
Abstract
In the last three decades, the scope of solid-state NMR has expanded to exploring complex biomolecules, from large protein assemblies to intact cells at atomic-level resolution. This diversity in macromolecules frequently features highly flexible components whose insoluble environment precludes the use of solution NMR to study their structure and interactions. While High-resolution Magic-Angle Spinning (HR-MAS) probes offer the capacity for gradient-based 1H-detected spectroscopy in solids, such probes are not commonly used for routine MAS NMR experiments. As a result, most exploration of the flexible regime entails either 13C-detected experiments, the use of partially perdeuterated systems, or ultra-fast MAS. Here we explore proton-detected pulse schemes probing through-bond 13C–13C networks to study mobile protein sidechains as well as polysaccharides in a broadband manner. We demonstrate the use of such schemes to study a mixture of microtubule-associated protein (MAP) tau and human microtubules (MTs), and the cell wall of the fungus Schizophyllum commune using 2D and 3D spectroscopy, to show its viability for obtaining unambiguous correlations using standard fast-spinning MAS probes at high and ultra-high magnetic fields.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Magic Angle Spinning (MAS) NMR is a powerful technique to study the structure, dynamics, and intermolecular interactions of insoluble biomolecules and materials. Over the last three decades, the nature of biomolecules that can be studied with MAS NMR have become more complex, from short peptides (Fu and Cross 1999; Griffin 1998; Jaroniec et al. 2002; Luca et al. 2003; Rienstra et al. 2000; Tycko 2001) to whole cells (Ghassemi et al. 2021; Narasimhan et al. 2019; Renault et al. 2012). Concomitant technological advances have brought fast- to ultra-fast spinning probes (Barbet-Massin et al. 2014; Penzel et al. 2019; Samoson 2021) for 1H-detection using progressively smaller rotors, which reduce linewidths in the case of homogeneous broadening (Maricq and Waugh 1979; Schledorn et al. 2020) while maintaining practical sensitivity due to the relatively large 1H gyromagnetic ratio (Agarwal et al. 2014). Such advances have led to the development of MAS 1H-detection techniques of fully protonated samples, circumventing the expense and loss of information of sample deuteration (Guo et al. 2014; Paulson et al. 2003; Zhou et al. 2007, 2012). As a result, a suite of 1H-detected experiments for de novo chemical shift assignments (Barbet-Massin et al. 2014; Fricke et al. 2017; Stanek et al. 2016) are increasingly commonplace and include frequent use of dipolar or scalar 13C–13C mixing schemes such as RFDR (Asami and Reif 2013; Bennett et al. 1992; Paluch et al. 2022; Sarkar et al. 2022), TOBSY (Baldus et al. 1997; Baldus and Meier 1996), WALTZ (Stanek et al. 2016), and DIPSI (Paluch et al. 2022; Shaka et al. 1988). Typical uses for these mixing techniques have occurred within the rigid part of biomolecular assemblies, as selected by cross-polarization at the start of the pulse sequence (Barbet-Massin et al. 2013).
However, for insoluble systems comprising flexible components, INEPT transfers select for nuclei in the fast-motion regime where dipolar self-averaging occurs. The INEPT-TOBSY experiment is frequently used to map 13C–13C connectivities in conjunction with dipolar-based counterparts, typically in a 13C-detected fashion (Andronesi et al. 2005; Baldus and Meier 1996; Damman et al. 2019; Heise et al. 2005). Heteronuclear 1H–13C-detected correlations can provide additional assignments for the flexible regime (Alam and Holland 2006; Damman et al. 2019; Elena et al. 2005; Siemer 2020). In the case of limited sample quantities, it is instead desirable to amplify the signal using 1H-detection, which requires an INEPT transfer back to the 1H channel following 13C evolution, mixing, and water suppression (Zhou and Rienstra 2008). Variants of a scalar 2D hCH J-HSQC experiment have been used to characterize complex systems including fungal cell walls (Safeer et al. 2023), human microtubules (Luo et al. 2020; Savastano et al. 2020), viral capsids (Callon et al. 2022), protein condensates (Damman et al. 2019) and membrane-bound proteins (Howarth and McDermott 2022). In addition, INEPT based homonuclear 1-bond transfers have been used to obtain sequential backbone correlations in highly mobile amyloid-fibrils (Falk and Siemer 2016; Zhang et al. 2023).
In the following, we explore the use of 1H detected experiments that involve an additional broadband through-bond CC transfer step to increase spectral dispersion in applications on complex biomolecular systems containing dynamic molecular (sub)species that are difficult to prepare in deuterated form. Such an INEPT-based 3D hCCH through-bond sequence with longitudinal mixing has previously been used at moderate spinning (20 kHz) on deuterated samples recrystallized in 90% D2O, to obtain residual protonated methyl group assignments while avoiding 1H2O suppression (Agarwal and Reif 2008). In addition, a 2D hC(c)H TOCSY experiment with DIPSI-2 mixing was used to obtain highly resolved spectra on a fully protonated peptidoglycan with an ultra-fast spinning probe (Bougault et al. 2019). However, in our hands, such usage of longitudinal homonuclear mixing is prone to producing antiphase artifacts generated by resonances with especially long coherence lifetimes. We found that these artifacts are resolved either by using long phase cycles that render extension to a 3D experiment impractical, or by instead using transverse mixing with a prior trim pulse (Bax et al. 1990) to purge unwanted coherences, similarly implemented as in solution-state NMR. We apply these schemes to study the flexible region of two complex insoluble systems using hCCH TOCSY, with DIPSI-3 (Shaka et al. 1988) and WALTZ (Shaka et al. 1983) homonuclear carbon mixing in gradient-free MAS probes. We demonstrate that such schemes provide broadband 13C–13C correlations at both high and ultra-high field NMR conditions.
Material and methods
Expression of tau K32
The gene encoding tau K32 inserted in a pNG2 plasmid was kindly gifted to us by the Mandelkow group of the German Center for Neurodegenerative Diseases, Bonn, Germany. The vector was transfected in E. coli BL21 (DE3) competent cells and expression was induced in M9 minimal media with 0.3 mM isopropyl–D-thiogalactopyranoside (IPTG) when the OD600 reached 0.6 for 16 h at 20 °C.
Purification of tau K32
[13C,15N] labeled tau K32 was purified according to the protocol described by Barghorn et al. (2005). Small adaptations to the protocol are described below. Firstly, 1 mM NaN3 was added to the buffers, to minimize the risk of growth in them. Moreover, cells were lysed either with a French pressure cell as described previously or by sonication. The cation exchange chromatography that was utilized was a HiTrap SP column of 5 mL and for gel filtration chromatography a HiLoad 26/60 Superdex 75 prep grade column. The protein concentration was measured by a BCA assay with a Thermo Scientific Pierce BCA Protein Assay Kit.
MT polymerization and preparation of solid-state NMR samples
Lyophilized tubulin (Cytoskeleton, Inc.) was solubilised in Brb80 buffer (80 mM PIPES, 2 mM MgCl2, 1 mM EGTA, pH 6.8, 1 mM NaN3, 1 mM DTT, pH 6.8), to a final concentration of 2 mg/mL. The polymerization was induced by adding 1 mM Guanosine-5′-triphosphate (GTP) and incubation for 15 min at 30 °C. Then, 20 μM paclitaxel (taxol, SIGMA) was used to stabilize the MT and incubation took place for another 15 min at 30 °C. The MT were spun down at 180.000 × g (Beckman TLA-55 rotor) for 30 min at 30 °C and the pellet was resuspended in warm Brb80 buffer with 20 μM paclitaxel. Subsequently, a 1:1 ratio of 13C15N tau K32 was added. The interaction partners were incubated for 30 min at 37 °C. In the following isotopically labelled tau K32 in complex with MT was separated from the unbound, non-polymerised fraction by centrifugation at 180.000 xg (Beckman TLA-55 rotor) for 30 min at 30 °C. Afterwards, the pellet was washed with 40 mM phosphate buffer, pH 7, with protease inhibitor (as described earlier) and 1 mM NaN3, without disturbing the pellet. A 1.3 mm rotor was packed with the pellet.
Basic experimental details for each sample used in this manuscript. Further details are provided in the materials and methods section, and Table S1.
MAS NMR experiments
MAS NMR experiments for the tau-microtubule mixture used a 3-channel HXY 1.3 mm probe at ω0H/2π = 700 MHz with a Bruker Avance 3 console. The MAS rate was set to 44 kHz, with a set temperature of 280 K, which results in a sample temperature of ~303 K based on calibration using KBr (Thurber and Tycko 2009). The pulse program corresponding to this sample is provided schematically in Fig. S1, with the full sequence in the supporting information. Pulse amplitudes for INEPT transfers and 15N J-refocusing were the following: ω1H/2π = 127 kHz, ω13C/2π = 83 kHz, and ω15N/2π = 75 kHz. MISSISSIPPI was used for water suppression (Zhou and Rienstra 2008). 2D experiments were collected by not evolving the t2 dimension of the pulse scheme shown in Fig. 1. For 0 ms mixing, neither the SLx nor the DIPSIy,-y blocks were executed, while in the case of mixing we used 2 ms of SLx at the same amplitude as the DIPSI strength (Bax et al. 1990; Clore et al. 1990). The 2D experiments were processed in Topspin: the data was zero-filled, and a QSine window function was applied with a sinebell shift of 2.5. For the 3D, non-uniform sampling was used with a Poisson Gap distribution schedule with 25% sampling density (Maciejewski et al. 2012), out to ~9 ms of acquisition time in both indirect 13C dimensions. The data was then reconstructed using the SMILE NUS reconstruction algorithm (Ying et al. 2017) in NMRBox (Maciejewski et al. 2017) using NMRPipe (Delaglio et al. 1995) for preparatory and post-processing. Each dimension was zero-filled, and sine bell-squared window processing was used for both indirect dimensions, with an offset of 0.5, endpoint of 0.95, and exponent 1.0. For SMILE processing, the noise factor for signal cutoff was set to 5, with an 80% threshold for signal detection. In both the 2D and 3D experiments, the 1H dimension was referenced using water at 4.7 ppm, while the 13C dimension was referenced indirectly with a correction factor determined from the gyromagnetic ratio. Chemical shift and linewidth analysis was performed in NMRFAM Sparky software (Lee et al. 2015).

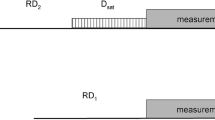
hCCH 3D used in this work. First, an INEPT block transfers magnetization through-bond from 1H to 13C prior to an evolution period to encode the 13C chemical shift. After the t1 period, mixing is applied according to Scheme 1 or Scheme 2. Afterwards, the chemical shift is again encoded during the t2 period. The carbon magnetization is then stored longitudinally during the water suppression period, prior to INEPT transfer back to protons for detection. Narrow and wide rectangles indicate 90°/180° pulses, respectively. The 3 hC(c)H experiments were collected by not evolving the t2 period. Scheme 1 was used for the tau-microtubule complex, while Scheme 2 was used for S. commune. More details regarding the experiments are available in the Supplementary information
The 13C/15N S. commune sample was prepared as discussed in previous publications (Ehren et al. 2020; Safeer et al. 2023). MAS NMR experiments used a 3-channel HXY 1.3 mm probe at ω0H/2π = 1.2 GHz with a Bruker NEO console. The MAS spin rate was set to 60 kHz, and the sample was cooled to 260 K leading to a sample temperature of ~20 °C. The pulse sequence outlined in Fig. S1b was used. The strength of the hard pulses on each channel were set as follows: ω1H/2π = 100 kHz, ω13C/2π = 67 kHz, and ω15N/2π = 50 kHz. The 1H dimension was referenced using water referenced to 4.7 ppm, while the 13C dimension was referenced to previously published work (Ehren et al. 2020). B0 field drift artifacts in the S. commune spectra were corrected using previously published scripts (Najbauer et al. 2019). Further information regarding data acquisition for both samples can be found in Table 1 and Table S1.
Results and discussion
In the following, we explore the flexible regions of two complex insoluble systems using hCCH TOCSY, with broadband DIPSI-3 (Shaka et al. 1988) and WALTZ-16 (Shaka et al. 1983) homonuclear carbon mixing in gradient-free probes using the pulse schemes given in Fig. 1. We tested pulse Scheme 1 in Fig. 1 on a sample of [13C,15N] labeled tau in complex with unlabeled microtubules (MTs). Tau is an intrinsically disordered protein that maintains microtubule stability (Brotzakis et al. 2021; Kadavath et al. 2015), and whose function is modulated by a series of posttranslational modifications including phosphorylation, nitration, and glycosylation (Martin et al. 2011). In the current context we used the K32 variant (~20.5 kDa of the full length), corresponding to the microtubule binding domain Q244-K369 (R1, R2, R3, R4), the N-terminal proline-rich region S198-Q244 (P2), and the C-terminal R’ region K369–Y394 (Brotzakis et al. 2021). For labeled tau bound to MTs, application of the 2D hC(c)H experiment shown in Fig. 1 Scheme 1 with 2 ms of preparatory Cx spin lock in addition to 25 ms of DIPSI-3 mixing, revealed multiple-bond CH correlations, to assign the protein backbone and non-aromatic sidechains (Fig. 2, blue). For reference, a 2D spectrum with 0 ms of mixing is shown in red.

2D hC(c)H spectral overlay of 0ms (red/green) and 25ms (blue/orange) of DIPSI mixing for the tau-microtubule complex at 700 MHz (1H Larmor frequency). In the dashed boxes we show examples of cross-peaks corresponding to complete intraresidual (along the side chain) scalar transfers within various residues, with unambiguous correlations as determined in the 3D shown in Fig. 3. In each vertical strip, the peaks in red are the resonance from which the first 1H→13C transfer originates before transferring to the blue crosspeaks
In line with previous work (El Mammeri et al. 2022) we expect that the observed correlations result from flexible tau residues not involved in strong binding to MTs. While typical chemical shift assignments allow for immediate recognition of some of these resonances (e.g. Ile), chemical shift degeneracy results in a heavy degree of spectral overlap, which was alleviated by adding a second dimension to the experiment after the DIPSI-3 mixing period. Figure 3 shows the resulting strips corresponding to Ile, Leu, Lys, Pro, and Val sidechains. We also identified additional Ile and Lys residues in the 3D spectrum (not shown), indicating its usefulness in distinguishing like residues whose chemical shift dispersion results in significant overlap in lower dimensional experiments. Finally, we observe several distinct Ala, Thr and Gly residues (not shown). Currently, our survey of identifiable residues is in agreement with those observed previously (El Mammeri et al. 2022), though we find that our 3D hCCH correlations lead to more complete chemical shift assignments from backbone and sidechain resonances. Amino-acid selective assignments and proton linewidths for the residues highlighted in Fig. 3 are provided in Table S2. While the average linewidth is less than 0.1 ppm, the observed variations did not correlate with backbone versus sidechain topology or degree of protonation.

Strips from a 3D hCCH TOCSY lengths spectrum of the tau-microtubule mixture at 700MHz using 25ms of DIPSI mixing, in which we show correlations from the backbone and sidechain of four example residues: Isoleucine, Leucine, Lysine, Proline and Valine. Asterisks indicate peaks along the diagonal of the CC plane. For Proline, we observe doubled peaks for protons originating from Cb/Cd diagonal peaks
Next, we tested the suitability of INEPT-based 13C–13C scalar transfers to explore the dynamic region of the cell wall of S. commune. Previous studies of 13C/15N-labeled S. commune reveal that it is composed of a complex mixture of proteins and various kinds of polysaccharides(Ehren et al. 2020; Safeer et al. 2023). Consequentially, the carbon spectrum features a larger protonated 13C chemical shift dispersion (~100 ppm) while the non-aromatic 1H shift range remains small (0–6 ppm). As a result, low-power irradiation for homonuclear CC transfer leads to shorter-range transfer, especially at ultra-high field.
For this system, we used a variant of the sequence as shown in Fig. 1 Scheme 2, in which τ1 = τ2 = 1/(4JHC), favoring the CH groups from polysaccharides and amino acids to alleviate spectral congestion. Figure 4 shows overlays of 3 spectra with 0 ms (red), 4.8 ms (cyan), and 11.2 ms (blue) of isotropic WALTZ-16 mixing, which has a larger bandwidth than DIPSI-3 and may thus be advantageous at ultra-high field (Shaka et al. 1988). In spite of these relatively short mixing times, low-power irradiation (15 kHz) resulted in efficient transfers among nuclei within the polysaccharide region (~ 65–105 ppm). We highlight unambiguous assignments corresponding to the reducing ends of α- (Ra, left) and β-glucan (Rb, right), which are both formed from a pyranose ring in chair conformation, in which C3 links the ring to C1 of the successive sugar subunit via a glycosidic bond. We observe nearly all correlations from the pyranose ring with 11.2 ms of mixing, with the exception of C3 and C6; the former is observed only weakly in INEPT-based experiments due to its reduced mobility, while the latter has been filtered out by our choice in τ2 INEPT delays. Notably, we also observe weak correlations between the amino acid backbone and sidechain resonances at 11.2 ms of mixing (Fig. 4, right). As shown for K32-tau, stronger correlations in this region may require longer mixing for complete transfer. Higher transfer efficiency might also be attained with the use of DIPSI instead of WALTZ mixing, which has higher transfer efficiency though at the cost of a narrower bandwidth (Shaka et al. 1988).

Comparing hC(c)H spectra of S. commune with varying TOCSY lengths, using WALTZ-16 at 1200 MHz (1H Larmor frequency). We specifically show peaks belonging to the reducing ends of -glucan (Ra, left) and -glucan (Rb, right) as they are relatively well-isolated and show prominent transfer. In red we show the spectrum with no mixing, so that all peaks correspond to directly bonded C–H pairs. The cyan (4.8 ms) and blue (11.2 ms) spectra instead show multiple bond correlations corresponding to various combinations of C–H pairs within the pyranose ring (structure shown). We also observe correlations among the amino acid region (dashed boxes). For fungal cell wall biological component analysis of 0 vs. 11.2 ms data, see Safeer et al. 2023
Notably, we found little overlap between peaks that appear in the INEPT-based spectrum in Fig. 4 and a CP-based hCH correlation experiment of the same sample of S. commune (Safeer et al. 2023). We thus expect that all observable correlations arise from the flexible domain of the fungal cell wall where chemical shift anisotropies and dipolar couplings are averaged out. Interestingly, when comparing the 1H linewidths between the tau 3D spectra (Table S1) and fungal 2Ds (Safeer et al. 2023), we observe slightly broader average 1H linewidths for the tau data (~0.093 ppm), which was collected at 44 kHz MAS vs. 60 kHz MAS for S. commune (~0.067 ppm). This improvement may be attributed to the nature of the sample or differences in effective temperature, and the larger magnetic field in the case of S. commune.
Conclusions
In this work we have introduced two and three-dimensional 13C–13C–1H correlation experiments that allowed us to characterize dynamic carbon networks of complex biomolecular systems without the need of deuteration, HR-MAS probes (Blicharski and Sobol 1982; Howarth and McDermott 2022; Li et al. 2005; Zupancic and Pirs 1976) or ultra-fast MAS in 1H-detected TOCSY experiments(Ikura et al. 1990; Kay et al. 1990). Our data show that we can indirectly determine through-bond 13C–13C connectivities with refocused INEPT-based pulse sequences in a selective manner as determined by the choice of J-coupling and delays. We obtained 2D hC(c)H spectra of exceptionally high resolution on a fully protonated mixture of 13C/15N-labeled K32-tau and unlabeled human microtubules as well as fungal cell wall preparations of Schizophyllum commune(Ehren et al. 2020). We extended the sequence to a 3D hCCH experiment to alleviate chemical shift degeneracy for obtaining unambiguous correlations. This experiment could be further tuned to additional selectivity, by adjusting the length of the τ2 delay. Combination with 1H-detected hNCOCA and hNCA experiments (Falk and Siemer 2016; Linser et al. 2008, 2010; Zhang et al. 2023) would lead to full sequential assignments of dynamic protein stretches. The hCCH experiment could also be expanded to a 4-dimensional hNCACH sequence, in which the N-edited transfer to CA’s selects for backbone resonances, expanding the information available in a single experiment.
This method can also be used in conjunction with CP-based 1H-detected schemes—which require fast-to-ultrafast MAS techniques for fully protonated samples—and also enable one to study biopolymers within native conditions. Adapting solution NMR methodology in this way will allow Magic Angle Spinning NMR to explore a wider scope of biomolecules with a large flexible domain, such as membrane proteins whose main functions lie in the extra membrane space e.g. integrins (Bergonzini et al. 2022), growth hormone receptors (Kaplan et al. 2016), and membrane-anchored lipoproteins (Xiang et al. 2022). In addition, such analysis is not restricted to biomolecules, but could also be used to identify soluble organic intermediates and byproducts in industrial catalysts (Chowdhury et al. 2018a, b). We thus predict widespread usage of such sequences using standard MAS probes to thoroughly characterize organic components across various applications.
References
Agarwal V, Reif B (2008) Residual methyl protonation in perdeuterated proteins for multi-dimensional correlation experiments in MAS solid-state NMR spectroscopy. J Magn Reson 194(1):16–24. https://doi.org/10.1016/j.jmr.2008.05.021
Agarwal V, Penzel S, Szekely K, Cadalbert R, Testori E, Oss A, Past J, Samoson A, Ernst M, Bockmann A, Meier BH (2014) De Novo 3D structure determination from sub-milligram protein samples by solid-state 100 kHz MAS NMR spectroscopy. Angew Chem-Int Edit 53(45):12253–12256. https://doi.org/10.1002/anie.201405730
Alam TM, Holland GP (2006) 1 H–13C INEPT MAS NMR correlation experiments with 1H–1H mediated magnetization exchange to probe organization in lipid biomembranes. J Magn Reson 180(2):210–221. https://doi.org/10.1016/j.jmr.2006.02.013
Andronesi OC, Becker S, Seidel K, Heise H, Young HS, Baldus M (2005) Determination of membrane protein structure and dynamics by magic-angle-spinning solid-state NMR spectroscopy. J Am Chem Soc 127(37):12965–12974. https://doi.org/10.1021/ja0530164
Asami S, Reif B (2013) Proton-detected solid-state NMR spectroscopy at aliphatic sites: application to crystalline systems. Accounts Chem Res 46(9):2089–2097. https://doi.org/10.1021/ar400063y
Baldus M, Meier BH (1996) Total correlation spectroscopy in the solid state. The use of scalar couplings to determine the through-bond connectivity. J Magn Reson Ser A 121(1):65–69. https://doi.org/10.1006/jmra.1996.0137
Baldus M, Iuliucci RJ, Meier BH (1997) Probing through-bond connectivities and through-space distances in solids by magic-angle-spinning nuclear magnetic resonance. J Am Chem Soc 119(5):1121–1124. https://doi.org/10.1021/ja9622259
Barbet-Massin E, Pell AJ, Jaudzems K, Franks WT, Retel JS, Kotelovica S, Akopjana I, Tars K, Emsley L, Oschkinat H, Lesage A, Pintacuda G (2013) Out-and-back 13C–13C scalar transfers in protein resonance assignment by proton-detected solid-state NMR under ultra-fast MAS. J Biomol NMR 56(4):379–386. https://doi.org/10.1007/s10858-013-9757-3
Barbet-Massin E, Pell AJ, Retel JS, Andreas LB, Jaudzems K, Franks WT, Nieuwkoop AJ, Hiller M, Higman V, Guerry P, Bertarello A, Knight MJ, Felletti M, Le Marchand T, Kotelovica S, Akopjana I, Tars K, Stoppini M, Bellotti V, Bolognesi M, Ricagno S, Chou JJ, Griffin RG, Oschkinat H, Lesage A, Emsley L, Herrmann T, Pintacuda G (2014) Rapid proton-detected NMR assignment for proteins with fast magic angle spinning. J Am Chem Soc 136(35):12489–12497. https://doi.org/10.1021/ja507382j
Barghorn S, Biernat J, Mandelkow E (2005) Purification of recombinant tau protein and preparation of alzheimer-paired helical filaments in vitro. In: Sigurdsson EM (ed) Amyloid Proteins: Methods and Protocols. NJ, Humana Press, Totowa, pp 35–51. https://doi.org/10.1385/1-59259-874-9:035
Bax A, Clore GM, Gronenborn AM (1990) 1H–1H correlation via isotropic mixing of 13C magnetization, a new 3-dimensional approach for assigning 1H and 13C spectra of 13C-enriched proteins. J Magn Reson 88(2):425–431. https://doi.org/10.1016/0022-2364(90)90202-k
Bennett AE, Ok JH, Griffin RG, Vega S (1992) Chemical-shift correlation spectroscopy in rotating solids—Radio frequency-driven dipolar recoupling and longitudinal exchange. J Chem Phys 96(11):8624–8627. https://doi.org/10.1063/1.462267
Bergonzini C, Kroese K, Zweemer AJM, Danen EHJ (2022) Targeting integrins for cancer therapy—Disappointments and opportunities. Front Cell Dev Biol. https://doi.org/10.3389/fcell.2022.863850
Blicharski JS, Sobol WT (1982) A new type of magnetic-field gradient coil for NMR measurements. J Magn Reson 46(1):1–8. https://doi.org/10.1016/0022-2364(82)90157-3
Bougault C, Ayala I, Vollmer W, Simorre J-P, Schanda P (2019) Studying intact bacterial peptidoglycan by proton-detected NMR spectroscopy at 100 kHz MAS frequency. J Struct Biol 206(1):66–72. https://doi.org/10.1016/j.jsb.2018.07.009
Brotzakis ZF, Lindstedt PR, Taylor RJ, Rinauro DJ, Gallagher NCT, Bernardes GJL, Vendruscolo M (2021) A structural ensemble of a tau-microtubule complex reveals regulatory tau phosphorylation and acetylation mechanisms. ACS Cent Sci 7(12):1986–1995. https://doi.org/10.1021/acscentsci.1c00585
Callon M, Malar AA, Lecoq L, Dujardin M, Fogeron ML, Wang SS, Schledorn M, Bauer T, Nassal M, Bockmann A, Meier BH (2022) Fast magic-angle-spinning NMR reveals the evasive hepatitis B Virus capsid C-terminal domain. Angew Chem Int Ed. https://doi.org/10.1002/anie.202201083
Chowdhury AD, Houben K, Whiting GT, Chung S-H, Baldus M, Weckhuysen BM (2018a) Electrophilic aromatic substitution over zeolites generates Wheland-type reaction intermediates. Nat Catal 1(1):23–31. https://doi.org/10.1038/s41929-017-0002-4
Chowdhury AD, Paioni AL, Houben K, Whiting GT, Baldus M, Weckhuysen BM (2018b) Bridging the gap between the direct and hydrocarbon pool mechanisms of the methanol-to-hydrocarbons process. Angew Chem Int Ed 57(27):8095–8099. https://doi.org/10.1002/anie.201803279
Clore GM, Bax A, Driscoll PC, Wingfield PT, Gronenborn AM (1990) Assignment of the side-chain 1H and 13C resonances of interleukin-beta using double-resonance and triple-resonance heteronuclear 3-dimensional nmr-spectroscopy. Biochemistry 29(35):8172–8184. https://doi.org/10.1021/bi00487a027
Damman R, Schütz S, Luo Y, Weingarth M, Sprangers R, Baldus M (2019) Atomic-level insight into mRNA processing bodies by combining solid and solution-state NMR spectroscopy. Nat Commun 10(1):4536. https://doi.org/10.1038/s41467-019-12402-3
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6(3):277–293. https://doi.org/10.1007/BF00197809
Ehren HL, Appels FVW, Houben K, Renault MAM, Wösten HAB, Baldus M (2020) Characterization of the cell wall of a mushroom forming fungus at atomic resolution using solid-state NMR spectroscopy. The Cell Surface 6:100046. https://doi.org/10.1016/j.tcsw.2020.100046
Elena B, Lesage A, Steuernagel S, Böckmann A, Emsley L (2005) Proton to carbon-13 INEPT in solid-state NMR spectroscopy. J Am Chem Soc 127(49):17296–17302. https://doi.org/10.1021/ja054411x
Falk AS, Siemer AB (2016) Dynamic domains of amyloid fibrils can be site-specifically assigned with proton detected 3D NMR spectroscopy. J Biomol NMR 66(3):159–162. https://doi.org/10.1007/s10858-016-0069-2
Fricke P, Chevelkov V, Zinke M, Giller K, Becker S, Lange A (2017) Backbone assignment of perdeuterated proteins by solid-state NMR using proton detection and ultrafast magic-angle spinning. Nat Protoc 12(4):764–782. https://doi.org/10.1038/nprot.2016.190
Fu R, Cross TA (1999) Solid-state nuclear magnetic resonance investigation of protein and polypeptide structure. Annu Rev Biophys Biomol Struct 28:235–268. https://doi.org/10.1146/annurev.biophys.28.1.235
Ghassemi N, Poulhazan A, Deligey F, Mentink-Vigier F, Marcotte I, Wang T (2021) Solid-state NMR investigations of extracellular matrixes and cell walls of algae, bacteria, fungi, and plants. Chem Rev. https://doi.org/10.1021/acs.chemrev.1c00669
Griffin RG (1998) Dipolar recoupling in MAS spectra of biological solids. Nat Struct Biol 5(Suppl):508–512. https://doi.org/10.1038/749
Guo CM, Hou GJ, Lu XY, O’Hare B, Struppe J, Polenova T (2014) Fast magic angle spinning NMR with heteronucleus detection for resonance assignments and structural characterization of fully protonated proteins. J Biomol NMR 60(4):219–229. https://doi.org/10.1007/s10858-014-9870-y
Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M (2005) Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc Natl Acad Sci U S A 102(44):15871–15876. https://doi.org/10.1073/pnas.0506109102
Howarth GS, McDermott AE (2022) High-resolution magic angle spinning NMR of KcsA in LIPOSOMES: the highly mobile C-terminus. Biomolecules 12(8):1122. https://doi.org/10.3390/biom12081122
Ikura M, Kay LE, Bax A (1990) A novel approach for sequential assignment of proton, carbon-13, and nitrogen-15 spectra of larger proteins—Heteronuclear triple-resonance 3-dimensional NMR-Spectroscopy—Application to calmodulin. Biochemistry 29(19):4659–4667. https://doi.org/10.1021/bi00471a022
Jaroniec CP, Filip C, Griffin RG (2002) 3D TEDOR NMR experiments for the simultaneous measurement of multiple carbon-nitrogen distances in uniformly 13C, 15N-labeled solids. J Am Chem Soc 124(36):10728–10742. https://doi.org/10.1021/ja026385y
Kadavath H, Hofele RV, Biernat J, Kumar S, Tepper K, Urlaub H, Mandelkow E, Zweckstetter M (2015) Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci U S A 112(24):7501–7506. https://doi.org/10.1073/pnas.1504081112
Kaplan M, Narasimhan S, de Heus C, Mance D, van Doorn S, Houben K, Popov-Čeleketić D, Damman R, Katrukha EA, Jain P, Geerts WJC, Heck AJR, Folkers GE, Kapitein LC et al (2016) EGFR dynamics change during activation in native membranes as revealed by NMR. Cell 167(5):1241-1251.e1211. https://doi.org/10.1016/j.cell.2016.10.038
Kay LE, Ikura M, Tschudin R, Bax A (1990) 3-Dimensional triple-resonance nmr-spectroscopy of isotopically enriched proteins. J Magn Reson 89(3):496–514. https://doi.org/10.1016/j.jmr.2011.09.004
Lee W, Tonelli M, Markley JL (2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31(8):1325–1327. https://doi.org/10.1093/bioinformatics/btu830
Li W, Lee REB, Lee RE, Li J (2005) Methods for acquisition and assignment of multidimensional high-resolution magic angle spinning NMR of whole cell bacteria. Anal Chem 77(18):5785–5792. https://doi.org/10.1021/ac050906t
Linser R, Fink U, Reif B (2008) Proton-detected scalar coupling based assignment strategies in MAS solid-state NMR spectroscopy applied to perdeuterated proteins. J Magn Reson 193(1):89–93. https://doi.org/10.1016/j.jmr.2008.04.021
Linser R, Fink U, Reif B (2010) Assignment of dynamic regions in biological solids enabled by spin-state selective NMR experiments. J Am Chem Soc 132(26):8891–8893. https://doi.org/10.1021/ja102612m
Luca S, Heise H, Baldus M (2003) High-resolution solid-state NMR applied to polypeptides and membrane proteins. Accounts Chem Res 36(11):858–865. https://doi.org/10.1021/ar020232y
Luo Y, Xiang S, Hooikaas PJ, van Bezouwen L, Jijumon AS, Janke C, Förster F, Akhmanova A, Baldus M (2020) Direct observation of dynamic protein interactions involving human microtubules using solid-state NMR spectroscopy. Nat Commun 11(1):18. https://doi.org/10.1038/s41467-019-13876-x
Maciejewski MW, Mobli M, Schuyler AD, Stern AS, Hoch JC (2011) Data Sampling in Multidimensional NMR: Fundamentals and Strategies. In: Billeter M, Orekhov V (eds) Novel Sampling Approaches in Higher Dimensional Nmr, vol 316. Springer, Berlin, pp 49–77. https://doi.org/10.1007/128_2011_185
Maciejewski MW, Schuyler AD, Gryk MR, Moraru II, Romero PR, Ulrich EL, Eghbalnia HR, Livny M, Delaglio F, Hoch JC (2017) NMRbox: a resource for biomolecular NMR computation. Biophys J 112(8):1529–1534. https://doi.org/10.1016/j.bpj.2017.03.011
El Mammeri N, Dregni AJ, Duan P, Wang HK, Hong M (2022) Microtubule-binding core of the tau protein. Sci Adv 8(29):eabo4459. https://doi.org/10.1126/sciadv.abo4459
Maricq MM, Waugh JS (1979) NMR in rotating solids. J Chem Phys 70(7):3300–3316. https://doi.org/10.1063/1.437915
Martin L, Latypova X, Terro F (2011) Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem Int 58(4):458–471. https://doi.org/10.1016/j.neuint.2010.12.023
Najbauer EE, Andreas LB (2019) Correcting for magnetic field drift in magic-angle spinning NMR datasets. J Magn Reson 305:1–4. https://doi.org/10.1016/j.jmr.2019.05.005
Narasimhan S, Scherpe S, Lucini Paioni A, van der Zwan J, Folkers GE, Ovaa H, Baldus M (2019) DNP-supported solid-state NMR spectroscopy of proteins inside mammalian cells. Angew Chem Int Ed Engl 58(37):12969–12973. https://doi.org/10.1002/anie.201903246
Paluch P, Augustyniak R, Org ML, Vanatalu K, Kaldma A, Samoson A, Stanek J (2022) NMR assignment of methyl groups in immobilized proteins using multiple-bond 13C Homonuclear transfers, proton detection, and very fast MAS. Front Mol Biosci. https://doi.org/10.3389/fmolb.2022.828785
Paulson EK, Morcombe CR, Gaponenko V, Dancheck B, Byrd RA, Zilm KW (2003) High-sensitivity observation of dipolar exchange and NOEs between exchangeable protons in proteins by 3D solid-state NMR spectroscopy. J Am Chem Soc 125(47):14222–14223. https://doi.org/10.1021/ja037559u
Penzel S, Oss A, Org ML, Samoson A, Bockmann A, Ernst M, Meier BH (2019) Spinning faster: protein NMR at MAS frequencies up to 126kHz. J Biomol NMR 73(1–2):19–29. https://doi.org/10.1007/s10858-018-0219-9
Renault M, Pawsey S, Bos MP, Koers EJ, Nand D, Tommassen-van Boxtel R, Rosay M, Tommassen J, Maas WE, Baldus M (2012) Solid-state NMR spectroscopy on cellular preparations enhanced by dynamic nuclear polarization. Angew Chem Int Ed 51(12):2998–3001. https://doi.org/10.1002/anie.201105984
Rienstra CM, Hohwy M, Hong M, Griffin RG (2000) 2D and 3D N-15-C-13-C-13 NMR chemical shift correlation spectroscopy of solids: assignment of MAS spectra of peptides. J Am Chem Soc 122(44):10979–10990. https://doi.org/10.1021/ja001092v
Safeer A, Kleijburg F, Bahri S, Beriashvili D, Veldhuizen EJA, van Neer J, Tegelaar M, de Cock H, Wösten HAB, Baldus M (2023) Probing cell-surface interactions in fungal cell walls by high-resolution 1H-detected solid-state NMR spectroscopy. Chem Eur J 29(1):e202202616. https://doi.org/10.1002/chem.202202616
Samoson, A. (2021). 0.2 MHz MAS. Experimental Nuclear Magnetic Resonance Conference, Virtual Conference
Sarkar S, Runge B, Russell RW, Movellan KT, Calero D, Zeinalilathori S, Quinn CM, Lu M, Calero G, Gronenborn AM, Polenova T (2022) Atomic-resolution structure of SARS-CoV-2 nucleocapsid protein N-terminal domain. J Am Chem Soc 144(23):10543–10555. https://doi.org/10.1021/jacs.2c03320
Savastano A, Jaipuria G, Andreas L, Mandelkow E, Zweckstetter M (2020) Solid-state NMR investigation of the involvement of the P2 region in tau amyloid fibrils. Sci Rep 10(1):21210. https://doi.org/10.1038/s41598-020-78161-0
Schledorn M, Malar AA, Torosyan A, Penzel S, Klose D, Oss A, Org ML, Wang SS, Lecoq L, Cadalbert R, Samoson A, Bockmann A, Meier BH (2020) Protein NMR spectroscopy at 150 kHz Magic-angle spinning continues to improve resolution and mass sensitivity. ChemBioChem 21(17):2540–2548. https://doi.org/10.1002/cbic.202000341
Shaka AJ, Keeler J, Frenkiel T, Freeman R (1983) An improved sequence for broadband decoupling: WALTZ-16 (1969). J Magn Reson 52(2):335–338. https://doi.org/10.1016/0022-2364(83)90207-X
Shaka AJ, Lee CJ, Pines A (1988) Iterative schemes for bilinear operators; application to spin decoupling (1969). J Magn Reson 77(2):274–293. https://doi.org/10.1016/0022-2364(88)90178-3
Siemer AB (2020) Advances in studying protein disorder with solid-state NMR. Solid State Nucl Magn Reson 106:101643. https://doi.org/10.1016/j.ssnmr.2020.101643
Stanek J, Andreas LB, Jaudzems K, Cala D, Lalli D, Bertarello A, Schubeis T, Akopjana I, Kotelovica S, Tars K, Pica A, Leone S, Picone D, Xu ZQ, Dixon NE, Martinez D, Berbon M, El Mammeri N, Noubhani A, Saupe S, Habenstein B, Loquet A, Pintacuda G (2016) NMR spectroscopic assignment of backbone and side-chain protons in fully protonated proteins: microcrystals, sedimented assemblies, and amyloid fibrils. Angew Chem-Int Ed 55(50):15503–15509. https://doi.org/10.1002/anie.201607084
Thurber KR, Tycko R (2009) Measurement of sample temperatures under magic-angle spinning from the chemical shift and spin-lattice relaxation rate of 79Br in KBr powder. J Magn Reson 196(1):84–87. https://doi.org/10.1016/j.jmr.2008.09.019
Tycko R (2001) Biomolecular solid state NMR: advances in structural methodology and applications to peptide and protein fibrils. Annu Rev Phys Chem 52(1):575–606. https://doi.org/10.1146/annurev.physchem.52.1.575
Xiang S, Pinto C, Baldus M (2022) Divide and conquer: a tailored solid-state NMR approach to study large membrane protein complexes. Angew Chem Int Ed Engl 61(33):e202203319. https://doi.org/10.1002/anie.202203319
Ying J, Delaglio F, Torchia DA, Bax A (2017) Sparse multidimensional iterative lineshape-enhanced (SMILE) reconstruction of both non-uniformly sampled and conventional NMR data. J Biomol NMR 68(2):101–118. https://doi.org/10.1007/s10858-016-0072-7
Zhang S, Li J, Xu Q, Xia W, Tao Y, Shi C, Li D, Xiang S, Liu C (2023) Conformational dynamics of an α-synuclein fibril upon receptor binding revealed by insensitive nuclei enhanced by polarization transfer-based solid-state nuclear magnetic resonance and cryo-electron microscopy. J Am Chem Soc. https://doi.org/10.1021/jacs.2c10854
Zhou DH, Rienstra CM (2008) High-performance solvent suppression for proton detected solid-state NMR. J Magn Reson 192(1):167–172. https://doi.org/10.1016/j.jmr.2008.01.012
Zhou DH, Shah G, Cormos M, Mullen C, Sandoz D, Rienstra CM (2007) Proton-detected solid-state NMR Spectroscopy of fully protonated proteins at 40 kHz magic-angle spinning. J Am Chem Soc 129(38):11791–11801. https://doi.org/10.1021/ja073462m
Zhou DHH, Nieuwkoop AJ, Berthold DA, Comellas G, Sperling LJ, Tang M, Shah GJ, Brea EJ, Lemkau LR, Rienstra CM (2012) Solid-state NMR analysis of membrane proteins and protein aggregates by proton detected spectroscopy. J Biomol NMR 54(3):291–305. https://doi.org/10.1007/s10858-012-9672-z
Zupancic I, Pirs J (1976) Coils Prodicing a magnetic field gradient for diffusion measurements with NMR. J Phys E-Scientific Instrum 9(1):79–80. https://doi.org/10.1088/0022-3735/9/1/025
Acknowledgements
We would like to thank Markus Weingarth, Andrei Gurinov, Johan van der Zwan and Shengqi Xiang for their technical NMR assistance and expertise. We are indebted to Fleur Kleijburg and Han Wösten for providing the labelled S. Commune sample. This work was funded by the Dutch Research Council (NWO, domain Applied and Engineering Sciences: MYCOAT- project number 18425 and a TOP-PUNT grant, number 718.015.001, to M.B.). In addition, this work was funded by the Horizon 2020 program of the European Union (FUNGAR; project 58132), while the high-field NMR experiments were supported by uNMR-NL, the National Roadmap Large-Scale NMR Facility of the Netherlands (NWO grant 184.032.207) and the uNMR-NL grid (NWO grant 184.035.002). This work was presented in part during the 18th EUROMAR at Utrecht, The Netherlands, 2022.
Author information
Authors and Affiliations
Contributions
SB, HvI and MB contributed to study conception and design. Material preparation was performed by AS, AA and HS. Data collection and analysis was performed by SB, AS, and HvI. The first draft of the manuscript was written by SB with contributions by AA, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors report no financial conflict of interest in the publication of this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bahri, S., Safeer, A., Adler, A. et al. 1H-detected characterization of carbon–carbon networks in highly flexible protonated biomolecules using MAS NMR. J Biomol NMR 77, 111–119 (2023). https://doi.org/10.1007/s10858-023-00415-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-023-00415-6