission. A centrally, but not peripherally, administered PAC1 receptor antagonist was able to inhibit dural nociceptive-evoked action potentials in central trigeminovascular neurons in rats, suggesting that the central PAC1 receptor is involved in PACAP-induced migraine [143].
Light aversion induced by CGRP and PACAP in mice
A shared activity of CGRP and PACAP is their ability to induce similar light aversive phenotypes in mice [144]. The light aversion assay serves as a surrogate for human photophobia [145, 146]. Central (intracerebroventricular, thalamic, and cerebellar) and peripheral (intraperitoneal) injection of CGRP induced light aversion in wildtype mice [144, 147,148,149,150]. CGRP-induced light aversion was accompanied by increased resting only in the dark zone, and a lack of light-independent anxiety in an open field assay [147, 151,152,153]. Likewise, intraperitoneal injection of PACAP caused light aversion coupled with increased resting in the dark and no anxiety in the open field [144]. These findings are consistent with a pioneering study by Helyes and colleagues who reported that peripheral injection of GTN and PACAP induced light aversive behavior in wildtype mice, but not in PACAP knockout mice [154]. It should be noted that compared to PACAP-38, injection of PACAP-27 caused only transient light aversion [144]. A pharmacokinetic explanation cannot be ruled out since the relative stability of the two PACAP isoforms is not clear [21]. However, it is possible that PACAP-38, but not PACAP-27, acts by mast cell degranulation, as shown for dilation of the MMA in rats [155]. In fact, in rats only PACAP-38 can degranulate mast cells and acts via the Mas-related GPCR B3 (MrgB3) receptor [156]. Studies exploring the role of the MrgB3 homologs in mice (MrgB2) and humans (MRGX2) may give insights to how PACAP-38 evokes symptoms of migraine.
Despite the similarities, CGRP and PACAP act independently in the light aversion assay. This was shown by the fact that CGRP and PACAP responses could not be blocked by monoclonal antibodies directed against the other peptide [144]. Hence, PACAP-induced responses could be blocked with a monoclonal anti-PACAP antibody, but not by an anti-CGRP antibody. Conversely, CGRP-induced responses could be blocked by an anti-CGRP antibody, but not by an anti-PACAP antibody. This result suggests that CGRP and PACAP do not act by sequential or dependent pathways. The possibility of dependent actions had been raised by the similar properties of CGRP and PACAP [131], co-expression in rat trigeminal ganglia neurons [99], and PACAP-38 causing CGRP release in the rat TNC (although not from the dura or ganglia) [107]. Contrary to the latter observation in rats, a clinical study did not detect increased CGRP levels after PACAP-38 infusion [24]. Furthermore, GTN increased the number of PACAP-responsive neurons in mouse trigeminal ganglia by a mechanism independent of CGRP [157]. In contrast, the parallel increase in CGRP-responsive neurons required CGRP. These data all suggest that PACAP and CGRP can act by distinct pathways that converge downstream of the receptors to cause migraine-like symptoms.
Further support for CGRP and PACAP acting by different pathways to cause light aversion is that PACAP was effective in only a subpopulation of CD-1 mice and their offspring, which was not seen with CGRP [144]. The CD-1 strain is a genetically diverse outbred strain of mice, which raised the possibility of genetic differences between the responder and nonresponder populations. An RNA-seq analysis of trigeminal ganglia gene expression between the two populations revealed a number of candidate genes, including pituitary hormones, receptors, and ion channels that are potential biomarkers and therapeutic targets. Whether these genes will provide clues for identifying human responder/nonresponder populations remains to be seen but this finding of heterogeneity reflects an advantage of using genetically diverse mice that may better model the variability observed in humans [158].
Allodynia induced by CGRP and PACAP in rodents
Subcutaneous injection of CGRP in the periorbital area in mice caused dose and time dependent mechanical allodynia [159]. This CGRP-induced periorbital allodynia was abolished by pretreatment with a CGRP receptor antagonist (olcegepant) or a monoclonal anti-CGRP antibody [159]. Similar allodynia was also induced by intraperitoneal and intrathecal injections of CGRP in mice [160, 161] and intraganglionic injections of CGRP into rat trigeminal ganglia [162]. Subcutaneous injection of PACAP in the periorbital area also caused dose and time dependent mechanical allodynia and was blocked by pretreatment with a PACAP receptor antagonist, PACAP6-38 [159]. Similarly, subcutaneous injection of PACAP induced plantar and periorbital hypersensitivity in wildtype mice [163].
Consistent with the light aversion findings, CGRP and PACAP-induced allodynia appears to act via independent pathways as reported by Christensen and colleagues. They observed PACAP responses in wildtype mice pretreated with anti-CGRP antibody, as well as in Ramp1 knockout mice lacking CGRP receptors [163]. For comparison, allodynic responses to GTN treatments were blocked by anti-CGRP antibodies in wildtype mice and not seen in the Ramp1 knockout mice [164]. This indicates that PACAP acts independently of CGRP signaling. Separate pathways were also suggested by pretreatment with the KATP channel inhibitor glibenclamide. Glibenclamide was able to block GTN-induced allodynia in mice, which involves CGRP [164, 165], but only partially attenuated PACAP-induced hypersensitivity, indicating that PACAP does not fully depend on this channel [163]. A follow up study showed that pretreatment with anti-PACAP antibody blocked PACAP-induced plantar hypersensitivity but was not able to block hypersensitivity caused by GTN or the KATP channel opener levcromakalin [166]. However, a caveat of these comparisons is that they did not directly test glibenclamide or PACAP antibodies against CGRP, but rather against GTN, which acts via CGRP, at least in rodents. Glibenclamide was also able to attenuate cephalic allodynia in spontaneous trigeminal allodynic rats and inhibited release of CGRP from dura mater and trigeminal ganglion [165]. Yet, translation of these rodent studies to migraine patients remains to be established, since as mentioned earlier, glibenclamide was unable to block CGRP or PACAP-induced headache in control subjects [72, 73]. While these findings suggest that CGRP and GTN act by pathways not shared with PACAP, other data link PACAP to nitric oxide pathways. Peripheral injection of GTN increased PACAP within the rat TNC [119], and increased the number of PACAP-responsive neurons in mouse trigeminal ganglia [157]. Also, GTN induced more vasodilation and neuronal activation in trigeminal ganglia and the TNC in wildtype mice compared to PACAP knockout mice [154]. Taken together, while there seems to be some cross-talk between CGRP, PACAP and nitric oxide in the trigeminovascular system, it appears that PACAP and CGRP can act by independent pathways to cause tactile and light sensitivities.
Signaling by multiple CGRP and PACAP receptors
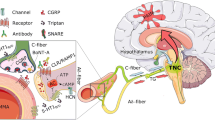
CGRP and PACAP receptors are both Gs-coupled and activate cAMP-dependent pathways [26, 123, 167,168,169,170] (Fig. 3). In addition, both peptides have been shown to activate MAP kinase pathways and are reported to couple to Gq, which signals via calcium pathways involving phospholipase C and inositol 1,4,5-triphosphate (IP3) activity [169, 171, 172]. However, conflicting results have been reported for the direct measurement of coupling of CGRP receptors to Gq [173, 174]. Furthermore, identification of Gq coupling has been mostly inferred from CGRP mediated calcium mobilization and IP3 signaling [175, 176]. Interestingly, in one report of CGRP signaling via Gq, the authors suggested that these cells did not have a cAMP response to CGRP [177]. Perhaps the absence of Gs and/or the high expression of Gq allows preferential Gq activation. In contrast, PACAP receptors have more robust evidence for Gq-mediated signaling [178], although direct comparisons between PACAP and CGRP receptors can be difficult to make due to differences in model systems. In a series of studies using the same transfected receptor model, CGRP mediated stimulation of IP accumulation (measured as IP1, a breakdown product of IP3) was > 200 fold less potent relative to cAMP [179, 180]. Whereas, PACAP- mediated stimulation of IP was only approximately 4–tenfold less potent relative to cAMP [181]. Hence, IP signaling appears to be more robust for PACAP receptors than CGRP receptors. Overall, it seems that the CGRP receptor can couple to Gq, but the coupling may not be as robust as Gs coupling. Whereas PACAP receptors appear to effectively couple both Gq and Gs.

Schematic of CGRP and PACAP signaling pathways. CGRP and PACAP can act via multiple receptors, as indicated. For simplicity, signaling pathways from generic receptors in a generic cell type are illustrated. In general, activation of both CGRP and PACAP receptors increase cAMP levels, which leads to protein kinase A (PKA) activation and EPAC1/2 activation. EPAC1/2 activation by PACAP is well-established, although activation by CGRP is less clear (dotted line). Canonical CGRP receptor, but not AMY1 CGRP receptor, and the PACAP receptor PAC1 can generate endosomal signals following β-arrestin-mediated receptor internalization. Activation of additional G protein pathways that elevate IP3 and calcium have been reported for PACAP and to a lesser extent CGRP (dotted lines). These pathways activate multiple downstream targets, including MAP kinases (MAPKs), ion channels, and genes, depending on the cell type. Created with BioRender.com
Thus, CGRP and PACAP receptors have the potential to activate similar intracellular signaling pathways that could lead to a host of cellular events, ranging from ion channel activation to mast cell degranulation (Fig. 3). Potential cellular targets relevant to migraine are likely both in the CNS, such as the hypothalamus and TNC, and in the periphery, such as in the meninges and trigeminal ganglia, where numerous cell types express both CGRP and PACAP receptors (Fig. 2) [28, 70, 99, 182]. CGRP and PACAP actions on these cells potentially activate similar intracellular signals leading to peripheral and central sensitization. Yet despite these similarities, the differences between CGRP and PACAP actions in people and rodents suggest divergent intracellular pathways and targets. However, before we can better understand these differences, a key step will be to identify the relevant receptors for each peptide.
For CGRP, there are two receptors with approximately equal affinities [26]. The canonical CGRP receptor is a heterodimer of the GPCR calcitonin receptor-like receptor (CLR) and receptor activity-modifying protein 1 (RAMP1). A second CGRP receptor, AMY1, is a heterodimer of the GPCR calcitonin receptor (CTR) and RAMP1. Both can activate cAMP pathways [183] (Fig. 3). However, a direct comparison is needed between the receptors given the heterogeneity of intracellular cAMP targets seen so far with the canonical receptor [169, 184]. While the relative contributions of the two receptors in migraine remain to be established, a role for AMY1 is supported by the ability of AMY selective ligands to cause migraine in people [40] and light aversion, touch sensitivity, and grimace in mice [40, 185]. While CGRP has a lower affinity for the adrenomedullin receptors (CLR/RAMP2 and CLR/RAMP3), given the ability of adrenomedullin to induce migraine-like attacks similar to CGRP [46], perhaps CGRP actions via these receptors should not be ignored.
For PACAP, the canonical receptors are the GPCRs VPAC1, VPAC2, and PAC1 [186], which all activate adenylate cyclase and increase intracellular cAMP levels analogous to both CGRP receptors [187, 188] (Fig. 3). However, one difference between PACAP and CGRP receptors may be the ability of PACAP receptors to recruit a noncanonical cAMP signaling pathway involving the Exchange Proteins directly Activated by cAMP (EPACs) [189, 190]. The EPACs are cAMP-activated guanine nucleotide exchange factors that activate small GTPases and thus expand the diversity of cAMP signaling pathways beyond the long-recognized canonical pathway involving protein kinase A [191]. Among these EPAC targets are MAP kinases [192], although PKA and endosomal β-arrestin complexes can also activate MAP kinases [193]. Whether CGRP receptors also use EPACs is not as well established. CGRP may recruit EPACs in macrophages [194], but in dendritic cells it seemingly does not [195]. In a study with primary cardiovascular cells, activation of ERK1/2 MAP kinase by CGRP acting at the adrenomedullin receptor (a low affinity member of the CGRP receptor family) was shown to be mediated by a Gi/o pathway, while adrenomedullin used a combination of Gq/11/14 signaling and EPAC activation not used by CGRP to activate ERK1/2 MAP kinase [196]. This example of biased agonism illustrates the diversity of different G protein couplings and their downstream signaling pathways for a receptor closely related to the canonical CGRP receptor. Interestingly, there is a long established connection between EPAC signaling and pain [197], although most evidence to date places EPAC signaling upstream of CGRP, leading to CGRP release from nociceptive neurons [191]. Thus, while both PACAP and CGRP signal via cAMP, there is the possibility that they may use different cAMP signaling pathways.
An intriguing difference between the two CGRP receptors is that they have distinct internalization kinetics from the plasma membrane. Cell culture data clearly show that CGRP binding to the canonical receptor causes β-arrestin complexes and internalization to endosomes, but not the AMY1 receptor [198,199,200]. Thus, the AMY1 receptor potentially has prolonged cell surface signaling, while the internalized canonical receptor could continue to signal from endosomes. Importantly, endosomal signaling has been reported to be responsible for CGRP-mediated nociception [201, 202]. Likewise, endosomal signaling has been reported from the PAC1 receptor [193] and VPAC1 and VPAC2 can also be internalized as β-arrestin complexes in endosomes [203]. As with the two CGRP receptors, the relative contributions of cell surface and internal signaling in migraine by the multiple PACAP receptors remains an open question.
A final consideration is that until recently, the dogma was that PACAP must be acting via PAC1 and not the VPAC1 or VPAC2 receptors, which bind both PACAP and VIP. The rationale was primarily based on a report that VIP could not induce migraine in patients [204]. Consequently, the first PACAP-based monoclonal antibody to be tested was an antagonist to the PAC1 receptor. Since the trial failed to meet primary and secondary endpoints [205], this suggests either poor target engagement or possibly involvement of PAC1 splice variants [181], or other receptors. Indeed, VPAC1 and VPAC2 should be considered as therapeutic targets since more recent studies have shown that prolonged VIP infusion can cause delayed headache in people [60] and that VIP can induce light aversive behavior in mice if measured immediately after administration [148]. Alternatively, it is possible that PACAP involvement in migraine may be independent of VPAC1, VPAC2 or PAC1 receptors. PACAP can act in the trigeminal nucleus via an unidentified mechanism [107] and MrgB3 can mediate PACAP actions on mast cells in rats [156]. Another less characterized candidate PACAP receptor may be GPR55 [206]. Hence, there are no shortage of candidate receptors for PACAP actions relevant to migraine, any of which has the potential for different intracellular signaling pathways from CGRP.
Conclusion
CGRP plays an integral role in migraine. However, CGRP alone cannot account for all cases of migraine. The neuropeptide PACAP is likely to play a related, but distinct role as CGRP based on similarities and differences observed in both clinical and preclinical studies. The PACAP pathway appears to be independent of the CGRP pathway in rodent models [144, 163] suggesting that CGRP and PACAP act by parallel paths that converge downstream of their receptors. The existence of multiple CGRP and PACAP receptors provides a plethora of potential diversity in signaling pathways for each peptide. Thus, we suggest that PACAP and its receptors provide ideal therapeutic targets to complement and augment the current CGRP-based migraine therapeutics.
Availability of data and materials
Not applicable.
Abbreviations
- AMY1 :
-
Amylin1 receptor
- CGRP:
-
Calcitonin gene-related peptide
- CLR:
-
Calcitonin receptor-like receptor
- CTR:
-
Calcitonin receptor
- CNS:
-
Central nervous system
- EPAC:
-
Exchange Proteins directly Activated by cAMP
- GPCR:
-
G protein-coupled receptor
- GTN:
-
Glyceryl trinitrate
- IP3 :
-
Inositol 1,4,5-triphosphate
- KATP :
-
ATP-sensitive potassium channel
- MCA:
-
Middle cerebral artery
- MMA:
-
Middle meningeal artery
- Mrg:
-
Mas-related GPCR
- PACAP:
-
Pituitary adenylate cyclase-activating polypeptide
- PAC1 :
-
PACAP1 receptor
- RAMP:
-
Receptor activity-modifying protein
- TNC:
-
Trigeminal nucleus caudalis
- VIP:
-
Vasoactive intestinal peptide
- VPAC:
-
VIP-PACAP receptor
References
Collaborators GBDH (2018) Global, regional, and national burden of migraine and tension-type headache, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 17(11):954–976
Headache Classification Committee of the International Headache Society (IHS) (2018) The International Classification of Headache Disorders, 3rd edition. Cephalalgia 38(1):1–211
Ramon C, Cernuda-Morollon E, Pascual J (2017) Calcitonin gene-related peptide in peripheral blood as a biomarker for migraine. Curr Opin Neurol 30(3):281–286
Goadsby PJ, Edvinsson L (1993) The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol 33(1):48–56
Goadsby PJ, Edvinsson L, Ekman R (1990) Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 28(2):183–187
Tesfay B et al (2022) Is calcitonin gene-related peptide a reliable biochemical marker of migraine? Curr Opin Neurol 35(3):343–352
Tvedskov JF et al (2005) No increase of calcitonin gene-related peptide in jugular blood during migraine. Ann Neurol 58(4):561–568
Lee MJ et al (2018) Feasibility of serum CGRP measurement as a biomarker of chronic migraine: a critical reappraisal. J Headache Pain 19(1):53
Dodick DW et al (2014) Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomised, double-blind, placebo-controlled, exploratory phase 2 trial. Lancet Neurol 13(11):1100–1107
Dodick DW et al (2014) Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene-related peptide, for the prevention of migraine: a phase 2, randomised, double-blind, placebo-controlled study. Lancet Neurol 13(9):885–892
Bigal ME et al (2015) Safety, tolerability, and efficacy of TEV-48125 for preventive treatment of chronic migraine: a multicentre, randomised, double-blind, placebo-controlled, phase 2b study. Lancet Neurol 14(11):1091–1100
Rissardo JP, Caprara ALF (2022) Gepants for acute and preventive migraine treatment: a narrative review. Brain Sci 12(12):1612
Charles A, Pozo-Rosich P (2019) Targeting calcitonin gene-related peptide: a new era in migraine therapy. Lancet 394(10210):1765–1774
Pavelic AR et al (2022) Monoclonal antibodies against calcitonin gene-related peptide for migraine prophylaxis: a systematic review of real-world data. Cells 12(1):143
Moreno-Ajona D, Villar-Martinez MD, Goadsby PJ (2022) New generation gepants: migraine acute and preventive medications. J Clin Med 11(6):1656
Russo AF (2017) Overview of neuropeptides: awakening the senses? Headache 57(Suppl 2):37–46
Amin FM et al (2014) Investigation of the pathophysiological mechanisms of migraine attacks induced by pituitary adenylate cyclase-activating polypeptide-38. Brain 137(Pt 3):779–794
Schytz HW et al (2009) PACAP38 induces migraine-like attacks in patients with migraine without aura. Brain 132(Pt 1):16–25
Tuka B et al (2013) Alterations in PACAP-38-like immunoreactivity in the plasma during ictal and interictal periods of migraine patients. Cephalalgia 33(13):1085–1095
Arimura A et al (1991) Tissue distribution of PACAP as determined by RIA: highly abundant in the rat brain and testes. Endocrinology 129(5):2787–2789
Bourgault S et al (2008) Novel stable PACAP analogs with potent activity towards the PAC1 receptor. Peptides 29(6):919–932
Zagami AS, Edvinsson L, Goadsby PJ (2014) Pituitary adenylate cyclase activating polypeptide and migraine. Ann Clin Transl Neurol 1(12):1036–1040
Ashina H et al (2017) PACAP38 in human models of primary headaches. J Headache Pain 18(1):110
Guo S et al (2017) Part II: Biochemical changes after pituitary adenylate cyclase-activating polypeptide-38 infusion in migraine patients. Cephalalgia 37(2):136–147
Ghanizada H et al (2020) PACAP27 induces migraine-like attacks in migraine patients. Cephalalgia 40(1):57–67
Russo AF, Hay DL (2022) CGRP physiology, pharmacology, and therapeutic targets: migraine and beyond. Physiol Rev 103(2):1565–1644
Christensen CE, Ashina M, Amin FM (2022) Calcitonin Gene-Related Peptide (CGRP) and Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) in migraine pathogenesis. Pharmaceuticals (Basel) 15(10):1189
Edvinsson L et al (2018) PACAP and its role in primary headaches. J Headache Pain 19(1):21
Ashina M (2020) Migraine. N Engl J Med 383(19):1866–1876
Guo S et al (2023) Role of PACAP in migraine: an alternative to CGRP? Neurobiol Dis 176:105946
Russo AF (2015) Calcitonin gene-related peptide (CGRP): a new target for migraine. Annu Rev Pharmacol Toxicol 55:533–552
Edvinsson L (2019) Role of CGRP in migraine. Handb Exp Pharmacol 255:121–130
Ashina H, Schytz HW, Ashina M (2019) CGRP in human models of migraine. Handb Exp Pharmacol 255:109–120
Lassen LH et al (2002) CGRP may play a causative role in migraine. Cephalalgia 22(1):54–61
Asghar MS et al (2011) Evidence for a vascular factor in migraine. Ann Neurol 69(4):635–645
Hansen JM et al (2010) Calcitonin gene-related peptide triggers migraine-like attacks in patients with migraine with aura. Cephalalgia 30(10):1179–1186
Guo S et al (2017) Calcitonin gene-related peptide induced migraine attacks in patients with and without familial aggregation of migraine. Cephalalgia 37(2):114–124
Guo S et al (2016) Premonitory and nonheadache symptoms induced by CGRP and PACAP38 in patients with migraine. Pain 157(12):2773–2781
Christensen CE et al (2018) Migraine induction with calcitonin gene-related peptide in patients from erenumab trials. J Headache Pain 19(1):105
Ghanizada H et al (2021) Amylin analog pramlintide induces migraine-like attacks in patients. Ann Neurol 89(6):1157–1171
Guo S et al (2017) Part I: Pituitary adenylate cyclase-activating polypeptide-38 induced migraine-like attacks in patients with and without familial aggregation of migraine. Cephalalgia 37(2):125–135
Wienholtz NKF et al (2021) Early treatment with sumatriptan prevents PACAP38-induced migraine: a randomised clinical trial. Cephalalgia 41(6):731–748
Falkenberg K et al (2020) Sumatriptan does not antagonize cgrp-induced symptoms in healthy volunteers. Headache 60(4):665–676
Rahmann A et al (2008) Vasoactive intestinal peptide causes marked cephalic vasodilation, but does not induce migraine. Cephalalgia 28(3):226–236
Pellesi L et al (2021) Effect of vasoactive intestinal polypeptide on development of migraine headaches: a randomized clinical trial. JAMA Netw Open 4(8):e2118543
Ghanizada H et al (2021) Effect of adrenomedullin on migraine-like attacks in patients with migraine: a randomized crossover study. Neurology 96(20):e2488–e2499
Sicuteri F et al (1987) Unmasking latent dysnociception in healthy subjects. Headache 27(4):180–185
Thomsen LL et al (1994) A nitric oxide donor (nitroglycerin) triggers genuine migraine attacks. Eur J Neurol 1(1):73–80
Christiansen I et al (1999) Glyceryl trinitrate induces attacks of migraine without aura in sufferers of migraine with aura. Cephalalgia 19(7):660–7 discussion 626
Afridi KS, Kaube H, Goadsby JP (2004) Glyceryl trinitrate triggers premonitory symptoms in migraineurs. Pain 110(3):675–680
Sances G et al (2004) Reliability of the nitroglycerin provocative test in the diagnosis of neurovascular headaches. Cephalalgia 24(2):110–119
Kruuse C et al (2003) Migraine can be induced by sildenafil without changes in middle cerebral artery diameter. Brain 126(Pt 1):241–247
Kruuse C et al (2006) Dipyridamole may induce migraine in patients with migraine without aura. Cephalalgia 26(8):925–933
Guo S, Olesen J, Ashina M (2014) Phosphodiesterase 3 inhibitor cilostazol induces migraine-like attacks via cyclic AMP increase. Brain 137(Pt 11):2951–2959
Al-Karagholi MA et al (2019) Opening of ATP-sensitive potassium channels causes migraine attacks: a new target for the treatment of migraine. Brain 142(9):2644–2654
Al-Karagholi MA et al (2021) Opening of ATP sensitive potassium channels causes migraine attacks with aura. Brain 144(8):2322–2332
Lassen LH, Thomsen LL, Olesen J (1995) Histamine induces migraine via the H1-receptor. Support for the NO hypothesis of migraine. Neuroreport 6(11):1475–9
Antonova M et al (2012) Prostaglandin E(2) induces immediate migraine-like attack in migraine patients without aura. Cephalalgia 32(11):822–833
Wienecke T, Olesen J, Ashina M (2010) Prostaglandin I2 (epoprostenol) triggers migraine-like attacks in migraineurs. Cephalalgia 30(2):179–190
Pellesi L et al (2020) Two-hour infusion of vasoactive intestinal polypeptide induces delayed headache and extracranial vasodilation in healthy volunteers. Cephalalgia 40(11):1212–1223
Lassen LH et al (2008) Involvement of calcitonin gene-related peptide in migraine: regional cerebral blood flow and blood flow velocity in migraine patients. J Headache Pain 9(3):151–157
Asghar MS et al (2010) Dilation by CGRP of middle meningeal artery and reversal by sumatriptan in normal volunteers. Neurology 75(17):1520–1526
Amin FM et al (2012) Headache and prolonged dilatation of the middle meningeal artery by PACAP38 in healthy volunteers. Cephalalgia 32(2):140–149
Dodick DW (2018) A phase-by-phase review of migraine pathophysiology. Headache 58(Suppl 1):4–16
Charles A (2013) The evolution of a migraine attack - a review of recent evidence. Headache 53(2):413–419
Banks WA et al (1993) Passage of pituitary adenylate cyclase activating polypeptide1-27 and pituitary adenylate cyclase activating polypeptide1-38 across the blood-brain barrier. J Pharmacol Exp Ther 267(2):690–696
Denuelle M et al (2007) Hypothalamic activation in spontaneous migraine attacks. Headache 47(10):1418–1426
Schulte LH, May A (2016) The migraine generator revisited: continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain 139(Pt 7):1987–1993
Gollion C et al (2022) The premonitory phase of migraine is due to hypothalamic dysfunction: revisiting the evidence. J Headache Pain 23(1):158
Vaudry D et al (2009) Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol Rev 61(3):283–357
Al-Karagholi MA et al (2017) The K(ATP) channel in migraine pathophysiology: a novel therapeutic target for migraine. J Headache Pain 18(1):90
Coskun H et al (2021) The Effect of K (ATP) CHANNEL Blocker Glibenclamide on CGRP-induced headache and hemodynamic in healthy volunteers. Front Physiol 12:652136
Kokoti L et al (2022) Effect of K(ATP) channel blocker glibenclamide on PACAP38-induced headache and hemodynamic. Cephalalgia 42(9):846–858
Al-Karagholi MA et al (2020) Effect of K(ATP) channel blocker glibenclamide on levcromakalim-induced headache. Cephalalgia 40(10):1045–1054
Warfvinge K, Edvinsson L (2019) Distribution of CGRP and CGRP receptor components in the rat brain. Cephalalgia 39(3):342–353
Warfvinge K, Edvinsson L (2020) Cellular distribution of PACAP-38 and PACAP receptors in the rat brain: relation to migraine activated regions. Cephalalgia 40(6):527–542
Oliver KR et al (2002) Immunohistochemical localization of calcitonin receptor-like receptor and receptor activity-modifying proteins in the human cerebral vasculature. J Cereb Blood Flow Metab 22(5):620–629
Knutsson M, Edvinsson L (2002) Distribution of mRNA for VIP and PACAP receptors in human cerebral arteries and cranial ganglia. NeuroReport 13(4):507–509
Chan KY et al (2011) Pharmacological characterization of VIP and PACAP receptors in the human meningeal and coronary artery. Cephalalgia 31(2):181–189
Levy D, Labastida-Ramirez A, MaassenVanDenBrink A (2019) Current understanding of meningeal and cerebral vascular function underlying migraine headache. Cephalalgia 39(13):1606–1622
Skofitsch G, Jacobowitz DM (1985) Calcitonin gene-related peptide: detailed immunohistochemical distribution in the central nervous system. Peptides 6(4):721–745
Tschopp FA et al (1985) Calcitonin gene-related peptide and its binding sites in the human central nervous system and pituitary. Proc Natl Acad Sci U S A 82(1):248–252
Hashimoto H et al (1996) Distribution of the mRNA for a pituitary adenylate cyclase-activating polypeptide receptor in the rat brain: an in situ hybridization study. J Comp Neurol 371(4):567–577
Steinberg A et al (2016) Expression of messenger molecules and receptors in rat and human sphenopalatine ganglion indicating therapeutic targets. J Headache Pain 17(1):78
Hensley K et al (2019) PAC1 receptor mRNA and protein distribution in rat and human trigeminal and sphenopalatine ganglia, spinal trigeminal nucleus and in dura mater. Cephalalgia 39(7):827–840
Csati A et al (2012) Calcitonin gene-related peptide and its receptor components in the human sphenopalatine ganglion – interaction with the sensory system. Brain Res 1435:29–39
Ivanusic JJ et al (2011) 5-HT(1D) receptor immunoreactivity in the sphenopalatine ganglion: implications for the efficacy of triptans in the treatment of autonomic signs associated with cluster headache. Headache 51(3):392–402
Kozicz T, Vigh S, Arimura A (1997) Axon terminals containing PACAP- and VIP-immunoreactivity form synapses with CRF-immunoreactive neurons in the dorsolateral division of the bed nucleus of the stria terminalis in the rat. Brain Res 767(1):109–119
Kruger L et al (1988) Calcitonin gene-related peptide (CGRP) in the rat central nervous system: patterns of immunoreactivity and receptor binding sites. Brain Res 463(2):223–244
Conti F, Sternini C (1989) Calcitonin gene-related peptide (CGRP)-positive neurons and fibers in the cat periaqueductal grey matter. Somatosens Mot Res 6(5–6):497–511
Eftekhari S et al (2016) Localization of CGRP receptor components and receptor binding sites in rhesus monkey brainstem: a detailed study using in situ hybridization, immunofluorescence, and autoradiography. J Comp Neurol 524(1):90–118
Castorina A et al (2019) PACAP and VIP expression in the periaqueductal grey of the rat following sciatic nerve constriction injury. Neuropeptides 74:60–69
Uddman R et al (2002) Neuropeptide expression in the human trigeminal nucleus caudalis and in the cervical spinal cord C1 and C2. Cephalalgia 22(2):112–116
Lennerz JK et al (2008) Calcitonin receptor-like receptor (CLR), receptor activity-modifying protein 1 (RAMP1), and calcitonin gene-related peptide (CGRP) immunoreactivity in the rat trigeminovascular system: differences between peripheral and central CGRP receptor distribution. J Comp Neurol 507(3):1277–1299
Zhang Q et al (2019) Dynamic changes in CGRP, PACAP, and PACAP receptors in the trigeminovascular system of a novel repetitive electrical stimulation rat model: Relevant to migraine. Mol Pain 15:1744806918820452
Hannibal J (2002) Pituitary adenylate cyclase-activating peptide in the rat central nervous system: an immunohistochemical and in situ hybridization study. J Comp Neurol 453(4):389–417
van Rossum D, Hanisch UK, Quirion R (1997) Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci Biobehav Rev 21(5):649–678
Mulder H et al (1994) Pituitary adenylate cyclase activating polypeptide expression in sensory neurons. Neuroscience 63(1):307–312
Eftekhari S et al (2015) Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood-brain barrier. Brain Res 1600:93–109
Chaudhary P, Baumann TK (2002) Expression of VPAC2 receptor and PAC1 receptor splice variants in the trigeminal ganglion of the adult rat. Brain Res Mol Brain Res 104(2):137–142
Mulderry PK et al (1988) Differential expression of alpha-CGRP and beta-CGRP by primary sensory neurons and enteric autonomic neurons of the rat. Neuroscience 25(1):195–205
Cottrell GS et al (2005) Localization of calcitonin receptor-like receptor and receptor activity modifying protein 1 in enteric neurons, dorsal root ganglia, and the spinal cord of the rat. J Comp Neurol 490(3):239–255
Nielsen KM et al (2004) PACAP promotes sensory neuron differentiation: blockade by neurotrophic factors. Mol Cell Neurosci 25(4):629–641
Cauvin A et al (1991) Properties and distribution of receptors for pituitary adenylate cyclase activating peptide (PACAP) in rat brain and spinal cord. Regul Pept 35(2):161–173
Edvinsson L et al (1987) Calcitonin gene-related peptide and cerebral blood vessels: distribution and vasomotor effects. J Cereb Blood Flow Metab 7(6):720–728
Rees TA et al (2022) CGRP and the calcitonin receptor are co-expressed in mouse, rat and human trigeminal ganglia neurons. Front Physiol 13:860037
Jansen-Olesen I et al (2014) PACAP-38 but not VIP induces release of CGRP from trigeminal nucleus caudalis via a receptor distinct from the PAC1 receptor. Neuropeptides 48(2):53–64
Tajti J et al (1999) Messenger molecules and receptor mRNA in the human trigeminal ganglion. J Auton Nerv Syst 76(2–3):176–183
Jansen-Olesen I, Hougaard Pedersen S (2018) PACAP and its receptors in cranial arteries and mast cells. J Headache Pain 19(1):16
Frederiksen SD et al (2018) Expression of pituitary adenylate cyclase-activating peptide, calcitonin gene-related peptide and headache targets in the trigeminal ganglia of rats and humans. Neuroscience 393:319–332
Csati A et al (2012) Distribution of vasoactive intestinal peptide, pituitary adenylate cyclase-activating peptide, nitric oxide synthase, and their receptors in human and rat sphenopalatine ganglion. Neuroscience 202:158–168
Eftekhari S et al (2010) Differential distribution of calcitonin gene-related peptide and its receptor components in the human trigeminal ganglion. Neuroscience 169(2):683–696
Uddman R et al (1999) Neuronal messengers and peptide receptors in the human sphenopalatine and otic ganglia. Brain Res 826(2):193–199
Schoenen J (2015) Sphenopalatine ganglion stimulation in neurovascular headaches. Prog Neurol Surg 29:106–116
Akerman S et al (2009) Oxygen inhibits neuronal activation in the trigeminocervical complex after stimulation of trigeminal autonomic reflex, but not during direct dural activation of trigeminal afferents. Headache 49(8):1131–1143
Akerman S et al (2012) A translational in vivo model of trigeminal autonomic cephalalgias: therapeutic characterization. Brain 135(Pt 12):3664–3675
Hostetler ED et al (2013) In vivo quantification of calcitonin gene-related peptide receptor occupancy by telcagepant in rhesus monkey and human brain using the positron emission tomography tracer [11C]MK-4232. J Pharmacol Exp Ther 347(2):478–486
Schytz HW, Olesen J, Ashina M (2010) The PACAP receptor: a novel target for migraine treatment. Neurotherapeutics 7(2):191–196
Tuka B et al (2012) Peripheral and central alterations of pituitary adenylate cyclase activating polypeptide-like immunoreactivity in the rat in response to activation of the trigeminovascular system. Peptides 33(2):307–316
Masuo Y et al (1992) Binding sites for pituitary adenylate cyclase activating polypeptide (PACAP): comparison with vasoactive intestinal polypeptide (VIP) binding site localization in rat brain sections. Brain Res 575(1):113–123
Tajti J, Uddman R, Edvinsson L (2001) Neuropeptide localization in the “migraine generator” region of the human brainstem. Cephalalgia 21(2):96–101
Narita M et al (1996) Hyperalgesia induced by pituitary adenylate cyclase-activating polypeptide in the mouse spinal cord. Eur J Pharmacol 311(2–3):121–126
Vaudry D et al (2000) Pituitary adenylate cyclase-activating polypeptide and its receptors: from structure to functions. Pharmacol Rev 52(2):269–324
Otto C et al (2001) Altered emotional behavior in PACAP-type-I-receptor-deficient mice. Brain Res Mol Brain Res 92(1–2):78–84
Hattori S et al (2012) Comprehensive behavioral analysis of pituitary adenylate cyclase-activating polypeptide (PACAP) knockout mice. Front Behav Neurosci 6:58
Ressler KJ et al (2011) Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature 470(7335):492–497
Hammack SE et al (2009) Chronic stress increases pituitary adenylate cyclase-activating peptide (PACAP) and brain-derived neurotrophic factor (BDNF) mRNA expression in the bed nucleus of the stria terminalis (BNST): roles for PACAP in anxiety-like behavior. Psychoneuroendocrinology 34(6):833–843
Hammack SE et al (2010) Roles for pituitary adenylate cyclase-activating peptide (PACAP) expression and signaling in the bed nucleus of the stria terminalis (BNST) in mediating the behavioral consequences of chronic stress. J Mol Neurosci 42(3):327–340
Hashimoto H, Shintani N, Baba A (2006) New insights into the central PACAPergic system from the phenotypes in PACAP- and PACAP receptor-knockout mice. Ann N Y Acad Sci 1070:75–89
Mason BN, Russo AF (2018) Vascular contributions to migraine: time to revisit? Front Cell Neurosci 12:233
Kaiser EA, Russo AF (2013) CGRP and migraine: could PACAP play a role too? Neuropeptides 47(6):451–461
Ashina M et al (2017) Human models of migraine - short-term pain for long-term gain. Nat Rev Neurol 13(12):713–724
Fahrenkrug J et al (2000) Immunohistochemical localization of the VIP1 receptor (VPAC1R) in rat cerebral blood vessels: relation to PACAP and VIP containing nerves. J Cereb Blood Flow Metab 20(8):1205–1214
Erdling A et al (2013) VIP/PACAP receptors in cerebral arteries of rat: characterization, localization and relation to intracellular calcium. Neuropeptides 47(2):85–92
Baun M et al (2012) Dural mast cell degranulation is a putative mechanism for headache induced by PACAP-38. Cephalalgia 32(4):337–345
Theoharides TC et al (2005) The role of mast cells in migraine pathophysiology. Brain Res Brain Res Rev 49(1):65–76
Zhang Y et al (1998) Pituitary adenylate cyclase-activating peptide is upregulated in sensory neurons by inflammation. NeuroReport 9(12):2833–2836
Helyes Z et al (2007) Inhibitory effect of PACAP-38 on acute neurogenic and non-neurogenic inflammatory processes in the rat. Peptides 28(9):1847–1855
Nemeth J et al (2006) Effect of pituitary adenylate cyclase activating polypeptide-38 on sensory neuropeptide release and neurogenic inflammation in rats and mice. Neuroscience 143(1):223–230
Sandor K et al (2009) Divergent peripheral effects of pituitary adenylate cyclase-activating polypeptide-38 on nociception in rats and mice. Pain 141(1–2):143–150
Sandor K et al (2010) Impaired nocifensive behaviours and mechanical hyperalgesia, but enhanced thermal allodynia in pituitary adenylate cyclase-activating polypeptide deficient mice. Neuropeptides 44(5):363–371
Robert C et al (2013) Paraventricular hypothalamic regulation of trigeminovascular mechanisms involved in headaches. J Neurosci 33(20):8827–8840
Akerman S, Goadsby PJ (2015) Neuronal PAC1 receptors mediate delayed activation and sensitization of trigeminocervical neurons: Relevance to migraine. Sci Transl Med 7(308):308ra157
Kuburas A et al (2021) PACAP induces light aversion in mice by an inheritable mechanism independent of CGRP. J Neurosci 41(21):4697–4715
Wang M, et al (2021) Investigating migraine-like behavior using light aversion in mice. J Vis Exp (174). https://doi.org/10.3791/62839
Vuralli D et al (2019) Behavioral and cognitive animal models in headache research. J Headache Pain 20(1):11
Mason BN et al (2017) Induction of migraine-like photophobic behavior in mice by both peripheral and central CGRP mechanisms. J Neurosci 37(1):204–216
Mason BN et al (2020) Vascular actions of peripheral CGRP in migraine-like photophobia in mice. Cephalalgia 40(14):1585–1604
Sowers LP et al (2020) Stimulation of posterior thalamic nuclei induces photophobic behavior in mice. Headache 60(9):1961–1981
Wang M et al (2022) CGRP administration into the cerebellum evokes light aversion, tactile hypersensitivity, and nociceptive squint in mice. Front Pain Res (Lausanne) 3:861598
Recober A et al (2009) Role of calcitonin gene-related peptide in light-aversive behavior: implications for migraine. J Neurosci 29(27):8798–8804
Recober A et al (2010) Induction of multiple photophobic behaviors in a transgenic mouse sensitized to CGRP. Neuropharmacology 58(1):156–165
Kaiser EA et al (2012) Modulation of CGRP-induced light aversion in wild-type mice by a 5-HT(1B/D) agonist. J Neurosci 32(44):15439–15449
Markovics A et al (2012) Pituitary adenylate cyclase-activating polypeptide plays a key role in nitroglycerol-induced trigeminovascular activation in mice. Neurobiol Dis 45(1):633–644
Bhatt DK et al (2014) PACAP-38 infusion causes sustained vasodilation of the middle meningeal artery in the rat: possible involvement of mast cells. Cephalalgia 34(11):877–886
Pedersen SH et al (2019) PACAP-38 and PACAP(6–38) degranulate rat meningeal mast cells via the orphan MrgB3-receptor. Front Cell Neurosci 13:114
Guo Z, et al (2020) Increase in trigeminal ganglion neurons that respond to both CGRP and PACAP in mouse models of chronic migraine and post-traumatic headache. Pain 162(5):1483–1499
Aldinger KA et al (2009) Genetic variation and population substructure in outbred CD-1 mice: implications for genome-wide association studies. PLoS ONE 4(3):e4729
De Logu F et al (2019) Migraine-provoking substances evoke periorbital allodynia in mice. J Headache Pain 20(1):18
Wattiez AS et al (2021) Different forms of traumatic brain injuries cause different tactile hypersensitivity profiles. Pain 162(4):1163–1175
Marquez de Prado B, Hammond DL, Russo AF (2009) Genetic enhancement of calcitonin gene-related Peptide-induced central sensitization to mechanical stimuli in mice. J Pain 10(9):992–1000
Araya EI et al (2020) Contribution of intraganglionic CGRP to migraine-like responses in male and female rats. Cephalalgia 40(7):689–700
Ernstsen C et al (2022) The PACAP pathway is independent of CGRP in mouse models of migraine: possible new drug target? Brain 145(7):2450–2460
Christensen SL et al (2021) CGRP-dependent signalling pathways involved in mouse models of GTN- cilostazol- and levcromakalim-induced migraine. Cephalalgia 41(14):1413–1426
Christensen SL et al (2020) ATP sensitive potassium (K(ATP)) channel inhibition: A promising new drug target for migraine. Cephalalgia 40(7):650–664
Guo S et al (2022) PACAP signaling is not involved in GTN- and levcromakalim-induced hypersensitivity in mouse models of migraine. J Headache Pain 23(1):155
Woolley MJ et al (2017) Receptor activity-modifying protein dependent and independent activation mechanisms in the coupling of calcitonin gene-related peptide and adrenomedullin receptors to Gs. Biochem Pharmacol 142:96–110
Zhang Z et al (2007) Sensitization of calcitonin gene-related peptide receptors by receptor activity-modifying protein-1 in the trigeminal ganglion. J Neurosci 27(10):2693–2703
Walker CS et al (2010) Regulation of signal transduction by calcitonin gene-related peptide receptors. Trends Pharmacol Sci 31(10):476–483
Hamelink C et al (2002) Coincident elevation of cAMP and calcium influx by PACAP-27 synergistically regulates vasoactive intestinal polypeptide gene transcription through a novel PKA-independent signaling pathway. J Neurosci 22(13):5310–5320
Harmar AJ et al (2012) Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: IUPHAR review 1. Br J Pharmacol 166(1):4–17
Langer I et al (2022) Signal transduction by VIP and PACAP Receptors. Biomedicines 10(2):406
Weston C et al (2016) Receptor activity-modifying protein-directed G protein signaling specificity for the calcitonin gene-related peptide family of receptors. J Biol Chem 291(49):25763
Parameswaran N et al (2000) Desensitization and resensitization of adrenomedullin-sensitive receptor in rat mesangial cells. Eur J Pharmacol 407(3):205–210
Wiley JW, Gross RA, MacDonald RL (1992) The peptide CGRP increases a high-threshold Ca2+ current in rat nodose neurones via a pertussis toxin-sensitive pathway. J Physiol 455:367–381
Aiyar N et al (1999) Calcitonin gene-related peptide receptor independently stimulates 3’,5’-cyclic adenosine monophosphate and Ca2+ signaling pathways. Mol Cell Biochem 197(1–2):179–185
Drissi H et al (1998) Activation of phospholipase C-beta1 via Galphaq/11 during calcium mobilization by calcitonin gene-related peptide. J Biol Chem 273(32):20168–20174
Blechman J, Levkowitz G (2013) Alternative splicing of the pituitary adenylate cyclase-activating polypeptide receptor PAC1: mechanisms of fine tuning of brain activity. Front Endocrinol (Lausanne) 4:55
Bower RL et al (2018) Molecular signature for receptor engagement in the metabolic peptide hormone amylin. ACS Pharmacol Transl Sci 1(1):32–49
Garelja ML et al (2020) Molecular mechanisms of class B GPCR activation: insights from adrenomedullin receptors. ACS Pharmacol Transl Sci 3(2):246–262
Tasma Z et al (2022) Characterisation of agonist signalling profiles and agonist-dependent antagonism at PACAP-responsive receptors: Implications for drug discovery. Br J Pharmacol 179(3):435–453
Messlinger K (2018) The big CGRP flood - sources, sinks and signalling sites in the trigeminovascular system. J Headache Pain 19(1):22
Hay DL et al (2018) Update on the pharmacology of calcitonin/CGRP family of peptides: IUPHAR review 25. Br J Pharmacol 175(1):3–17
Walker CS et al (2018) CGRP receptor antagonist activity of olcegepant depends on the signalling pathway measured. Cephalalgia 38(3):437–451
Rea BJ et al (2022) Automated detection of squint as a sensitive assay of sex-dependent calcitonin gene-related peptide and amylin-induced pain in mice. Pain 163(8):1511–1519
Miyata A et al (1989) Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem Biophys Res Commun 164(1):567–574
Dickson L, Finlayson K (2009) VPAC and PAC receptors: From ligands to function. Pharmacol Ther 121(3):294–316
Walker CS, Sundrum T, Hay DL (2014) PACAP receptor pharmacology and agonist bias: analysis in primary neurons and glia from the trigeminal ganglia and transfected cells. Br J Pharmacol 171(6):1521–1533
Emery AC, Eiden LE (2012) Signaling through the neuropeptide GPCR PAC(1) induces neuritogenesis via a single linear cAMP- and ERK-dependent pathway using a novel cAMP sensor. FASEB J 26(8):3199–3211
Ravni A et al (2008) A cAMP-dependent, protein kinase A-independent signaling pathway mediating neuritogenesis through Egr1 in PC12 cells. Mol Pharmacol 73(6):1688–1708
Robichaux WG 3rd, Cheng X (2018) Intracellular cAMP sensor EPAC: physiology, pathophysiology, and therapeutics development. Physiol Rev 98(2):919–1053
Stork PJ, Schmitt JM (2002) Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol 12(6):258–266
May V et al (2021) PAC1 receptor internalization and endosomal MEK/ERK activation is essential for PACAP-mediated neuronal excitability. J Mol Neurosci 71(8):1536–1542
Hartopo AB et al (2013) Endothelin-converting enzyme-1 gene ablation attenuates pulmonary fibrosis via CGRP-cAMP/EPAC1 pathway. Am J Respir Cell Mol Biol 48(4):465–476
Mikami N et al (2014) Calcitonin gene-related peptide regulates type IV hypersensitivity through dendritic cell functions. PLoS ONE 9(1):e86367
Clark AJ et al (2021) CGRP, adrenomedullin and adrenomedullin 2 display endogenous GPCR agonist bias in primary human cardiovascular cells. Commun Biol 4(1):776
Hucho TB, Dina OA, Levine JD (2005) Epac mediates a cAMP-to-PKC signaling in inflammatory pain: an isolectin B4(+) neuron-specific mechanism. J Neurosci 25(26):6119–6126
Gingell JJ et al (2020) Distinct patterns of internalization of different calcitonin gene-related peptide receptors. ACS Pharmacol Transl Sci 3(2):296–304
Bhakta M et al (2021) Migraine therapeutics differentially modulate the CGRP pathway. Cephalalgia 41(5):499–514
Dal Maso E et al (2018) Characterization of signalling and regulation of common calcitonin receptor splice variants and polymorphisms. Biochem Pharmacol 148:111–129
Yarwood RE et al (2017) Endosomal signaling of the receptor for calcitonin gene-related peptide mediates pain transmission. Proc Natl Acad Sci U S A 114(46):12309–12314
De Logu F et al (2022) Schwann cell endosome CGRP signals elicit periorbital mechanical allodynia in mice. Nat Commun 13(1):646
Langer I (2012) Mechanisms involved in VPAC receptors activation and regulation: lessons from pharmacological and mutagenesis studies. Front Endocrinol (Lausanne) 3:129
Hansen JM et al (2006) Vasoactive intestinal polypeptide evokes only a minimal headache in healthy volunteers. Cephalalgia 26(8):992–1003
Ashina M et al (2021) A phase 2, randomized, double-blind, placebo-controlled trial of AMG 301, a pituitary adenylate cyclase-activating polypeptide PAC1 receptor monoclonal antibody for migraine prevention. Cephalalgia 41(1):33–44
Foster SR et al (2019) Discovery of human signaling systems: pairing peptides to G protein-coupled receptors. Cell 179(4):895-908.e21
Acknowledgements
The authors acknowledge extensive advice on signaling pathways from Dr. Christopher Walker (University of Auckland, New Zealand) and helpful comments from Dr. Levi Sowers and Brandon Rea.
Funding
AFR was supported by grants from the NIH (RF1 NS113839; R01 NS075599) and VA Medical Center (I01 RX003523-01). The contents do not represent the views of the VA or the United States Government.
Author information
Authors and Affiliations
Contributions
Both authors contributed equally to writing and editing the manuscript. Both authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
AFR is a consultant for Lundbeck, Abbvie, Eli Lilly, and Schedule One Therapeutics. AK declares no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kuburas, A., Russo, A.F. Shared and independent roles of CGRP and PACAP in migraine pathophysiology. J Headache Pain 24, 34 (2023). https://doi.org/10.1186/s10194-023-01569-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10194-023-01569-2