
Abstract
Background
In migraineurs pituitary adenylate cyclase activating peptide1–38 (PACAP1–38) is a potent migraine provoking substance and the accompanying long lasting flushing suggests degranulation of mast cells. Infusion of the closely related vasoactive intestinal peptide (VIP) either induces headache or flushing. This implicates the pituitary adenylate cyclase activating peptide type I receptor (PAC1) to be involved in the pathophysiology of PACAP1–38 provoked headaches. Here we review studies characterizing the effects of mainly PACAP but also of VIP on cerebral and meningeal arteries and mast cells.
Discussion
PACAP1–38, PACAP1–27 and VIP dilate cerebral and meningeal arteries from several species including man. In rat cerebral and meningeal arteries the dilation seems to be mediated preferably via vasoactive intestinal peptide receptor type 1 (VPAC1) receptors while, in human, middle meningeal artery dilation induced via vasoactive intestinal peptide receptor type 2 (VPAC2) receptors cannot be ruled out. PACAP1–38 is a strong degranulator of peritoneal and dural mast cells while PACAP1–27 and VIP only have weak effects. More detailed characterization studies suggest that mast cell degranulation is not mediated via the known receptors for PACAP1–38 but rather via a still unknown receptor coupled to phospholipase C.
Conclusion
It is suggested that PACAP1–38 might induce migraine via degranulation of dural mast cells via a yet unknown receptor.
Similar content being viewed by others


Review
Migraine is number six in WHO list of all diseases causing disability [1] and it is the third most costly neurological disorder in Europe [2]. Even though the triptans revolutionized the acute treatment of migraine, a huge unmet need for better or different acute treatments exists [3]. An interesting molecule in this aspect is pituitary adenylate cyclase activating peptide (PACAP), which exists in the body as 38- and 27- amino acid peptides [4, 5]. These peptides partly share receptors with their family member vasoactive intestinal peptide (VIP) [6]. In migraineurs, elevated levels of PACAP1–38 were found in blood sampled from the external jugular vein [7] and cubital vein [8] during migraine attacks. Infusion of PACAP1–38 provokes immediate headache in 11 out of 12 migraine patients, 7 of these patients develop delayed migraine attacks. In all 12 healthy subjects an immediate headache was experienced, two of these subsequently reporting migraine-like symptoms [9, 10]. Interestingly, VIP only induces a mild headache and no migraine-like attacks in migraineurs [11]. These findings point towards the PAC1 receptor, which is targeted by PACAP with much higher affinity than VIP, as a key target for migraine treatment. In this review we describe studies characterizing the receptors upon which PACAP and VIP mediate dilation of intracranial arteries and degranulation of peritoneal and dural mast cells.
Pituitary adenylate cyclase activating peptide
Pituitary adenylate cyclase activating peptide (PACAP) is a highly conserved signaling peptide of identical structure in mammals including human, sheep, rat and mouse [12]. It is a member of the glucagon/secretin superfamily of peptides [6, 13, 14] and is endogenously present in two isoforms namely; PACAP1–38 and the C-terminal truncated version PACAP1–27. High concentrations of PACAP1–38 are found in brain and in testis. Especially the hypothalamus but also other brain regions contain considerable amounts of PACAP1–38. PACAP1–27 is considerably less abundant in these regions as compared to PACAP1–38 [4]. A related member of the glucagon/secretin superfamily is the 28 amino acid peptide, VIP that shares 68% homology with PACAP1–27 from the N-terminal end. PACAP and VIP are signaling molecules widely distributed throughout the central and peripheral nervous system [6, 13] involved in e.g. regulation of circadian rhythm [15], neuroprotection [16, 17], inflammation and pain perception [18, 19].
PACAP-immunoreactivity (−IR) and VIP-IR co-localize in nerve fibers innervating cerebral vessels and parasympathetic ganglia [20,21,22,23,24] and in dura mater where it occasionally co-localizes with calcitonin gene-related peptide (CGRP) [25]. In trigeminal ganglion, PACAP-IR co-localizes with CGRP-IR neurons, while only PACAP-IR is found in satellite glial cells [26,27,28]. In the spinal trigeminal nucleus PACAP-IR co-localizes with CGRP-IR in nerve fibers in laminae I and II [26, 29].
PACAP receptors
PACAP and VIP partially share receptors and PACAP signal transduction is mediated through three high-affinity G protein-coupled receptors namely pituitary adenylate cyclase activating peptide type I receptor (PAC1), vasoactive intestinal peptide receptor type 1 (VPAC1), and vasoactive intestinal peptide receptor type 2 (VPAC2). The affinities of PACAP1–38 and PACAP1–27 are equal to that of VIP for VPAC1- and VPAC2- receptors, whereas the affinity of PACAP1–38 and PACAP1–27 for the PAC1 receptor (PAC1-R) is about 1000-fold higher than that of VIP [6, 19, 30] (Fig. 1). The potent headache provoking property of PACAP1–38 [10] in comparison with the poor effect of VIP [11], suggests PAC1- R to be an interesting target for migraine treatment.

Schematic overview of selectivity of receptors for pituitary adenylate cyclase activating polypeptide (PACAP) and vasoactive intestinal peptide (VIP). Pituitary adenylate cyclase activating polypeptide receptor 1 (PAC1) has a 1000-fold greater affinity for PACAP1–27 (red) and PACAP1–38 (yellow) than for VIP (light blue). Vasoactive intestinal peptide receptor (VPAC)1 and VPAC2 bind VIP (blue) and PACAP1–27(red) and PACAP1–38 (yellow) with equal affinity. pKi (negative logarithm of the concentration that occupies half the receptor population at equilibrium) values given in the figure is adapted from [19]. No difference in receptor selectivity between PACAP1–38 and PACAP1–27 is described
In human cerebral and middle meningeal arteries, messenger RNA (mRNA) for VPAC1, VPAC2 and PAC1 receptors has been identified [31, 32]. In rat, mRNA of the same three receptors was shown by qPCR in middle meningeal arteries [33] and by in situ hybridization to be localized in smooth muscle cells of middle cerebral arteries, basilar arteries and middle meningeal arteries [34]. Immunohistochemistry with antibodies for the VPAC1 receptor shows its presence in the smooth muscle cells of rat cerebral arteries [24]. In rat trigeminal ganglion and spinal trigeminal nucleus all three receptors are detected at the mRNA level [26, 33].
Cranial arteries and migraine
In the 1940’s the genesis of migraine pain was attributed to meningeal and cerebral arteries, as it was reported that electrical stimulation of these arteries evoked nausea and ipsilateral pain, localized to the area in and around the eye, including the forehead and temple [35, 36]. The perivascular proximity of nociceptive afferents [37, 38], the pulsating nature of migraine headache (in 80% of patients) aggravating with physical activity [39] as well as pain and nausea induction during arterial stimulation [36], have all been interpreted as strong indicators of a vascular component of migraine pathogenesis. However, accumulating evidence has challenged the theory of migraine as a vascular disease. Migraine-provoking substances are strong vasodilators [10, 40,41,42,43]. However, not all vasodilatory compounds provoke accompanying headache [11, 44]. It was recently shown that spontaneous migraine attacks in patients are accompanied by dilation of the pain sensitive middle cerebral and internal carotid arteries whereas no dilation of dural and extracranial arteries are observed [45].
The effect of PACAP on cerebral arteries
In vitro
The relaxant effect of PACAP has been studied on isolated cerebral arteries from several species including humans. The potency of PACAP1–38 and PACAP1–27 given as pD2 values (the negative logarithm to base 10 of the concentration of a drug that gives half-maximal response) are in most specimens around 8 (Table I). In cat the potency and efficacy for VIP were somewhat higher than for PACAP1–38 and PACAP1–27 [46], while no difference in potency was found between PACAP1–27 and VIP in rabbit [23]. PACAP1–27 is less potent as a dilator of human cerebral arteries than calcitonin gene-related peptide (CGRP) and VIP (Fig. 2). Comparing data from two different studies performed in human cerebral arteries, one with PACAP1–38 and the other using PACAP1–27, the relaxations were of the same potency, but PACAP1–38 has a lower efficacy than PACAP1–27 (Table 1) [47, 48]. This observation was also made in rat using pressurized arteriography [47] but not in a wire myography study [34]. However, a direct comparison of PACAP1–38 and PACAP1–27 induced effects on human cerebral arteries in parallel experiments has yet to be performed. Blockade experiments suggest VPAC1 receptors to be of importance for PACAP and VIP induced relaxation of rat middle cerebral and basilar arteries [34].

Relaxant responses to PACAP1–27 (n = 4), VIP (n = 7) and CGRP (n = 10), expressed as % of pre-contraction induced by prostaglandin F2α in human cerebral arteries. Mean values ± S.E.M. are given. n = number of experiments, one from each patient. Modified from Jansen-Olesen et al. [48]
In vivo
No studies describe the in vivo effect of PACAP on cerebral arteries after i.v. infusion to laboratory animals. The reason for this is most probably due to the fact that PACAP has to cross the blood–brain barrier to reach its receptors in the smooth muscle cells of cerebral arteries. A transport mechanism for PACAP1–38 has been described, which is dependent on the peptide transport system-6 (PTS-6) [49]. However, only a small percentage (0.053%) of PACAP-38 enters the brain after intravenous administration [50]. If a dilation of cerebral arteries is achieved together with a fall in mean arterial blood pressure the interpretation of the results is made complicated due to activation of autoregulatory mechanisms leading to dilation of cerebral arteries [51]. To avoid confusion about dilation of cerebral arteries, pharmacological substances can be infused via an indwelling catheter in the common carotid artery (i.c.), which allows cerebral arteries to be studied without systemic effects [52]. However, no studies has to date been performed to investigate the effect of PACAP1–38 on cerebral arteries after i.c. infusion. In human experimental studies PACAP1–38 infusion in healthy volunteers [53] and migraine patients [54] showed a minor short-lasting dilation of middle cerebral arteries. The measurement of middle cerebral artery diameter in these studies was calculated from blood velocity in the middle cerebral artery and was therefore indirect. In another study, no change in mean circumference of middle cerebral artery was found after infusion of PACAP1–38. Here magnetic resonance angiography was used, which is a more direct way to measure the artery diameter and is superior to measurement of blood velocity [9].
The effect of PACAP on middle meningeal arteries
In vitro
To the best of our knowledge only two studies have been published describing vascular responses of isolated middle meningeal arteries from animals. In the first study, administration of PACAP1–38, PACAP1–27, and VIP to pre-contracted rat arterial segments did not cause any significant effect. Confirming the viability of the preparation, treatment with CGRP of the same arterial segments caused a 100% relaxation of the pre-contraction [34]. In the second study, rat middle meningeal arteries were mounted in a pressurized myograph system. In concentrations as low as 1–1000 pM, PACAP1–38 caused dilation of middle meningeal arteries that were blocked by the PAC1 receptor antagonist PACAP6–38 [55] (Fig. 3). It was suggested that PACAP1–38 affected middle meningeal artery tone by acting on a combination of two splice variants of the PAC1 receptor, namely the PAC1null and PAC1Hop1 receptor isoforms. Stimulation of PAC1 receptor causes in turn activation of the cyclic adenosine monophosphate/protein kinase A pathway leading to the opening of adenosine triphosphate sensitive potassium channels [56].

Low picomolar concentrations of PACAP, but not VIP, dilate isolated pressurized rat middle meningeal arteries. Cumulative concentrations of PACAP and VIP were administered to arterial segments pressurized to 40 mmHg ex vivo. Arteries were exposed to aCSF containing each concentration of PACAP1–38 or VIP for 20 min. Dilation to PACAP1–38 or VIP are expressed as percentage of maximum dilation obtained in the presence of Ca2+ −free artificial CSF containing 100 μM of the calcium channel blocker diltiazem and 1 μM of the adenylyl cyclase activator forskolin. p < 0.05 by unpaired t test, n = 4. From Syed et al. [55]
In man, PACAP1–38 and VIP induced only a weak relaxation of isolated middle meningeal arteries [31, 57]. VIP had a somewhat more potent effect on dilation than PACAP1–38. Neither the PAC1 antagonist PACAP6–38 nor the VPAC1 antagonist PG97–269 were able to block the PACAP1–38 induced relaxation suggesting the effect to be mediated via VPAC2 receptors [31].
In vivo
The genuine closed cranial window model has been used to study the effect of PACAP1–38, PACAP1–27, and VIP on rat middle meningeal artery in vivo. When given as a bolus i.v. infusion to anaesthetized rats a maximal dilation of ~60% was found for VIP and PACAP1–27, while the efficacy of PACAP1–38 was somewhat lower with a dilation of ~45%. Interestingly, the pD2 value of ~6 (in g/kg) for PACAP1–38 indicated a higher sensitivity of the middle meningeal artery as compared to PACAP1–27 with a pD2 value of ~5.5 [33]. In the presence of VPAC1 receptor antagonist (PG97–269) the response to PACAP1–38 but not to VIP was significantly decreased [33]. When given alone PACAP6–38 induced a slight dilation, but no significant inhibition of PACAP1–38 induced dilation of middle meningeal arteries was observed [33]. All together suggesting PACAP1–38 induced dilation of middle meningeal artery to be mediated via VPAC1 receptors. In another study, PG97–269 did not inhibit VIP and PACAP1-38 induced dilation of middle meningeal arteries. On the other hand, the VPAC1/VPAC2 antagonist VIP6–28 significantly inhibited VIP and PACAP1–38 induced dilation, suggesting VPAC2 receptors to be responsible [58]. This is in support of the findings in human meningeal arteries [31]. Thus, controversy exists whether VIP and PACAP induced dilation of rat meningeal arteries are mediated via VPAC1 or VPAC2 receptors.
Intra carotid artery administration of PACAP1–38 induces an ED50 (the dose of a drug that gives half-maximal response) response in dural arteries at ten times lower concentrations of PACAP1–38 than after i.v. infusion [52]. Also, the maximum change in artery diameter from baseline was around 75% when given i.c. and 50% when given i.v. [52]. Increasing doses of PACAP1–38, PACAP1–27, and VIP administered as bolus i.c. infusion exhibited pD2-values of 6.7, 6.5, and 6.2, respectively. The maximum responses to PACAP1–38 and PACAP1–27 were around 105% (change from baseline) and VIP around 75% (Fig. 4) [59]. Because of variations between animals no significant differences between PACAP1–38, PACAP1–27 and VIP responses were observed.

Effects of increasing doses (i.c.) of PACAP1–38, PACAP1–27 and VIP on middle meningeal artery diameter in the genuine closed cranial window model. Mean values ± SEM from 5 to 7 animals. Adapted from Bhatt et al. [59]
Mast cells and migraine
Mast cells
Mast cells were first described in the late nineteenth century, but it was not until the 1950’s that part of their biological contribution to inflammatory allergic diseases became known through the discovery of histamine release. Mast cells contain vesicles comprising numerous inflammatory and vasodilatory substances (Fig. 5) and undergo degranulation upon activation by exogenous allergens or endogenous stimuli [60]. Mast cells are derived from pluripotent hematopoietic CD34+ stem cells in the bone marrow and circulate in the blood as progenitors before they acquire a mature phenotype in the microenvironment of their target tissue [61]. They are embedded in various tissues throughout the body and derive into either of two subtypes as referred to as mucosal or connective tissue type mast cells. The local cytokine environment conditions their subtype, but they hold an ability to adapt and change phenotype if needed [62, 63]. Mast cells embedded in skin, peritoneum, and dura mater are all of the connective tissue type, and thus peritoneal mast cells can potentially be used as a model for dura mater mast cells [64].

Toluidine blue stained intact and degranulated mast cell are shown together with a list of mast cell mediators [91]
Two different signaling pathways leading to degranulation have been identified, namely the antigen and the basic secretagogue. The antigen pathway comprises stimulation through cross-linking with the high-affinity immunoglobulin E (IgE) receptors, FcεRI, and mast cells release their mediators to the local environment. Basic secretagogues stimulate mast cells to degranulate via G protein-dependent activation of phospholipase C. However, they can also be stimulated to degranulate via mechanical, thermal, or even receptor-independent mechanisms [65].
Clinical implications of mast cell-involvement in migraine
A correlation between mast cell function and migraine has been clinically implicated by significantly elevated plasma histamine levels in migraine patients, both during attacks and in interictal periods [66, 67]. For migraineurs there is a high comorbidity to histamine-driven conditions like allergic rhinitis, asthma, and food allergy [68,69,70,71] as compared to the general population [72]. Histamine-infusion to migraineurs induced an immediate headache during infusion, followed by a genuine migraine attack several hours later. This can be abolished by pretreatment with the histamine-receptor 1 (H1) antagonist, mepyramine [73]. However, histamine release alone is not responsible for spontaneous migraine attacks, as histamine-receptor H1 and H2 blockade is a poor prophylaxis for migraine sufferers [73, 74], indicating a discrepancy between genuine migraine attacks as compared to histamine-provoked attacks. Stimulation of histamine H3 receptors have been suggested to be involved in a negative feedback loop causing inhibition of histamine release from mast cells and C-fiber nerve endings [75]. The histamine catabolite Nα-methylhistamine, that is about 3 times more active as an agonist on the H3 receptor, was found to be significantly better than placebo after prophylactic treatment twice a week for 12 weeks [75]. These findings are somewhat surprising, considering that H1- and H2 receptor antihistamines have not been effective in treating migraine [76].
In addition to histamine, mast cells release several chemical mediators such as prostaglandin I2 (PGI2), that has been shown to cause activation and sensitization of meningeal sensory afferents [77, 78] and to induce immediate headache in migraine patients and non-migraineurs as well as migraine-like attacks in migraineurs [43, 79]. Glyceryl trinitrate (GTN) is a potent migraine provoking substance that in low doses causes degranulation of dural mast cells after i.v. infusion to awake as well as anaesthetized rats [80, 81]. PACAP, but not VIP, has been shown to induce migraine headache as well as mast cell degranulation [10, 11, 82]. Thus, given their pro-inflammatory properties and their dense population in dura mater, mast cells are suggested to be involved in the pathophysiological processes leading to migraine [83,84,85].
Characterization of PACAP-induced mast cell degranulation
The mast cell degranulating effect of PACAP was first shown in human skin biopsies [86]. Single challenges with PACAP1–38, PACAP1–27, and VIP caused significant release of histamine peaking at 4 min after skin challenge. The release of histamine was significantly higher for VIP and PACAP1–27 as compared to PACAP1–38 [86]. In mice an intradermal injection of PACAP1–38 induced oedema and significant degranulation of mast cells [87]. In a more detailed study, mast cell degranulation induced by PACAP analogues, including both PAC1 receptor agonists and antagonists, was characterized in isolated rat peritoneal mast cells. PACAP1–38, PACAP1–27, VIP, PACAP6–38, PACAP16–38, and PACAP28–38 induced a concentration-dependent degranulation of the mast cells (Fig. 6). The compounds tested divided in two distinct groups, the efficient degranulators being PACAP1–38, PACAP6–38, and PACAP16–38 with pEC50 values between 6.6 and 6.2; interestingly, the PAC1 receptor antagonist PACAP6–38 is a member of this group. The other group consisted of weaker degranulators being PACAP1–27, VIP, and PACAP28–38 with pEC50 values between 5.5 and 4.8. Furthermore both, the PAC1 receptor agonist maxadilan, a 61–amino acid peptide with no significant sequence homology to PACAP [23], and the PAC1 receptor selective antagonist max.d.4, a modified fragment of maxadilan, showed no mast cell degranulating effects when applied at a concentration of up to 10−5 M [82]. These findings all suggest a PAC1 receptor independent mast cell degranulation and are further supported by a still unpublished study from our group where the PAC1 receptor antagonist M65 (another modified fragment of Maxadilan) failed to inhibit PACAP1–38 induced mast cell degranulation. Inhibition of intracellular mechanisms demonstrated that the phospholipase C inhibitor U-73122 significantly inhibited PACAP1–38- but not PACAP1–27- and VIP-induced mast cell degranulation (Fig. 7). The adenylyl cyclase inhibitor SQ 22536 has no effect on mast cell degranulation induced by either of the peptides. When taken together, the difference in potency between mast cell degranulating effects of PACAP1–38 and PACAP1–27 known to be equipotent on PAC1 receptors, the potent mast cell degranulating properties of the PAC1 receptor antagonist PACAP6–38 and the lack of inhibitory effect of M65 on PACAP1–38 induced mast cell degranulation, all suggest that degranulation is not mediated via the PAC1 receptor in rat [82].

Degranulation of rat peritoneal mast cells expressed as percentage of PACAP1–38, which is the strongest mast cell degranulator tested. a Shows the effect of the endogenous peptides PACAP1–38, PACAP1–27, and VIP. b Shows the effect of PACAP1–38 and the fragments PACAP6–38, PACAP16–38, and PACAP28–38. Values are given as means ± SEM of 4–8 experiments. From Baun et al. [82]

Degranulation of peritoneal mast cells induced by a PACAP1–38, b PACAP1–27 and c VIP in the presence of the adenylyl cyclase inhibitor SQ 22536 and the phospholipase C inhibitor U-73122 alone or in combination. Values are presented as amount of degranulation expressed as percentage of degranulation with each peptide alone. Values are given as mean ± SEM, n = 5; **p < 0.01 Mann Whitney U-test as compared to the vehicle group [82]
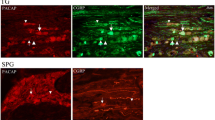
The role of PACAP1–38 induced mast cell degranulation on dural artery dilation
In healthy human volunteers PACAP1–38 was given as a 20 min infusion leading to vasodilation of the middle meningeal artery for up to five hours after infusion [10]. PACAP1–38 has an elimination half-life of 3.5 to 10 min [53, 88], hence the delayed effect cannot be attributed to a direct vascular effect of PACAP1–38, but rather to a cascade of events triggered by PACAP1–38. The strong degranulatory effect of PACAP1–38 on rat mast cells [82] and the dense population of mast cells found in apposition to dural arteries (Fig. 8) inspired our group to perform a set of experiments investigating the role of mast cell degranulation in middle meningeal artery dilation using the rat closed cranial window model. In these experiments one group of rats received repeated treatment with the secretagogue Compound 48/80, while the other group received vehicle. At the time of experiment, 4–5 days after the treatment, the mast cells were depleted of their granules (Fig. 8) [59]. In control rats a 20 min infusion of PACAP1–38, PACAP1–27, and CGRP but not VIP caused a significant increase in middle meningeal artery diameter. The response to CGRP returned to normal within 10 min after the end of infusion, while vasodilation induced by PACAP1–38 and PACAP1–27 showed a slower recovery. Fifty minutes after PACAP1–38 infusion, but not after PACAP1–27 infusion, the middle meningeal artery was still significantly dilated (Fig. 9) [59].

Toluidine blue staining revealed the presence of intact mast cells in dura mater from control rats (a) and the depletion of mast cells in dura mater from compound 48/80 treated rats (b)

Middle meningeal artery (MMA) response to 20 min i.v. infusion of CGRP (0.25 μg kg−1 min−1), PACAP1–38 (0.4 μg kg−1 min−1), PACAP1–27 (0.4 μg kg−1 min−1) and PACAP6–38 (0.4 μg kg−1 min−1). The darker color represents experiments performed on control rats while experiments represented with the lighter color are performed in mast cell depleted (MCD) rats. Mean values ± SEM are given as percentage increase in MMA diameter from the pre-stimulation baseline. Statistical analysis by ANOVA (Kruskal-Wallis test) followed by Dunn’s comparison test to compare differences from baseline values (0) ***p < 0.001; **p < 0.01; *p < 0.5. ## p < 0.01; # p < 0.05 compared to the corresponding time point in MCD rats [59]
The PAC1 receptor antagonist PACAP6–38 exhibits potent mast cell degranulating properties [82], but without direct vascular effects. When infused over 20 min no significant change in middle meningeal artery diameter is observed. However, after termination of the infusion the artery starts to dilate and dilation reaches significance at 30 min, lasting until the end of experiment 50 min after the infusion. Chronic depletion of mast cells attenuates the responses to PACAP1–38 and PACAP1–27 and abolishes the delayed PACAP6–38 induced dilation (Fig. 9) [59]. This suggests that PACAP1–38 causes dilation of middle meningeal arteries partly due to mast cell degranulation. These effects might be responsible for long-lasting flushing and delayed migraine attacks observed after PACAP1–38 infusion.
Taking the results of all the described studies together, it is interesting to note that the PAC1 receptor antagonist PACAP6–38 is as potent a mast cell degranulator as PACAP1–38 and that the effect seems to be mediated via a non-PAC1 receptor. Furthermore, the weak mast cell degranulating effects of VIP suggest that VPAC1 and VPAC2 receptors are not involved. Though, PACAP6–38 is widely used as a PAC1 receptor antagonist it should be kept in mind that it has agonistic mast cell degranulating properties similar to that of PACAP1–38 [82] and thus hypothetically PACAP6–38 might cause hypersensitivity via this mechanism. The PAC1 receptor antagonists M65 and max.d.4 don’t share the mast cell depleting properties of PACAP6–38 and therefore should be preferred in studies characterizing the effects of PACAP on durally evoked hypersensitivity. The stimulatory effect of PACAP6–38 on a non-PAC1 receptor is supported by a study performed in a primary culture of trigeminal ganglion neurons from rat and mice in which, PACAP6–38, act as an agonist [89]. However, in this study the antagonists M65 (PAC1) and VIP6–28 (VPAC1 and VPAC2) share the agonistic features with PACAP6–38. The mast cell degranulation and migraine provoking effects of PACAP6–38 have not been investigated in humans. Presuming that the rank order of potency for these compounds to induce mast cell degranulation in humans equals that in rats, such a study would disclose if PACAP1–38 and PACAP6–38 have the same order of potency in headache provocation and if mast cell degranulation is involved in migraine pathophysiology. Such a study would also reveal if PACAP provoked migraine is induced by PAC1 receptors or via a yet unknown PACAP receptor.
Conclusion
The few studies involving pharmacological characterization of PACAP- and VIP-induced relaxant responses of cerebral arteries from animals suggest the involvement of VPAC1 receptors. The mechanism for PACAP1–38 to cross the blood–brain barrier seems to be insufficient for transporting PACAP into the smooth muscle layer of the cerebral arteries in concentrations high enough to induce vasodilation after i.v. infusion of PACAP1–38.
Though isolated rat dural arteries do not respond to PACAP or VIP in a wire myograph system, PACAP1–27, PACAP1–38, and VIP show equipotent effects in studies performed on human middle meningeal arteries in vitro and rat dural arteries in vivo. In man, blockade experiments with VPAC1 and PAC1 receptor antagonists, suggests the dilation to be mediated via VPAC2 receptors. However, this assumption has not been confirmed by the use of selective antagonists for VPAC2 receptors. In rat, controversy exists weather VPAC1 or VPAC2 receptors are involved in PACAP1–38 induced meningeal artery vasodilation. As the PAC1 receptor has been suggested to be responsible for PACAP1–38 induced headache/migraine, the above described findings suggest PACAP1–38 induced headache/migraine not to be mediated via vascular responses. However, the extremely potent PAC1 receptor mediated effect of PACAP1–38 on middle meningeal arteries in a pressurized myograph system suggests a mechanism that can be involved in migraine pathophysiology. This finding was however, not observed in vivo after bolus or long term infusion with PACAP1–38 to rat or in vitro in wire myograph studies of human middle meningeal arteries.
Neurogenic inflammation involving degranulation of dural mast cells has been proposed to be part of the pathophysiological mechanisms of migraine. In rat, PACAP induces degranulation of peritoneal and dural mast cells via receptors coupled to phospholipase C. Long-term PACAP infusion causes middle meningeal artery dilation that partly is caused by degranulation of dural mast cells. Characterization of the responses suggests that the effect on mast cells is mediated via non-VPAC and –PAC1 receptors. Identifying such a receptor and a subsequent development of substances with selective antagonistic/inhibitory effect on this receptor, will open doors for more detailed studies on the role of mast cells in migraine pathophysiology. Another question to be answered is whether it is the PAC1 receptor or a still not identified receptor(s) that is (are) responsible for migraines provoked by PACAP.
Abbreviations
- CGRP:
-
Calcitonin gene-related peptide
- ED50:
-
The dose of a drug that gives half-maximal response
- FcεRI:
-
Immunoglobulin E (IgE) receptor
- i.c.:
-
Intra carotid artery
- i.v.:
-
Intravenous
- mRNA:
-
Messenger RNA
- PAC1:
-
Pituitary adenylate cyclase activating polypeptide type I receptor
- PACAP:
-
Pituitary adenylate cyclase activating polypeptide
- pD2:
-
Negative logarithm of the molar concentration producing the half maximum response
- VIP:
-
Vasoactive intestinal peptide
- VPAC1:
-
Vasoactive intestinal peptide (VIP) receptor type 1
- VPAC2:
-
Vasoactive intestinal peptide (VIP) receptor type 2
References
Vos T, Barber RM, Bell B, Bertozzi-Villa A, Biryukov S, Bolliger I et al (2013) Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the global burden of disease study 2013. Lancet 386(9995):743–800
Olesen J, Gustavsson A, Svensson M, Wittchen HU, Jonsson B (2012) The economic cost of brain disorders in Europe. Eur J Neurol 19(1):155–162
Tfelt-Hansen P, Olesen J (2012) Taking the negative view of current migraine treatments: the unmet needs. CNS Drugs 26(5):375–382
Arimura A, Somogyvári-Vigh A, Miyata A, Mizuno K, Coy DH, Kitada C (1991) Tissue distribution of PACAP as determined by RIA: highly abundant in the rat brain and testes. Endocrinology 129(5):2787–2789
Miyata A, Jiang L, Dahl RD, Kitada C, Kubo K, Fujino M et al (1990) Isolation of a neuropeptide corresponding to the N-terminal 27 residues of the pituitary adenylate cyclase activating polypeptide with 38 residues (PACAP38). Biochem Biophys Res Commun 170(2):643–648
Vaudry D, Gonzalez BJ, Basille M, Yon L, Fournier A, Vaudry H (2000) Pituitary adenylate cyclase-activating polypeptide and its receptors: from structure to functions. Pharmacol Rev 52(2):269–324
Zagami AS, Edvinsson L, Goadsby PJ (2014) Pituitary adenylate cyclase activating polypeptide and migraine. Ann Clin Transl Neurol 1(12):1036–1040
Tuka B, Helyes Z, Markovics A, Bagoly T, Szolcsányi J, Szabó N et al (2013) Alterations in PACAP-38-like immunoreactivity in the plasma during ictal and interictal periods of migraine patients. Cephalalgia 33(13):1085–1095
Amin FM, Asghar MS, Guo S, Hougaard A, Hansen AE, Schytz HW et al (2012) Headache and prolonged dilatation of the middle meningeal artery by PACAP38 in healthy volunteers. Cephalalgia 32(2):140–149
Schytz HW, Birk S, Wienecke T, Kruuse C, Olesen J, Ashina M (2008) PACAP38 induces migraine-like attacks in patients with migraine without aura. Brain 132(1):16–25
Rahmann A, Wienecke T, Hansen J, Fahrenkrug J, Olesen J, Ashina M (2008) Vasoactive intestinal peptide causes marked cephalic vasodilation, but does not induce migraine. Cephalalgia 28(3):226–236
Sherwood NM, Krueckl SL, McRory JE (2000) The origin and function of the pituitary adenylate cyclase-activating polypeptide (PACAP)/glucagon superfamily. Endocr Rev 21(6):619–670
Fahrenkrug J (2006) PACAP—A multifacetted neuropeptide. Chronobiol Int 23(1–2):53–61
Vaudry D, Falluel-Morel A, Bourgault S, Basille M, Burel D, Wurtz O et al (2009) Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol Rev 61(3):283–357
Hannibal J, Fahrenkrug J (1995) Expression of pituitary adenylate cyclase activating polypeptide (PACAP) gene by rat spermatogenic cells. Regul Pept 55(1):111–115
Chen Y, Samal B, Hamelink CR, Xiang CC, Chen Y, Chen M et al (2006) Neuroprotection by endogenous and exogenous PACAP following stroke. Regul Pept 137(1):4–19
Vaudry D, Hamelink C, Damadzic R, Eskay RL, Gonzalez B, Eiden LE, Endogenous PACAP (2005) Acts as a stress response peptide to protect cerebellar neurons from ethanol or oxidative insult. Peptides 26(12):2518–2524
Dickinson T, Fleetwood-Walker S, Mitchell R, Lutz E (1997) Evidence for roles of vasoactive intestinal polypeptide (VIP) and pituitary adenylate cyclase activating polypeptide (PACAP) receptors in modulating the responses of rat dorsal horn neurons to sensory inputs. Neuropeptides 31(2):175–185
Harmar AJ, Arimura A, Gozes I, Journot L, Laburthe M, Pisegna JR et al (1998) International Union of Pharmacology. XVIII. Nomenclature of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide. Pharmacol Rev 50(2):265–270
Edvinsson L, Elsås T, Suzuki N, Shimizu T, Jer-Fu Lee T (2001) Origin and co-localization of nitric oxide synthase, CGRP, PACAP, and VIP in the cerebral circulation of the rat. Microsc Res Tech 53(3):221–228
Mulder H, Uddman R, Moller K, Elsås T, Ekblad E, Alumets J et al (1995) Pituitary adenylate cyclase activating polypeptide is expressed in autonomic neurons. Regul Pept 59(1):121–128
Uddman R, Goadsby P, Jansen I, Edvinsson L (1993) PACAP, a VIP-like peptide: immunohistochemical localization and effect upon cat pial arteries and cerebral blood flow. J Cereb Blood Flow Metab 13(2):291–297
Dalsgaard T, Hannibal J, Fahrenkrug J, Larsen CR, Ottesen BVIP (2003) PACAP display different vasodilatory effects in rabbit coronary and cerebral arteries. Regul Pept 110(3):179–188
Fahrenkrug J, Hannibal J, Tams J, Georg B (2000) Immunohistochemical localization of the VIP1 receptor (VPAC1R) in rat cerebral blood vessels: relation to PACAP and VIP containing nerves. J Cereb Blood Flow Metab 20(8):1205–1214
Eftekhari S, Warfvinge K, Blixt FW, Edvinsson L (2013) Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J Pain 14(11):1289–1303
Jansen-Olesen I, Baun M, Amrutkar DV, Ramachandran R, Christophersen DV, Olesen J (2014) PACAP-38 but not VIP induces release of CGRP from trigeminal nucleus caudalis via a receptor distinct from the PAC 1 receptor. Neuropeptides 48(2):53–64
Mulder H, Uddman R, Moller K, Zhang Y-Z, Ekblad E, Alumets J et al (1994) Pituitary adenylate cyclase activating polypeptide expression in sensory neurons. Neuroscience 63(1):307–312
Tajti J, Uddman R, Möller S, Sundler F, Edvinsson L (1999) Messenger molecules and receptor mRNA in the human trigeminal ganglion. J Auton Nerv Syst 76(2):176–183
Uddman R, Tajti J, Hou M, Sundler F, Edvinsson L (2002) Neuropeptide expression in the human trigeminal nucleus caudalis and in the cervical spinal cord C1 and C2. Cephalalgia 22(2):112–116
Ciccarelli E, Svoboda M, De Neef P, Di Paolo E, Bollen A, Dubeaux C et al (1995) Pharmacological properties of two recombinant splice variants of the PACAP type I receptor, transfected and stably expressed in CHO cells. Eur J Pharmacol 288(3):259–267
Chan KY, Baun M, Vries RD, van den Bogaerdt AJ, Dirven CM, Danser AH et al (2011) Pharmacological characterization of VIP and PACAP receptors in the human meningeal and coronary artery. Cephalalgia 31(2):181–189
Knutsson M, Edvinsson L (2002) Distribution of mRNA for VIP and PACAP receptors in human cerebral arteries and cranial ganglia. Neuroreport 13(4):507–509
Boni L, Ploug KB, Olesen J, Jansen-Olesen I, Gupta S (2009) The in vivo effect of VIP, PACAP-38 and PACAP-27 and mRNA expression of their receptors in rat middle meningeal artery. Cephalalgia 29(8):837–847
Baun M, Hay-Schmidt A, Edvinsson L, Olesen J, Jansen-Olesen I (2011) Pharmacological characterization and expression of VIP and PACAP receptors in isolated cranial arteries of the rat. Eur J Pharmacol 670(1):186–194
Kelman L (2005) Migraine pain location: a tertiary care study of 1283 migraineurs. Headache 45(8):1038–1047
Ray BS, Wolff HG (1940) Experimental studies on headache: pain-sensitive structures of the head and their significance in headache. Arch Surg 41(4):813–856
Fricke B, Andres KH, Von Düring M (2001) Nerve fibers innervating the cranial and spinal meninges: morphology of nerve fiber terminals and their structural integration. Microsc Res Tech 53(2):96–105
Mayberg M, Langer RS, Zervas NT, Moskowitz MA (1981) Perivascular meningeal projections from cat trigeminal ganglia: possible pathway for vascular headaches in man. Science 213(4504):228–230
Headache Classification Committee of the International Headache Society (2013) The international classification of headache disorders, 3rd edition (beta version). Cephalalgia 33(9):629–808
Iversen HK, Olesen J, Tfelt-Hansen P (1989) Intravenous nitroglycerin as an experimental model of vascular headache. Basic characteristics. Pain 38(1):17–24
Lassen L, Haderslev P, Jacobsen V, Iversen HK, Sperling B, Olesen JCGRP (2002) May play a causative role in migraine. Cephalalgia 22(1):54–61
Thomsen LL, Iversen HK, Brinck TA, Olesen J (1993) Arterial supersensitivity to nitric oxide (nitroglycerin) in migraine sufferers. Cephalalgia 13(6):395–399
Wienecke T, Olesen J, Oturai PS, Ashina M (2008) Prostacyclin (epoprostenol) induces headache in healthy subjects. Pain 139(1):106–116
Kruuse C, Thomsen LL, Birk S, Olesen J (2003) Migraine can be induced by sildenafil without changes in middle cerebral artery diameter. Brain 126(1):241–247
Amin FM, Asghar MS, Hougaard A, Hansen AE, Larsen VA, de Koning PJ et al (2013) Magnetic resonance angiography of intracranial and extracranial arteries in patients with spontaneous migraine without aura: a cross-sectional study. Lancet Neurol 12(5):454–461
Jansen-Olesen I, Goadsby PJ, Uddman R, Edvinsson L (1994) Vasoactive intestinal peptide (VIP) like peptides in the cerebral circulation of the cat. J Auton Nerv Syst 49:97–103
Grände G, Nilsson E, Edvinsson L (2013) Comparison of responses to vasoactive drugs in human and rat cerebral arteries using myography and pressurized cerebral artery method. Cephalalgia 33(3):152–159
Jansen-Olesen I, Gulbenkian S, Engel U, Cunha e Sá M, Edvinsson L (2004) Peptidergic and non-peptidergic innervation and vasomotor responses of human lenticulostriate and posterior cerebral arteries. Peptides 25(12):2105–2114
Banks WA, Kastin AJ, Komaki G, Arimura A (1993) Passage of pituitary adenylate cyclase activating polypeptide1-27 and pituitary adenylate cyclase activating polypeptide1-38 across the blood–brain barrier. J Pharmacol Exp Ther 267(2):690–696
Dogrukol-Ak D, Tore F, Tuncel N (2004) Passage of VIP/PACAP/secretin family across the blood–brain barrier: therapeutic effects. Curr Pharm Des 10(12):1325–1340
Barzo P, Bari F, Doczi T, Jancso G, Bodosi M (1993) Significance of the rate of systemic change in blood pressure on the short-term autoregulatory response in normotensive and spontaneously hypertensive rats. Neurosurgery 32(4):611–618
Gupta S, Bhatt D, Boni L, Olesen J (2010) Improvement of the closed cranial window model in rats by intracarotid infusion of signalling molecules implicated in migraine. Cephalalgia 30(1):27–36
Birk S, Sitarz JT, Petersen KA, Oturai PS, Kruuse C, Fahrenkrug J et al (2007) The effect of intravenous PACAP38 on cerebral hemodynamics in healthy volunteers. Regul Pept 140(3):185–191
Schytz HW, Birk S, Wienecke T, Kruuse C, Olesen J, Ashina M (2009) PACAP38 induces migraine-like attacks in patients with migraine without aura. Brain 132(Pt 1):16–25
Syed AU, Koide M, Braas KM, May V, Wellman GC (2012) Pituitary Adenylate Cyclase-activating polypeptide (PACAP) potently dilates middle Meningeal arteries: implications for migraine. J Mol Neurosci 48(3):574–583
Syed AU, Koide M, May V, Wellman GC. PACAP regulation of vascular tone: differential mechanism among vascular beds. In: Reglodi D, Tamas A, editors. Pituitary Adenylate Cyclase activating polypeptide — PACAP. Curr Top Neurotox. 2016 11:617–630. Springer International Publishing
Grände G, Labruijere S, Haanes KA, MaassenVanDenBrink A, Edvinsson L (2014) Comparison of the vasodilator responses of isolated human and rat middle meningeal arteries to migraine related compounds. J Headache Pain 15(1):22
Akerman S, Goadsby PJ (2015) Neuronal PAC1 receptors mediate delayed activation and sensitization of trigeminocervical neurons: relevance to migraine. Sci Transl Med 7(308):308ra157
Bhatt DK, Gupta S, Olesen J, Jansen-Olesen I (2014) PACAP-38 infusion causes sustained vasodilation of the middle meningeal artery in the rat: possible involvement of mast cells. Cephalalgia 34(11):877–886
Gri G, Frossi B, D’Inca F, Danelli L, Betto E, Mion F, et al (2012) Mast cell: an emerging partner in immune interaction. Front Immunol 3:120
Brown JM, Wilson TM, Metcalfe DD (2008) The mast cell and allergic diseases: role in pathogenesis and implications for therapy. Clin Exp Allergy 38(1):4–18
Nakano T, Sonoda T, Hayashi C, Yamatodani A, Kanayama Y, Yamamura T et al (1985) Fate of bone marrow-derived cultured mast cells after intracutaneous, intraperitoneal, and intravenous transfer into genetically mast cell-deficient W/Wv mice. Evidence that cultured mast cells can give rise to both connective tissue type and mucosal mast cells. J Exp Med 162(3):1025–1043
Okayama Y, Kawakami T (2006) Development, migration, and survival of mast cells. Immunol Res 34(2):97–115
Warbrick EV, Taylor AM, Botchkarev VA, Coleman JW (1995) Rat connective tissue-type mast cells express MHC class II: up-regulation by IFN-gamma. Cell Immunol 163(2):222–228
Zhang D, Spielmann A, Wang L, Ding G, Huang F, Gu Q et al (2012) Mast-cell degranulation induced by physical stimuli involves the activation of transient-receptor-potential channel TRPV2. Physiol Res 61(1):113
Gazerani P, Pourpak Z, Ahmadiani A, Hemmati A, Kazemnejad AA (2003) Correlation between migraine, histamine and immunoglobulin E. Scand J Immunol 57(3):286–290
Heatley R, Denburg J, Bayer N, Bienenstock J (1982) Increased plasma histamine levels in migraine patients. Clinical & Experimental. Allergy 12(2):145–149
Kemper R, Meijler W, Korf J, Ter Horst G (2001) Migraine and function of the immune system: a meta-analysis of clinical literature published between 1966 and 1999. Cephalalgia 21(5):549–557
Mansfield LE, Vaughan T, Waller SF, Haverly RW, Ting S (1985) Food allergy and adult migraine: double-blind and mediator confirmation of an allergic etiology. Ann Allergy 55(2):126–129
Peng Y-H, Chen K-F, Kao C-H, Chen H-J, Hsia T-C, Chen C-H et al (2016) Risk of migraine in patients with asthma: a nationwide cohort study. Medicine 95:9
Wilson C, Kirker J, Warnes H, O'Malley M (1980) The clinical features of migraine as a manifestation of allergic disease. Postgrad Med J 56(659):617–621
Ku M, Silverman B, Prifti N, Ying W, Persaud Y, Schneider A (2006) Prevalence of migraine headaches in patients with allergic rhinitis. Ann Allergy Asthma Immunol 97(2):226–230
Lassen LH, Thomsen LL, Olesen J (1995) Histamine induces migraine via the H1-receptor. Support for the NO hypothesis of migraine. Neuroreport 6(11):1475–1479
Anthony M, Lord G, Lance J (1978) Controlled trials of cimetidine in migraine and cluster headache. Headache 18(5):261–264
Millan-Guerrero RO, Isais-Millan R, Benjamin TH, Tene CE (2006) Nalpha-methyl histamine safety and efficacy in migraine prophylaxis: phase III study. The Canadian journal of neurological sciences. J Can Sci Neurol 33(2):195–199
Alstadhaug KB (2014) Histamine in migraine and brain. Headache 54(2):246–259
Strassman A, Raymond S, Burstein R (1996) Sensitization of meningeal sensory neurons and the origin of headaches. Nature 384(6609):560
Zhang X-C, Strassman AM, Burstein R, Levy D (2007) Sensitization and activation of intracranial meningeal nociceptors by mast cell mediators. J Pharmacol Exp Ther 322(2):806–812
Wienecke T, Olesen J, Ashina M (2011) Discrepancy between strong cephalic arterial dilatation and mild headache caused by prostaglandin D2 (PGD2). Cephalalgia 31(1):65–76
Pedersen SH, Ramachandran R, Amrutkar DV, Petersen S, Olesen J, Jansen-Olesen I (2015) Mechanisms of glyceryl trinitrate provoked mast cell degranulation. Cephalalgia 35(14):1287–1297
Reuter U, Bolay H, Jansen-Olesen I, Chiarugi A, del Rio MS, Letourneau R et al (2001) Delayed inflammation in rat meninges: implications for migraine pathophysiology. Brain 124(12):2490–2502
Baun M, Pedersen MHF, Olesen J, Jansen-Olesen I (2012) Dural mast cell degranulation is a putative mechanism for headache induced by PACAP-38. Cephalalgia 32(4):337–345
Levy D (2009) Migraine pain, meningeal inflammation, and mast cells. Curr Pain Headache Rep 13(3):237–240
Levy D, Burstein R, Kainz V, Jakubowski M, Strassman AM (2007) Mast cell degranulation activates a pain pathway underlying migraine headache. Pain 130(1):166–176
Levy D, Burstein R, Strassman AM (2006) Mast cell involvement in the pathophysiology of migraine headache: a hypothesis. Headache 46:s1
Ødum L, Petersen L, Skov P, Ebskov L (1998) Pituitary adenylate cyclase activating polypeptide (PACAP) is localized in human dermal neurons and causes histamine release from skin mast cells. Inflamm Res 47(12):488–492
Schmidt-Choudhury A, Furuta GT, Galli SJ, Schmidt WE, Wershil BK (1999) Mast cells contribute to PACAP-induced dermal oedema in mice. Regul Pept 82(1):65–69
Li M, Maderdrut JL, Lertora JJ, Batuman V (2007) Intravenous infusion of pituitary adenylate cyclase-activating polypeptide (PACAP) in a patient with multiple myeloma and myeloma kidney: a case study. Peptides 28(9):1891–1895
Saghy E, Payrits M, Helyes Z, Reglodi D, Banki E, Toth G et al (2015) Stimulatory effect of pituitary adenylate cyclase-activating polypeptide 6–38, M65 and vasoactive intestinal polypeptide 6–28 on trigeminal sensory neurons. Neuroscience 308:144–156
Erdling A, Sheykhzade M, Maddahi A, Bari F, Edvinsson LVIP (2013) PACAP receptors in cerebral arteries of rat: characterization, localization and relation to intracellular calcium. Neuropeptides 47(2):85–92
Sismanopoulos N, Delivanis D-A, Alysandratos K-D, Angelidou A, Therianou A, Kalogeromitros D et al (2012) Mast cells in allergic and inflammatory diseases. Curr Pharm Des 18(16):2261–2277
Acknowledgements
This work was supported by Candys Foundation, Novo Nordisk Foundation and Lundbeck Foundation.
Author information
Authors and Affiliations
Contributions
IJ-O conceived and designed the review, drafted the manuscript and revised it for intellectual content. SHP revised the manuscript for intellectual content and provided essential comments to finalize the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jansen-Olesen, I., Hougaard Pedersen, S. PACAP and its receptors in cranial arteries and mast cells. J Headache Pain 19, 16 (2018). https://doi.org/10.1186/s10194-017-0822-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10194-017-0822-2