
Abstract
Background
To review the distribution and function of KATP channels, describe the use of KATP channels openers in clinical trials and make the case that these channels may play a role in headache and migraine.
Discussion
KATP channels are widely present in the trigeminovascular system and play an important role in the regulation of tone in cerebral and meningeal arteries. Clinical trials using synthetic KATP channel openers report headache as a prevalent-side effect in non-migraine sufferers, indicating that KATP channel opening may cause headache, possibly due to vascular mechanisms. Whether KATP channel openers can provoke migraine in migraine sufferers is not known.
Conclusion
We suggest that KATP channels may play an important role in migraine pathogenesis and could be a potential novel therapeutic anti-migraine target.
Similar content being viewed by others

Introduction
Adenosine 5′-triphosphate-sensitive K+ (KATP) channel openers have been used in clinical trials for the treatment of hypertension and asthma. The most common side effect mentioned during treatment with KATP channel openers was headache (62, 64, 66–79) (Tables 2 and 3). However, only little attention has been focused on the role of KATP channels in migraine pathophysiology.
KATP channels were originally identified in cardiomyocytes [1], but have also been found in several tissues, including pancreatic α- and ß-cells, smooth muscle, skeletal muscle and central neurons [2, 3]. The channels belong to the family of inwardly rectifying K+ channels that are inhibited at physiological intracellular levels ATP/ADP ratio. When intracellular ATP is reduced under conditions of metabolic challenges they open. KATP channels are critical in regulating insulin secretion, controlling vascular tone, and protecting cells against metabolic stress [2, 4, 5].
Over the past three decades, some preclinical evidence has emerged indicating that KATP channels may play an important role in migraine pathophysiology. In particular, the vasodilation effect of KATP channels is relevant, since it is has been established that endogenous neurotransmitters that trigger migraine attacks are often associated with dilation of cranial arteries [6].
Here we review preclinical and clinical studies on KATP channels and discuss the KATP channel as a novel therapeutic target for migraine treatment.
Molecular structure and isoforms
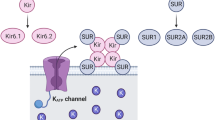
The KATP channel is a hetero-octameric complex that consists of four pore-forming K+ inwardly rectifying (Kir) subunits and four regulatory sulfonylurea receptor (SUR) subunits [7].
The Kir6.x subunit exists in two isoforms, Kir6.1 and Kir6.2. The SUR subunit belongs to the ATP-binding cassette (ABC) transporter family, regulated by sulfonylurea, with three isoforms, SUR1, SUR2A, and SUR2B [7, 8].
KATPchannels have specific tissue expression with different compositions of Kir6.x and SUR subunits which lead to distinct functional properties (Figs. 1 and 2 and Table 1).

Molecular structure and isoforms. a Two major Kir6.x isoforms (Kir6.1 and Kir 6.2) and three major SUR isoforms (SUR1, SUR2A and SUR 2B) have been identified. b Kir.x subunits combine tissue-specifically with different SUR subunits to form various native KATP channels. Pancreatic, cardiac and smooth muscle KATP channels are made up of Kir6.2/SUR1, Kir6.2/SUR2A and Kir6.1 (or Kir6.2)/SUR2B, respectively [2]. Kir, inwardly rectifying K+ channels; SUR, sulfonylurea receptor

Schematic diagram of the KATP channel. Kir6.x subunits have two transmembrane domains, and a large cytoplasmic domain including an inhibitory binding site for ATP [8, 84]. SUR subunits have many transmembrane domains and two intracellular nucleotide binding domains (NBD1 and NBD2), which stimulate opening of the channel after binding to MgADP [85]
Channel function
KATP channel activity is controlled by changes in concentrations of intracellular ATP and magnesium adenosine diphosphate (Mg-ADP). KATP channels couple the metabolic state of the cell to the membrane potential and thus play a crucial role in many tissues under both physiological and pathological conditions [9]. K+ channels participate in the regulation of vascular tone, including cerebral arteries [10]. When intracellular ATP is reduced, KATP channels become activated; K+ efflux hyperpolarize the membrane and close voltage-operated Ca2+-channels (VOCC). The result is a decrease in cytosolic Ca2+ concentration followed by relaxation of vascular smooth muscle cells and an increase in blood flow [11]. The same applies if cells are exposed to metabolic stress such as ischemia or hypoglycemia [12]. Closure of K+ channels leads to membrane depolarization and constriction of the vessels [11]. In addition an increase in intracellular cAMP and cGMP levels activate KATP channels to produce vasodilation [11]. Synthetic KATP channel openers (like levcromakalim and cromakalim) and blockers (like glibenclamide, second generation of sulfonylurea and PNU37883A) directly activate or inhibit the vascular KATP channels, respectively [9] (Fig. 3).

Opening of vascular ATP sensitive K channels. Endogenous molecules (ATP, cAMP and cGMP) and exogenous pharmacological agents (cromakalim and glibenclamide) regulate the activity of KATP channels, which help controlling the vascular tone
Distribution of KATP channels in migraine related structures
Intracranial arteries
KATP channels are present and functional in intracranial arteries [13,14,15]. They are found in vascular smooth muscle cells and vascular endothelial cells [16, 17]. In rat cerebral arteries, the distribution of KATP channels varies with vessel size and brain region [18]. Real time polymerase chain reaction (RT-PCR) analysis revealed Kir6.1 and SUR2B subunits in middle meningeal artery (MMA) and middle cerebral artery (MCA) in rats and pigs [19, 20]. This profile of KATP channels is also identified in human MMA [21] (Table 1).
Trigeminal ganglion and trigeminal nucleus caudalis
Kir6.1, Kir6.2, SUR1 and SUR2 are expressed in the trigeminal ganglion and trigeminal nucleus caudalis [22] (Table 1). In trigeminal neurons Kir 6.1 and Kir 6.2 immunoreactivity were expressed in cells with all soma sizes in all three divisions of the trigeminal ganglion [23].
KATP channels openers and migraine signaling pathways
A number of endogenous vasoactive signaling molecules have been implicated in migraine [6], and KATP channels may interact with these molecules.
Nitric oxide (NO)
In humans, infusion of the NO donor, glyceryl trinitrate, and inhibition of the breakdown of cGMP by sildenafil [24] provoke migraine attacks in migraineurs [25,26,27]. The NO-cGMP signaling pathway is involved in the relaxation of vascular smooth muscle [28]. In vitro studies with cerebral arteries isolated from rat and piglet and extra-cerebral arteries from rabbit reported that activation (opening) of KATP channels contributed to both cAMP- and cGMP-mediated vasodilation [29,30,31]. Yuan et al. [32] reported that sildenafil-induced vasodilation in porcine retinal arterioles was significantly inhibited by glibenclamide and suggested that cGMP signaling triggers opening of KATP channels. In contrast, NO-induced dural and pial artery dilation in rats was not attenuated by the KATP channel blocker, glibenclamide [33]. Together, these data suggest that interspecies differences are likely to explain the discrepancy in findings of the role of KATP channels in NO-induced vasodilation.
Calcitonin gene-related peptide (CGRP)
CGRP is one of the most potent endogenous vasodilators and major arteries in the intracranial circulation of man and animals are innervated by CGRP-containing nerve fibers [34,35,36]. Efficacy of CGRP antagonism is established in acute [37, 38] and preventive treatment of migraine [39]. CGRP activates vascular smooth muscle KATP channels indirectly through adenylate cyclase and protein kinase A (PKA) phosphorylation (Fig. 4) [40,41,42,43]. In rats, CGRP-induced dilation of the dural and pial arteries in vivo was shown to be inhibited by glibenclamide [33], but KATP channel openers do not interact with CGRP release in trigeminal ganglion and trigeminal nucleus caudalis [22]. This suggests that KATP channels are involved in CGRP-induced intracranial vasodilation.

Signaling pathways through vascular smooth muscle KATP channels. Numerous endogenous vasodilators activate vascular smooth muscle KATP channels through adenylate cyclase and PKA phosphorylation. Conversely, endogenous vasoconstrictors inhibit vascular smooth muscle KATP channels through DAG and PKC phosphorylation. CGRP, calcitonin gene-related peptide; PGI2, prostaglandin I2; VIP, vasoactive intestinal peptide; AngII, angiotensin II; NPY, neuropeptide Y; NA, noradrenaline; 5-HT, 5-hydroxytryptamine; Gs, G-protein-coupled receptor alpha stimulation; Gi, G-protein-coupled receptor alpha i/q; DAG, diacylglycerol; PKA and PKC, protein kinase A and C, respectively
Pituitary adenylate cyclase activating polypeptide (PACAP)
Pituitary adenylate cyclase activating polypeptide (PACAP) is a potent endothelium independent vasodilator of various vascular beds, including cerebral arteries [44, 45]. In vivo and in vitro studies have demonstrated that PACAP dilates cranial arteries in different species, e.g. human cerebral arteries [34, 46, 47], pig pia artery, canine basilar artery, cat cerebral arteries, rabbit posterior cerebral arteries and rat middle cerebral arteries [48,49,50,51,52]. Emerging data suggest that PACAP or it receptors are a promising target for migraine therapeutics [53]. PACAP has three types of receptors; Pituitary adenylate cyclase PAC1 (pituitary adenylate cyclase receptor 1), VPAC1 (vasoactive intestinal peptide and pituitary adenylate cyclase receptor 1) and VPAC2 (vasoactive intestinal peptide and pituitary adenylate cyclase receptor 2) [54] the two latter ones are also activated by vasoactive intestinal peptide and all three receptors are found in cerebral artery smooth muscle cells [55]. Through these receptors, PACAP leads to an increase in intracellular cAMP, which activates PKA and produces vasodilation by several mechanisms including activation of KATP channels (Fig. 4) [45]. Interestingly, glibenclamide could partially inhibit PACAP induced vasodilation in cerebral, coronary and pulmonary arteries, suggesting that PACAP may also activate KATP channels [44, 45].
Prostaglandins
Prostacyclin (PGI2) activates and sensitizes meningeal sensory afferents, and provokes immediate migraine-like attacks in migraine sufferers [56]. PGI2 also increases KATP channel activity in vascular smooth muscle preparations by cAMP-dependent PKA activation [57] (Fig. 4).
Headache induced by KATP channels openers
In the late 80’s there was a tremendous interest in developing novel KATP channel openers for hypertension, angina pectoris and asthma. Three pharmacological drugs were developed, pinacidil, nicorandil and levcromakalim. One of most common adverse events after treatment reported in these studies was headache [58,59,60,61,62,63].
Six clinical trials with pinacidil have been published for treatment of essential hypertension. Between 7% and 21% of the patients reported headache as an adverse effect (Table 2).
Nicorandil was tested for the treatment of angina pectoris and ischemic heart disease. 23% to 88% of the patients reported headache as an adverse event (Table 3). The high incidence of headache is likely due to the mixed KATP channel opener and NO donor properties of nicorandil which thus cause vasodilation via two separate mechanisms.
Levcromakalim was investigated for the treatment of asthma and essential hypertension. In these studies between 29% and 76% of the patients reported headache as an adverse event (Table 4).
The selective synthetic KATP channel openers levcromakalim and pinacidil have been shown to induce dilation in rat cranial arteries [13, 15, 19] and in isolated human cerebral arteries [64]. Moreover, the arterial dilation can be inhibited by synthetic KATP channel blockers like glibenclamide [10, 33] and PNU37883A [21, 65] (Fig. 3). These findings suggest that high incidences of headache could be due to vasoactive effect of the KATP channel openers in pain-sensitive extra- and/or intracerebral arteries.
Discussion and future perspectives
KATPchannels are expressed in migraine-related structures such as the cranial arteries, TG and TNC [18,19,20,21,22, 66]. KATP channels are also connected to a number of key molecules in migraine pathogenesis, particularly nitric oxide, CGRP, PACAP and PGI2 known to provoke migraine attacks [56, 67,68,69,70,71]. Therefore, the KATP channels are interesting in migraine context.
Human experimental models have demonstrated that the activation of the cAMP and cGMP pathways can trigger headache in healthy volunteers and migraine attacks in migraine sufferers [6, 71, 72]. The cAMP and cGMP signaling pathways are crucial in the activation of KATP channels, which result in the relaxation of smooth muscle [29,30,31]. Furthermore, synthetic KATP channel openers like levcromakalim and pinacidil trigger headache in non-migraine patients [58,59,60,61,62,63]. Although a detailed description of levcromakalim- and pinacidil-induced headache and accompanying symptoms are lacking, these data support a role of KATP channels in migraine headache. Because KATP channel openers were tested for other indications, there are no available data on the potential migraine-inducing effects of pinacidil and levcromakalim in migraine patients. It is conceivable that both headache and migraine are underreported as adverse events, as was found for the phosphodiesterase inhibitors, cilostazol and sildenafil [73, 74].
In addition to the vasoactive effects, the KATP channels might also tap into other parts of the migraine cascade. For a number of patients, migraine attacks are associated with transient focal neurological symptoms called the aura [75], possibly caused by cortical spread depression (CSD) [76]. During CSD K+ conductance is increased, and CSD may be inhibited by Kir antagonist [77]. The fact that KATP channels open under cellular stress, as seen during long lasting depolarizations, could provide a link between KATP channels, CSD and migraine aura.
With regard to the migraine pain, it is worth noting that KATP channels are also found in peripheral nociceptive fibers [78] and activation of these channels play a crucial role in anti-nociception at both spinal and supra-spinal levels [23, 79]. The exact role of these findings in the headache induced by KATP channel openers is unknown.
If KATP channel openers are in fact able to trigger migraine, the next step to consider is whether KATP channel antagonists can relieve migraine. KATP blockers for the treatment of migraine should be selective for the Kir6.1/SUR2B subtype because of its dominant presence in vascular tissue (Table 1). The necessity of a subtype specific blocker is unavoidable because of occurrence of different subtypes in different tissues. Glibenclamide cannot be used due to its high affinity to the Kir6.2/SUR1 subtype of KATP channels present in the pancreas with hypoglycemia as a side effect [80]. PNU-37883A is a Kir6.1 selective KATP channel blocker that was originally developed as a diuretic drug [81, 82]. The drug was not approved to human studies because of its cardiac depressant activity in animal studies [83]. This precludes further clinical development of PNU-37883A due to possible serious adverse events but may not exclude further investigations in other blockers against Kir6.1 subunit because it is not clear if all blockers against Kir6.1 subunit have non-favorable effects. These findings indicate that the SUR2B subunit and the Kir6.1 subunit should be a potential target for the treatment of migraine, but proof of concept studies are needed to examine this hypothesis.
Conclusion
Emerging evidence suggests that KATP channels could be involved in the pathophysiology of migraine. KATP channels exist in structures which are believed to be linked to the pathophysiology of migraine, including cerebral and meningeal arteries and the trigeminal system [19,20,21,22]. It is established that the cAMP signaling pathway and possibly cGMP signaling pathway are involved in the activation of KATP channels [29,30,31]. This is interesting in migraine contexts, as the two signaling pathways are likely to be crucial in the development of a migraine attack.
We suggest that the presented clinical and theoretical evidence support further studies of KATP channel openers in migraine context. Future human studies will help clarify the role of KATP channels in the pathophysiology of migraine.
Abbreviations
- ABC transporter:
-
ATP-binding cassette transporter
- BA:
-
Basilar artery
- CGRP:
-
Calcitonin gene-related peptide
- CSD:
-
Cortical spread depression
- DRG:
-
Dorsal root ganglia
- KATP channel:
-
Adenosine 5′-triphosphate-sensitive K+ channel
- Kir:
-
K+ inwardly rectifying
- MCA:
-
Middle cerebral artery
- Mg-ADP:
-
Magnesium adenosine diphosphate
- MMA:
-
Middle meningeal artery
- NBD:
-
Nucleotide binding domains
- NO:
-
Nitric oxide
- PACAP:
-
Pituitary adenylate cyclase activating polypeptide
- PGI2 :
-
Prostacyclin
- SUR:
-
Sulfonylurea receptor
- TG:
-
Trigeminal ganglion,
- TNC:
-
Trigeminal nucleus caudatus
- VOCC:
-
Voltage-operated Ca2+-channels
References
Noma A (1983) ATP-regulated K+ channels in cardiac muscle. Nature 305:147–148. doi:10.1038/305147a0
Aguilar-Bryan L, Bryan J (1999) Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev 20:101–135. doi:10.1210/er.20.2.101
Dunn-Meynell AA, Rawson NE, Levin BE (1998) Distribution and phenotype of neurons containing the ATP-sensitive K+ channel in rat brain. Brain Res 814:41–54. doi:10.1016/S0006-8993(98)00956-1
Yamada K, Inagaki N (2005) Neuroprotection by KATP channels. J Mol Cell Cardiol 38:945–949. doi:10.1016/j.yjmcc.2004.11.020
Saito T, Fujiwara Y, Fujiwara R, et al (2002) Experimental biology 2001 symposium potassium channels that regulate vascular tone : which are the important players ? ROLE OF AUGMENTED EXPRESSION OF INTERMEDIATE- CONDUCTANCE CA 2 + −ACTIVATED K + CHANNELS IN. 324–329
Ashina M, Hansen JM, Olesen J (2013) Pearls and pitfalls in human pharmacological models of migraine: 30 years’ experience. Cephalalgia 33:540–553. doi:10.1177/0333102412475234
Clement JP, Kunjilwar K, Gonzalez G et al (1997) Association and stoichiometry of K(ATP) channel subunits. Neuron 18:827–838. doi:10.1016/S0896-6273(00)80321-9
Shyng S-L, Nichols CG (1997) Octameric stoichiometry of the K ATP Channel complex. J Gen Physiol 110:655–664. doi:10.1085/jgp.110.6.655
Rubaiy HN (2016) The therapeutic agents that target ATP-sensitive potassium channels. Acta Pharma 66:23–34. doi:10.1515/acph-2015-0040
Faraci FM, Sobey CG (1998) Role of potassium channels in regulation of cerebral vascular tone. J Cereb Blood Flow Metab 18:1047–1063. doi:10.1097/00004647-199810000-00001
Chrissobolis S, Sobey CG (2003) Inwardly rectifying potassium channels in the regulation of vascular tone. Curr Drug Targets 4:281–289
Henn MC, Janjua MB, Zhang H et al (2016) Increased tolerance to stress in cardiac expressed gain-of-function of adenosine triphosphate–sensitive potassium channel subunit Kir6.1. J Surg Res 206:460–465. doi:10.1016/j.jss.2016.08.043
Gozalov A, Petersen KA, Mortensen C et al (2005) Role of KATP channels in the regulation of rat dura and pia artery diameter. Cephalalgia 25:249–260. doi:10.1111/j.1468-2982.2004.00848.x
Kitazono T, Faraci FM, Taguchi H, Heistad DD (1995) Role of potassium channels in cerebral blood vessels. Stroke 26:1713–1723
Jansen-Olesen I, Mortensen CH, El-Bariaki N, Ploug KB (2005) Characterization of KATP-channels in rat basilar and middle cerebral arteries: studies of vasomotor responses and mRNA expression. Eur J Pharmacol. 523:109–118. doi:10.1016/j.ejphar.2005.08.028
Janigro D, West GA, Gordon EL, Winn HR (1993) ATP-sensitive K+ channels in rat aorta and brain microvascular endothelial cells. Am J Phys. 265:C812–C821
Faraci FM, Heistad DD (1998) Regulation of the Cerebral Circulation: Role of Endothelium and Potassium Channels. Physiol Rev. 78:53–97
McPherson GA, Stork AP (1992) The resistance of some rat cerebral arteries to the vasorelaxant effect of cromakalim and other K+ channel openers. Br J Pharmacol 105:51–58. doi:10.1111/j.1476-5381.1992.tb14209.x
Ploug KB, Edvinsson L, Olesen J, Jansen-Olesen I (2006) Pharmacological and molecular comparison of KATP channels in rat basilar and middle cerebral arteries. Eur J Pharmacol 553:254–262. doi:10.1016/j.ejphar.2006.09.053
Ploug KB, Baun M, Hay-Schmidt A et al (2010) Presence and vascular pharmacology of KATP channel subtypes in rat central and peripheral tissues. Eur J Pharmacol 637:109–117. doi:10.1016/j.ejphar.2010.03.027
Ploug KB, Sørensen MA, Strøbech L et al (2008) KATP channels in pig and human intracranial arteries. Eur J Pharmacol 601:43–49. doi:10.1016/j.ejphar.2008.10.041
Ploug KB, Amrutkar DV, Baun M et al (2012) K(ATP) channel openers in the trigeminovascular system. Cephalalgia 32:55–65. doi:10.1177/0333102411430266
Niu K, Saloman JL, Zhang Y, Ro JY (2011) Sex differences in the contribution of ATP-sensitive K+ channels in trigeminal ganglia under an acute muscle pain condition. Neuroscience 180:344–352. doi:10.1016/j.neuroscience.2011.01.045
Leoni LAB, Leite GS, Wichi RB, Rodrigues B (2013) Sildenafil: two decades of benefits or risks? Aging Male 16:85–91. doi:10.3109/13685538.2013.801952
Kruuse C, Thomsen LL, Birk S, Olesen J (2003) Migraine can be induced by sildenafil without changes in middle cerebral artery diameter. Brain 126:241–247. doi:10.1093/brain/awg009
Olesen J, Thomsen LL, Lassen LH, Olesen IJ (1995) The nitric oxide hypothesis of migraine and other vascular headaches. Cephalalgia 15:94–100. doi:10.1046/j.1468-2982.1995.015002094.x
Olesen J, Iversen HK, Thomsen LL (1993) Nitric oxide supersensitivity: a possible molecular mechanism of migraine pain. Neuroreport 4:1027–1030
Niehaus L, Gottschalk S, Weber U (1998) Effect of drug-induced vasodilatation of basal brain arteries with nitroglycerin on blood flow velocity and volume flow in the middle cerebral artery. Ultraschall Med 19:225–229. doi:10.1055/s-2007-1000495
Armstead WM (1996) Role of ATP-sensitive K+ channels in cGMP-mediated pial artery vasodilation. Am J Phys 270:H423–H426
Hempelmann RG, Ziegler A, Mehdorn HM (2001) Role of potassium channels in the relaxation induced by the nitric oxide ( NO ) donor DEA / NO in the isolated rat basilar artery. Neurosci Lett 313:21–24
Murphy ME, Brayden JE (1995) Nitric oxide hyperpolarizes rabbit mesenteric arteries via ATP-sensitive potassium channels. J Physiol 486 ( Pt 1:47–58. doi:10.1113/jphysiol.1995.sp020789
Yuan Z, Hein TW, Rosa RH, Kuo L (2008) Sildenafil (viagra) evokes retinal arteriolar dilation: dual pathways via NOS activation and phosphodiesterase inhibition. Investig Ophthalmol Vis Sci 49:720–725. doi:10.1167/iovs.07-1208
Gozalov A, Jansen-Olesen I, Klaerke D, Olesen J (2008) Role of KATP channels in cephalic vasodilatation induced by calcitonin gene-related peptide, nitric oxide, and transcranial electrical stimulation in the rat. Headache 48:1202–1213. doi:10.1111/j.1526-4610.2008.01205.x
Jansen-Olesen I, Gulbenkian S, Engel U et al (2004) Peptidergic and non-peptidergic innervation and vasomotor responses of human lenticulostriate and posterior cerebral arteries. Peptides 25:2105–2114. doi:10.1016/j.peptides.2004.08.002
Edvinsson L, Gulbenkian S, Barroso CP et al (1998) Innervation of the human middle meningeal artery: immunohistochemistry, ultrastructure, and role of endothelium for vasomotility. Peptides 19:1213–1225. doi:10.1016/S0196-9781(98)00066-7
Edvinsson L, Ekman R, Jansen I et al (1987) Calcitonin gene-related peptide and cerebral blood vessels: distribution and vasomotor effects. J Cereb Blood Flow Metab 7:720–728. doi:10.1038/jcbfm.1987.126
Olesen J, Diener H, Husstedt IW, et al (2004) Calcitonin gene–related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. Society 1104–1110
Ho TW, Ferrari MD, Dodick DW et al (2008) Efficacy and tolerability of MK-0974 (telcagepant), a new oral antagonist of calcitonin gene-related peptide receptor, compared with zolmitriptan for acute migraine: a randomised, placebo-controlled, parallel-treatment trial. Lancet 372:2115–2123. doi:10.1016/S0140-6736(08)61626-8
Hou M, Xing H, Cai Y et al (2017) The effect and safety of monoclonal antibodies to calcitonin gene-related peptide and its receptor on migraine: a systematic review and meta-analysis. J Headache Pain 18:42. doi:10.1186/s10194-017-0750-1
Nelson MT, Huang Y, Brayden JE et al (1990) Arterial dilations in response to calcitonin gene-related peptide involve activation of K+ channels. Nature 344:770–773. doi:10.1038/344770a0
Quayle JM, Bonev AD, Brayden JE, Nelson MT (1994) Calcitonin gene-related peptide activated ATP-sensitive K+ currents in rabbit arterial smooth muscle via protein kinase a. J Physiol 475:9–13. doi:10.1113/jphysiol.1994.sp020045
Nakaya Y, City T (1995) Calcitonin gene-related peptide activates the K + channels of vascular smooth muscle cells via adenylate cyclase. 332–336
Wellman GC, Quayle JM, Standen NB (1998) ATP-sensitive K+ channel activation by calcitonin gene-related peptide and protein kinase a in pig coronary arterial smooth muscle. J Physiol. 507(1):117–129
Bruch L, Rubel S, Kästner A, et al (1998) Pituitary adenylate cyclase activating peptides relax human pulmonary arteries by opening of K ATP and K Ca channels. 586–587
Chalovich JM, Eisenberg E (2005) NIH public access. Biophys Chem 257:2432–2437. doi:10.1016/j.immuni.2010.12.017 Two stage
Amin FM, Asghar MS, Guo S, et al (2011) Headache and prolonged dilatation of the middle meningeal artery by PACAP38 in healthy volunteers. 32:140–149. doi:10.1177/0333102411431333
Amin FM, Hougaard A, Schytz HW et al (2014) Investigation of the pathophysiological mechanisms of migraine attacks induced by pituitary adenylate cyclase-activating polypeptide-38. Brain 137:779–794. doi:10.1093/brain/awt369
Erdling A, Sheykhzade M, Maddahi A et al (2013) VIP/PACAP receptors in cerebral arteries of rat: characterization, localization and relation to intracellular calcium. Neuropeptides 47:85–92. doi:10.1016/j.npep.2012.12.005
Dalsgaard T, Hannibal J, Fahrenkrug J et al (2003) VIP and PACAP display different vasodilatory effects in rabbit coronary and cerebral arteries. Regul Pept 110:179–188. doi:10.1016/S0167-0115(02)00205-7
Uddman R, Goadsby PJ, Jansen I, Edvinsson L (1993) PACAP, a VIP-like peptide: immunohistochemical localization and effect upon cat pial arteries and cerebral blood flow. J Cereb Blood Flow Metab 13:291–297. doi:10.1038/jcbfm.1993.36
Tong S, Parfenova H, Shibata M et al (1993) Pituitary adenylate cyclase-activating polypeptide dilates cerebral arterioles of newborn pigs. Proc Soc Exp Biol Med 203:343–347
Seki Y, Suzuki Y, Baskaya MK et al (1995) The effects of pituitary adenylate cyclase-activating polypeptide on cerebral arteries and vertebral artery blood flow in anesthetized dogs. Eur J Pharmacol 275:259–266
Zagami AS, Edvinsson L, Goadsby PJ (2014) Pituitary adenylate cyclase activating polypeptide and migraine. Ann Clin Transl Neurol 1:1036–1040. doi:10.1002/acn3.113
Vaudry D, Falluel-morel A, Bourgault S et al (2009) Pituitary Adenylate Cyclase-activating polypeptide and its receptors : 20 years after the discovery. Pept Res 61:283–357. doi:10.1124/pr.109.001370.283
Syed AU, Koide M, Braas KM et al (2012) Pituitary adenylate cyclase-activating polypeptide (PACAP) potently dilates middle meningeal arteries: implications for migraine. J Mol Neurosci 48:574–583. doi:10.1007/s12031-012-9851-0
Wienecke T, Olesen J, Ashina M (2010) Prostaglandin I2 (epoprostenol) triggers migraine-like attacks in migraineurs. Cephalalgia 30:179–190. doi:10.1111/j.1468-2982.2009.01923.x
Ray CJ, Marshall JM (2006) The cellular mechanisms by which adenosine evokes release of nitric oxide from rat aortic endothelium. J Physiol 570:85–96. doi:10.1113/jphysiol.2005.099390
Thomas P, Dixon MS, Winterton SJ, Sheridan DJ (1990) Acute haemodynamic effects of cromakalim in patients with angina pectoris. Br J Clin Pharmacol 29:325–331
Antihypertensive Effect of Levcromakalim in patients with essential hypertension.pdf
Williams AJ, Lee TH, Vyse T et al (1990) Attenuation of nocturnal asthma by cromakalim. Lancet 336:334–336. doi:10.1016/0140-6736(90)91877-D
von Nguyen P, Holliwell DL, Davis A et al (1991) Effects of the potassium channel activator, cromakalim, on arterial and cardiac responses to norepinephrine, angiotensin II, and isoproterenol in normotensive men. J Cardiovasc Pharmacol 18:797–806
Kidney JC, Fuller RW, Worsdell YM et al (1993) Effect of an oral potassium channel activator, BRL 38227, on airway function and responsiveness in asthmatic patients: comparison with oral salbutamol. Thorax 48:130–133. doi:10.1136/thx.48.2.130
Fox JS, Whitehead EM, Shanks RG (1991) Cardiovascular effects of cromakalim (BRL 34915) in healthy volunteers. Br J Clin Pharmacol 32:45–49
Hempelmann RG, Barth HL, Mehdorn HM et al (1995) Effects of potassium channel openers in isolated human cerebral arteries. Neurosurgery 37:1146–1153
Ploug KB, Boni LJ, Baun M et al (2008) K(ATP) channel expression and pharmacological in vivo and in vitro studies of the K(ATP) channel blocker PNU-37883A in rat middle meningeal arteries. Br J Pharmacol 154:72–81. doi:10.1038/bjp.2008.86
Zoga V, Kawano T, Liang M-Y et al (2010) KATP channel subunits in rat dorsal root ganglia: alterations by painful axotomy. Mol Pain 6:6. doi:10.1186/1744-8069-6-6
Olesen J, Jansen-Olesen I (2000) Nitric oxide mechanisms in migraine. Pathol Biol (Paris) 48:648–657
Villalón CM, Olesen J (2009) The role of CGRP in the pathophysiology of migraine and efficacy of CGRP receptor antagonists as acute antimigraine drugs. Pharmacol Ther 124:309–323. doi:10.1016/j.pharmthera.2009.09.003
Schytz HW, Birk S, Wienecke T et al (2009) PACAP38 induces migraine-like attacks in patients with migraine without aura. Brain 132:16–25. doi:10.1093/brain/awn307
Kaiser EA, Russo AF (2013) CGRP and migraine: could PACAP play a role too? Neuropeptides 47:451–461. doi:10.1016/j.npep.2013.10.010
Schytz HW, Schoonman GG, Ashina M (2010) What have we learnt from triggering migraine? Curr Opin Neurol 23:259–265. doi:10.1097/WCO.0b013e328337b884
Garthwaite J, Charles SL, Chess-Williams R (1988) Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 336:403–405. doi:10.1038/332141a0
Guo S, Olesen J, Ashina M (2014) Phosphodiesterase 3 inhibitor cilostazol induces migraine-like attacks via cyclic AMP increase. Brain 137:2951–2959. doi:10.1093/brain/awu244
Dinn RB, Wall M (2006) Tadalafil associated with typical migraine aura without headache. Cephalalgia 26:1344–1346. doi:10.1111/j.1468-2982.2006.01188.x
Road C (2013) The international classification of headache disorders, 3rd edition (beta version). Cephalalgia 33:629–808. doi:10.1177/0333102413485658
Lauritzen M (1994) Pathophysiology of the migraine aura. The spreading depression theory. Brain 117(Pt 1):199–210
Wendt S, Wogram E, Korvers L, Kettenmann H (2016) Experimental cortical spreading depression induces NMDA receptor dependent potassium currents in microglia. J Neurosci 36:6165–6174. doi:10.1523/JNEUROSCI.4498-15.2016
Staurengo-Ferrari L, Zarpelon AC, Longhi-Balbinot DT et al (2014) Nitroxyl inhibits overt pain-like behavior in mice: role of cGMP/PKG/ATP-sensitive potassium channel signaling pathway. Pharmacol Reports 66:691–698. doi:10.1016/j.pharep.2014.04.003
North RA, Williams JT, Surprenant A, Christie MJ (1987) Mu and delta receptors belong to a family of receptors that are coupled to potassium channels. Neurobiology 84:5487–5491
Riefflin A, Ayyagari U, Manley SE et al (2015) The effect of glibenclamide on insulin secretion at normal glucose concentrations. Diabetologia 58:43–49. doi:10.1007/s00125-014-3399-1
Perricone SC, Humphrey SJ, Skaletzky LL, et al (1994) In rats and dogs. 3693–3700
Meisheri KD, Humphrey SJ, Khan SA et al (1993) 4-morpholinecarboximidine-N-1-adamantyl-N’-cyclohexylhydrochloride (U-37883A): pharmacological characterization of a novel antagonist of vascular ATP-sensitive K+ channel openers. J Pharmacol Exp Ther 266:655–665
Humphrey SJ, Smith MP, Cimini MG et al (1996) Cardiovascular effects of the K-ATP channel blocker U-37883A and structurally related morpholinoguanidines. Methods Find Exp Clin Pharmacol 18:247–260
Shyng SL, Nichols CG (1998) Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 282(80):1138–1141. doi:10.1126/science.282.5391.1138
Davies GC, Thornton MJ, Jenner TJ et al (2005) Novel and established potassium channel openers stimulate hair growth in vitro: implications for their modes of action in hair follicles. J Invest Dermatol 124:686–694. doi:10.1111/j.0022-202X.2005.23643.x
Muiesan G, Fariello R, Muiesan ML, Christensen OE (1985) Effect of pinacidil on blood pressure, plasma catecholamines and plasma renin activity in essential hypertension. Eur J Clin Pharmacol 28:495–499
Laher MS, Hickey MP (1985) Pharmacokinetics and bioavailability of pinacidil capsules in human volunteers. J Int Med Res 13:159–162. doi:10.1177/030006058501300302
D’Arcy V, Laher M, McCoy D et al (1985) Pinacidil, a new vasodilator, in the treatment of mild to moderate essential hypertension. Eur J Clin Pharmacol 28:347–349
Zachariah PK, Sheps SG, Schirger A et al (1986) Antihypertensive efficacy of pinacidil--automatic ambulatory blood pressure monitoring. Eur J Clin Pharmacol 31:133–141
Sterndorff B, Johansen P (1988) The antihypertensive effect of pinacidil versus prazosin in mild to moderate hypertensive patients seen in general practice. Acta Med Scand 224:329–336
Goldberg MR (1988) Clinical pharmacology of pinacidil, a prototype for drugs that affect potassium channels. J Cardiovasc Pharmacol 12(Suppl 2):S41–S47
Camm AJ, Maltz MB (1989) A controlled single-dose study of the efficacy, dose response and duration of action of nicorandil in angina pectoris. Am J Cardiol 63:61J–65J
Raftery EB, Lahiri A, Hughes LO, Rose EL (1993) A double-blind comparison of a beta-blocker and a potassium channel opener in exercise induced angina. Eur heart J 14(Suppl B):35–39
Roland E (1993) Safety profile of an anti-anginal agent with potassium channel opening activity: an overview. Eur heart J 14(Suppl B):48–52
Wolf DL, Ferry JJ, Hearron AE et al (1993) The haemodynamic effects and pharmacokinetics of intravenous nicorandil in healthy volunteers. Eur J Clin Pharmacol 44:27–33
Witchitz S, Darmon JY (1995) Nicorandil safety in the long-term treatment of coronary heart disease. Cardiovasc Drugs Ther 9(Suppl 2):237–243
Dunn N, Bm MA, Freemantle S, et al (1999) Safety pro ® le of Nicorandil Ð prescription-event monitoring ( PEM ) study. 205:197–205
Singer DR, Markandu ND, Miller MA et al (1989) Potassium channel stimulation in normal subjects and in patients with essential hypertension: an acute study with cromakalim (BRL 34915). J Hypertens Suppl 7:S294–S295
Suzuki S, Yano K, Kusano S, Hashimoto T (1995) Antihypertensive effect of levcromakalim in patients with essential hypertension. Study by 24-h ambulatory blood pressure monitoring. Arzneimittelforschung 45:859–864
Funding
This article was supported by the Lundbeck Foundation.
Author information
Authors and Affiliations
Contributions
MMK designed and performed the review, with the help of JMH, JS, IJO and MA. MMK drafted the manuscript with the help of JMH, JS, IJO and MA. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Al-Karagholi, M.AM., Hansen, J.M., Severinsen, J. et al. The KATP channel in migraine pathophysiology: a novel therapeutic target for migraine. J Headache Pain 18, 90 (2017). https://doi.org/10.1186/s10194-017-0800-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10194-017-0800-8