
Abstract
Solid-state NMR spectroscopy represents a powerful method for the investigation of diatom biosilica but detailed studies regarding its chemical composition and structural organization can be prohibited by insufficient spectroscopic sensitivity. Here, we used two-dimensional (2D) Dynamic Nuclear Polarization (DNP)-supported solid-state NMR experiments to obtain information about the molecular composition and supramolecular organization of proteins and carbohydrates in 13C,15N,29Si-labeled biosilica of C. cryptica. As a reference, we conducted DNP experiments on isotope-labeled biosilica of T. pseudonana. DNP-enhancement factors for different NMR signals, and thus, for different organic compounds, provide information about the supramolecular architecture of the biosilica. In addition, DNP-supported heteronuclear nitrogen-carbon correlation experiments allowed us to prove the presence of different structural elements of long chain polyamines (LCPAs) and revealed the occurrence of amine-nitrogen moieties exhibiting a correlation with carbonyl carbons that may indicate cross-linking of LCPAs to proteins as previously seen in studies on proteins extracted from other diatoms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Solid-state NMR spectroscopy is a powerful method for the investigation of biomaterials [1,2,3,4,5,6,7] including diatom biosilica [8]. Diatoms are unicellular algae. They are frequently studied model organisms for biomineralization research and also of significant interest in the context of designing bioinspired materials [3, 8,9,10,11]. Their ability to build up micro- and nano-structured cell walls out of amorphous silica makes them an interesting model organism. Since various biomolecules show proximity to the silica phase, this biosilica may be understood as composite material. The silica-attached biomolecules include different proteins, e.g., sillafins, carbohydrates and long-chain polyamines (LCPAs). Especially different proteins and LCPAs are able to precipitate silica and are assumed to be involved in cell wall synthesis [12,13,14,15,16] A mixture of different LCPAs which differ in chain-length and degree of methylation is usually found in diatom biosilica sample. Structure and amount of LCPAs are also species-specific [17, 18]. Note that LCPAs could even be found in sedimented diatomite samples from the ocean floor [19]. Previous studies show that especially LCPAs are embedded within the silica phase while proteins and carbohydrates can be mainly found on the surface of silica [3, 10]. The silica phase of the cell walls can be characterized using 29Si solid-state NMR spectroscopy and the organic compounds by 13C and 15 N solid-state NMR spectroscopy [3, 8,9,10,11]. The sensitivity of such experiments is strongly enhanced by isotope-labeling which, as shown by Brunner et al. [8] can be elegantly achieved by diatom growth in isotope-enriched culture medium. In recent years, high-frequency Dynamic Nuclear Polarization [20] (DNP), in which polarization is transferred from free electrons to atomic nuclei has become a powerful method to enhance solid-state NMR signal intensities by up to two orders of magnitude. These advancements have greatly expanded the use of ssNMR to study complex biomolecular systems including membrane [21,22,23,24,25,26] and amyloid [27, 28] proteins as well as cellular preparations [29,30,31,32,33] and biomaterials [2,3,4, 34]. Such experiments not only enhance the prospects to study local molecular structure but also offer a spectroscopic means to probe local as well as supramolecular arrangements [27] as we have previously shown for diatom biosilica from Stephanopyxis turris [3].
In the present work, we have used DNP-ssNMR to examine isotope-labeled diatoms of Cyclotella cryptica biosilica [35], a centric diatom which has a massive insoluble organic matrix. This diatom species forms external β-chitin fibrils as well as silica-attached chitin. In particular, the use of DNP allowed us to study the presence of silica-attached chitin. In addition, we could spectroscopically probe LCPAs and we examined the organic–inorganic interface in C. cryptica. We compare our findings to experimental results obtained on Thalassiosira pseudonana, a centric diatom, which is one of the most studied diatoms and our previously published results on S. turris biosilica [3]. Interestingly, the amount of silica-attached biomolecules in S. turris is remarkably smaller compared to those of T. pseudonana and especially C. cryptica. The higher amount of biomolecules in the samples studied here in combination with the strong sensitivity gain provided by DNP allowed us to investigate the silica-attached biomolecules in additional detail.
2 Methods
2.1 DNP sample preparation of 13C,15N,29Si-enriched and 13C-enriched C. cryptica & T. pseudonana biosilica
Diatom species of both C. cryptica and T. pseudonana were cultivated as described previousl [35]. Likewise, (13C,15N, 29Si) isotope labeling, cell pellet cleaning and preparation of the biosilica samples using lysis buffer solution [0.1 M EDTA (ethylenediaminetetraacetic acid), 2 wt-% SDS (sodium dodecyl sulfate)] followed earlier procedures [10, 35, 36].
2.2 DNP-supported NMR experiments
2.2.1 NMR sample preparation using the incipient wetness method
3.2 mm sapphire rotors were filled with isotope-labeled C. cryptica or T. pseudonana biosilica which were wetted directly before measurements. The wetting of the biosilica was performed using the method of incipient wetness impregnation with the radical solution at room temperature [21]. The radical stock solution with a concentration of 15 mM was prepared by dissolving AMUPol in D2O/H2O 9:1. The solution was stored at -20 °C. 10.2 mg of dry biosilica were wetted with 50 µL AMUPol solution and immediately packed into the MAS rotor. Directly afterwards, the filled rotors were transferred to the NMR probe which was cooled to 92 K.
2.2.2 DNP-supported NMR experiments
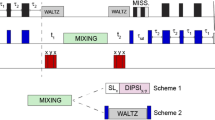
DNP experiments were acquired using a 1H–13C–15N triple resonance probe at a static magnetic field of 9.4T corresponding to proton/electron resonance frequencies of 400 MHz/263 GHz (Bruker BioSpin). The temperature was set to 100 K and an MAS spinning speed of 9 kHz was used. The recycle delay was set to 2 s for all experiments (see, e.g. Ref. [37]). For the 1D cross-polarization (CP) spectra, 90 degree 1H, 13C and 15N pulses were applied using field strengths of 77 kHz, 43 kHz and 40 kHz, respectively. H-C CP matching was achieved by using a 62 kHz, 70–100% ramped 1H pulse and a 45 kHz r.f. 13C pulse for a contact time of 700 µsec. HN CP conditions were optimized using a 70–100% ramped pulse on protons with a 41 kHz field, a matching 15N pulse of 33 kHz and a transfer time of 1.4 ms. High power proton decoupling at 85 kHz was achieved using SPINAL-64 [38] during evolution and acquisition in all experiments. For 2D proton-driven spin diffusion (PDSD) experiments, Carbon–carbon mixing was established with a mixing time of 50 ms. For the isotope-enriched C. cryptica biosilica sample, 224 scans were acquired with acquisition times of 15 ms and 6 ms for the direct and indirect dimensions, respectively. For the isotope-enriched T. pseudonana biosilica sample, 64 scans were acquired with acquisition times of 15 ms and 7 ms for the direct and indirect dimensions, respectively. In both cases two spectra were recorded, with and without DNP, and 1D slices were used to calculate the DNP enhancement. The 15N–13C correlation experiment was performed using a H–N CP step of 1 ms and a SPECIFIC-CP [39] transfer step of 2.5 ms. Acquisition times were 8 ms and 7.5 ms for the direct and indirect dimensions, respectively. 256 scans were acquired and the spectrum was processed using a 0.5 π shifted sine squared window function in both dimensions The 15N-edited PDSD experiment [40] was recorded using a SPECIFIC-CP transfer step of 2.5 ms which is typical for one-bond NC correlations experiments [39] and a carbon–carbon mixing time of 50 ms. 1728 scans were acquired with acquisition times of 10 ms and 4 ms for the direct and indirect dimensions, respectively. The center frequencies were set to 50 ppm for 15N and 13C dimensions. The spectrum was processed using a 0.33 π shifted sine squared window function in both dimensions.
2.3 Scanning electron microscopy
SEM samples were prepared on an aluminum sample holder with an aluminum tape on top. A small amount of the biosilica sample was suspended in water and 50 µL of the suspension were transferred to the sample holder. After drying overnight at 40 °C, the sample was sputtered with 5 nm gold using a Q150R ES sputter coater from Quorum Technologies. SEM images were acquired on a Hitachi SU 8020. An acceleration voltage of 5 kV was used for acquisition. Secondary electrons were used for detection of surface topography. Images with a magnification of 20,000 and 90,000 were acquired.
3 Results and discussion
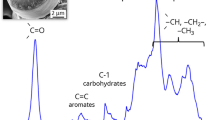
In our previous study [35] of C. cryptica using conventional 1D and 2D ssNMR spectroscopy, we observed that a high amount of proteins is associated to the biosilica and we detected two different chitin conformations (Fig. 1). In the following, we used DNP-supported NMR experiments to further enhance ssNMR signal intensities. As mentioned above, DNP relies on spin polarization transfer from electron spins to nuclei. In our experiments, samples were impregnated with a “DNP juice” (see methods) containing the biradical AMUPol [41]. First, we conducted one-dimensional 13C and 15N detected CP experiments (Fig. 2) under DNP conditions and, as in our previous study [3], defined the DNP enhancement factor by the ratio of signal intensities with and without microwave irradiation. In 13C CP experiments, we observed enhancement factors that (except for carbohydrates and possibly some methyl signals, see also Additional file 1: Figures S1–4) ranged between 18 and 26 (Fig. 2, left). In the 15N CP measurement, we observed an enhancement of 26 for both amide and amine 15 N signals (Fig. 2, right).

Left: SEM images of the 13C,15N,29Si-enriched C. cryptica biosilica samples prepared for the ssNMR experiments (magnification: 20,000 and 90,000). The fibrils shown in these images are believed to consist of β-chitin and have a diameter of ca. 50 nm. Right: Schematic figure of a centric diatom cell with its siliceous cell wall and a zoom into the cell wall region. Biomolecules are assumed to be embedded in (LCPAs) or attached to (proteins and carbohydrates) diatom silica. The drawn chitin fibrils are characteristic for C. cryptica. These chitin fibrils are located at the so-called portulae

1D CP data of C. cryptica with and without DNP enhancement. Left: The 13C CP spectrum shows DNP enhancement factors ranging from 9 (carbohydrates) to 18–28 for other carbon moieties. Right: In the 15N CP spectrum an enhancement of 26 for both amide and amine signals is observed. See also supporting Fig. 1
As we have discussed elsewhere [35], C. cryptica contains a massive organic matrix and produces extracellular chitin fibrils of ca. 50 nm thickness [42, 43] (see also Fig. 1). To further investigate molecule-specific DNP enhancements, we performed a two-dimensional DNP-enhanced PDSD (Proton-Driven Spin Diffusion) experiment (Fig. 3). We observed similar 13C–13C correlations as in the standard PDSD experiment [35], however with considerably higher signal intensities. This improvement enabled the detection of new signals which are not observable in the conventional experiment. In particular, two spin systems were observable (Fig. 3, blue box). One spin system shows correlations from 92 to 81 ppm, 75 ppm, and 72 ppm. The other system exhibits connectivties between 95 and 81 ppm, 75 ppm, 73 ppm, as well as 70 ppm. These correlations are characteristic for carbohydrates, where the C1 is not connected. These unconnected anomeric carbon atoms characteristically give rise to peaks between 90 and 100 ppm [44]. Both spin systems also contain a shift higher than 80 which indicates a substitution on the carbocycle [45] by e.g. another sugar. This points towards the presence of reducing ends of polysaccharides [46] or would be consistent with sugar–protein linkages which often yield lower chemical shifts for the anomeric carbon than a connection to another sugar(see, e.g. Ref. [47]). The different line widths of the resonance peaks of the sugar spin systems compared to the much narrower chitin peaks can be explained by two independent effects. Firstly, the relative position and exposure of the molecules/sugars towards the radical increased T2 relaxation rates due to paramagnetic relaxation enhancement (PRE), resulting in broader NMR lines. Secondly, it could be an indication of different morphologies of the sugar networks, where there is crystalline chitin present with narrow line widths due to the increased isotropy in crystals as well as amorphous parts of chitin and potentially other sugars, which then exhibit broader line widths due to their anisotropy especially under freezing conditions. A monosaccharide analysis which provides information about the total carbohydrate composition revealed glucose and mannose as major part of the monosaccharides fraction. Furthermore, xylose, ribose, fucose, galactose and glucosamine are present in significant concentrations [35]. However, a differentiation between monosaccharides and monosaccharide units from polysaccharides is not possible due to the performed hydrolysis step.

DNP-supported Proton-Driven Spin Diffusion (PDSD) experiment of isotope-enriched C. cryptica biosilica using a mixing time of 50 ms and an MAS rate of 9 kHz. For better resolution, the enhancements were determined from 1D slices in the fingerprint region for protein C-alpha (slice A, 53 ppm, enhancement = 26) and characteristic for carbohydrates (Slice B, 105 ppm, Enhancement = 5–8)
Moreover, the determination of DNP enhancement factors for different signals, which are characteristic for different organic compounds, can help to probe the supramolecular architecture of the biosilica [3]. To distinguish DNP enhancements of protein and carbohydrate (including chitin) signals, we analyzed different 1D slices of the 2D spin diffusion experiment (Fig. 3, right). For the protein signals, we examined a 1D slice at 53 ppm which is typical for protein backbone Cα resonances and observed a strong enhancement factor of 26 in line with our 1D 15N CP studies (Fig. 2). Thus, the proteins should be located at the solvent-accessible surface of the biosilica, where the radical solution (AMUPol) can polarize nuclei located in the biomolecules [22]. In contrast, for the carbohydrates, e.g. the chitin signals at 105 ppm, we determined significantly lower enhancement factors between 5 and 8 which is close to the 13C enhancements seen in 1D CP MAS for carbohydrates (Fig. 2). Interestingly, when analyzing the carbohydrates with diagonal peaks at 92.4 and 95.7 (see also Additional file 1: Figure S2) we observed enhancement factors of 19 and 22–26. Their relatively high enhancement, especially when compared to the chitin signals, can -similar to the case of proteins- be explained by their location on the surface of the frustule where sugar moieties that are close to the radical are more strongly enhanced than the ones that are further away from it or buried/shielded like parts of the chitin.
As discussed elsewhere [3, 27, 48], the DNP enhancement in complex biomolecules depends on the internuclear distances between hydrogens responsible for the polarization transfer via proton-driven spin diffusion and the macromolecular layer size. In addition, the DNP enhancement for biomolecules close to the radical, may itself be modulated by the details of the local electron-nucleus geometry. This geometry determines the spin-diffusion barrier [49], influences the DNP magnetic field dependence of the DNP effect [50] and leads to paramagnetic line broadening [22, 51] within a sphere of about 1 nm around the DNP radical [22]. We expect this effect to be particularly strong for biosilica such as from S. turris which are characterized by small protein/organic surface layers [3]. Indeed, DNP signal enhancements for polysaccharides and polyamines amounted to about 40% to the protein signal enhancements for S. turris [3]. In contrast, the organic layer for C. cryptica is significantly larger [35] which results in almost uniform DNP enhancements in 1D 13C CP MAS data and is possibly the reason for the larger (relative) decrease in DNP enhancement from 26 to 5–8 for chitin.
Note that the increased line broadening often observed at low-temperature DNP conditions [22] prevents the differentiation between the α-like chitin- and β-chitin signals as it was possible without DNP and at ambient sample temperatures [35]. On the other hand and as mentioned earlier, C. cryptica contains a massive organic matrix and produces extracellular chitin fibrils of ca. 50 nm thickness [42, 43]. Hence, a more detailed analysis employing a classical spin-diffusion approximation to correlate relative DNP enhancements with biosilica layer thickness as performed earlier for labeled diatom biosilica of S. turris [3] was not attempted here. Instead we can draw the following general conclusions. Firstly, we would expect from previous theoretical work from our laboratory [48] only minor changes for the DNP enhancement for chitin embedded in the extracellular chitin fibrils compared to the protein layer. On the other hand, fluorescence spectroscopy [52] has shown that chitin in C. cryptica biosilica can also be directly associated to the siliceous cell walls. For this species, that is potentially surrounded by a low density of hydrogens that limit the spin-diffusion process, low DNP enhancements as found in Fig. 2 may be possible. Since ssNMR detects the entire ensembles of chitin moieties in our sample, the latter species may hence represent a prominent fraction in our C. cryptica preparations.
To further investigate the carbohydrate association to the siliceous cell walls, we conducted similar experiments on isotope-enriched T. pseudonana biosilica (Fig. 4) for which the presence of a chitin-based meshwork is already known [11]. Again, we analyzed 1D slices typical for protein and carbohydrate signals and observed a strong enhancement factor of 32 for the protein Cα signals (signal at 53 ppm). In line with our earlier observations, the proteins are thus predominately located at the solvent-accessible surface of the biosilica, where the radical solution (AMUPol) can polarize nuclei located in the biomolecules. Remarkably, for the chitin signals resonating at 105 ppm, a lower enhancement compared to the protein signals is again observed. However, the relative enhancement of about 15 is significantly larger than in the case of C. cryptica biosilica (amounting to 5–8). These findings would be consistent with the notion that the organic matrix of T. pseudonana biosilica is smaller than in the case of C. cryptica [35] but larger than for S. turris. Such an interpretation would be consistent with previous findings [17, 18, 53].

DNP-supported Proton-Driven Spin Diffusion (PDSD) experiment of isotope-enriched T. pseudonana biosilica using a mixing time of 50 ms and a MAS rate of 9 kHz. For better resolution, enhancements were again determined from 1D slices in the fingerprint region for protein C-alpha (slice A, 53 ppm, enhancement = 32) and characteristic for carbohydrates (Slice B, 105 ppm, Enhancement = 14–15)
Moreover, our DNP experiments revealed the presence of additional carbohydrate species as seen before for C. cryptica (Fig. 3). Interestingly, one of the observed new sugar spin systems exhibits a correlation of the anomeric carbon to a signal around 165 ppm (Additional file 1: Figure S5). Sp2-configured carbon atoms of nitrogen containing functional groups including imines [54] and guanidiniums [55] as well as amide carbonyls give rise to peaks in this region. This correlation could hence indicate a cross linking between a carbohydrate C1 and an amino acid such as the guanidinium containing arginine, which has rarely been described as part of N-glycans. Another possibility would be the presence of citrulline as its carboxamide gives rise to resonances at around 164 ppm and it has been described as being linked to polysaccharides before [56]. This link could represent the interface of the sugar layers with the residual organic matrix.
Finally, we used DNP-supported 15N–13C correlation experiments to further investigate proteins as well as the presence of LCPA in our preparations. Long chain poly amines are based on putrescine, spermidine or 1,3-diaminopropane with a number of propylenediamine repeats ranging from 6–20. They are often positively charged and thereby able to attract negatively charged molecules, by which they are able to facilitate rapid silica precipitation. Since they are species specific their involvement in silica patterning is highly likely [57]. Again, DNP greatly facilitated such experiments because of the increased spectroscopic sensitivity, thereby reducing measurement times. To study nitrogen containing 13C moieties, we performed 15N filtered experiments [39, 40]. Figure 5 (red spectrum) shows a 15N–13C correlation spectrum. The observed correlations are characteristic for those of primary, secondary and tertiary amines to their directly adjacent carbon atom. The introduction of a spin diffusion (mixing) step between carbons allowed us to study correlations of the amines to more distant 13C atoms (blue spectrum). An increasing number of correlations occur, e.g., correlations to alkyl groups at lower 13C chemical shift between 15 and 30 ppm which would be consistent with corrrelations for both Lysine sidechains as well as the above mentioned structural elements of LCPAs. Due to the expected manifold of LCPAs [57] as well as the increased spectral line width under DNP conditions, we were however, not able to isolate individual spin systems or deduct exact structures for different LCPAs. Interestingly, we even observed a correlation to carbonyl-carbons at 174 ppm. This signal occurs probably due to relay transfer between close-by 13C labeled atoms to a carbonyl atom as usually seen in fully labeled protein samples under MAS conditions (see e.g. Ref. [58]) Such a correlation between the amine of a LCPA and a carbonyl group was not observed for other diatom biosilica, including diatom biosilica from S. turris [3]. However, it is well known from the model organism T. pseudonana [11] that LCPAs can be covalently attached to lysine residues or posttranslational modified hydroxylysine residues that may give rise to the correlation seen at 174 ppm. Additionally, the chemical shift of this carbonyl group is more typical for carbonyl groups of proteins. The above discussed chitin should give rise to a signal at a higher chemical shift of ca. 176 ppm. Thus, proximity between amine nitrogens and protein carbonyl groups is more likely here.

DNP-supported NC and NCC NMR correlation experiments of isotope-enriched C. cryptica biosilica. In both experiments, 15N- > 13C transfer was established using a SPECIFIC-CP transfer step of 3 ms. In the NCC correlation experiment (blue) a mixing time of 50 ms was used for proton-driven spin diffusion (PDSD)
4 Conclusions
Our DNP-supported solid-state NMR studies provide further insight into the composition and architecture of C. cryptica biosilica and allow us to draw the following conclusions:
-
(i)
We could confirm the presence of chitin seen in ssNMR conducted at ambient temperatures [35] and we were also able to isolate two additional carbohydrate spin systems using PDSD experiments performed under DNP conditions. Their chemical shifts point towards the presence of either terminal reducing ends of polysaccharides or monomers that form a cross-connection between polysaccharides and proteins.
-
(ii)
We also observed a reduced DNP efficiency for chitin compared to signals from proteins and other carbohydrates in preparations of T. pseudonana biosilica. However, this reduction was lower than for the case of C. cryptica biosilica. We attribute these variations to an increased size of the organic layer in the case of C. cryptica biosilica. Their different enhancements can be further correlated to their relative position on the diatom surface and their structural properties. Paramagnetic quenching due to the presence or binding of DNP radicals has the smallest influence on the observed protein signal under DNP conditions.
-
(iii)
DNP-supported 15N–13C 2D correlation experiments are consistent with the presence of different structural elements of LCPAs. Interestingly, a correlation of amine-nitrogen to a carbonyl carbon can be observed which may reflect chemical linkage to proteins as previously seen in studies on T. Pseudonana [59, 60].
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional information files.
Abbreviations
- 2D:
-
Two-dimensional
- AMUPol:
-
15-{[(7-Oxyl-3,11-dioxa-7-azadispiro[5.1.5.3]hexadec-15-yl)carbamoyl][2-(2,5,8,11-tetraoxatridecan-13-ylamino)}-[3,11-dioxa-7- azadispiro[5.1.5.3]hexadec-7-yl])oxidanyl
- ASW:
-
Artificial seawater
- C. cryptica :
-
Cyclotella cryptica
- CP:
-
Cross polarization
- DNP:
-
Dynamic nuclear polarization
- EDTA:
-
Ethylenediaminetetraacetic acid
- LCPA:
-
Long-chain polyamine
- MAS:
-
Magic angle spinning
- NC:
-
15N–13C correlation spectrum
- NCC:
-
15N–13C–13C correlation spectrum
- NMR:
-
Nuclear magnetic resonance
- PDSD:
-
Proton-driven spin-diffusion
- ppm:
-
Parts per million
- SPECIFIC-CP:
-
Spectrally induced filtering in combination with cross polarization
- RF:
-
Radio frequency
- SPINAL64:
-
Small phase incremental alternation, with 64 steps
- ssNMR:
-
Solid-state NMR
- S. turris :
-
Stephanopyxis turris
- T. pseudonana :
-
Thalassiosira pseudonana
References
Akiva-Tal A, Kababya S, Balazs YS, Glazer L, Berman A, Sagi A, Schmidt A. In situ molecular NMR picture of bioavailable calcium stabilized as amorphous CaCO 3 biomineral in crayfish gastroliths. Proc Natl Acad Sci USA. 2011;108(36):14763–8.
Koers EJ, Lopez-Deber MP, Weingarth M, Nand D, Hickman DT, Ndao DM, Reis P, Granet A, Pfeifer A, Muhs A, Baldus M. Dynamic Nuclear Polarization NMR spectroscopy: revealing multiple conformations in lipid-anchored peptide vaccines. Angewandte Chemie-Int Ed. 2013;52(41):10905–8.
Jantschke A, Koers E, Mance D, Weingarth M, Brunner E, Baldus M. Insight into the supramolecular architecture of intact diatom biosilica from DNP-supported solid-state NMR spectroscopy. Angewandte Chemie-Int Ed. 2015;54(50):15069–73.
Chakraborty A, Deligey F, Quach J, Mentink-Vigier F, Wang P, Wang T. Biomolecular complex viewed by dynamic nuclear polarization solid-state NMR spectroscopy. Biochem Soc Trans. 2020;48(3):1089–99.
Kang X, Kirui A, Muszyński A, Widanage MCD, Chen A, Azadi P, Wang P, Mentink-Vigier F, Wang T. Molecular architecture of fungal cell walls revealed by solid-state NMR. Nat Commun. 2018;9(1):2747.
Roehrich A, Drobny G. Solid-state NMR studies of biomineralization peptides and proteins. Acc Chem Res. 2013;46(9):2136–44.
Wang T, Phyo P, Hong M. Multidimensional solid-state NMR spectroscopy of plant cell walls. Solid State Nucl Magn Reson. 2016;78:56–63.
Groger C, Lutz K, Brunner E. NMR studies of biomineralisation. Prog Nucl Magn Reson Spectrosc. 2009;54(1):54–68.
Tesson B, Masse S, Laurent G, Maquet J, Livage J, Martin-Jézéquel V, Coradin T. Contribution of multi-nuclear solid state NMR to the characterization of the Thalassiosira pseudonana diatom cell wall. Anal Bioanal Chem. 2008;390(7):1889–98.
Wisser D, Brückner SI, Wisser FM, Althoff-Ospelt G, Getzschmann J, Kaskel S, Brunner E. 1H–13C–29Si triple resonance and REDOR solid-state NMR—a tool to study interactions between biosilica and organic molecules in diatom cell walls. Solid State Nucl Magn Reson. 2015;66–67:33–9.
Sumper M, Brunner E. Silica biomineralisation in diatoms: the model organism Thalassiosira pseudonana. ChemBioChem. 2008;9(8):1187–94.
Kröger N, Deutzmann R, Sumper M. Polycationic peptides from diatom biosilica that direct silica nanosphere formation. Science. 1999;286(5442):1129.
Kröger N, Lorenz S, Brunner E, Sumper M. Self-assembly of highly phosphorylated silaffins and their function in biosilica Morphogenesis. Science. 2002;298(5593):584.
Hildebrand M. Diatoms, biomineralization processes, and genomics. Chem Rev. 2008;108(11):4855–74.
Tesson B, Hildebrand M. Dynamics of silica cell wall morphogenesis in the diatom Cyclotella cryptica: substructure formation and the role of microfilaments. J Struct Biol. 2010;169(1):62–74.
Sumper M, Kröger N. Silica formation in diatoms: the function of long-chain polyamines and silaffins. J Mater Chem. 2004;14(14):2059–65.
Kröger N, Deutzmann R, Bergsdorf C, Sumper M. Species-specific polyamines from diatoms control silica morphology. Proc Natl Acad Sci. 2000;97(26):14133.
Sumper M, Brunner E. Learning from diatoms: nature’s tools for the production of nanostructured silica. Adv Func Mater. 2006;16(1):17–26.
Bridoux MC, Ingalls AE. Structural identification of long-chain polyamines associated with diatom biosilica in a Southern Ocean sediment core. Geochim Cosmochim Acta. 2010;74(14):4044–57.
Ni QZ, Daviso E, Can TV, Markhasin E, Jawla SK, Swager TM, Temkin RJ, Herzfeld J, Griffin RG. High frequency dynamic nuclear polarization. Acc Chem Res. 2013;46(9):1933–41.
Bajaj VS, Mak-Jurkauskas ML, Belenky M, Herzfeld J, Griffin RG. Functional and shunt states of bacteriorhodopsin resolved by 250 GHz dynamic nuclear polarization-enhanced solid-state NMR. Proc Natl Acad Sci. 2009;106(23):9244–9.
Koers EJ, van der Cruijsen EAW, Rosay M, Weingarth M, Prokofyev A, Sauvee C, Ouari O, van der Zwan J, Pongs O, Tordo P, Maas WE, Baldus M. NMR-based structural biology enhanced by dynamic nuclear polarization at high magnetic field. J Biomol NMR. 2014;60(2–3):157–68.
Reggie L, Lopez JJ, Collinson I, Glaubitz C, Lorch M. Dynamic nuclear polarization-enhanced solid-state NMR of a C-13-labeled signal peptide bound to lipid-reconstituted sec translocon. J Am Chem Soc. 2011;133(47):19084–6.
Linden AH, Lange S, Franks WT, Akbey U, Specker E, van Rossum B-J, Oschkinat H. Neurotoxin II bound to acetylcholine receptors in native membranes studied by dynamic nuclear polarization NMR. J Am Chem Soc. 2011;133(48):19266–9.
Pinto C, Mance D, Sinnige T, Daniels M, Weingarth M, Baldus M. Formation of the beta-barrel assembly machinery complex in lipid bilayers as seen by solid-state NMR. Nat Commun. 2018;9:1–10.
Joedicke L, Mao J, Kuenze G, Reinhart C, Kalavacherla T, Jonker HRA, Richter C, Schwalbe H, Meiler J, Preu J, Michel H, Glaubitz C. The molecular basis of subtype selectivity of human kinin G-protein-coupled receptors. Nat Chem Biol. 2018;14:284.
van der Wel PCA, Hu K-N, Lewandowski J, Griffin RG. Dynamic nuclear polarization of amyloidogenic peptide nanocrystals: GNNQQNY, a core segment of the yeast prion protein Sup35p. J Am Chem Soc. 2006;128(33):10840–6.
Frederick KK, Michaelis VK, Corzilius B, Ong T-C, Jacavone AC, Griffin RG, Lindquist S. Sensitivity-enhanced NMR reveals alterations in protein structure by cellular milieus. Cell. 2015;163(3):620–8.
Kaplan M, Cukkemane A, van Zundert GCP, Narasimhan S, Daniëls M, Mance D, Waksman G, Bonvin AMJJ, Fronzes R, Folkers GE, Baldus M. Probing a cell-embedded megadalton protein complex by DNP-supported solid-state NMR. Nat Methods. 2015;12(7):649–52.
Kaplan M, Narasimhan S, de Heus C, Mance D, van Doorn S, Houben K, Popov-Celeketic D, Damman R, Katrukha EA, Jain P, Geerts WJC, Heck AJR, Folkers GE, Kapitein LC, Lemeer S, Henegouwen P, Baldus M. EGFR dynamics change during activation in native membranes as revealed by NMR. Cell. 2016;167(5):1241–51.
Narasimhan S, Scherpe S, Lucini Paioni A, van der Zwan J, Folkers GE, Ovaa H, Baldus M. DNP-supported solid-state nmr spectroscopy of proteins inside mammalian cells. Angew Chem Int Ed. 2019;58(37):12969–73.
Damman R, Lucini Paioni A, Xenaki KT, Beltrán Hernández I, van Bergen en Henegouwen PMP, Baldus M. Development of in vitro-grown spheroids as a 3D tumor model system for solid-state NMR spectroscopy. J Biomol NMR. 2020;74:401–12.
Narasimhan S, Pinto C, Lucini Paioni A, van der Zwan J, Folkers GE, Baldus M. Characterizing proteins in a native bacterial environment using solid-state NMR spectroscopy. Nat Protoc. 2021;16(2):893–918.
Wang T, Park YB, Caporini MA, Rosay M, Zhong L, Cosgrove DJ, Hong M. Sensitivity-enhanced solid-state NMR detection of expansin’s target in plant cell walls. Proc Natl Acad Sci. 2013;110(41):16444–9.
Kolbe F, Ehren HL, Kohrs S, Butscher D, Reiß L, Baldus M, Brunner E. Solid-state NMR spectroscopic studies of 13C,15N,29Si-enriched biosilica from the marine diatom Cyclotella cryptica. Discov Mater. 2020;1(1):3.
Spinde K, Pachis K, Antonakaki I, Paasch S, Brunner E, Demadis KD. Influence of polyamines and related macromolecules on silicic acid polycondensation: relevance to “Soluble Silicon Pools”? Chem Mater. 2011;23(21):4676–87.
Zhai W, Lucini Paioni A, Cai X, Narasimhan S, Medeiros-Silva J, Zhang W, Rockenbauer A, Weingarth M, Song Y, Baldus M, Liu Y. Postmodification via thiol-click chemistry yields hydrophilic trityl-nitroxide biradicals for biomolecular high-field dynamic nuclear polarization. J Phys Chem B. 2020;124(41):9047–60.
Fung BM, Khitrin AK, Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J Magn Reson. 2000;142(1):97–101.
Baldus M, Petkova AT, Herzfeld J, Griffin RG. Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Mol Phys. 1998;95(6):1197–207.
Baker LA, Daniels M, van der Cruijsen EAW, Folkers GE, Baldus M. Efficient cellular solid-state NMR of membrane proteins by targeted protein labeling. J Biomol NMR. 2015;62(2):199–208.
Sauvee C, Rosay M, Casano G, Aussenac F, Weber RT, Ouari O, Tordo P. Highly efficient, water-soluble polarizing agents for dynamic nuclear polarization at high frequency. Angewandte Chemie-Int Ed. 2013;52(41):10858–61.
Herth W, Zugenmaier P. Ultrastructure of the chitin fibrils of the centric diatom Cyclotella cryptica. J Ultrastruct Res. 1977;61(2):230–9.
Herth W. The site of β-chitin fibril formation in centric diatoms II The chitin-forming cytoplasmic structures. J Ultrastruct Res. 1979;68(1):16–27.
Ehren HL, Appels FVW, Houben K, Renault MAM, Wösten HAB, Baldus M. Characterization of the cell wall of a mushroom forming fungus at atomic resolution using solid-state NMR spectroscopy. Cell Surf. 2020;6:100046.
Synytsya A, Novák M. Structural diversity of fungal glucans. Carbohydr Polym. 2013;92(1):792–809.
Fontaine T, Simenel C, Dubreucq G, Adam O, Delepierre M, Lemoine J, Vorgias CE, Diaquin M, Latgé J-P. Molecular organization of the alkali-insoluble fraction of Aspergillus fumigatus cell wall. J Biol Chem. 2000;275(36):27594–607.
Shukla R, Medeiros-Silva J, Parmar A, Vermeulen BJA, Das S, Paioni AL, Jekhmane S, Lorent J, Bonvin AMJJ, Baldus M, Lelli M, Veldhuizen EJA, Breukink E, Singh I, Weingarth M. Mode of action of teixobactins in cellular membranes. Nat Commun. 2020;11(1):2848.
Mance D, Weingarth M, Baldus M. Solid-state NMR on complex biomolecules: methods and applications. Mod Magn Resonan. 2018;13:487–503.
Tan KO, Mardini M, Yang C, Ardenkjær-Larsen JH, Griffin RG. Three-spin solid effect and the spin diffusion barrier in amorphous solids. Sci Adv. 2019;5(7):eaax2743.
Mance D, Gast P, Huber M, Baldus M, Ivanov KL. The magnetic field dependence of cross-effect dynamic nuclear polarization under magic angle spinning. J Chem Phys. 2015;142(23):234201.
Corzilius B, Andreas LB, Smith AA, Ni QZ, Griffin RG. Paramagnet induced signal quenching in MAS–DNP experiments in frozen homogeneous solutions. J Magn Reson. 2014;240:113–23.
Pawolski D, Heintze C, Mey I, Steinem C, Kröger N. Reconstituting the formation of hierarchically porous silica patterns using diatom biomolecules. J Struct Biol. 2018;204(1):64–74.
Kotzsch A, Pawolski D, Milentyev A, Shevchenko A, Scheffel A, Poulsen N, Shevchenko A, Kröger N. Biochemical composition and assembly of biosilica-associated insoluble organic matrices from the diatom Thalassiosira pseudonana. J Biol Chem. 2016;291(10):4982–97.
Look GC, Murphy MM, Campbell DA, Gallop MA. Trimethylorthoformate: a mild and effective dehydrating reagent for solution and solid phase imine formation. Tetrahedron Lett. 1995;36(17):2937–40.
Huang M-J, Lee KS. 1H and 13C NMR chemical shift assignments of agmatine analogues, (3-aminopropyl)guanidine and (trans-4-aminocyclohexyl)guanidine. Mol Phys. 2005;103(15–16):2229–37.
Sietsma JH, Wessels JGH. Evidence for Covalent Linkages between Chitin and β-glucan in a fungal wall. Microbiology. 1979;114(1):99–108.
Lechner CC, Becker CFW. Silaffins in silica biomineralization and biomimetic silica precipitation. Mar Drugs. 2015;13(8):5297–333.
Bayro MJ, Huber M, Ramachandran R, Davenport TC, Meier BH, Ernst M, Griffin RG. Dipolar truncation in magic-angle spinning NMR recoupling experiments. J Chem Phys. 2009;130(11):114506.
Poulsen N, Sumper M, Kröger N. Biosilica formation in diatoms: characterization of native silaffin-2 and its role in silica morphogenesis. Proc Natl Acad Sci. 2003;100(21):12075–80.
Poulsen N, Kröger N. Silica morphogenesis by alternative processing of silaffins in the diatom Thalassiosira pseudonana. J Biol Chem. 2004;279(41):42993–9.
Acknowledgements
We thank Prof. Nils Kroeger and Dr. Marie Renault for scientific discussions and Johan v.d. Zwan for excellent technical support. We are indebted to P. Tordo and O. Ouari (Aix-Marseille Université) for providing AMUPol for the DNP experiments.
Funding
Financial support from the DFG (FOR2038: Nanopatterned Organic Matrices in Biological Silica Mineralization) is gratefully acknowledged. A.L.P. was supported by the Dutch Research Council (NWO, Grant numbers 718.015.001 to M.B.) This work was also supported the infrastructure project I.Next (Horizon 2020 program of the European Union, Grant # 653706).
Author information
Authors and Affiliations
Contributions
HLE, FK and ALP performed and analyzed the DNP solid-state NMR experiments. All authors contributed to writing the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ehren, H.L., Kolbe, F., Lucini Paioni, A. et al. DNP-supported solid-state NMR studies of 13C,15N,29Si-enriched biosilica of Cyclotella cryptica and Thalassiosira pseudonana. Discov Mater 1, 9 (2021). https://doi.org/10.1007/s43939-021-00009-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s43939-021-00009-9