
Abstract
Diatoms are algae producing micro- and nano-structured cell walls mainly containing amorphous silica. The shape and patterning of these cell walls is species-specific. Herein, the biosilica of Cyclotella cryptica, a centric marine diatom with a massive organic matrix, is studied. Solid-state NMR spectroscopy is applied to gain deeper insight into the interactions at the organic–inorganic interface of the cell walls. The various organic compounds like polysaccharides as well as proteins and long-chain polyamines (LCPAs) are detected by observation of heteronuclei like 13C and 15N whereas the silica phase is studied using 29Si NMR spectroscopy. The sensitivity of the NMR experiments is strongly enhanced by isotope-labeling of the diatoms during cultivation with 13C, 15N and 29Si. The presence of two different chitin species in the biosilica is demonstrated. This observation is supported by a monosaccharide analysis of the silica-associated organic matrix where a high amount of glucosamine is found. Moreover, the Rotational Echo Double Resonance (REDOR) experiment provides distance information for heteronuclear spins. 13C{29Si} REDOR experiments reveal proximities between different organic compounds and the silica phase. The closest contacts between silica and organic compounds appear for different signals in the 13C-chemical shift range of 40–60 ppm, the typical range for LCPAs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Diatoms are unicellular algae and frequently studied model organisms in biomineralization research. Their micro- and nano-structured cell walls with species-specific patterning mainly contain amorphous silica [1,2,3,4]. Insights into the biomineralization process, the involved biomolecules as well as the properties of the formed materials are of special interest also for bioinspired materials syntheses because diatoms form minerals under relatively mild conditions. The process of biomineralization is not fully understood until today although various involved compounds could be identified meanwhile [5,6,7,8,9,10]. Apart from the identification of relevant biomolecules, the investigation of the formed mineral phases is interesting for materials science and technology [11, 12]. Studying the organic–inorganic interface of diatom biosilica combines identification of tightly silica-associated biomolecules and determination of their spatial arrangement in the biomineral phase. Solid-state NMR spectroscopy is a powerful method for the investigation of biominerals [13,14,15,16]. The silica phase of the diatom cell walls can be characterized using 29Si solid-state NMR spectroscopy whereas the organic compounds are investigated by 13C and 15N solid-state NMR spectroscopy [17,18,19,20,21]. The sensitivity of the experiments is strongly enhanced by isotope-labeling. This is achieved by diatom growth in isotope-enriched culture medium. Furthermore, the application of distance-sensitive NMR techniques like the Rotational Echo Double Resonance (REDOR) experiment [22] help to gain a deeper insight into the organic–inorganic interface of the biosilica. This solid-state NMR experiment (cf. Figure S6) is used to determine the distance between hetero-spins I and S by the re-introduction of the dipolar coupling, which is usually averaged by the MAS (magic angle spinning) technique [22, 23]. This is achieved by application of π-pulses in the I-spin channel. A spectrum with reduced signal intensity Sr is then acquired and compared with the signal intensity S0 measured without dephasing of I spins. The so-called REDOR fraction (S0–Sr)/S0 = ΔS/S0 depends on the echo time (NTr) where Tr is the rotor period and N the number of rotor cycles within the echo time. The heteronuclear dipolar second moment M2 can be extracted directly from the experimentally determined REDOR curve using the first order approximation, which is especially interesting for materials of unknown spin system geometry like glasses or the amorphous biosilica studied here. This first order approximation can only be used for the initial REDOR curve up to a REDOR fraction of 0.3: [23,24,25].
This calculated second moment contains information about the distance between the heteronuclear spins. Thus, proximity between 13C and 29Si spins of different organic compounds and the silica phase in diatom biosilica can be determined. Herein, the biosilica of Cyclotella cryptica, a centric marine diatom with a massive insoluble organic matrix is studied. The cell wall morphogenesis and some characteristics of silica-associated biomolecules in C. cryptica were studied before [9, 26, 27]. It is well-known, that these algae form extracellular β-chitin fibers, which are supposed to be important for cell separation processes and buoyancy [28,29,30]. Interestingly, recent studies provided first hints for the presence of chitin also in the directly silica-attached organic matrix [27]. The presence of chitin attached to or incorporated into the biosilica was already demonstrated for the diatom Thalassiosira pseudonana, where a chitin-based meshwork was found [31]. The presence of silica-attached chitin in C. cryptica would thus show that the observations made for T. pseudonana are not a peculiarity of this singular species. Moreover, it should be noted that the carbohydrate composition of the silica-attached organic matrix is less well-characterized than the protein composition. The present investigation of C. cryptica biosilica thus serves to answer the following questions: (i) How does the carbohydrate fraction of C. cryptica compare with other diatoms? Is silica-attached chitin part of this carbohydrate fraction? (ii) Does the organic–inorganic interface in C. cryptica also involve LCPAs similar like in the two previously studied species T. pseudonana and Stephanopyxis turris?
2 Results and discussion
The extraction and purification of biosilica from diatom cells is necessary for the selective investigation of the silica and its tightly associated biomolecules. To this end, the studied C. cryptica cells were treated with an SDS/EDTA containing buffer solution. After this lysis buffer treatment, only tightly silica-associated organic material remains. A comparably massive insoluble organic matrix is characteristic for C. cryptica [32]. It makes up about 25–40 wt.-% of the biosilica as could be determined by measuring the mass difference between the purified biosilica and the organic matrix remaining after de-silicification of the cell walls. This amount of organic matrix is up to ten times higher than the values reported previously for other diatom species like S. turris or T. pseudonana [17, 33].
The solid-state 13C cross polarization (CP)/MAS NMR spectrum of isotope-enriched diatom biosilica reveals various, strongly overlapping and relatively broad signals (Fig. 1) because the silica-associated organic matrix is a mixture of various components (see above). Nevertheless, different functional groups can be identified by their characteristic 13C chemical shifts. The intense signal at 175 ppm is due to the carbonyl groups of proteins and carbohydrates. The signals between 120–140 ppm are characteristic for aromatic carbons especially in the aromatic amino acids of silica-attached proteins. An amino acid analysis was performed additionally to characterize the overall composition of biosilica-attached proteins (cf. ESI, Figure S12). Most alkyl signals appear in a chemical shift range of 10–40 ppm. N- and O-alkyl signals are found between 40–90 ppm mainly due to the presence of long-chain polyamines (LCPAs) and carbohydrates. One prominent signal group appears around 105 ± 5 ppm. This region is characteristic for C-1 atoms of polysaccharides including chitin. In general, the recorded spectrum shows similarity to the spectra of biosilica from other diatom species [17]. However, the massive organic matrix of the studied species results in high signal intensities for different organic compounds, and especially for the carbohydrates.

13C CP/MAS NMR spectrum of 13C,15 N,29Si-enriched C. cryptica biosilica. CP contact time: 4 ms. The insert shows a SEM image of one diatom cell
The directly excited 29Si MAS NMR spectrum of isotope-enriched diatom biosilica shows three characteristic signals (Figure S1). They are assigned to Si(OSi)n(OH)4-n moieties with n = 2,3,4, denoted as Qn-groups [34]. The strong signal intensity of Q3- and especially Q4-groups confirms the high condensation state of the silica phase, as it is characteristic for biosilica and other amorphous silica compounds [17]. In addition, a 15 N CP/MAS NMR spectrum was recorded (Figure S2). An intense, broad signal at 120 ppm is assigned to amide nitrogen atoms characteristic for proteins. The signals between 20–60 ppm are due to amines in LCPAs and the nitrogen atoms of lysine side chains.
As also indicated in the above-discussed 13C NMR spectrum, chitin is present in the biosilica extracted from C. cryptica as reported previously [27] similar like in T. pseudonana [31]. The presence of silica-associated chitin was shown in C. cryptica using a GFP (green fluorescent protein)-tagged fusion protein with a chitin-binding domain [27]. To isolate chitin, the biosilica was dissolved and the remaining insoluble organic matrix (Figure S9) was treated with NaOH. This treatment removes the other biomolecules whereas the alkali-resistant chitin remains unchanged. Solid-state NMR spectroscopy was used previously to characterize isolated chitin as well as chitin in biological samples. It is demonstrated in these papers that various chitin structures can be differentiated and characterized by chemical shift differences of the sensitive 13C signal [35,36,37]. The recorded 13C NMR spectrum shows all characteristic NMR signals for chitin (Fig. 2) [38, 39]. Small but significant differences in chemical shift and line width of several signals confirm the presence of two chitin conformations in the sample (Table 1, Figures S3 and S4). The extremely narrow signals and their chemical shifts are characteristic for the highly ordered β-chitin. It is well-known that C. cryptica forms extracellular β-chitin fibers [28,29,30]. Minor amounts of such chitin fibers were indeed found in our samples (Figures S8 and S9) and are probably responsible for the set of narrow 13C NMR signals. In addition, a second set of signals is observed. These signals exhibit clearly different chemical shifts especially for C-1 and C-4 (cf. inserts in Fig. 2). The C-1 signal (around 105 ppm) splits into two signals. The narrow signal at higher chemical shift is due to β-chitin. The broader signal exhibits a lower chemical shift indicative for a conformation similar to α-chitin. Both signals split into doublets due to 13C–13C spin–spin coupling in this isotope-enriched sample (cf. Figure S4). The coupling constant amounts to 44 Hz for both C-1 signals. The C-4 signal splits into a triplet with 36 Hz coupling constant for both chitin conformations. In summary, the second set of signals differs from β-chitin and exhibits chemical shifts and linewidths similar like α-chitin. However, the partial transformation of β-chitin fibers into another conformation by the used extraction procedure can be ruled out [40]. Significantly higher NaOH concentrations and temperatures would be necessary to modify β-chitin. The α-chitin-like set of signals is thus attributed to the silica-associated chitin in analogy to the chitin-based meshworks observed for T. pseudonana previously [31].

13C CP/MAS NMR spectra (contact time: 4 ms) of the 13C-enriched biosilica of C. cryptica (black) and the NaOH-treated insoluble organic matrix (blue). The main signals after treatment are characteristic for chitin. Enlarged sections of C-1 and C-4 signals are shown. The insert shows the chitin structure
Some additional signals with relatively low signal intensity can be observed, e.g., at 34 ppm or 128 ppm. These signals remaining after NaOH treatment indicate biomolecules cross-linked to the chitin, most likely proteins. Such cross-linking between chitin and other biomolecules is well-known in nature and was also found for chitin in different biominerals previously [41,42,43,44].
A monosaccharide analysis was performed to further characterize the carbohydrate composition of the silica-associated organic matrix using gas chromatography coupled with mass spectrometry (GC–MS, Table 2). Note that these monosaccharides will mainly be present as polysaccharides in the native samples. Sample pretreatment for analysis cleaves polysaccharides into the monomer units. Mannose and glucose are the main monosaccharide components in C. cryptica and its insoluble organic matrix. This is similar to previous observations on other diatom species where high amounts of both monosaccharides were found as well [45, 46]. For the isolated insoluble organic matrix, the monosaccharide glucosamine shows a similarly high concentration as mannose and glucose. Note, that N-acetyl-glucosamine, the monosaccharide unit of chitin, is not stable under the chosen hydrolysis conditions. It is de-acetylated and contributes to the glucosamine signal which is the monomer unit of chitosan. To prove the presence of N-acetyl-glucosamine, chitinase from Trichoderma viride is used for bond cleavage of chitin instead of hydrochloric acid. The detection of the N-acetyl-glucosamine as monosaccharide unit of chitin was performed with GC–MS (Figure S10). High amounts of N-acetyl-glucosamine are detected what supports the above-discussed NMR results. This observation is remarkable because significant amounts of glucosamine and N-acetyl-glucosamine are unusual in diatom biosilica and were just found for the diatom species Triceratium dubium before [45].
Moreover, some additional monosaccharides can be observed at high retention times especially for the insoluble organic matrix. These monosaccharides are pyranoses which cannot be assigned to a specific monosaccharide with our detection method and spectra library. Phosphorylation of these monosaccharides as it was observed for S. turris previously [46] can be excluded here by the recorded mass spectra. As an example, one of those mass spectra is shown in Figure S11. The mass spectra show some indication for cross-linking of the monosaccharide components. This meets the NMR results very nicely, where even after NaOH treatment some additional signals confirm the presence of chitin-bound biomolecules different from N-acetyl-glucosamine (cf. Fig. 2).
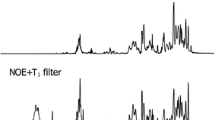
Two-dimensional (2D) NMR experiments can visualize homonuclear as well as heteronuclear correlations. Proton-driven spin-diffusion (PDSD) experiments show 13C–13C correlations between neighboring carbon atoms. PDSD experiments were recorded at mixing times of 30 ms and 120 ms (Fig. 3). The application of a longer mixing time visualizes correlations over longer distances. For example, correlations extended over a whole monosaccharide unit are then observable. In the chemical shift region typical for most proteins, numerous correlations between different amino acids are found. A high amount of proteins is obviously associated to the biosilica. Following the results of the NMR experiments on the insoluble organic matrix, a closer look to the chemical shifts characteristic for carbohydrates clearly shows the presence of the characteristic chitin spin system (e.g. for C-1 at 105 ppm) in the biosilica sample. The occurrence of two signals with only minor differences in the chemical shift for example at the carbon positions C-1 and C-4 confirms the presence of two chitin conformations discussed above (cf. Table 1). Since the presence of a chitin-based meshwork is already known for T. pseudonana, the chitin spin systems for both species are compared in the ESI (Figure S5).

Proton-driven spin-diffusion (PDSD) experiment of 13C,15 N,29Si-enriched C. cryptica biosilica at two mixing times and a spinning speed of 13 kHz. Top: The correlations for the chitin spin system are marked. Bottom: A zoom into the carbohydrate chemical shift region shows the presence of two types of chitin, which is nicely visible for the C-1, C-4, and C-5 signals (correlation signals of the amorphous chitin are marked in pink). Correlation signals at 78 ppm and 83 ppm indicate a cross-link of carbohydrates (marked by green crosses)
Moreover, a correlation between the signals at 83 ppm and 78 ppm is observed which cannot be assigned to a known spin system. However, this correlation within the chemical shift range of carbohydrates supports the possibility of cross-linking between different carbohydrates or other biomolecules as discussed in the chitin spectrum above. In studies of T. pseudonana carbohydrates cross-linked to the protein silaffin-2 were found previously [47, 48]. Taken together, the discussed experiments as well as a comparison to literature show strong indication for carbohydrate cross-linking which may be a topic of future analyses.
Information on possible silica-biomolecule contacts can be obtained from the 13C{29Si} REDOR experiment. Figure 4 displays 13C spectra acquired at 10 ms echo time. One spectrum represents the overall signal intensity S0 while the other spectrum shows the reduced signal intensity Sr acquired with 29Si dephasing. For two signals at 43 ppm and at 56 ppm, remarkable differences between both spectra, i.e., high REDOR fractions are observed. The signal at 43 ppm can be assigned to LCPAs and/or lysine sidechains. The lysine can significantly contribute to this signal especially because we used a CP-transfer from neighboring 1H nuclei [20]. However, inspection of the results of the amino acid analysis (cf. Figure S12) reveals only low amounts of lysine present in the sample in comparison to other amino acids. The relatively low overall amount of lysine gives rise to the assumption that LCPAs should be the major component contributing to this signal. For the signal at 56 ppm, overlap of different signals is likely. The C-2 position of carbohydrates and the C-α atoms of proteins can be found at this shift. However, LCPAs can also contribute to the 56 ppm-signal significantly. This is further confirmed in 15 N–13C correlation experiments which are discussed in a related paper published in the same journal issue. The analysis of the REDOR data for all echo times between 2 and 20 ms yields the REDOR curves shown in Fig. 4 and Figure S7. These curves allow to calculate heteronuclear dipolar second moments (Eq. 1). For the signal at 43 ppm, data up to 10 ms echo time are used for the first-order approximation resulting in a second moment of 1.3·104 s−2. For the signal at 56 ppm, data points up to 14 ms echo time can be used for the first-order approximation providing a second moment of 0.8·104 s−2. These observations indicate a close proximity between 29Si spins and the 13C spins at 43 and 56 ppm. These second moments are smaller but comparable to the values determined for synthetic silica-LCPA composites containing one defined LCPA. A second moment of ca. 2·105 s−2 is then detected [21]. The smaller second moment calculated for the 56 ppm signal is not necessarily due to a weaker dipolar coupling between 13C spins and neighboring 29Si spins. As already mentioned, an overlap of different signals at this chemical shift is very likely. Since some of the corresponding compounds may be far away from silica, this would lead to an underestimation of the calculated second moment. That means, the calculated second moments are probably underestimated for both signals at 43 and 56 ppm.

13C{29Si} REDOR experiments on 13C,15N,29Si-enriched C. cryptica biosilica. Left: 13C spectra at an echo time of 10 ms. Right: REDOR curves and calculated heteronuclear dipolar second moments of the signals at 43 ppm and 56 ppm using the first-order approximation. The error bars for the data points of the REDOR curve were calculated on the basis of the corresponding signal-to-noise ratio and a statistical security of P = 95%
For the amorphous biosilica and its unknown spin system geometry, distance calculation is not possible. However, a comparison with former results shows similarities between the different diatom species. High REDOR fractions and thus a proximity between 13C and 29Si were observed in the same typical chemical shift range of LCPAs for T. pseudonana biosilica [19]. Higher REDOR fractions at smaller echo times were observed for T. pseudonana than for C. cryptica. These differences are probably due to the different organic matrix of both species. The massive organic matrix of C. cryptica biosilica can cause stronger signal overlap and thus lead to a more pronounced underestimation of the heteronuclear dipolar second moment. However, the occurrence of REDOR effects in the same chemical shift range for the different diatom species indicates that the close contact between LCPAs and silica may be a general property of diatom biosilica.
3 Conclusions
Our studies provide insight into the composition and architecture of C. cryptica biosilica and allow to draw the following conclusions:
-
(i)
13C solid-state NMR spectra show the presence of different silica-attached biomolecules, mainly proteins, carbohydrates and LCPAs as it was found for different diatom species before. The observed high signal intensities indicate a high amount of organic matter in comparison to other diatom species. Intense signals are especially observed for carbohydrates.
-
(ii)
The presence of two different types of chitin in biosilica extracted from C. cryptica is shown based on 13C solid-state NMR spectroscopy. These experiments revealed two sets of chitin signals. One signal set with narrower lines is assigned to highly ordered β-chitin which is probably present in the form of remaining external chitin fibers. The signal set with broader lines shows spectroscopic similarity to α-chitin. This “α-like” chitin is assumed to be directly associated to the siliceous cell walls as detected previously in C. cryptica biosilica by fluorescence spectroscopy [27]. However, this is the first time that the structure of this silica-attached chitin is investigated.
The monosaccharide analysis further confirms the presence of silica-attached carbohydrates. The detection of a high amount of N-acetyl-glucosamine, the monosaccharide unit of chitin confirms the presence of chitin in C. cryptica.
-
(iii)
The 13C{29Si} REDOR experiment reveals close silica contact for signals at 13C chemical shifts of ca. 43 and 56 ppm. This is typical for LCPAs. A comparison with former experiments on T. pseudonana biosilica shows similar results. Furthermore, DNP-supported studies of S. turris even concluded that the LCPA in this species should be embedded into the silica phase [20]. Taking further into account that previous studies revealed the presence of LCPAs even in aged oceanic sediments, [49] the embedment of LCPAs into the silica phase can be hypothesized as a general construction principle of diatom cell walls.
4 Methods
4.1 Cultivation of 13C,15N,29Si-enriched and 13C-enriched Cyclotella cryptica
Cultivation of the diatoms was performed in a sealed 20 L plastic container in an artificial seawater (ASW) corresponding to a recipe of the North East Pacific Culture Collection [50]. For isotope-labeling the enriched salts NaH13CO3 (Sigma Aldrich, 98 at.%), Na15NO3 (Sigma Aldrich, 98 at.%) and Na229SiO3 were added. 29Si-enriched Na229SiO3 was prepared using a well-established protocol for a solid phase reaction of 29SiO2 (CortecNet, 99.9 at.%) and Na2CO3 (Grüssing) [19, 51]. The algae grew at a temperature of 18 °C and a day/night-lighting cycle of 12 h/12 h. The pH was set to 8.0–8.2. The cells were harvested by centrifugation.
4.2 Purification steps
4.2.1 SDS/EDTA treatment—preparation of the biosilica sample
The received cell pellet was cleaned using a lysis buffer solution containing 0.1 M EDTA (ethylenediaminetetraacetic acid, isocommerz) and 2 wt-% SDS (sodium dodecyl sulfate, Merck KGaA) at pH 8.0. The suspension was heated to 95 °C for 10 min. Then the cells were centrifuged (2870 g, 10 min) and the supernatant was decanted. The cells were re-suspended in new lysis buffer solution and the procedure was repeated up to 5 times, until the supernatant was nearly colorless and the cell walls were nearly white. The sample was washed three times with ultrapure water and freeze-dried.
The purification results in a removal of a great part of organic matter. Only tightly to the silica cell walls associated organic molecules should be left.
4.2.2 NH4F/HF treatment—preparation of the insoluble organic matrix
The biosilica sample was de-silicified using an 8 M NH4F-solution (VWR) which contains 2 M HF (prepared from 38–40% HF, Merck) [52]. 1 mL of this freshly prepared solution was added per 10 mg biosilica. The suspension was treated in an ultrasonic bath for 20 min. The received insoluble organic matrix was separated using centrifugal concentrators (Vivaspin® 20, 100000 Da, PES). The sample was washed with ultrapure water five times and freeze-dried. The yield varies between 25–40%.
4.2.3 NaOH treatment—preparation of the chitin sample
The insoluble organic matrix was treated with 2.5 M NaOH solution for 48 h at 37 °C. 1 mL NaOH solution was used per 10 mg of the sample. Afterwards the suspension was centrifuged (10600 g, 10 min) and washed with ultrapure water three times. Then the sample was freeze-dried. The yield is around 5%.
4.3 Solid-state NMR experiments
The shown one-dimensional NMR spectra as well as the REDOR experiments were carried out on a Bruker Ascend 800 spectrometer at resonance frequencies of 800.18 MHz for 1H, 201.21 MHz for 13C and 158.97 MHz for 29Si equipped with a 1H-13C–29Si triple resonance probe. The measurements were performed at room temperature. The 13C and 29Si chemical shifts were referenced relative to TMS (tetramethylsilane). The 15 N chemical shift is referenced relative to liquid NH3.
For the 13C{29Si} REDOR experiments, RF (Radio Frequency) field strengths of 120 kHz for 1H, 45 kHz for 13C, and 39 kHz for 29Si were applied. 1H–13C CP (Cross Polarization) transfer was applied before recording the 13C rotational echo with and without 29Si dephasing. A ramp of 70–100% was applied to the proton channel while the RF field on the 13C channel was kept constant at 45 kHz during the initial 1H–13C CP transfer. The contact time was set to 2 ms. Proton decoupling was realized using the SPINAL-64 sequence [53]. The recycle delay was set to 3 s. Experiments with and without dephasing of magnetization were acquired alternating for each echo time. The exact pulse sequence is shown in Figure S6. The spinning speed was set to 17 kHz.
The 2-dimensional PDSD experiments were acquired on a wide bore magnet with a Bruker AVANCE-III console equipped with a 3.2 mm HNC probe using the PARIS [54] (Phase Alternated Recoupling Irradiation Schemes) pulse sequence for proton recoupling during the homonuclear 13C–13C magnetization transfer. The experiment was performed at two different mixing times of 30 ms and 120 ms, at a spinning speed of 13 kHz, and at 270 K.
4.4 Monosaccharide analysis of C. cryptica biosilica and its insoluble organic matrix
The monosaccharide analysis was performed using an established protocol for GC–MS (gas chromatography coupled with mass spectrometry) measurements [46].
4.4.1 Hydrolysis
For the analysis of monosaccharides one sample of diatom biosilica and one sample of the insoluble organic matrix were hydrolyzed. 5 mg of a sample were sealed under vacuum in a glass ampoule after adding 0.7 mL 2 N HCl and 0.1 mL internal standard solution (0.4 mg/mL myo-inositol, Sigma Aldrich, in 2 N HCl). The closed glass ampoules were put into a thermomixer and hydrolysis was performed at 110 °C and shaking of 250 rpm for 2 h. Afterwards the ampoules were opened and the hydrolysates were transferred to centrifuge tubes with 0.2 mL of ultrapure water. The remaining solid components were separated by centrifugation (20800 g, 25 min) and washed with 0.15 mL of ultrapure water two times. The supernatant was collected in a GC vial and HCl was removed at room temperature under vacuum.
4.4.2 Chitinase treatment
1 mL chitinase solution (chitinase from Trichoderma viride, Sigma Aldrich, 1 unit/mL in 0.05 M KH2PO4, pH = 6.0) and 250 µL of the 0.05 M KH2PO4 buffer solution (pH = 6.0) were added to 5 mg of the insoluble organic matrix. The sample was incubated for 43 h at room temperature and 600 rpm. Afterwards, the residue was separated using centrifugal concentrators (Vivaspin® 500, 5000 Da, PES) The supernatant was transferred to a GC vial and freeze-dried.
4.4.3 Derivatization
The hydrolyzed samples and the samples treated with chitinase were derivatized using the trimethylsilyl (TMS)-donor N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA, Merck KGaA). 75 µL dry pyridine and 50 µL MSTFA were added to the samples in the vials under argon atmosphere. For the chitinase treated sample 50 µL of an internal standard solution (2.5 mg myo-inositol in 50 µL MSTFA and 75 µL pyridine, 1 h in an ultrasonic bath at 70 °C, dilution 1:20) were added, additionally. The closed GC vials were treated in an ultrasonic bath at 70 °C for 1 h and the derivatized samples were measured directly afterwards.
4.4.4 GC–MS analysis
For the gas chromatography a capillary column (Supleco SPB-5, 30 m × 25 mm, 0.25 µm film) in an Agilent 6890 N gas chromatograph was used. For detection an Agilent 5973 mass spectrometer with electron impact (EI) ion source was connected. GC separation was performed using a temperature program from 130 °C (1 min) to 195 °C (10 min) to 250 °C (5 min) at a heat rate of 3.5 °C/min and 10 °C/min, respectively. The injector and transfer line temperature were set to 250 °C. A gas flow of 1 mL/min was applied for the carrier gas helium. For each measurement 2 µL were injected with a split ratio of 1:10. The ionization energy of the EI was 70 eV. The mass spectra were acquired with a speed of 2.36 scans/s and with a mass scan ranged from m/z 70 to 700. Mass spectra were detected after a solvent delay of 5 min. The chromatogram was displayed as retention time-dependent reconstructed ion current (RIC). The retention time (tR) and the intensity were normalized to the internal standard myo-inositol. The peaks were assigned by comparing retention times and mass spectra with standard substances and the use of the mass spectra library MassLib [55].
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- 2D:
-
Two-dimensional
- ASW:
-
Artificial seawater
- C. cryptica :
-
Cyclotella cryptica
- CP:
-
Cross polarization
- EDTA:
-
Ethylenediaminetetraacetic acid
- EI:
-
Electron impact
- ESI:
-
Electronic supporting information
- GC:
-
Gas chromatography
- GFP:
-
Green fluorescent protein
- LC:
-
Liquid chromatography
- LCPA:
-
Long-chain polyamine
- MAS:
-
Magic angle spinning
- MS:
-
Mass spectrometry
- MSTFA:
-
N-Methyl-N-(trimethylsilyl)trifluoroacetamide
- NaOH:
-
Sodium hydroxide
- NMR:
-
Nuclear magnetic resonance
- PARIS:
-
Phase alternated recoupling irradiation schemes
- PDSD:
-
Proton-driven spin-diffusion
- PES:
-
Polyester
- Ppm:
-
Parts per million
- RIC:
-
Reconstructed ion current
- REDOR:
-
Rotational echo double resonance
- RF:
-
Radio frequency
- Rpm:
-
Rounds per minute
- S. turris :
-
Stephanopyxis turris
- S 0 :
-
Overall signal intensity
- SDS:
-
Sodium dodecylsulfate
- SEM:
-
Scanning electron microscopy
- S r :
-
Reduced signal intensity
- T. pseudonana :
-
Thalassiosira pseudonana
- TMS:
-
Tetramethylsilane
References
Round FE, Crawford RM, Mann DG. The Diatoms; 1990.
Mann S. Biomineralization. Oxford, New York: Principles and concepts in bioinorganic materials chemistry; Oxford University Press; 2001.
Hildebrand M. Diatoms, biomineralization processes, and genomics. Chem Rev. 2008;108(11):4855–74.
Armbrust EV. The life of diatoms in the world’s oceans. Nature. 2009;459(7244):185–92.
Kröger N, Deutzmann R, Sumper M. Polycationic peptides from diatom biosilica that direct silica nanosphere formation. Science. 1999;286(5442):1127–32.
Kröger N, Lorenz S, Brunner E, Sumper M. Self-Assembly of highly phosphorylated silaffins and their function in biosilica morphogenesis. Science. 2002;298(5593):584–6.
Wenzl S, Hett R, Richthammer P, Sumper M. Silacidins: highly acidic phosphopeptides from diatom shells assist in silica precipitation in vitro. Angew Chem Int Ed. 2008;47(9):1729–32.
Scheffel A, Poulsen N, Shian S, Kröger N. Nanopatterned protein microrings from a diatom that direct silica morphogenesis. Proc Natl Acad Sci U S A. 2011;108(8):3175–80.
Tesson B, Hildebrand M. Dynamics of silica cell wall morphogenesis in the diatom cyclotella cryptica: substructure formation and the role of microfilaments. J Struct Biol. 2010;169(1):62–74.
Sumper M, Kröger N. Silica formation in diatoms: the function of long-chain polyamines and silaffins. J Mater Chem. 2004;14(14):2059–65.
Jeffryes C, Campbell J, Li H, Jiao J, Rorrer G. The potential of diatom nanobiotechnology for applications in solar cells, batteries, and electroluminescent devices. Energy Environ Sci. 2011;4(10):3930–41.
Ragni R, Cicco SR, Vona D, Farinola GM. Multiple routes to smart nanostructured materials from diatom microalgae: a chemical perspective. Adv Mater. 2018;30(19):1–23.
Ben SI, Kababya S, Amitay-Rosen T, Balazs YS, Schmidt A. Molecular level characterization of the inorganic-bioorganic interface by solid state NMR: alanine on a silica surface, a case study. J Phys Chem B. 2010;114(18):5989–96.
Kababya S, Gal A, Kahil K, Weiner S, Addadi L, Schmidt A. Phosphate-water interplay tunes amorphous calcium carbonate metastability: spontaneous phase separation and crystallization vs stabilization viewed by solid state NMR. J Am Chem Soc. 2015;137(2):990–8.
Hu YY, Rawal A, Schmidt-Rohr K. Strongly bound citrate stabilizes the apatite nanocrystals in bone. Proc Natl Acad Sci U S A. 2010;107(52):22425–9.
Roehrich A, Drobny G. Solid-state NMR studies of biomineralization peptides and proteins. Acc Chem Res. 2013;46(9):2136–44.
Gröger C, Lutz K, Brunner E. NMR studies of biomineralisation. Prog Nucl Magn Reson Spectrosc. 2009;54(1):54–68.
Tesson B, Masse S, Laurent G, Maquet J, Livage J, Martin-Jézéquel V, Coradin T. Contribution of multi-nuclear solid state nmr to the characterization of the Thalassiosira pseudonana diatom cell wall. Anal Bioanal Chem. 2008;390(7):1889–98.
Wisser D, Brückner SI, Wisser FM, Althoff-Ospelt G, Getzschmann J, Kaskel S, Brunner E. 1H–13C–29Si triple resonance and REDOR solid-state NMR—a tool to study interactions between biosilica and organic molecules in diatom cell walls. Solid State Nucl Magn Reson. 2015;66–67:33–9.
Jantschke A, Koers E, Mance D, Weingarth M, Brunner E, Baldus M. Insight into the supramolecular architecture of intact diatom biosilica from DNP-supported solid-state NMR spectroscopy. Angew Chem Int Ed. 2015;54(50):15069–73.
Abacilar M, Daus F, Haas C, Brückner SI, Brunner E, Geyer A. Synthesis and NMR analysis of 13C and 15N-labeled long-chain polyamines (LCPAs). RSC Adv. 2016;6(96):93343–8.
Gullion T, Schaefer J. Rotational-echo double-resonance NMR. J Magn Reson. 1989;81:196–200.
Bertmer M, Eckert H. Dephasing of spin echoes by multiple heteronuclear dipolar interactions in rotational echo double resonance NMR experiments. Solid State Nucl Magn Reson. 1999;15(3):139–52.
Bertmer M, Züchner L, Chan JCC, Eckert H. Short and medium range order in sodium aluminoborate glasses. 2. Site connectivities and cation distributions studied by rotational echo double resonance NMR spectroscopy. J Phys Chem B. 2000;104(28):6541–53.
Nizamutdinova A, Uesbeck T, Grammes T, Brauer DS, Van Wüllen L. Structural role of phosphate in metaluminous sodium aluminosilicate glasses as studied by solid state NMR spectroscopy. J Phys Chem B. 2020;124(13):2691–701.
Reimann BEF, Lewin JMC, Guillard RRL. Cyclotella Cryptica, a new brackish-water diatom species. Phycologica. 1963;3(2):75–84.
Pawolski D, Heintze C, Mey I, Steinem C, Kröger N. Reconstituting the formation of hierarchically porous silica patterns using diatom biomolecules. J Struct Biol. 2018;204(1):64–74.
Herth W, Zugenmaier P. Ultrastrucure of the chitin fibrils of the centric diatom cyclotella cryptica. J Ultrastruct Res. 1977;61:230–9.
Herth W. The site of β-chitin fibril formation in centric diatoms II. The chitin-forming cytoplasmic structures. J Ultrastruct Res. 1979;68:16–27.
Morin LG, Smucker RA, Herth W. Effects of two chitin synthesis inhibitors on thalassiosira fluviatilis and cyclotella cryptica. FEMS Microbiol Lett. 1986;37(3):263–8.
Brunner E, Richthammer P, Ehrlich H, Paasch S, Simon P, Ueberlein S, Van Pée KH. Chitin-based organic networks: an integral part of cell wall biosilica in the diatom Thalassiosira pseudonana. Angew Chem Int Ed. 2009a;48(51):9724–7.
Pawolski D. Structures and silica forming properties of insoluble organic matrices from diatoms, thesis, TU Dresden 2018.
Brunner E, Ehrlich H, Schupp P, Hedrich R, Hunoldt S, Kammer M, Machill S, Paasch S, Bazhenov VV, Kurek DV, Arnold T, Brockmann S, Ruhnow M, Born R. Chitin-based scaffolds are an integral part of the skeleton of the marine demosponge ianthella basta. J Struct Biol. 2009;168(3):539–47.
Engelhardt G. Michel D. High-resolution solid-state NMR of silicates and zeolites; 1987.
Kono H. Two-dimensional magic angle spinning NMR investigation of naturally occurring chitins: precise 1H and 13C resonance assignment of α- and β-chitin. Biopolymers. 2004;75(3):255–63.
Chatterjee S, Prados-Rosales R, Itin B, Casadevall A, Stark RE. Solid-state NMR reveals the carbon-based molecular architecture of cryptococcus neoformans fungal eumelanins in the cell wall. J Biol Chem. 2015;290(22):13779–90.
Kang X, Kirui A, Muszyński A, Widanage MCD, Chen A, Azadi P, Wang P, Mentink-Vigier F, Wang T. Molecular architecture of fungal cell walls revealed by solid-state NMR. Nat Commun. 2018;9(1):1–12.
Ehrlich H, Maldonado M, Spindler K-D, Eckert C, Hanke T, Born R, Goebel C, Simon P, Heinemann S, Worch H. First evidence of chitin as a component of the skeletal fibers of marine sponges. Part I. Verongidae (Demospongia: Porifera). J Exp Zool (Mol Dev Evol). 2007;308B:347–56.
Heux L, Brugnerotto J, Desbrières J, Versali MF, Rinaudo M. Solid state NMR for determination of degree of acetylation of chitin and chitosan. Biomacromol. 2000;1(4):746–51.
Noishiki Y, Takami H, Nishiyama Y, Wada M, Okada S, Kuga S. Alkali-induced conversion of β-chitin to α-chitin. Biomacromol. 2003;4(4):896–9.
Schaefer J, Kramer KJ, Garbow JR, Jacob GS, Stejskal EO, Hopkins TL, Speirs RD. Aromatic cross-links in insect cuticle: detection by solid-state 13C and 15N NMR. Science. 1987;235(4793):1200–4.
Vincent JFV, Wegst UGK. Design and mechanical properties of insect cuticle. Arthropod Struct Dev. 2004;33(3):187–99.
Iconomidou VA, Willis JH, Hamodrakas SJ. Unique features of the structural model of “hard” cuticle proteins: implications for chitin-protein interactions and cross-linking in cuticle. Insect Biochem Mol Biol. 2005;35(6):553–60.
Weiss IM, Kaufmann S, Heiland B, Tanaka M. Covalent modification of chitin with silk-derivatives acts as an amphiphilic self-organizing template in nacre biomineralisation. J Struct Biol. 2009;167(1):68–75.
Tesson B, Hildebrand M. Characterization and localization of insoluble organic matrices associated with diatom cell walls: insight into their roles during cell wall formation. PLoS ONE. 2013;8(4):e61675.
Hedrich R, Machill S, Brunner E. Biomineralization in diatoms—phosphorylated saccharides are part of stephanopyxis turris biosilica. Carbohydr Res. 2013;365:52–60.
Poulsen N, Sumper M, Kröger N. Biosilica formation in diatoms: characterization of native silaffin-2 and its role in silica morphogenesis. Proc Natl Acad Sci U S A. 2003;100(21):12075–80.
Poulsen N, Kröger N. Silica morphogenesis by alternative processing of silaffins in the diatom Thalassiosira pseudonana. J Biol Chem. 2004;279(41):42993–9.
Bridoux MC, Ingalls AE. Structural identification of long-chain polyamines associated with diatom biosilica in a southern ocean sediment core. Geochim Cosmochim Acta. 2010;74(14):4044–57.
Harrison PJ, Waters RE, Taylor FJR. A broad spectrum artificial medium for coastal and open ocean phytoplankton. J Phycol. 1980;16(1):28–35.
Spinde K, Pachis K, Antonakaki I, Paasch S, Brunner E, Demadis KD. Influence of polyamines and related macromolecules on silicic acid polycondensation: relevance to “soluble silicon pools”? Chem Mater. 2011;23(21):4676–87.
Brunner E, Richthammer P, Ehrlich H, Paasch S, Simon P, Ueberlein S, Van Pée KH. Chitin-based organic networks: an integral part of cell wall biosilica in the diatom Thalassiosira Pseudonana. Angew Chem Int Ed. 2009b;48(51):9724–7.
Fung BM, Khitrin AK, Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J Magn Reson. 2000;142(1):97–101.
Weingarth M, Demco DE, Bodenhausen G, Tekely P. Improved magnetization transfer in solid-state NMR with fast magic angle spinning. Chem Phys Lett. 2009;469(4–6):342–8.
MassLib V8.7B (MPI Mülheim/Ruhr, Germany).
Acknowledgements
We would further like to thank A. Brünner (TU Dresden) for carrying out the SEM measurements.
Funding
Financial support from the DFG (FOR2038: Nanopatterned Organic Matrices in Biological Silica Mineralization) is gratefully acknowledged. Felicitas Kolbe thanks the infrastructure project I.Next (Horizon 2020 program of the European Union, Grant # 653706) for funding the visit at Utrecht University.
Author information
Authors and Affiliations
Contributions
FK prepared the diatom samples, performed and analyzed the 1-dimensional solid-state NMR and REDOR experiments. HLE and FK performed and analyzed the 2-dimensional solid-state NMR experiments (Proton-Driven Spin-Diffusion, PDSD). SK and FK performed and interpreted the monosaccharide analysis. DB performed the amino acid analysis and LR established the used LC–MS protocol. FK wrote the manuscript mainly. EB and MB supervised the project and data interpretation and participated in manuscript writing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kolbe, F., Ehren, H.L., Kohrs, S. et al. Solid-state NMR spectroscopic studies of 13C,15N,29Si-enriched biosilica from the marine diatom Cyclotella cryptica. Discov Mater 1, 3 (2021). https://doi.org/10.1007/s43939-020-00003-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s43939-020-00003-7