
Abstract
Proton exchange membrane fuel cells (PEMFCs) have demonstrated their viability as a promising candidate for clean energy applications. However, performance of conventional PEMFC electrodes, especially the cathode electrode, suffers from low catalyst utilization and sluggish mass transport due to the randomly distributed components and tortuous transport pathways. Development of alternative architectures in which the electrode structure is controlled across a range of length scales provides a promising path toward overcoming these limitations. Here, we provide a comprehensive review of recent research and development of advanced electrode structures, organized by decreasing length-scale from the millimeter-scale to the nanometer-scale. Specifically, advanced electrode structures are categorized into five unique architectures for specific functions: (1) macro-patterned electrodes for enhanced macro-scale mass transport, (2) micro-patterned electrodes for enhanced micro-scale mass transport, (3) electrospun electrodes with fiber-based morphology for enhanced in-plane proton transport and through-plane O2 transport, (4) enhanced-porosity electrodes for improved oxygen transport through selective inclusion of void space, and (5) catalyst film electrodes for elimination of carbon corrosion and ionomer poisoning. The PEMFC performance results achieved from each alternative electrode structure are presented and tabulated for comparison with conventional electrode architectures. Moreover, analysis of mechanisms by which new electrode structures can improve performance is presented and discussed. Finally, an overview of current limitations and future research needs is presented to guide the development of electrode structures for next generation PEMFCs.
Graphical Abstract
Development of improved electrode architectures with the control of structure on length scales ranging from millimeters to nanometers could enable a new generation of fuel cells with increased performance and reduced cost. This paper presents an in-depth review and critical analysis of recent developments and future outlook on the design of advanced electrode structures.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Our society must rapidly reduce anthropogenic greenhouse gas emissions to avoid a major climate disaster [1]. Transportation-related energy consumption is a principal contributor to global CO2 emissions. According to the U.S. Energy Information Administration, transportation is projected to remain as the dominant source of CO2 emission from the energy sector through 2050, contributing ~ 27% of the total carbon emissions from this sector [2]. An attractive alternative to CO2-producing internal combustion engines is the proton exchange membrane fuel cell (PEMFC), an electrochemical conversion device that produces on-demand electricity with zero local CO2 emissions and typical energy efficiency around 60%. If coupled with hydrogen produced from renewable energy sources (e.g., wind and solar power), PEMFCs could enable a complete decarbonization of the transportation sector. A PEMFC utilizes two half-cell electrochemical reactions, the hydrogen oxidation reaction (HOR) at the anode and the oxygen reduction reaction (ORR) at the cathode, to convert energy stored in hydrogen into electricity, with water as the only product. Specifically, the HOR and ORR are expressed as follows:
The ORR suffers from significantly slower kinetics than the HOR. Furthermore, the higher molecular weight of O2 compared to H2, along with the relatively lower concentration (21% in air), causes O2 transport to catalytically active sites to be significantly slower compared to H2 transport. As a result, PEMFC cathodes typically suffer much higher overpotentials (> 300 mV) compared to anodes (< 10 mV). Therefore, most research work devoted to development of novel PEMFC electrode structures has focused on the cathode [3, 4].
For PEMFCs to become commercially competitive at large scales, PEMFC system cost must be significantly reduced. Specifically, the U.S. Department of Energy (DOE) set the ultimate target cost of PEMFC systems for automotive applications to be $30 kW−1, whereas the current cost is projected to be $45 kW−1 for a production rate of 500 000 systems per year [5]. The largest contributor to the total cost of PEMFC systems stems from the platinum group metal (PGM)-based catalysts used in the electrode; at high volume, PGM catalysts are projected to contribute to 42% of the total stack cost for light-duty vehicles, and 53% for medium- and heavy-duty vehicles [6]. Areal PGM loading targets remain at 0.125 and 0.3 mgPt cm−2 for light-duty and medium-/heavy-duty vehicles, respectively [6]. PGM loadings used in the cathode are typically higher than those in the anode due to the need to overcome the slow ORR kinetics and sluggish O2 transport. Due to the significant cost of PGMs, PEMFC research and development has primarily focused on reducing PGM loading, or even completely eliminating PGMs by replacement with PGM-free catalysts [7,8,9,10,11]. However, to date, PGM-free catalysts still exhibit relatively poor performance and durability, preventing their deployment in PEMFC systems.
Conventional PEMFC electrode structures consist of an ionomer/catalyst/support mixture applied to the fuel cell membrane or diffusion media using wet ink deposition techniques such as ultrasonic spray coating (in the research scale) or slot-die coating (in the manufacturing scale). The random nature of the ink mixing and deposition results in electrodes with limited catalyst utilization, as well as tortuous proton, electron, and oxygen transport pathways, limiting performance. To overcome these challenges and increase power density and efficiency, recent efforts have focused on development of improved electrode structures, enabled by new micro-/nano-fabrication techniques, which could provide mass transport pathways with reduced tortuosity as well as improved catalyst utilization. The implementation of improved electrode structures could reduce the overall costs of PEMFC systems, since achievement of higher power density would enable the use of smaller, less expensive stacks without sacrificing power or efficiency. Furthermore, as fuel cell durability is becoming increasingly important with emerging applications in heavy-duty vehicles [6, 12,13,14,15], the ability of alternative electrode structures to reduce or eliminate key degradation modes may be critical to enabling larger-scale PEMFC deployment. For example, novel design approaches can enable complete elimination of unstable materials (e.g. carbon supports) from the electrode. Overall, the benefits of increased power density and efficiency, reduced cost, and enhanced durability make development of alternative electrode structures a promising path to the wider adoption of PEMFCs.
Recently, excellent reviews have summarized progress on the design of membrane/electrode interfaces [16], nanowire- and nanotube-type electrodes [17], and electrode nanostructure [18, 19]. However, comprehensive analysis and comparison of alternative electrode designs across all relevant length scales (including micro and macro-scale features) are needed to facilitate informed research and enable wider-scale PEMFC deployment. Here, we provide an in-depth review of alternative electrode structures covering all relevant length scales from nanometers to millimeters, including analysis of the advantages and disadvantages of each structure. For the purpose of this review, “electrode” is understood to mean the part of the cathode that includes ORR electrocatalyst, except where indicated otherwise. Alternative gas diffusion layer (GDL) structures are outside the scope of this review; readers are referred to other review works related to the GDL [20,21,22]. The organization of this review is summarized in Fig. 1. Section 2 presents and justifies the need for alternative electrode structures by discussing conventional electrode structures and their limitations. Section 3 reviews and discusses alternative electrode structures reported in literature. We have categorized these electrode structures into the following five types based on their functions and architectures, arranged by the length scale from largest to smallest. (1) Macro-patterned electrodes fabricated by using techniques such as inkjet printing to achieve structural control on length scales ranging from millimeters to hundreds of microns. These electrodes feature selective electrode geometries inspired by flow channel configurations to improve the catalyst utilization and provide effective H2O management. (2) Micro-patterned electrodes produced by using microfabrication techniques such as lithography to achieve structural control on the micron scale by patterning the membrane surface or the electrode. These electrodes feature non-tortuous H+ and/or O2 transport pathways to promote a uniform reaction rate across the electrode thickness. (3) Electrospun electrodes based on fiber morphologies with structural control at the micron to sub-micron scales. These electrodes feature excellent fiber connectivity for enhanced e− and H+ transport and macropores for improved O2 and H2O transport. (4) Enhanced-porosity electrodes characterized by controlled inclusion of macropores (> 50 nm) engineered by using pore formers and other methods of crack/void formation to control structure at the micron to sub-micron scales. These electrodes feature improved O2 and H2O transport enabled by the macropores. (5) Catalyst film electrodes fabricated by using techniques such as chemical and physical vapor deposition and electrodeposition to generate distinctive film-type catalyst morphology with structure controlled at the sub-micron to nanometer scales. These electrodes are carbon- and ionomer-free, enhancing O2 transport and durability. Section 4 outlines our perspective on advantages and disadvantages of each electrode structure and summarizes the remaining challenges and the path forward for commercial application of these alternative PEMFC electrodes. Our discussions are focused on performance improvements at 0.67 V; the U.S DOE power density target of 1.0 W cm−2 at 0.67 V is based on the need for effective heat removal in transportation applications (defined as Q/ΔT = 1.45 kW °C−1)] and an expected maximum operating temperature of 94 °C.

An illustration summarizing the organization of this review, with electrode types arranged clockwise from largest to smallest length scales. The electrode structures are categorized based on their functions and architectures: macro-patterned electrodes for macro-scale mass transport; micro-patterned electrodes for micro-scale mass transport; electrospun electrodes with fiber-based morphology for enhanced in-plane proton transport and through-plane O2 transport; enhanced-porosity electrodes for improved O2 transport through selective inclusion of void space; and catalyst film electrodes for elimination of carbon corrosion and ionomer poisoning. Reprinted with permission from Ref. [23]. Copyright ©2020, American Chemical Society. Reprinted with permission from Ref. [24]. Copyright © 2014, Elsevier. Reprinted with permission from Ref. [25]. Copyright © 2012, IOP Science. Reprinted with permission from Ref. [26]. Copyright © 2013, IOP Science. Reprinted with permission from Ref. [27]. Copyright © 2019, IOP Science. Reprinted with permission from Ref. [28]. Copyright ©2017, Elsevier. Reprinted with permission from Ref. [29]. Copyright © 2014, IOP Science. Reprinted with permission from Ref. [30]. Copyright ©2021, American Chemical Society
2 Conventional Electrodes
2.1 Conventional Electrode Structures
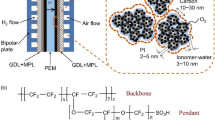
Conventional PEMFC electrodes are typically fabricated by using a catalyst ink that contains a mixture of Pt-based catalyst nanoparticles supported on carbon (Pt/C) and a proton-conducting ionomer (e.g., perfluorosulfonic acid (PFSA)) dispersed in a solvent that typically contains a mixture of water and alcohols (e.g., isopropanol). This ink can be deposited and dried to form an electrode (Fig. 2a, b), as originally demonstrated by Wilson and Gottesfeld [31, 32]. These early works developed the two standard types of electrodes that have now been used in PEMFCs for more than 30 years: (1) the gas diffusion electrode (GDE), in which the catalyst layer coats a GDL [33]; and (2) the catalyst-coated membrane (CCM), in which the catalyst layer coats a PEM, either by direct deposition on the PEM or by transfer from a catalyst-coated decal substrate. Conventional electrodes typically have a thickness of about 5–10 μm, depending on the catalyst composition, loading, and carbon morphology [34, 35]. These electrodes include thin layers of ionomer that bind the electrode and provide proton transport, but that also cover Pt nanoparticles and block larger mesopores and macropores (Fig. 2c, d). Therefore, triple-phase boundaries for both PEMFC electrochemical reactions, ORR and HOR, in a conventional electrode are produced by: (1) high surface area Pt nanoparticles as catalysts, and a carbon support providing electrical conductivity and allowing for high dispersion of the Pt nanoparticles; (2) an ionomer providing pathways for proton conduction; and (3) void spaces for transport of H2, O2, and H2O (Fig. 2b) [36]. Fabrication of conventional electrodes involves the use of catalyst inks, with ionomer-to-catalyst ratios typically ranging from 0.5 to 1.1, to provide proton conductivity and connectivity between the carbon-supported catalyst particles [37, 38]. Ink solvents based on mixtures of isopropanol, water, and other solvents are used to control the ionomer morphology and to enable uniform dispersion and favorable fluid properties for the selected fabrication method. The ink mixture is typically stirred, mixed, or sonicated to uniformly disperse ionomer and break apart large particle agglomerates. The final structure of the conventional electrode is then fabricated by depositing the catalyst ink onto either a PEM, GDL, or decal through painting [39], air spraying [40,41,42], ultrasonic spraying [39], doctor blade [39], screen printing [41], gravure coating [43], slot-die coating [44], Mayer rod coating [45], bar coating [41], and inkjet printing [46].

a Fabrication of conventional electrodes using spray deposition. Reprinted with permission from Ref. [40]. Copyright ©2017, American Chemical Society. b Conventional electrode structure and mass transport pathways. Reprinted with permission from Ref. [51]. Copyright © 2014, Royal Society of Chemistry. Reprinted with permission from Ref. [52]. Copyright © 2006, IOP Science. c Ionomers on a carbon-supported catalyst surface. Reprinted with permission from Ref. [36]. Copyright © 2020, American Association for the Advancement of Science. d Transmission electron microscopy (TEM) image showing the structure of Pt/C/ionomer. Reprinted with permission from Ref. [52]. Copyright © 2006, IOP Science. e Current densities of conventional electrodes at 0.67 V before, during, and after 30 000 catalyst accelerated stress test (AST) cycles between 0.60 and 0.95 V. Reprinted with permission from Ref. [47]. Copyright © 2019, IOP Science
According to the U.S. DOE 2025 targets for light duty vehicle (LDV) applications (Table 1), the PEMFC power density should be greater than 1.0 W cm−2 with a total PGM loading of 0.125 mgPt cm−2; in practice, this loading is likely to be achieved with 0.1 mgPt cm−2 on the cathode and 0.025 mgPt cm−2 on the anode [5]. Technology status reported by General Motors (GM) has demonstrated PEMFC performance approaching or achieving this DOE target (Table 1), with power densities of 0.94 W cm−2 achieved by using PtCo alloys. Moreover, LANL also reported up to 0.84 W cm−2 at 0.67 V under typical operating conditions (H2/air, 80 °C, 100% RH, 150 kPa) using conventional electrodes with a commercial Pt/C catalyst (0.1 mgPt cm−2 at both cathode and anode electrodes) as shown in Fig. 2e [47]. Recently, state-of-the-art power density with conventional electrodes was reported to be up to 1.0 W cm−2 at 0.67 V under typical operating conditions (H2/air, 80 °C, 100% RH, 150 kPa) [48]. While most literature reports on alternative electrode designs have been oriented toward LDV applications, recently growing interest in PEMFCs for heavy duty vehicles (HDVs) has resulted in new challenges and opportunities for design of alternative electrode structures. HDV applications require higher system efficiency and significantly longer lifetime; therefore, HDV membrane electrode assemblies (MEAs) use significantly higher PGM loading, and thus require thicker electrodes, with greater transport challenges. U.S. DOE targets for HDV PEMFCs are listed in Table 2 [6, 49]. Most of the recent works highlighted in this review were focused on LDV applications, so the current state-of-the-art fuel cell performance in LDVs (Table 1) can effectively serve as the starting point and benchmark for next-generation HDVs. Power densities at 0.67 and 0.7 V are mainly summarized and presented in this review, in conformance with the US Department of Energy target metrics for LDV and HDV applications, respectively, which were determined based on system models that simulate drive cycles [49,50,51].
2.2 Challenges Associated with Conventional Electrodes
Substantial efforts have been undertaken to improve conventional electrodes, including development of Pt alloy catalysts [54], tuning of ink formulas [35, 55], and improvement of electrode fabrication methods [19, 56]. However, to date, DOE targets for cost, performance, and durability have yet to be realized in a single system. Current conventional electrodes still require a high total PGM loading ranging from 0.1–0.4 mgPGM cm−2 for both light and heavy duty PEMFCs [5]. Challenges associated with the requirement of high PGM loadings are mainly due to the intrinsic limitations of conventional electrodes, including the random distribution of catalyst and ionomer in catalyst layers, and challenges associated with material properties of the catalyst and support. These challenges can be briefly summarized as: (1) insufficient catalytic activity of carbon-supported Pt-based catalysts [54, 57]; (2) limited catalyst utilization due to the agglomeration of carbon particles and ionomer, and blockage of catalyst in the ionomer and pores of the carbon support [36, 42, 58]; (3) tortuous mass transport pathways and low protonic conductivity resulting from disordered conventional electrode structures [59]; and (4) electrode degradation caused by dissolution and agglomeration/coalescence of Pt nanoparticles as well as oxidation and corrosion of the carbon support [52, 60, 61]. More detailed discussion of these challenges is provided in subsequent sections.
Insufficient ORR activity of conventional Pt-based catalysts is due to strong adsorption of O-containing species, as well as adsorption of ionomer and contaminants. Despite considerable efforts made to develop new Pt-based catalysts with high intrinsic activity [62], these newly developed Pt-based catalysts have not been able to meet performance targets due to low catalyst utilization once incorporated into the fabricated electrode structure [63]. This low catalyst utilization is mainly caused by the agglomeration of ionomer-coated carbon particles, forming 30–300 nm aggregates, often covered by up to 30 nm thick ionomer films that create highly disordered electrode structures (Fig. 2d, Fig. 3) [64]. For reference, pores inside these aggregates and pores between aggregates are named primary and secondary pores, respectively [42]. Consequently, Pt nanoparticles embedded within the carbon support, or without contact with the ionomer (catalyst in primary pores, center Fig. 3), or covered with a thick ionomer film, will lose electron, proton, or oxygen transport paths, limiting the formation of triple-phase boundaries and resulting in low catalyst utilization [42, 59].Additionally, Pt nanoparticles may detach from the carbon support surface during the electrode fabrication processes, causing them to be isolated and electronically disconnected within the ionomer, further reducing catalyst utilization [52]. The combination of a few or all of these challenges in conventional PEMFC electrodes will cause low Pt utilization and require high Pt loadings (0.1–0.4 mg cm−2) to achieve practical performance [65].

Schematic of gas, proton, and electron transport in a conventional electrode. (Top) Qualitative illustration of micro-scale reactant transport. (Bottom-left) Enlarged image of agglomerates coated by ionomer thin-film. The image highlights the active and inactive regions of the agglomerate at high current density. (Bottom-right) The ionomer induced resistance includes a series of ionomer and Pt nanoparticle interface resistances and permeation resistance within the ionomer thin-film
The tortuous and random structure of conventional electrodes can limit O2 and water transport. In PEMFCs, O2 transport barriers can result in significant losses due to the need for reactant O2 to navigate a complicated network of secondary pores, primary pores, and catalyst/ionomer and ionomer/gas interfaces (right Fig. 3) [51, 55]. O2 transport resistance stems from both pressure-dependent limitations associated with gas–gas collisions in secondary pores, and pressure-independent limitations associated with transport through narrow pores, ionomer films, and interfaces [64,65,66]. The local resistance mainly includes four parts (as shown in the rightmost image in Fig. 3): (1) resistance associated with diffusion through small pores to reach the ionomer surface; (2) resistance at the interface between the gas and ionomer; (3) bulk gas permeation resistance through the ionomer film; and (4) resistance at the interface between the ionomer and catalyst surface. Quantification of each local resistance is needed to guide efforts to minimize the overall mass transport resistance [66]. The need for effective water management presents another mass transport challenge in PEMFCs. Insufficient water transport rates can lead to liquid water flooding of the electrode and/or diffusion media, blocking pathways for O2 transport. Water transport into, out of, and within PEMFC electrodes can include: electro-osmotic transport from the PEM to the catalyst layer, back-diffusion from the electrode to the PEM, water evaporation and condensation, and removal of liquid water from the electrode by wicking through the GDL [67]. During PEMFC operation, water flooding is more severe at low operating temperatures and high current densities [67, 68]. Modeling results have shown that the wettability and structure of the primary pores (3–10 nm) favor large reaction and evaporation rates, while secondary pores with a diameter of 10–40 nm play an important role in gaseous transport and in reducing water flooding [67, 69]. Most water transport studies have focused on liquid water transport in PEMs and GDLs, with only a few theoretical studies on water transport within the electrode; better understanding of multiphase water transport within the electrode is urgently needed.
The degradation of carbon-supported PGM catalysts is the major factor controlling the long-term durability of conventional electrodes. Degradation mechanisms, shown in Fig. 4a, mainly comprise 5 processes: Pt dissolution, Ostwald ripening, agglomeration/coalescence, Pt detachment, and carbon corrosion [70]. Pt dissolution mainly affects small Pt nanoparticles due to their higher surface energy [60, 71] Ostwald ripening occurs when dissolved Pt2+ redeposits on other Pt nanoparticles, leading to loss of small Pt particles and growth of large ones (bottom graphs in Fig. 4b), resulting in net loss of catalyst surface area. Another process for Pt surface area loss is the growth of Pt particles due to agglomeration/coalescence. This process stems from migration of Pt particles or corrosion of the carbon support, causing multiple small Pt particles to agglomerate and coalesce into a single large particle [52]. Particle detachment refers to whole Pt nanoparticles detaching from the support, and is mainly caused by weak interaction between the Pt nanoparticles and the support [72]. Finally, the corrosive PEMFC environment and the ability of Pt to catalyze carbon oxidation lead to corrosion of the carbon support, especially during transient operation (startup/shutdown). Support corrosion can rapidly degrade the structural integrity of the electrode (top micrographs in Fig. 4b) [73], leading to local Pt non-uniformity, loss of electrochemical surface area electrochemcial active surface area (ECSA), and collapse of the electrode pore structure [60]. The degradation of conventional Pt/C electrodes under real-world use or accelerated stress testing can lead to more than 70% ECSA loss and more than 80% cell power density loss [60, 74, 75].

a Schematic of degradation mechanisms for conventional electrodes. Reprinted with permission from Ref. [70]. Copyright ©2017, Beilstein Journal of Nanotechnology. b TEM images of Pt/C before (left) and after (center) durability testing (top figures show the carbon support accelerated stress test (AST), and bottom figures show the catalyst AST). Reprinted with permission from Ref. [73]. Copyright ©2014, Elsevier
Overall, conventional PEMFC electrode structures face significant challenges with respect to catalyst utilization, mass transport, and durability. Alternative electrode architectures are urgently needed to address these challenges. Current approaches to design of alternative electrode structures with specific functions focus on improved control of electrode porosity, patterning of electrodes, development of alternative morphologies such as electrospun fibers, and development of catalyst film electrodes. In particular, macro/micro patterned electrodes can enhance macro/micro scale mass transport, respectively; electrospun electrodes with fiber-based morphology and high porosity can improve in-plane proton transport and through-plane O2 transport; enhanced-porosity electrodes can increase oxygen transport through selective inclusion of void space; and catalyst film electrodes can eliminate the problems of carbon corrosion and ionomer poisoning. More details about different alternative electrodes are introduced in the next section.
3 Alternative Electrodes
3.1 Macro-Patterned Electrodes
The function of macro-scale patterned electrodes is to provide an alternative electrode structure designed for optimization of fuel cell performance through improved macro-scale gas transport and water management. These electrodes are characterized by patterning on length scales in the hundreds of microns, with patterns in some cases selected to match the land-channel structure of the flow field. These electrodes were first developed by using inkjet printing via depositing catalyst ink droplets layer-by-layer onto GDLs to give desired shapes and graded distributions of the catalyst [76]. Since then, inkjet printing has become a widely used technique for preparation of macro-scale patterned electrodes with the advantages of low cost, easy scale-up, and precise controllability. Localized inclusion of additives such as hydrophobic polytetrafluoroethylene (PTFE) into parts of the electrode to create patterned hydrophobic and hydrophilic areas has been explored for improvement of water management [77]. Therdthianwong et al. observed some performance improvement using PTFE patterning to create alternating hydrophobic and hydrophilic electrode regions, though an electrode with PTFE used throughout the active area also showed significantly higher performance than the control electrode (PTFE-free Pt/C and PFSA). With the advantages of pico-liter resolution of droplets, Shukla et al. prepared patterned electrodes with sub-millimeter scale pores in the shape of patterned squares, as well as continuous pores shaped as gas flow channels, via inkjet printing. However, fuel cell testing did not show any performance benefit from the electrode patterns [24, 29].
A similar study of inkjet-printed electrodes patterned to include catalyst only under the gas channels was performed by Song and Park [78]. Results from fuel cell testing, along with comparison to spray-coated electrodes covering the entire active area, are shown in Fig. 5. The inkjet-printed electrodes featured serpentine and interdigitated channel shapes following the pattern of the flow field, with the intention of localizing the catalyst in the channel areas where the gas transport is maximized. The authors demonstrated higher performance with the patterned inkjet-printed electrodes compared to a uniform spray-coated electrode; direct comparison to a uniform inkjet-printed control sample is still needed to determine whether the differences in performance stemmed from electrode patterning or from differences between inkjet printing and spray coating.

a–c Schematic illustrations (left and right) and photographs (middle) of three different patterns of electrodes: a square pattern spray coated electrode (SMEA), b gas channel pattern with rib part uncoated (GMEA), and c gas channel pattern with Pt/C on gas channels and bare Vulcan carbon on ribs; d polarization curves of the three above prepared electrodes operated at 70 °C and 100% relative humidity. Reprinted with permission from Ref. [78]. Copyright © 2017, The Chemical Society of Japan
In principle, macro-scale patterning of electrodes could be used to modulate hydrophobicity and porosity for enhanced water management. It could also be used to localize Pt in regions of the electrode where it can be utilized more effectively, enabling higher performance with lower catalyst loading. Although several preliminary studies have attempted to realize these advantages, rigorous demonstration of fuel cell performance enhancement via macro-scale patterning is still needed. Existing studies have been limited by lack of relevant baseline/control samples. In cases where performance enhancement over baseline levels has been reported, the performance has remained below the state of the art. Therefore, further work is needed to establish the value of macro-scale electrode patterning. Notably, much of the work on macro-scale patterning has taken an extreme approach of placing all catalyst under the flowfield channels, while leaving the area under the lands devoid of catalyst; such catalyst-free areas are not expected to provide any contribution to the fuel cell performance. These concerns explain why current macro-patterned electrodes are not able to provide significant improvements compared to conventional electrodes, resulting in fewer researchers working on this topic nowadays. Moreover, extreme cases in which all catalyst is localized under the flowfield channels can lead to counterproductive results, especially in HDV fuel cells with higher catalyst loading, due to the greatly increased electrode thickness in these regions resulting in longer mass transport paths. Investigation of less extreme distributions is recommended to meet the US DOE’s target for HDV fuel cells. In particular, modeling studies would be helpful to guide selection of the optimal apportionment of catalyst between land and channel areas.
3.2 Micro-Patterned Electrodes
Patterned electrodes with ordered structures in the micro-scale could provide improved proton conductivity, better interfacial contact between the catalyst layer and membrane, increased electron conductivity (through a conductive support or the catalyst itself), and faster oxygen and water transport through reduced pore tortuosity. Published reports of micro-patterned electrodes are mainly based on two approaches: (1) patterned membranes and (2) patterned catalyst layers. Micro-patterned electrodes based on patterned membranes can be prepared by modification of a flat ionomer membrane or casting of an ionomer solution on a patterned substrate, followed by catalyst deposition. Micro-patterned electrodes based on patterned catalyst layers can be prepared by using templates to control the catalyst distribution on the membrane surface or by deposition of catalyst nanoparticles on an ordered catalyst support structure.
3.2.1 Patterned Membranes
Modification of ionomer membranes has been studied widely for improving fuel cell performance, including development of various surface modification methods for membrane patterning [76, 79]. Initial research started with roughening the PEM surface by abrasion [80], stamping [81], plasma etching [82, 83], and then advanced into periodic patterns such as line-patterned [84, 85], pillar-patterned [86], and prism-patterned membranes [87]. Most recently, PEM modification methods such as templated pressing, solution casting, pulsed laser micromachining, and lithography have been reported [88, 89]. These methods are covered individually in this section.
Hard template molded ionomer arrays Patterned membranes can be fabricated through stamping of hard templates with desired patterns under high pressure and/or high temperature on the PEM surface to replicate the patterns of the master mold [89, 90]. Templates with tunable pore diameters and thicknesses such as anodic aluminum oxide (AAO) [89, 90] and porous silicon templates [88], stainless steel meshes [91], or prism array molds [92] have been reported in the literature. The parameters associated with these patterns include the line width, pillar length, array density, and aspect ratio, all of which may affect the resulting fuel cell performance.
Illustrative examples of the use of hard templates to control electrode structure via membrane patterning are provided by the work of Cho et al. [93] and Jang et al. [94], who exploited multiplex lithography with creep-assisted imprinting to produce hierarchically patterned PFSA membranes using polyurethane acrylate (PUA) molds (Fig. 6a–b). Spraying of Pt/C catalyst ink onto the patterned membranes yielded MEAs with a catalyst loading of 0.12 mg cm−2. These MEAs produced power densities up to 0.80 W cm−2 at 0.67 V, (80 °C, H2/air, 100% RH, 150 kPa), which was marginally higher than the baseline MEA; larger performance enhancement was observed at lower voltages (Fig. 6c–d), where the micro-scale electrode patterning enabled a maximum power density of 1.04 W cm−2, a 10.7% improvement in performance compared to the baseline MEA. The authors attributed the enhancement to the presence of cone-shaped voids at the electrode surface that provided improved water expulsion, along with improved Pt utilization with the patterned membrane. Effects of membrane patterning on cell performance were also studied by Paul et al. [95], who used thermal embossing to prepare membranes with arrays of micro-scale cylindrical holes. Membrane patterning was observed to produce a decrease in Ohmic resistance of the membrane and electrode, with greater pattern depth providing greater reductions in resistance. However, the decreased membrane thickness under the holes, along with microscopic damage produced during the embossing procedure, was observed to increase gas cross-over and shorting current. In addition, oxygen transport resistance measurements suggested a role of membrane patterning in enhancing water management, with improved pressure-independent resistance observed under some conditions. The improved transport behavior enabled an increase in power density to 0.60 W cm−2 at 0.67 V, compared to 0.49 W cm−2 for a flat baseline MEA (cathode loadings of 0.15 mgPt cm−2, 70 °C, H2/air, 75% RH, 250 kPa).

a Schematic illustration of sequential imprinting to pattern a PFSA film. b Scanning electron microscopy (SEM) images of the resulting patterned PFSA membrane. c and d Polarization curves of MEAs based on patterned and conventional membranes under H2/air and H2/O2 at ambient pressure. Reprinted with permission from Ref. [94]. Copyright ©2016, American Chemical Society
Stainless steel meshes have also been used for patterning of membrane surfaces. Kang et al. [96] used a stainless-steel mesh as a template to introduce mesh patterns on PFSA, yielding a surface area more than double that of a flat membrane. A similar work with mesh-patterned PFSA membranes was reported by Chen et al. [91], which involved hot-pressing a stainless steel mesh to both sides of a PFSA membrane. The authors achieved depths ranging from 30 to 43 μm by controlling the pressing pressure. These works focused on electrode design for direct methanol fuel cells, but in principle, a similar approach could be used to enhance PEMFC performance.
Solution-cast patterned PFSA membranes Patterned PFSA membranes can also be produced from the casting of liquid PFSA solution through a micromolding process (Fig. 7a). In this process, PFSA forms a patterned film derived from the negative of a master mold after evaporation of the solvent [94, 97]. Solution-based methods offer the advantage of avoiding hot-pressing or plasma treatment during the preparation of a patterned membrane, which can protect the patterned membrane from degrading [26, 86, 98,99,100]. Such methods can use either disposable or reusable templates. For instance, Chi et al. [99] prepared a surface-patterned PFSA membrane via solution-casting onto ZnO nanorod templates, followed by template dissolution. These templates offer several advantages, since the ZnO nanorods can be easily fabricated via hydrothermal methods, and the diameter of the rods can be controlled by the precursor concentration. An MEA fabricated from a patterned PFSA film after removal of the ZnO template achieved a power density of 0.44 W cm−2 at 0.67 V (cathode loadings of 0.4 mgPt cm−2, 75 °C, H2/O2, 100% RH, ambient pressure), compared to a power density of 0.32 W cm−2 for a baseline MEA. Similar membrane surface patterning can also be achieved with reusable templates. For instance, Koh et al. [100] utilized reusable poly(dimethyl siloxane) (PDMS) molds to fabricate line-patterned membranes with micron to sub-micron feature size by casting a PFSA solution onto the mold. Electrodes were fabricated on the patterned membranes by spraying with Pt/C catalyst. The resulting patterned electrodes exhibited a significantly higher ECSA (up to 58 m2 g−1) compared to a baseline flat MEA (40 m2 g−1). The power density at 0.67 V increased from 0.52 W cm−2 for the baseline MEA to up to 0.83 W cm−2 for the patterned MEA (cathode loadings of 0.4 mgPt cm−2, 75 °C, H2/O2, 100% RH, ambient pressure). When comparing membranes with micron-scale patterns, performance was found to increase with decreasing feature size, but excessively small (sub-micron) features exhibited lower performance owing to the poor infiltration of the Pt/C/PFSA mixture into the grooved structures. In a follow-up study, Jeon et al. [101] applied the same solution-casting strategy to prepare membrane pillars rather than grooves, with different shapes and sizes (Fig. 7a–b). An MEA with circle-shaped patterns 2 μm in diameter produced the highest power density at 0.67 V of 1.14 W cm−2 (cathode loadings of 0.4 mgPt cm−2, 75 °C, H2/O2, 100% RH, ambient pressure), exceeding the previous work with the line-patterned MEA (Fig. 7c) [100]. The authors attributed the enhanced performance with the circle-shaped pattern to the increased membrane surface area (Fig. 7d–e), which allows shorter proton transport pathways to the reaction sites. Further investigation on the effect of pattern shape on performance (with identical membrane surface area) would strengthen the rationale for selecting circles as the optimal shape. This work presents an attractive method for patterning membranes with morphology control, since reusable PDMS templates can readily be reproduced from casting on the silicon master even if the PDMS templates become worn out and non-usable.

a Schematic illustration of patterned membrane preparation using a PDMS mold and catalyst deposition. b SEM and 3D atomic force microscopy (AFM) images (insets) of patterned membranes in different shapes. The scale bar 10 μm. c Performance of MEAs based on patterned and plain membranes. d Impedance analysis of patterned and plain MEAs at 0.6 V. e Maximum power density and charge transfer resistance of patterned and plain MEAs at various voltages as a function of membrane specific areas. Reprinted with permission from Ref. [101]. Copyright ©2015, Nature publishing group
Direct patterning of membranes In addition to the template-based approaches discussed in the previous sections, direct surface treatments, including plasma and electron beam etching, can also be used to produce patterned membrane surfaces [102,103,104,105]. For instance, Omosebi and Besser [106] employed dry etching with an electron beam on masked PFSA membranes to produce square and line patterns, while Yakovlev et al. [104] used direct sputtering of CeO2 in Ar and O2 atmosphere to produce nanopillar arrays and vertical pores on the membrane surface. Recently, membranes with micro-hole arrays were fabricated by Seol et al. [105] by plasma-etching a PFSA membrane with a patterned PUA mask to make holes of depth 4, 8, and 12 μm (Fig. 8). The power density at 0.67 V was 0.50 W cm−2 for a patterned-membrane MEA with 8 μm hole depth, compared to 0.32 W cm−2 for the baseline (cathode loadings of 0.2 mgPt cm−2, 100% RH, H2/Air, ambient pressure). The performance improvement was attributed to enhanced mass transport and a larger membrane/electrode interfacial area. Interestingly, a deeper pattern of 12 μm resulted in a worse performance, which was attributed to increased crossover affecting the charge transfer kinetics. Further evaluation of the effect of hole spacing on performance would be valuable, since the spacing would also have a strong effect on the membrane/electrode interfacial area.

a SEM image of a 40 μm thick PUA stencil with microholes. b SEM image of a plasma-etched membrane with 4 μm hole depth. c and d SEM images of a plasma-etched membrane spray-coated with PFSA + Pt/C mixture. e HFR-free polarization curves with fully humidified H2/air. f EIS measurements of MEAs with untreated and patterned PFSA membrane at 0.5 V under 70 °C and 100% RH. Reprinted with permission from Ref. [105]. Copyright ©2021, American Chemical Society
3.2.2 Patterned Catalyst Arrays
Template-based catalyst arrays Templating methods can also be applied to pattern the catalyst distribution within an electrode. Such methods can yield electrodes that incorporate a range of shapes including ridges, pillars, holes, and channels. For instance, Paul et al. [23] developed electrodes in which the catalyst was patterned via molding or printing to create hexagonally aligned cylindrical pillars or holes. Poly(dimethylsiloxane) (PDMS) elastomeric templates with hexagonally arranged arrays were used to fabricate patterned electrodes. Each of the patterned electrodes yielded a power density at 0.67 V of 0.50 W cm−2, compared to 0.25 W cm−2 for a baseline electrode (cathode loadings of 0.15 mg cm−2 for patterned and 0.2 mg cm−2 for baseline, 80 °C, 100% RH, H2/O2, ambient pressure). The improvement was attributed to enhanced water removal due to a higher void fraction in the patterned electrodes, as well as enhanced mass activity, which may have been related to the inadequate mass transport in the thick baseline electrode. Both molded and printed electrodes yielded similar performance under relevant operating potentials, but the molded electrodes showed relatively high levels of hydrogen crossover and short-circuiting, possibly due to deformation of the membrane during the fabrication process. At high current densities, the electrodes with cylindrical pillars provided higher performance than the electrodes with holes, which was attributed to the larger void fraction in the electrode with pillars. Further studies are recommended to understand the mechanism of enhanced water removal.
A substrate-assisted decal transfer technique for the fabrication of electrodes with in-plane channels was developed by Lee et al. [107]. Channel-patterned PUA substrates were coated with Pt/C/PFSA catalyst ink via spin-coating, followed by transfer to a PFSA membrane by hot-pressing (Fig. 9a–c). Effects of electrode channels oriented parallel to and perpendicular to the rib/channel structure (Fig. 9b) were investigated. Both channel orientations provided some improvement in performance compared to the baseline electrode (Fig. 9d–e), suggesting a possible effect of enhanced through-plane transport, but larger performance enhancements provided by the perpendicular channel orientation were interpreted as indicating enhanced in-plane oxygen transport, enabling improved utilization of regions of the electrode underneath the ribs. Oxygen gain measurements further supported the proposed enhancement in oxygen transport. Limiting current oxygen transport resistance measurements indicated that enhancements in both molecular diffusion and Knudsen diffusion (due to shorter effective diffusion distances) were responsible for the observed performance increase. While the inclusion of electrode channels would, in principle, be expected to provide enhanced in-plane oxygen transport, clarification of the degree to which in-plane electrode transport helps performance is needed. Specifically, the macroporous GDL substrate would be expected to provide a lower-resistance pathway for oxygen transport under ribs, calling the role of electrode-level channels into question. Analysis of the role of water flooding in both the GDL and electrode is needed to fully assess the significance of in-plane transport.

a Fabrication of channel-patterned electrodes using a PUA template prepared by soft lithography. b Configurations of three different MEAs: the FCL (flat catalyst layer), PA-PCL (channel direction of the CL in parallel with the direction of flow field), and PE-PCL (perpendicular relationship between the channel of the CL and flow field). c SEM images of the patterned CL. Left—the top view, middle—the surface and cross section views. d and e Polarization curves at 50% and 100% RH. Error bars showed voltage fluctuation during galvanostatic measurements. Reprinted with permission from Ref. [107]. Copyright © 2018, American Chemical Society
1D nanostructure support-based catalyst arrays Vertically aligned 1D-nanostructured supports have also been used to provide a framework for heterogeneously-distributed catalyst arrays. Such supports can incorporate a range of 1D materials including carbon nanotubes [108, 109], metal oxide [110] and metal carbide nanorods [111], and conductive polymer nanofibers [113, 114]. Electrode preparation approaches based on 1D-nanostructures share a common procedure: making a one-dimensional array on a substrate as the first step, followed by deposition of catalyst particles onto the nanostructures, and lastly, transfer of the catalyst-coated nanostructures to the membrane to complete the electrode assembly.
Bio-inspired electrode structures with catalyst arrays were designed by Xia et al. [115] using PFSA-decorated polypyrrole (PPy) nanowires as a support for Pt nanoparticles. Briefly, PFSA-decorated PPy nanowires were grown on a GDL through the electrochemical oxidation of pyrrole in a PFSA ionomer dispersion, followed by electrostatic adsorption of Pt cations and reduction in hydrogen gas. PEMFC performance reached power densities of 0.49 and 0.37 W cm−2 at 0.67 V for the catalyst array and a baseline electrode, respectively (cathode loadings of 0.13 and 0.40 mgPt cm−2 for array and baseline, respectively; 70 °C, H2/air, 100% RH, 150 kPa). The improved performance was attributed to enhanced ORR activity and improved proton transport enabled by the sulfonate group clusters surrounding the Pt particles, as well as the rapid oxygen and water transport through the voids between the nanowires. Instead of using conductive PPy as the support, Fu et al. [116] reported a follow-up study using conductive polyaniline and creating catalyst layers by oxidation of the aniline monomer onto the GDL, forming polyaniline (PANI) nanorods, followed by spray coating with Pt black. The power densities at 0.67 V were 0.079 and 0.036 W cm−2 for the array and baseline electrode, respectively (cathode loadings of ~ 0.1 and 0.11 mgPt cm−2 for array and baseline respectively, 70 °C, H2/O2, 100% RH, ambient pressure) [110, 114, 116]. Although these results provide insights into designing 1D conductive support-based electrodes, a deeper understanding of the effects of the 1D structure on electrode performance could aid efforts to optimize the design of these electrodes.
Metal oxides and hybrid materials can also provide 1D nanostructures for patterned catalyst arrays. For instance, Chen et al. developed electrodes based on PANI-coated TiO2 nanowires [117]. While PANI suffers from mechanical instability and TiO2 suffers from poor electronic conductivity, the combination of these materials in a core–shell structure can produce robust and conductive nanowires, while also providing good acid and oxidation resistance. Electrodes based on these PANI-coated TiO2 nanowires loaded with Pt nanoparticles (Pt-TiO2@PANI) provided a power density at 0.67 V of 0.38 W cm−2, compared to 0.34 cm−2 for a baseline electrode (cathode loadings of 0.2 mgPt cm−2, 70 °C, H2/O2, ambient pressure). The higher performance of the Pt-TiO2@PANI electrode was attributed to improved Pt utilization and reduced tortuosity of transport pathways, based on the higher ECSA and lower charge transport resistance. Additionally, durability testing demonstrated lower degradation for the Pt-TiO2@PANI electrode, which still provided 0.23 W cm−2 at 0.67 V after 3 000 cycles between 0.6 and 1.2 V at 50 mV s−1, compared to 0.056 W cm−2 for the baseline electrode.
3.2.3 Summary
Micro-patterned electrodes with carefully designed structures can provide several potential benefits, including increased interfacial area, enhanced Pt utilization, and reduced resistance to proton and oxygen transport. Micro-scale patterning can provide significant benefits compared to the macro-patterned electrodes discussed in the previous section, since reaction and mass transport limitations tend to occur more at the micro-scale than the macro-scale, leading to a more rational way to meet the fuel cell reaction requirements on electrodes. Numerous innovative approaches to micro-scale patterning of the membrane-electrode interface and the catalyst distribution within the electrode have been reported in recent years, including a wide range of direct patterning, templating, and substrate-based techniques. This rational design of electrodes with improved microstructure, in which electrode components are positioned and arranged to maximize catalyst utilization while minimizing transport resistances, represents a promising path toward improved PEMFC performance for HDV applications, in which incorporation of micro-scale mass transport pathways could be crucial in overcoming the larger transport resistances resulting from increased electrode thickness. Several studies reviewed in the previous sections have indeed demonstrated relative performance improvements compared to flat baseline electrodes. However, absolute performance still lags behind that of the best conventional electrodes, highlighting the need for further work to realize the promise of micro-patterned electrodes.
3.3 Electrospun Electrodes
Electrospinning is an attractive technique for the manufacturing of nanofibrous electrodes with enhanced in-plane proton transport and through-plane O2 transport. This technique involves extrusion of a polymer solution from a positively charged syringe tip to a grounded substrate. Electrostatic forces cause ejection of solution from the resulting Taylor cone in the form of a thin jet, which dries to form a nanofiber (Fig. 10a). Since the first use of electrospinning for PEMFC membranes [118, 119] and electrodes [120, 121], numerous electrospun electrode designs reported in the literature have demonstrated competitive performance and durability when compared to conventional PEMFC electrodes [122]. Nanofibrous mats fabricated via electrospinning exhibit a macroporous structure and excellent connectivity of fibers, making them ideal for PEMFC electrode applications. Additionally, electrospinning enables direct control over the electrode structure through modification of various steps of the fabrication process [123].

a Electrospinning apparatus used to fabricate electrospun electrodes. Reprinted with permission from Ref. [122]. Copyright © 2020, Elsevier. b SEM images of an electrospun electrode. Reprinted with permission from Ref. [27]. Copyright © 2019, IOP science. c Performance at beginning of life (BOL) and end of test (EOT) compared to a sprayed electrode. The cell was operated at 80 °C, 100% RH reactant gases (H2 on the anode, air on the cathode), 200 kPa pressure, and 30 000 accelerated stress test cycles from 0.6 to 0.95 V. d 3D reconstruction of scanning transmission electron microscopy (STEM) tomography images of a single nanofiber, where blue particles are PtCo, C and ionomer are gray, and void space is white
A wide range of electrospun polymers have been used for enhanced connectivity to improve electron or proton transport in PEMFCs. For example, polymer precursor solutions such as polyacrylonitrile (PAN), when further pyrolyzed, can create a conductive carbon-support network [113, 120, 123,124,125,126,127]. Alternative electrospinning solutions (e.g., TiO2, NbC, ATO) have been used to create conducting non-carbon-support networks [128,129,130,131,132,133,134]. Ionomer inks have also been electrospun to create proton conductive networks [135,136,137,138,139]. Lastly, ionomer-catalyst inks have been used to directly create electrospun nanofiber electrode mats [27, 28, 121, 140,141,142,143,144,145].
In a conventional electrospinning setup (i.e., extrusion from a single syringe tip onto a rotating drum), operating parameters can be altered to control the resultant electrode structure. For instance, a higher rotation speed of the grounded drum has been shown to produce more aligned nanofibers [146]. Further, the electrospinning apparatus can also be modified to fabricate unique electrode structures. A lot of modified apparatus reported in the literature includes (1) parallel electrode substrates for orthogonal fiber orientation [147], (2) co-electrospinning of two syringes on a single drum for generation of a dual fiber mat [132], (3) co-axial electrospinning to produce hollow nanofibers [127], and (4) co-electrospinning (ionomer ink)-electrospraying (Pt/C ink) on a single drum to independently control ionomer and catalyst content [136, 137].
After electrospinning, the resulting nanofiber mat can be further treated to create an electrode with a desired structure. For instance, the mat can be pulverized to create nanofibers that can be subsequently mixed into a catalyst ink [120, 125, 135]. Additionally, catalyst can be deposited within the nanofibrous mat using techniques including underpotential deposition [148], sputter coating [147], and electroless plating methods [147]. Recently, electrospun mats have also been used as sacrificial templates to fabricate catalyst structures with a nanotrough-like structure [149].
In this section, we discuss three different types of electrospun electrode structures. Firstly, we discuss publications associated with direct electrospinning of electrodes, where an ionomer/catalyst/support ink is electrospun to create a desired structure. These electrodes can be directly used as an electrode with minimal post-processing (e.g., water soak) after the electrospinning process. Secondly, we discuss electrospinning of catalyst supports, in which a carbon-precursor polymer is electrospun and then carbonized, followed by catalyst deposition on the resulting fibrous carbon support. Thirdly, we discuss electrospinning of ionomer structures, which are mainly fabricated to provide an improved proton-conductive network.
3.3.1 Direct Electrospinning of Electrodes
Electrospinning can be used to produce electrodes composed of ionomer (e.g., PFSA) and catalyst (e.g., Pt/C) in a porous and nanofibrous morphology. However, conventional catalyst inks cannot be electrospun directly, since the low degree of polymer chain entanglement in PFSA solutions tends to favor spray of small droplets instead of jet-based spinning [150]. Therefore, electrospinning of catalyst inks typically requires inclusion of a carrier polymer such as poly(acrylic acid) (PAA). Slack et al. [27] developed electrospun electrodes by spinning an ink based on commercial PtCo/C catalyst and PFSA, with PAA as a carrier polymer, using a conventional electrospinning apparatus (Fig. 10a). The resulting fibers had uniform catalyst and binder distribution, and a rough fiber surface with an intra-fiber porosity of 31% (Fig. 10b, d). The diameter of the fibers was 600 nm, which is larger than the agglomerates in a conventional PEMFC electrode (30–300 nm [64]), but high intra-fiber porosity enabled effective gas transport with high mass activity. The authors reported power densities of 0.71 and 0.77 W cm−2 at 0.67 V for electrospun and sprayed electrodes, respectively (cathode loadings of 0.1 mgPt cm−2, H2/Air, 80 °C, 100% RH, 200 kPa). On the other hand, the electrospun electrode achieved a higher maximum power density of 1.05 W cm−2 at 0.55 V, compared to 0.87 W cm−2 at 0.62 V for the sprayed electrode. This difference in power density was attributed to a higher ECSA (48 m2 gPt−1 vs. 44 m2 gPt−1) and lower gas transport resistance (35 s m−1 vs. 52 s m−1). The authors suggested that these properties were due to the high porosity (both in-between and within fibers) and a thinner, more uniform ionomer film covering catalyst particles. Additionally, the large porosity of the produced electrode mats could enable formation of preferential paths for the removal of liquid water within the electrode with lower tortuosity than that of conventional electrodes. The PtCo/C electrode only showed an 8% decrease in maximum power density (compared to the 32% loss observed for sprayed electrodes) after durability testing (Fig. 10c). The increased durability may be at least partially explained by improved Co retention in the PtCo catalyst for the electrospun electrode compared to the sprayed electrode.
Inclusion of carrier polymer is required to enable electrospinning of catalyst inks; in general, some or all of this carrier polymer is expected to remain in the resulting electrode. Therefore, understanding of the role of the carrier polymer is critical to enabling effective design of electrospun electrodes. Kabir et al. [151] used a carrier polymer/ionomer/catalyst-based ink to create electrospun electrodes, but with different carrier polymer concentrations (5%–20% (by weight) PAA). While inks with lower PAA content (10% (by weight) or less) tended to yield uniform distribution of ionomer within the fibers, higher PAA content inks yielded fibers with ionomer-rich coatings, leading to undesired proton transport limitations to the catalyst particles in the ionomer-poor core. A follow-up work reported more details of the PAA interaction with the catalyst [152]. The observed ionomer segregation was attributed to a higher affinity of PAA to the Pt catalyst relative to that of the ionomer; this causes more PAA to coat on the catalyst surface, which leads to the excess ionomer depositing on the exterior of the fiber. The electrode with 10% (by weight) PAA achieved a similar power density of 0.32 W cm−2 at 0.67 V compared to an ultrasonic sprayed electrode without PAA (cathode loadings of 0.05 mgPt cm−2, H2/Air, 80 °C, 100% RH, 200 kPa), but achieved improved performance below 0.67 V (0.48 and 0.37 W cm−2 at 0.6 V). The authors explained the improved performance based on (1) uniform ionomer coverage, (2) macroporous structure, and (3) hydrophilic PAA promoting water retention in the ionomer phase. This work demonstrates the need for improved understanding of how carrier polymers affect fiber and electrode properties, including structure/morphology, water management, proton and oxygen transport, and catalyst performance. In this regard, investigation of how PAA and other carrier polymer additives affect conventional electrodes would help to clarify the effects of carrier polymer and elucidate how the nanofibrous electrospun morphology affects the PAA-catalyst interaction.
While electrospun electrodes can be used in a fuel cell directly after electrospinning, it is also possible to post-process the electrospun electrodes for further modification of the electrode properties. Hong et al. [28] reported the electrospinning of an ink based on Pd/C, PFSA, and PAA, followed by deposition of a Pt skin via replacement of a Cu UPD layer to create a Pd/C@Ptskin core–shell catalyst. While improvements in performance compared to a sprayed Pt/C electrode were reported at low cell voltages (~ 0.4 V), performance was lower than the baseline at voltages relevant for applications (~ 0.7 V), highlighting the need for further development.
Although the electrospun electrodes mentioned here exhibited excellent durability post-AST, additional studies regarding the chemical and mechanical stability are suggested. A recent study by Gao et al. [140] showed that after a durability test performed by using Fenton’s reagents (i.e. soaking the electrode in 15% (by weight) H2O2 with 16 μmol mol−1 FeSO4 at 60 °C), an electrospun electrode containing PAA lost a significant amount of Pt (~ 32%, from 0.13 to 0.09 mgPt cm−2). The authors attributed this loss to PAA nanofibers being prone to radical attack and chemical decomposition, leading to release of Pt/C particles. Additionally, water formation within the pores can create carbon corrosion conditions which could be severer for electrospun electrodes with higher surface areas [142]. Furthermore, a recent work by Slack et al. [145] showed that incorporating polyvinylidene fluoride (PVDF) as a carrier polymer rendered the electrode more resistant to carbon corrosion, owing to the hydrophobic nature of PVDF and its role as a mechanical stabilizer for the nanofibrous structure. These results highlight the need for thorough durability testing, including analysis of stressors for catalyst, support, ionomer, and carrier polymers, when conducting research on electrospun electrodes.
3.3.2 Electrospun Catalyst Supports
While conventional carbon supports suffer from undesired agglomeration and subsequent reduction in Pt utilization, electrospun carbon fiber supports can provide a robust connected electrical conductivity network while maintaining a mechanically stable and mesoporous structure [123]. Additionally, the carbon fiber structure can be controlled by tuning electrospinning parameters that affect the resulting fiber diameter, density, and orientation. Chan et al. [147] reported electrospun carbon nanofibers (CNFs) with two unique fiber orientations, random and orthogonal, to investigate the potential benefits of tuning the nanofibrous carbon support orientation. The authors hypothesized that orthogonally-oriented fibers would feature through-plane pores (i.e. pores in the electrode-thickness direction) that could provide non-tortuous transport pathways. Thus, orthogonally-aligned fibers were fabricated by electrospinning a carbon precursor (polyacrylonitrile co-methylacrylate) with a parallel electrode set-up, illustrated in Fig. 11a. SEM imaging confirmed that the parallel electrode setup successfully produced orthogonal fiber alignment (Fig. 11b). However, the orthogonally oriented CNFs produced worse fuel cell performance than randomly oriented fibers. Specifically, at 0.65 V, the power density of the electrode with orthogonally oriented CNF was 0.54 W cm−2, compared to 0.65 W cm−2 (Pt loading of 0.15 mgPt cm−2 (H2/Air, 65 °C, 105% RH, 150 kPa)). The limiting current density was also lower for the electrode with orthogonally oriented CNF (3.00 A cm−2 vs. 3.19 A cm−2), attributed to its lower porosity. Despite the lower performance of the aligned fibers reported here, the work presents a unique opportunity to understand and potentially enhance CNF support structures through optimizing the fiber orientation.

a Schematic of the electrospinning apparatus for producing orthogonally aligned carbon nanofibers. Reprinted with permission from Ref. [147]. Copyright ©2018, Elsevier. b SEM images of randomly oriented nanofibers (left) and orthogonally oriented nanofibers (right). c SEM image of a pulverized carbon nanofiber-incorporated electrode (top), and polarization curves of electrodes with and without hydrophilic and hydrophobic nanofibers (bottom). The cell was operated at 80 °C, 80% RH H2/air, 150/200 kPa. Reprinted with permission from Ref. [125]. Copyright ©2018, Elsevier; 2017, Elsevier
Rather than using the electrospun carbon mat directly as a catalyst support, Chung et al. [125] pulverized the mat and mixed the CNFs with commercial Pt/C and PFSA to produce an electrode in which CNF hydrophobicity was tuned to control performance. This tuning was accomplished by varying the pyrolysis temperature from 1 000 to 2 500 °C using polyacrylonitrile as the carbon precursor (Fig. 11c), with higher temperature providing increased graphitization and higher hydrophobicity. Electrodes prepared with hydrophobic CNFs pyrolyzed at 2 500 °C exhibited a power density of 0.70 W cm−2 at 0.67 V, compared to 0.59 W cm−2 at 0.67 V for the electrode using hydrophilic CNFs pyrolyzed at 1 000 °C (both with cathode loadings of 0.15 mgPt cm−2, 80 °C, 80% RH, H2/Air, 150/200 kPa) (Fig. 11c). The improved performance was attributed to improved gas transport and creation of water-free regions enabled by the hydrophobic CNFs, similar to the role of PTFE in microporous layers. Another important factor that should be considered is the higher thermal conductivity associated with higher graphitization, which may have also contributed to the improved performance.
3.3.3 Electrospun Ionomer Structures
Electrospun ionomer fibers can provide enhanced proton conductivity due to the interconnected fiber morphology, as well as the presence of anisotropic ionic aggregates that favorably enhance conductivity compared to PFSA films [153]. Such proton conductivity enhancement could enable improvements in PEMFC performance, since proton transport resistance in the electrode is one of the main contributors to performance loss [154]. To fabricate an electrode with uniformly distributed catalyst and ionomer film across the electrospun ionomer network, Hwang and Elabd [136] fabricated nanofibrous electrodes by simultaneously electrospinning ionomer nanofibers and electrospraying a catalyst ink (Pt/C and ionomer), enabling control of the ionomer film thickness around the catalyst particles without compromising the overall porous structure of the electrode. With an optimized ionomer content of 48% (by weight), a maximum power density of 0.57 W cm−2 was achieved with 0.05 mgPt cm−2 (H2/O2, 80 °C, 100% RH, 100 kPa), which was similar to that of a baseline electrode with twice the Pt loading. The combined electrospin-electrospray approach provides a powerful platform to engineer electrode structure, and further examination of how the optimal ionomer content in the electrode changes with the PFSA nanofiber morphology would be informative for future studies.
In addition to combined electrospin-electrospray approaches, electrospun PFSA fibers can also be incorporated into electrodes by pulverizing electrospun fiber mats and dispersing them in a catalyst ink for subsequent deposition. This approach was used by Sun et al. [135] to produce electrodes with hierarchical proton transport pathways based on PFSA nanofibers (Fig. 12a). From SEM characterization, the authors identified ionomer films preferentially covering Pt/C aggregates, while individual nanofibers were uniformly distributed across the electrode (Fig. 12b); these nanofibers provided improved proton conductivity relative to that of a sprayed electrode without electrospun nanofibers (Fig. 12c). This improved proton transport resulted in improved performance compared to the baseline, with a power density of 0.67 W cm−2 at 0.67 V (H2/O2, 70 °C, 100% RH, 100 kPa) (Fig. 12c). Further studies on the trade-off between pore volume provided by the nanofibers and the water retention in the electrode would be valuable, since, intuitively, increased hydrophilic PFSA content is expected to promote water retention and consequently increase gas transport resistance.

a Schematic of the process for fabricating hierarchical proton transport pathways via nanofibrous PFSA. Reprinted with permission from Ref. [135]. Copyright © 2019, American Chemical Society. b SEM image of the resulting mixture of PFSA ionomer, nanofibers, and Pt/C. c The ionic conductivities of the electrode with (NNF) and without (NNI) electrospun PFSA nanofibers (x-axis shows varying PFSA ionomer content) (right) and polarization curves of the cell (left). The cell was operated at 70 °C, 100% RH reactant gases (H2 on the anode, O2 on the cathode), and ambient pressure. d 3D schematic of a rugged electrode achieved by electrospinning PFSA nanofibers onto a PFSA membrane substrate. e SEM image of the electrode. Reprinted with permission from Ref. [139]. Copyright ©2018, Elsevier
Electrospun ionomer has also been used to provide an improved electrode/membrane interface by a direct deposition of PFSA nanofibers onto a membrane, followed by spraying a Pt/C and ionomer ink to produce a rugged PFSA fiber-supported electrode (Fig. 12d, e) [139]. Fuel cell testing showed that this alternative electrode structure provided enhanced performance across the entire current density range compared to a baseline electrode without PFSA nanofibers, with power densities at 0.67 V of 0.29 and 0.25 W cm−2 for the rugged structure and baseline, respectively (both with cathode loadings of 0.15 mgPt cm−2, 80 °C, 80% RH, H2/Air, 180 kPa). The improved performance was attributed to improved oxygen transport within the electrode, enabled by the expanded gas/solid interface provided by the rugged electrode surface, along with enhanced water removal.
3.3.4 Summary
Electrospinning of electrodes and electrode components, as presented in this section, presents a promising method to engineer electrode structure and tune performance and durability characteristics. Electrospun electrodes can provide two major benefits: (1) excellent connectivity of e− and H+ transport pathways enabled by the nanofibrous network, and (2) higher porosity enabling enhanced oxygen and liquid water transport. These benefits, particularly the first, can potentially become key drivers for enhancing the performance of PEMFCs at cell voltages relevant for transportation applications. Electrospun electrodes are particularly promising for HDV applications, since thicker electrodes resulting from higher catalyst loadings will exacerbate mass transport resistances. However, challenges remain, such as the predominantly in-plane fiber orientation suppressing through-plane proton transport and dependence on carrier polymers for the electrospinning process. Thus, further work is needed to evaluate effects of electrode characteristics such as fiber orientation and carrier polymer distribution on performance and durability. In addition, despite the promise, most of the demonstrated work on electrospinning does not provide performance competitive with state-of-the-art PEMFC electrodes. Future work should focus on directly addressing the limitations of electrospun electrodes, such as addition of carrier polymers, to achieve high PEMFC performance.
3.4 Enhanced-Porosity Electrodes
Although conventional PEMFC electrodes are inherently porous, augmentation of electrode porosity can facilitate transport of oxygen to the reaction sites, enabling higher power density and efficiency. In the following sub-sections, we highlight recent advances in electrode porosity enhancement.
3.4.1 Enhanced-Porosity Electrodes via Pore Formers
Macropores can be introduced into the electrode via inclusion of pore-forming agents (pore formers), which are subsequently removed via dissolution or evaporation. Previously reported enhanced-porosity electrodes used pore formers including polystyrene (PS) [155,156,157,158,159], ammonium bicarbonate [160], lithium carbonate [161], ortho-dichlorobenzene [162], and fluorocarbon oil [30]. The resulting pore diameters range from hundreds of nanometers to a few microns, which are larger than the typical average pore diameter of conventional electrodes (i.e., < 100 nm) [163]. The introduced pores tend to have narrow size distribution and spherical morphology when particle-based pore formers are used, and wider size distribution with random morphology when emulsion-based methods are used. These electrodes also become thicker due to the extra pore volume [158], although the extent to which this extra thickness persists after cell assembly and compression will vary based on cell characteristics.
Pore formers based on PS have been widely used to impart porosity in electrodes, as well as in other microfabrication applications. Mixing PS beads into a catalyst ink, followed by ink drying and removal of the PS pore former, can provide enhanced porosity. For instance, Zlotorowicz et al. [157] used monodisperse PS beads to fabricate a porous electrode in which the pore sizes were consistent with the 0.5 μm diameter of the PS beads (Fig. 13a and b). The resulting MEA exhibited a power density of 0.14 W cm−2 at 0.67 V, slightly higher than the 0.11 W cm−2 provided by the baseline MEA made without pore formers, despite having a 60% lower Pt loading than the baseline MEA (H2/Air, 60 °C, 100% RH, 100 kPa). The improved performance was attributed to the enhanced liquid and gas transport enabled by the mixture of meso and macropores, although the physical mechanism of enhanced transport remains unknown. Liquid water is expected to preferentially flow from a smaller (i.e., mesopore) to larger (i.e., macropore) hydrophobic pore when the flow is governed by capillary forces. Thus, liquid water is expected to mainly transport via the macropores, while maintaining the mesopores relatively dry for effective reactant gas transport. Similarly, Kim et al. [159] used monodispersed PS pore-formers, but used them to create a highly ordered catalyst thin film with macroporous structure, which is discussed in Sect. 3.5.1.

a SEM images of modified electrodes via PS beads directly mixed within catalyst ink, before (left) and after dissolution (right). Reprinted with permission from Ref. [157]. Copyright ©2015, Elsevier. b Schematic of the fabrication procedure of a multiscale porous electrode based on a fluorocarbon oil pore former, where a high internal phase emulsion is created to engineer an interconnected porous structure. Reprinted with permission from Ref. [30]. Copyright © 2021, American Chemical Society. c SEM image of the multiscale porous electrode and d its performance (e-CL) relative to the baseline electrode (c–CL)
Emulsion-based approaches to pore formation provide an alternative to the use of solid-phase pore formers, with the advantage of eliminating the sacrificial pore former dissolution step from the electrode processing procedure. For instance, Choi et al. [30] used an emulsion template method (mixture of fluorocarbon oil and catalyst ink) to introduce meso/macropores that only requires one additional step of mixing the oil in the ink (Fig. 13b); from a manufacturing standpoint, this approach may be preferable compared to methods that require pore former removal via dissolution in solvents. When a high internal phase emulsion (emulsion exceeding 0.74 internal phase volume fraction and exhibiting highly viscoelastic flow behavior [164]) is stabilized with a polymer (e.g., ionomer), an interconnected porous structure is formed after evaporation. Choi et al. applied this approach to modify a conventional catalyst ink (with carbon nanofibers for mechanical stability) by mixing with a fluorocarbon oil, followed by deposition and drying to obtain a multiscale porous electrode (Fig. 13c) with a porosity almost double that of a conventional sprayed electrode. At 0.67 V, the power density of the resulting electrode was 0.30 W cm−2 (cathode loadings of 0.1 mgPt cm−2, H2/Air, 65 °C, 100% RH, 180 kPa), which was similar to the baseline electrode fabricated without the emulsion template method (Fig. 13d). However, improvements were observed below 0.67 V, with a maximum power density of 0.76 W cm−2 at 0.4 V (compared to 0.60 W cm−2 at 0.4 V for the baseline). Impedance analysis suggested that the improvement at low voltages resulted from lower mass transport resistance, which was attributed to improved liquid water removal through the macropores. The authors also noted the difficulty in generating reproducible pore size distribution with this technique, since the emulsion properties were highly sensitive to the applied thermal and mechanical energy. The work presents a promising approach of using a simple method to engineer a multiscale porous electrode, which requires further optimization to fabricate reproducible electrode structures with improved performance at voltages higher than 0.67 V.
3.4.2 Enhanced-Porosity Electrodes with Cracks
Intentional crack formation provides an alternative means to create additional electrode pores [92, 165, 166]. Although cracks in electrodes have traditionally been identified as undesired results of degradation induced by carbon corrosion [167] or freeze–thaw cycling [168, 169], recent studies have shed new light on cracks as preferential liquid water removal pathways that could enable increased power density. Cracks can be induced in electrodes via several fabrication methods, such as varying the droplet size during catalyst ink spraying [166], changing the catalyst ink composition [170,171,172], and modifying the ink drying conditions [172]. Cracks formed during electrode fabrication or during degradation can be as wide as tens of microns, and their patterns can also vary depending on the mechanism of crack nucleation and the direction of stress development [170]. Additionally, the cracks can feature straight vertical walls [173] or more random variations in topology [174], depending on how the cracks were introduced.
Recently, Ahn et al. [92] introduced cracks in electrodes through inducing plastic deformation of a prism-patterned membrane via stretching in the direction perpendicular to the pattern (Fig. 14a, b). The prism-patterned membrane promoted preferential nucleation of cracks in the valleys of the prism pattern, in contrast to the randomly generated cracks produced by stretching a flat membrane (Fig. 14c) [165]. Crack size and density were controlled by varying the applied stress and the pitch of the prism pattern, respectively. Environmental scanning electron microscopy observations revealed that water droplets first condensed within the cracks, suggesting that electrode cracks may act as primary water passages during PEMFC operation. The potentially positive role of cracks was further supported by fuel cell testing. The cracked electrode outperformed the baseline electrode with identical loadings of 0.3 mgPt cm−2 at 0.67 V, where the power densities were 0.59 and 0.50 W cm−2, respectively (H2/air, 70 °C, 100% RH, 100 kPa) (Fig. 14d). The cracked electrode also achieved a maximum power density of 0.85 W cm−2. The improved performance was attributed to the lower oxygen transport resistance, based on impedance analysis (Fig. 14d). Further work is recommended to assess how much of the observed performance increase can be attributed to electrode cracks, and how much to the prism-patterned membrane. Notably, previous work by the same group [87] demonstrated that an MEA based on a prism-patterned membrane without electrode cracks had performance close to that of the MEA with electrode cracks. Partitioning the roles of crack and the prism-patterned membrane would provide a deeper understanding of how the cracks lead to improved performance.

a Schematic of electrode fabrication using membrane stretching to produce guided cracks, and b the resulting SEM images of the guided cracks with a strain of 0.5 (left) and 1.0 (right). Reprinted with permission from Ref. [92]. Copyright © 2018, Nature Publishing Group. c Electrodes with cracks without patterned membrane, showing a more random crack formation behavior. Reprinted with permission from Ref. [165]. Copyright © 2016, Nature Publishing Group. d Polarization curve recorded at 70 °C, 100% RH, H2/air, and ambient pressure for a conventional electrode and for cracked electrodes with two different strains (left). Nyquist plot at 1.6 A cm−2 for a conventional electrode and for electrodes with cracks spaced 20 μm (P-20) and 50 μm (P-50) apart (right)
3.4.3 Summary
As evident from the summarized literature, there are a variety of ways to increase the porosity of electrodes; however, all works agree that the main benefit of the enhanced-porosity electrodes is the improved liquid water removal and O2 gas transport. Although electrodes with engineered macropores have demonstrated advantageous transport properties, the commercial applicability of these electrodes remains unclear. Lingering questions associated with the ability of enhanced-porosity electrodes to provide meaningful performance enhancement under commercially-relevant operating conditions, as well as uncertainty over the effect of increased macropore content on durability, remain to be resolved. Most enhanced-porosity electrodes reported in literature provide improvements at low operating voltages, but fail to provide significant advantages at voltages targeted for transportation and other applications (i.e., higher than 0.67 V). Future research should focus on how enhanced-porosity electrodes can enhance performance at higher voltages; examples include: (1) engineering the pore structure to enhance liquid water retention under dry conditions, and (2) providing enhanced O2 diffusion in electrodes that suffer from high O2 transport resistances (such as thick electrodes). Additionally, cracks in electrodes were observed to propagate during carbon corrosion accelerated stress test (AST) cycles due to erosion, leading to electrode failure [167]. Electrode cracks have also been shown to influence crack formation in the membrane during cyclic open circuit voltage AST cycles, leading to membrane failure [175]. Therefore, development of deliberately cracked electrodes must involve careful analysis of the trade-off between desired transport enhancement and undesired durability issues. On the other hand, Kim et al. [165] reported that electrodes with cracks fabricated via stretching of a flat CCM exhibited improved durability during extended polarization curve measurements. This apparently contradictory result highlights the need for further study of the effects of electrode cracks and other macroporous structures on both performance and durability. Durability becomes particularly important for HDV applications, since HDVs require driving lifetime ~ 7.5 times longer than that of LDVs, necessitating careful consideration of the structural design of enhanced-porosity electrodes.
3.5 Catalyst Film Electrodes
Catalyst film electrodes consisting of a thin film of Pt or Pt alloy deposited directly onto a structured PEM or GDL can eliminate carbon corrosion and ionomer poisoning of the catalyst. By removing the dispersed carbon and ionomer phases of the electrode, they provide a simpler interface with fewer degradation modes. They can also enable ultralow catalyst loadings (< 0.05 mg cm−2)
Electrodes based on tubular Pt film structures have been used to achieve lower tortuosity compared to conventional Pt/C electrode structures and to avoid the problem of catalyst agglomeration, resulting in high surface-specific activity [15, 176]. Moreover, the elimination of carbon support and/or ionomer can provide enhanced corrosion resistance and mass transport in catalyst film electrodes [177, 178]. Decreased electrode thickness can also help with decreasing the O2 mass transport resistance and ohmic resistance. Catalyst film deposition typically involves sputtering, but other techniques such as chemical vapor deposition (CVD), electrodeposition, galvanic displacement, and etching methods have also been employed to fabricate structured catalyst film electrodes with or without supports. Based on the type of support structures and materials utilized, catalyst film electrodes can be divided into 4 sub-categories: (1) catalyst film electrodes on flat membranes and GDLs, (2) catalyst film electrodes on structured ionomer-free support, (3) supportless catalyst film electrodes, and (4) catalyst film electrodes on structured ionomer. A typical supportless catalyst film electrode fabrication process is shown in Fig. 15.

Schematic of fabrication process of structured catalyst film electrodes
3.5.1 Catalyst Film Electrodes on Flat Membranes and GDLs
Several attempts to directly deposit Pt or Pt-alloy films on flat membranes or GDLs in order to achieve ultra-low Pt catalyst loadings have been reported [179,180,181]. However, none of them have yet provided a comparable performance to the conventional CCM. Directly depositing Pt on the membrane results in a Pt nanolayer that can break and delaminate from the membrane surface when it swells in water, resulting in poor interfacial integration and low catalyst utilization. The typically low ECSA of such films and the lack of conductive ionomer pathways to parts of the film surface result in high barriers to both O2 and proton transport, leading to poor performance [80, 180]. O’Hayre et al. showed that deposition of Pt thin films on a roughened membrane surface provided improved performance compared to a flat membrane. This improvement was attributed to the surface morphology of Pt thin films, increased Pt surface area, and improved thickness of the catalyst layer [80].
In a related approach, Pt thin films can be deposited by sputtering directly onto the MPL. This approach can provide a low Pt loading (lower than 0.1 mg cm−2) and nanoscale thickness, but with increased surface area, roughness, and volume for mass transport [179, 182]. Sputter deposition of Pt thin films onto both anode and cathode MPLs has been demonstrated to yield functional MEAs with a low Pt loading of only 0.054 mg cm−2, though the reported power density of 0.14 W cm−2 at 0.67 V (H2/O2, 21 °C, 100% RH, ambient pressure) was modest [183]. A similar approach reported by Ostroverkh et al. used simultaneous magnetron sputtering of Pt and C onto the MPL to produce thin films of Pt-doped C [184]. However, the sputtered cathode yielded less than 0.2 W cm−2 at 0.67 V (H2/O2, 70 °C, 425 kPa), while the maximum power density was 0.93 W cm−2.
Low surface area has constrained the performance of most thin film electrodes reported to date. To increase the surface area, Kim et al. [159] fabricated a Pt thin film electrode with ordered macropores via Pt electrodeposition on a self-assembled PS bead substrate (Fig. 16a), followed by PS dissolution in toluene. The resulting 1.5–2 μm thick inverse opal (IO) electrode structure contained 0.12 mg cm−2 Pt loading with Pt wall thickness of 10–15 nm. Fuel cell testing revealed similar performance for the IO electrode and a conventional electrode with similar loading at low current densities, with both yielding a power density of 0.47 W cm−2 at 0.67 V (H2/Air, 80 °C, 100% RH, 150 kPa). However, the IO electrode provided a higher maximum power density of 0.84 W cm−2 (Fig. 16c). The improved performance at the higher current density region was attributed to the favorable electrode structure with low tortuosity, interconnected pores, and large surface area. Additionally, the IO electrode outperformed the conventional electrode in dead-ended operation under ambient humidity (Fig. 16c), which was attributed to improved liquid water transport in the IO electrode.

a Fabrication of a self-assembled inverse opal electrode. b SEM images of the packed polystyrene beads (left) and the electrode surface after dissolution of the beads (right). The scale bars show 2 μm. c Polarization performance for the inverse opal electrode and a conventional electrode. The cells were operated at 80 °C, 100% RH H2/air, 150 kPa (left) and room temperature and ambient humidity, under the dead-ended cathode mode (right). Reprinted with permission from Ref. [159]. Copyright © 2013, Nature Publishing Group
Most studies of catalyst films on GDLs and flat membranes have focused on the anode side due to the challenge of applying this approach to the cathode. These studies have generally produced electrodes with low ECSA, resulting in similar or worse performance compared to conventional electrodes. Use of a substrate with higher surface roughness could address this limitation and enable higher performance. Therefore, development of structured supports that enable higher surface area films appears to be the most promising path to achieve high performance in catalyst thin film electrodes.
3.5.2 Catalyst Film Electrodes on Structured Ionomer-Free Supports
A well-known structured catalyst film electrode was developed by 3M, the nanostructured-thin-film (NSTF) electrode (Fig. 17a) [25, 178, 185]. The NSTF electrode was fabricated from a perylene-based heterocyclic organic solid (PR-149 pigment) deposited using sublimation vapor deposition, followed by heating above 250 °C to produce a disorder-order transition that yields a thin monolayer of crystalline organic whiskers. Pt sputtering was used to catalyze the whiskers, followed by transfer to a PFSA membrane by hot-pressing. The rectangular lath-like whiskers were 30 nm × 55 nm in cross-section, and 300–1 500 nm long with a density of 3–5 billion cm−2. The whole NSTF fabrication process can be performed by using roll-to-roll manufacturing.

NSTF electrodes provide several advantages over conventional Pt/C electrodes, including minimal O2 mass transport resistance as a result of the reduced thickness and absence of ionomer and carbon particles, improved ORR kinetics as a result of the absence of ionomer adsorption on the catalyst surface, short transport distances for protons and electrons, and elimination of support corrosion since the perylene red substrate has higher oxidative stability than conventional carbon supports. These advantages contribute to the high mass activity (> 0.5 A mgPt−1), power density (0.84 W cm−2 at 0.67 V), maximum power density (1.2 W cm−2), and durability (only 14% loss in ECSA after 30 000 catalyst AST cycles between 0.6 and 1.0 V) for a PtCoMn NSTF with Pt loadings of 0.1 mg cm−2 at the cathode and 0.05 mg cm−2 at the anode (H2/air, 80 °C, 67% RH, 150 kPa) [25]. Use of advanced PtNi alloys has been shown to further improve the performance of NSTF electrodes. Cullen et al. linked the nanoscale morphology of PtNi to the surface area and activity in an NSTF electrode [185]. Their study found that PtNi changed from Pt3Ni7 to Pt7Ni3 during testing, and the Ni loss resulted in the formation of nanopores within the NSTF electrode, yielding higher performance.
Although NSTF electrodes with low Pt loadings can provide high performance at elevated temperatures, they suffer from performance limitations at low temperature, since the extremely thin electrode structure is vulnerable to water flooding. This limitation can be mitigated by using anode GDL design strategies for improved water management [186]. Use of GDLs that were compressed to form fiber-density modulations (regions of higher and lower fiber contents), along with a proprietary hydrophobization process, resulted in faster water removal through the anode, enabling 4 times higher current density at 0.5 V and 50 °C. While NSTF electrodes show promising properties, a full understanding of the mechanism of electron and proton transport in this unique electrode architecture remains elusive. Retaining a free-standing structure after transferring to a PEM might be another challenge for NSTF electrodes, because the whiskers are significantly compressed and deformed during the transfer process; the extent to which this deformation affects performance is not clear (Fig. 17b).
Metal oxide nanoarrays (e.g., TiO2 and SnO2), metal nitride nanoarrays (TiN), and SiO2 nanofibers have also been developed to support catalyst thin films for better corrosion resistance and higher surface area; however, these nanostructures may be limited in electrochemical applications by their low electrical conductivity [187,188,189]. To solve this issue, conductive polymer arrays have been used to provide electron transport, but structure optimization for lower loadings and higher ECSAs is still required to improve performance. Also, the low durability of the polymer arrays remains a problem [115, 190]. Jiang et al. developed a GDL with TiN nanorod arrays and electronically conductive polypyrrole (PPy) nanowires, and a thin PtPd catalyst layer [188, 190]. However, these innovative structures yielded only about 0.1 W cm−2 at 0.67 V on H2/air, suggesting that improved fabrication and integration will be required to achieve competitive performance.
Pt/CNTs can provide higher catalyst utilization, enhanced corrosion resistance, and smaller mass transport resistance compared to conventional Pt/C electrodes. However, the disordered stacked carbon nanostructures in a spray-coated Pt/CNT electrode result in reduced electrode conductivity and tortuous mass transport paths. In principle, catalyst film electrodes based on vertically aligned CNTs (VACNTs) could provide improved transport compared to disordered Pt/CNT electrodes. For instance, a novel electrode based on Pt nanorods/aligned CNTs deposited directly on 16 cm2 GDLs achieved 0.45 W cm−2 at 0.67 V (H2/air, 80 °C, 50% RH/30% RH, 230 kPa) as a result of the low thickness and the small proton migration path [191]. During durability testing at high potentials (0.6–1.2 V vs. RHE), this electrode showed a much smaller surface area loss of 41% compared to a 76% decrease for Pt/C. Tian et al. further reported fabrication of VACNTs with 1.3 μm in height and less than 10 nm in diameter using plasma-enhanced chemical vapor deposition (PECVD) on an aluminum foil, with a Pt catalyst layer deposited via sputtering [192]. The high mechanical strength of the VACNTs enabled the electrode to be transferred onto a PFSA membrane without deforming the vertically aligned structure. The resulting MEA achieved a maximum power density of 1.02 W cm−2 and a power density of 0.89 W cm−2 at 0.67 V (H2/O2, 80 °C, 100% RH/72% RH, 200 kPa) with a Pt loading of 0.035 mg cm−2 at both anode and cathode sides. However, the performance of the Pt/VACNTs under air needs to be evaluated to assess the viability of this approach under typical fuel cell operating conditions.
As more ionomer-free thin film electrode structures continue to be invented, development of improved understanding of proton transport along these new types of nano/micro-scale catalyst films is becoming increasingly important. Two mechanisms have been proposed to explain proton transport across a catalyst thin film: (1) adsorbed H atoms diffuse atop the catalyst thin film surface; and (2) protons are transported at the interface between the catalyst and a water film that coats the catalyst [193, 194]. Proton conductivity on an ionomer-free Pt surface with water has been reported to be 1–2 orders of magnitude lower than that of an ionomer-containing electrode, which may explain the lower performance of most catalyst thin film electrodes reported to date [192]. More exploration of methods to improve proton conductivity is needed to fully realize the promise of catalyst thin film electrodes. Issues associated with flooding during low temperature operation, as well as deformation of the electrode during compressing, also remain to be resolved.
3.5.3 Supportless Catalyst Film Electrodes
Recently, considerable effort has been devoted to the development of nanostructured catalyst film electrodes that do not require a support. Most of these electrode structures consist of catalyst nanotubes without any substrate. These types of electrode structures have demonstrated resistance to corrosion and surface area loss. Additionally, well-ordered nanostructures of supportless catalyst film electrodes can be fabricated perpendicular to the plane of the PEM or GDL substrate, providing non-tortuous transport of reactants and products. Lastly, the hollow structure of these nanotubular catalyst films enables the formation of 3-phase boundaries in the inner surface, increasing the available surface area for catalysis.
Among synthesis methods for producing supportless Pt-based nanostructures, wet chemical methods are appealing due to their simplicity, tunability, and potential scalability. For instance, formic acid reduction of H2PtCl6 provides a facile method for growing 1D nanostructures such as nanowires and nanorods [197, 198]. Such structures can be further improved through alloying with other metals. For instance, a Pt-alloy (PtAg–Au) nanorod array with an average length of 20 nm grown directly on a GDL surface provided a peak power density of up to 0.61 W cm–2 and a power density 0.37 W cm−2 at 0.67 V (H2/air, 80 °C, 100% RH, 230 kPa) [197]. This electrode also achieved a low performance loss of 37% via integration of 2% (by atom number) Au, which may be due to the stabilizing role of Au in preventing the surface oxidation and atomic segregation in fuel cells. Thin film electrodes with supportless Pt nanotube arrays have also been prepared by using atomic layer deposition (ALD) and electron beam (e-beam) evaporation of Pt onto AAO templates, followed by transfer to a PFSA membrane and AAO etching (Fig. 18a) [195, 198, 199]. The Pt nanotubes prepared by ALD had a density of 1 billion nanotubes cm−2, an external diameter of 200 nm, and a wall thickness of around 20 nm (Fig. 18a). These tubes had an average length of 2 μm, resulting in a loading of 0.27 mgPt cm−2. Conversely, using e-beam evaporation, the length of Pt nanotubes ranged from 150–200 nm due to the limited Pt transport length in AAO templates, resulting in a Pt loading of 0.1 mgPt cm−2. The high stiffness of the Pt nanotube arrays provided vertically-aligned structures with non-tortuous transport pathways, enabling good tolerance to low RH in fuel cell testing. However, the ECSA (around 20 m2 g−1) and power density of 0.27 W cm−2 at 0.65 V (H2/O2, 80 °C, 0% RH, 150 kPa) were still low. In addition, bundling of the nanotubes was observed (Fig. 18a), potentially impeding transport to the interior of the bundles and reducing Pt utilization. Possible causes of the observed bundling include electrostatic interaction among Pt nanotubes, movement during the etching and rinsing processes, and capillary forces pulling the tubes together during drying, all of which are exacerbated by the high aspect ratio of the nanotubes. To further improve the structure and performance of thin film electrodes with catalyst nanotubes, Galbiati et al. proposed a simple galvanic displacement method to fabricate self-standing PtCu bimetallic nanotube arrays on a PFSA membrane with the help of AAO templates [200]. The resulting PtCu nanotubes had a density of 32 billion nanotubes cm−2, a diameter of 200 nm, a length of 2 μm, and a wall thickness of 12 nm, resulting in an ultra-low catalyst loading of 0.045 mg cm−2 Pt. However, they provided relatively low power density compared to conventional electrodes. The low specific area may have limited the performance during fuel cell evaluation. In addition, the steep decline in voltage with increasing current appears to indicate high ohmic resistance, suggesting the need for further study of proton and electron transport in these unique electrode structures.

a Supportless Pt nanotubes array by atomic layer deposition (ALD). Reprinted with permission from Ref. [195]. Copyright © 2014, Elsevier. b Supportless Pt skin@PdCo nanotube arrays on the GDL. Reprinted with permission from Ref. [148]. Copyright © 2018, Royal Society of Chemistry. c 3D scaffold Pt nanostructure with multiscale Pt nanowires. Reprinted with permission from Ref. [196]. Copyright © 2021, American Association for the Advancement of Science
In contrast to the close-walled nanotube electrode previously mentioned, fabrication of PtCo nanotubes that are split to produce an open-walled geometry has been achieved by using a combination of hydrothermal synthesis, magnetron sputtering, and wet etching [201]. During the etching process, the evolution of H2 resulted in increased pressure within the nanowire core that led to rupture of the PtCo nanotubes, resulting in open-walled structures. An MEA with this electrode architecture achieved an ECSA of 35 m2 g−1, a peak power density of 0.76 W cm−2, and a power density of 0.57 W cm−2 at 0.67 V (H2/O2, 80 °C, 80% RH, 200 kPa). This novel approach was further developed through fabrication of an ultrathin Pt skin on an open-walled PdCo nanotube [148]. The fabricated Ptskin@PdCo nanotubes (Fig. 18b) were vertically aligned on the GDL, with the open-walled structure enabling exposure of both interior and exterior walls. This structure consisting of a 1.3 nm Pt skin coating PdCo nanotubes that were 3 μm in length and 100 nm in diameter. The resulting Pt and Pt + Pd loadings were 0.003 5 and 0.036 mgPGM cm–2, respectively. An MEA containing Ptskin@PdCo nanotubes at both the anode and cathode achieved a power density of 0.70 W cm−2 at 0.67 V (H2/O2, 80 °C, 100% RH, 200 kPa), while also showing improved durability compared to a Pt/C-based MEA. These open-walled nanotube approaches provide a potential advantage over close-walled nanotubes due to the improved accessibility to both sides of the tube, though further work is needed to achieve increased power density.
Most supportless catalyst film electrodes reported to date have relied on vertically aligned nanotube arrays. In contrast, Kim et al. demonstrated a unique electrode structure based on a 3D Pt scaffold (Fig. 18c) [196]. This scaffold was produced by glancing angle deposition of Pt onto nanopatterned polymer substrates to form nanowires, followed by transfer onto a Cu foil using nanotransfer printing. This process was repeated to build a multiscale 3D scaffold Pt nanostructure consisting of alternating layers of narrower and wider Pt nanowires. The resulting MEA provided a power density of 0.30 W cm−2 at 0.67 V (H2/Air, 80 °C, 100% RH, 150 kPa). While this performance is lower than that of conventional Pt/C-based electrodes, it compares favorably with other supportless catalyst film electrodes, which have mainly been tested in H2/O2. Notably, the relatively low ECSA of the 3D scaffold Pt nanostructure (19.7 m2 gPt−1) suggests that strategies to increase ECSA could yield improved performance.
While supportless catalyst film electrodes could in principle provide improved transport, attempts to translate this improved transport into increased performance have thus far had limited success at least in part due to electrode flooding caused by the combination of hydrophilic materials and low electrode volume. Therefore, incorporation of hydrophobic materials or coatings could be investigated to improve performance. Additionally, it is known that proton conductivity on ionomer-free Pt surfaces is 1–2 orders of magnitude lower than that of ionomer-containing electrodes [202, 203]. Thus, enhancing proton transport along supportless catalyst films remains as a challenge. Incorporation of proton-conducting materials could provide a better proton transport path.
3.5.4 Catalyst Film Electrode on Structured Ionomer
High performing PEMFCs electrodes typically require the presence of triple-phase boundaries (Pt, ionomers, voids) to enable effective transport to and from catalytically active sites. Notably, many of the catalyst thin film electrodes reviewed in the previous sections have omitted the ionomer component, and have instead relied on relatively slow proton transport along the catalyst surface or through adsorbed water films, resulting in relatively low performance. Structured ionomer supports could address this problem by providing effective proton transport paths between the catalyst film and the PEM, enlarging triple-phase boundaries, and providing improved performance compared to other types of catalyst film electrodes [202, 203]. Electron beam lithography, laser or plasma etching, abrasion, or imprinting techniques used to produce patterned PEMs to support the catalyst film electrode are discussed in this section.
Several template-based approaches have been used to create structured ionomer supports. In one such approach, Babu et al. used track-etched polycarbonate as a sacrificial template to produce a vertically-aligned PFSA nanofiber array, which was subsequently coated with a Pt nanofilm by CVD [176]. The resulting ionomer-supported porous Pt films had high ECSA (55 m2 g−1) and were rich in microcracks, providing pathways for transport of oxygen and protons (Fig. 19a). Despite incorporating a high Pt loading of 0.44 mgPt cm−2, the resulting MEA provided only 0.090 W cm−2 at 0.67 V (H2/air, 80 ºC, 100% RH, 150 kPa). The electrode performance may have suffered from excessive Pt film thickness (more than 100 nm), along with non-uniform PFSA nanofiber distribution.

a Schematic of species transport in a catalyst film electrode with structured ionomer-containing support. Reprinted with permission from Ref. [176]. Copyright © 2015, Wiley. b Selectively plasma etched PFSA membrane for catalyst film electrodes. Reprinted with permission from Ref. [104]. Copyright © 2019, Elsevier. c Catalyst film electrode on PFSA support with three fast pathways for mass, electron and proton transfer, based on a highly dense, well-ordered, and cone-shaped PFSA array. Reprinted with permission from Ref. [90]. Copyright © 2020, Royal Society of Chemistry
Structured ionomer supports can also be produced by using plasma etching, as reported by Omosebi and Besser [103], who etched PFSA 212 membranes in an O2 plasma to form nanopillars with characteristic roughness of 140 nm. Sputtering the coating of a 10 nm Pt layer onto the PFSA nanopillars resulted in an electrode with a loading of 0.020 mgPt cm−2. This catalyst film electrode yielded a low power density of 0.062 W cm−2 at 0.67 V (H2/ O2, 60 ºC, 100% RH, 100 kPa), but high mass-specific density of 8.5 W mg−2, due to the ultra-low Pt loading. However, the resulting roughness and fiber length of the electrode structure is too low for realistic applications. To solve this problem, Yakovlev et al. developed a two-step process to fabricate a catalyst film electrode on a selectively plasma etched membrane using the CeO2 nuclei as a protective mask to increase roughness and fiber length [104]. Figure 19b displays the resulting array of vertically-aligned PFSA nanopillars with lengths of 500 nm (the left figure) or 3 μm (the right figure). After Pt sputter coating, the 500-nm-high PFSA nanoarrays resulted in an electrode with Pt loading of 0.032 mgPt cm−2, which yielded an improved power density of 0.34 W cm−2 at 0.67 V (H2/O2, 80 ºC, 100% RH, 150 kPa). Achievement of higher power density may require addressing the low Pt loading and discontinuous Pt film, which could limit electrical connectivity and reduce Pt utilization.
Use of templates can provide similar PFSA nanopillar geometries. For instance, Ning et al. developed a cone-shaped PFSA array catalyst film electrode for use as an anode (Fig. 19c) [90]. Arrays of cone-shaped PFSA pillars with a diameter of 400 nm and a length ranging from 0.5 to 2.5 μm were produced by using AAO templates, followed by sputtering a thin Pt layer and depositing a slurry of graphene nanosheets for increased electronic conductivity (Fig. 19c). A freeze-drying method was developed to address the problem of collapse of cone pillars [204]. The resulting MEAs showed improved performance compared to an MEA with a Pt anode sputtered directly on the membrane. Komini Babu et al. [205, 206] recently used AAO templates with Pt ALD coating to develop a coaxial nanowire electrode (CANE) comprising ionomer nanowires wrapped in a uniform Pt film. The nanowires had an aspect ratio of more than 25. During AST, the CANE showed an ECSA loss of 20% after 30 000 voltage cycles, demonstrating excellent durability. Furthermore, the high proton conductivity from membrane to the catalyst surface was also enhanced. The uniform Pt catalyst film provided a good connection between Pt clusters, leading to high electronic conductivity, and resulting in high catalyst utilization. Templates other than AAO, such as laser-ablated stainless steel templates [207], can also be used to create PFSA pillars to fabricate a catalyst film electrode with a similar geometry.
3.5.5 Summary
In the recent works presented here, catalyst film electrodes have been fabricated by using uniform Pt and Pt alloy nanofilms instead of nanoparticles, with mass and charge transport occurring through several mechanisms: (1) adsorbed H atoms diffuse atop the catalyst thin film surface, while protons are transported in the ionomer support or at the interface between the catalyst and a water film that coats the catalyst; (2) O2 is transported through pore space between nanofilms; and (3) electrons are transported along the conductive structured support or catalyst layer itself. Because the electrodes are typically free of carbon supports, and in some cases are ionomer-free, these electrodes can eliminate several performance and durability limitations associated with conventional Pt/C based electrodes, including carbon support corrosion, Pt nanoparticle agglomeration, and blockage of mass transport by ionomer, potentially enabling improved durability and mass transport. Considering the shift in focus from LDVs to HDVs, catalyst film electrodes provide a promising path to achieve the requisite ultrahigh durability. However, further efforts are required to achieve practical catalyst film electrodes that realize these potential benefits in high-performance and durable MEAs. In particular, better understanding of proton transport along ionomer-free Pt thin films, and development of techniques to increase proton conductivity, are needed. Catalyst film electrodes typically have low electrode volume and high hydrophilicity, making them vulnerable to water flooding. Modified film architectures that enable greater electrode thickness are needed to address this issue, along with development of hydrophobic materials or coatings that can facilitate water removal from the electrode, especially for HDV fuel cell applications with high catalyst loading, high water generation rates, and long mass transport pathways. The lack of a conductive carbon support means that most catalyst film electrode approaches have relied on the Pt catalyst to provide electronic conductivity, which can result in electronic conductivity limitations in electrodes with ultralow Pt loading and discontinuous Pt films. Development of inexpensive and durable conductive sublayers for beneath the Pt films may be considered to improve the surface continuity and electrical conductivity. Finally, retention of the open electrode structure after transfer to the membrane is also a challenge for catalyst film electrodes due to compression and crushing in the cell assembly process. Development of more robust structures is needed to reduce this deformation and maintain open architectures for effective mass transport.
4 Conclusions and Outlook
4.1 Conclusions
In this review, we have presented recent research and development on advanced electrode architectures for improved PEMFC performance. Five different categories of alternative electrodes were analyzed based on their functions and architectures: (i) macro-patterned electrodes, (ii) micro-patterned electrodes, (iii) electrospun electrodes, (iv) enhanced-porosity electrodes, and (v) catalyst film electrodes. Their working mechanisms are summarized in Fig. 20a. In principle, each of these alternative electrode architectures can provide improved transport and higher performance. Macro-patterned electrodes can more efficiently distribute catalyst to the regions where transport is most facile (e.g., under flow-field channels), while also enabling improved water management through manipulation of local hydrophobicity to facilitate liquid water expulsion. These characteristics can lead to decreased oxygen transport resistance, resulting in higher performance particularly at low voltages. Micro-patterned electrodes can provide ordered structures with larger interfacial areas, while facilitating proton and oxygen transport via improved distribution of ionomer. Decreases in both proton and oxygen transport resistances can significantly enhance the performance at high current density. Enhanced-porosity electrodes can similarly improve oxygen transport through selective inclusion of void space (resulting in enhanced low voltage operation), while electrospun electrodes provide high porosity along with favorable ionomer connectivity, enabling enhanced transport of oxygen and protons (and higher performance under high current conditions). Catalyst film electrodes can reduce effective transport distances by enabling reduced electrode thickness (albeit at the cost of higher vulnerability to flooding), while also providing a support-free catalyst geometry that eliminates degradation mechanisms associated with carbon corrosion and Pt nanoparticle coalescence. Use of ionomer-free catalyst film electrodes can also eliminate issues associated with oxygen transport through the ionomer, potentially enabling design of PEMFCs with improved performance and durability.

a Working mechanisms of alternative electrodes. b Summary of power density at 0.67 V of alternative electrodes compared to conventional electrodes. The black line and red line indicate the DOE power density target of 1.0 W cm−2 with H2/air, and the targeted cathode loadings of 0.100 mg cm–2 (which is required to achieve the DOE total Pt loadings target of 0.125 mg cm–2), respectively
The performance of PEMFCs based on novel electrode structures is summarized in Table 3, along with selected conventional electrodes for comparison. From this table, a graphical representation of PEMFC performance (power density at 0.67 V) versus cathode Pt loading was extracted and is shown in Fig. 20b. The DOE power density target of 1.0 W cm−2 at 0.67 V is based on the need for effective heat removal in transportation applications and an expected maximum operating temperature of 94 °C. While all five types of alternative electrode structures have attributes that, in principle, should enable enhanced performance, these electrodes have yet to demonstrate performance higher than that of the best conventional electrodes, as illustrated in Fig. 20b. This surprising observation leads to an obvious question: if alternative electrode structures possess clear advantages in principle, then why are these advantages not realized in practice? Our answer to this question is twofold:
-
1.
Design, fabrication, and testing of MEAs are hard. Relatively few research groups have demonstrated competitive MEA performance with conventional electrodes. Use of complicated alternative structures leads to additional MEA integration challenges, making achievement of competitive performance even more difficult.
-
2.
Conventional electrodes are already highly optimized. Automotive companies, MEA developers, and the research community have invested billions of dollars during decades of R&D on electrode optimization. While development of alternative electrodes could produce new architectures capable of outperforming conventional electrodes, one should not expect this task to be easy.
Most reports of novel electrode structures covered in this review include comparison to an in-house baseline electrode, and in most cases, the novel electrode structure provided higher performance than the baseline. Therefore, there is reason for optimism, and substantial evidence that alternative electrode structures can provide a path toward advancing the state of the art and enabling faster deployment of PEMFCs in clean energy applications. While these improvements in relative performance are promising, advancement to the next stage of development requires demonstration of absolute performance improvements. Notably, most in-house baseline MEAs have exhibited performance below that of the state-of-the-art conventional electrodes included in Fig. 20b. Therefore, our advice to groups pursuing R&D on novel electrode structures is that they should develop and validate their MEA fabrication and testing procedures using conventional electrodes. Research groups that can achieve competitive performance with conventional MEA technology will have a better chance of advancing the state of the art through development of novel electrode structures.
4.2 Challenges and Outlook
While recent progress in development of alternative electrode structures for PEMFCs has demonstrated the potential to improve catalyst utilization, reduce Pt loading, and improve mass transport, further development is necessary to provide significant improvements beyond state-of-the-art conventional electrodes. For example, micro-patterned and macro-patterned electrodes could provide enhanced performance and stability through improved Pt utilization, improved water management, and expedited proton and oxygen transport, but fabrication challenges, including significant compressibility and deformation, have thus far limited progress. Electrospun electrodes could provide enhanced transport of both protons and oxygen, but improved understanding of transport directionality (i.e., in-plane vs. through-plane transport) and the role of carrier polymers are needed to guide further development. Electrodes with engineered macropores can provide improved transport, but the commercial applicability of these electrodes remains unclear, with lingering questions about the durability of these structures. For example, electrode cracks have been observed to propagate during carbon corrosion AST cycles, eventually causing MEA failure [167, 175]. Catalyst film electrode structures have shown significant promise, but better understanding of ionomer-free proton transport is needed to realize performance improvements. Water flooding at low temperatures is another challenge for the application of catalyst film electrodes due to the hydrophilicity and low volume of these electrodes, which may require development of improved water management structures and techniques. Strategies for improving the electronic conductivity and ensuring adequate connectivity are also needed to enable high performance in catalyst thin film electrodes with ultra-low Pt loadings.
Increasing interest in application of PEMFCs in heavy duty vehicles, which require higher energy efficiency and significantly longer lifetime, presents new challenges and opportunities for design of alternative electrode structures [49]. To achieve these performance and durability requirements, HDVs require higher catalyst loadings (and thicker electrodes) compared to LDVs. These thicker electrodes suffer from longer proton and oxygen transport pathways, while longer operating lifetimes result in even more severe degradation [49]. While most previous work on novel electrode design has focused on LDV applications, the beneficial features of these novel electrodes can extend to and address key challenges in HDV applications. Indeed, the greater transport and durability challenges associated with the use of higher catalyst loadings and the need for longer lifetimes in HDV applications make improved electrode design even more important for these applications. For example, durability issues related to carbon corrosion can be resolved by eliminating carbon support from the electrode using thin film catalysts, while higher oxygen and proton transport resistances stemming from thicker electrodes can be addressed by introducing macropores in combination with a fiber-based proton conducting network in electrospun electrodes. Alternative electrodes are promising candidates that can potentially be the key to enabling PEMFCs for HDV applications via enhanced efficiency and durability.
Each of the five categories of novel electrode structures covered in this review presents real advantages that could enable enhanced MEA performance (and, in some cases, enhanced durability), but strategically targeted R&D is required to realize these advantages. This observation presents a call to action. Research efforts on novel electrode structures should be redoubled, but this research should be carried out with careful attention to MEA best practices. Specifically, the following three areas merit attention:
-
1.
Careful baselining. Reports on enhanced MEA performance and durability enabled by novel electrode structures can only be valuable to the community if they are accompanied by rigorous comparison to a relevant baseline MEA; ideally, this baseline MEA should provide performance close to the state of the art.
-
2.
Use of relevant operating conditions. Testing efforts should be focused on use of H2/air under relevant pressures (typically considered to be 150–250 kPa absolute pressure). While H2/O2 testing can be useful as a diagnostic, and is directly relevant to a few applications, H2/air testing is required to evaluate electrode performance for typical applications in transportation and stationary power.
-
3.
Focusing on relevant operating voltages. While maximum power density is perhaps the most frequently cited figure of merit, we consider maximum power density to have little real-world relevance. Maximum power density is typically achieved at voltages lower than 0.6 V, but the use of such low operating voltages implies a low energy efficiency, negating one of the principal advantages of fuel cell technology. In addition to sacrificing efficiency, operation at low voltages results in significant waste heat generation; the rejection of this waste heat presents system engineering challenges, and significant cost and packaging requirements for larger radiators. Therefore, we recommend that testing and analysis focusing on operating voltages of 0.6 V or higher for all applications. For LDV-focused research, we recommend 0.65 V or higher; for HDV work, we recommend 0.7 V or higher. These operating voltages are consistent with the U.S. Department of Energy target metrics for LDV and HDV applications, determined based on system models that simulate drive cycles [49, 50]. Operating voltages of 0.7 V and above are also consistent with other governing agencies across the globe, as highlighted in a recently published perspective [208]. In particular, PEMFCs for HDVs need to be operated above 0.7 V in order to maintain high fuel economy [49]. Thus, a harmonized effort on enhancing performance and durability focused on relevant operating voltages will maximize the impact of our research.
Accelerating climate change represents an existential problem for human society, which can only be addressed through a transition to clean energy technologies with reduced or eliminated emissions of CO2 and other greenhouse gases. Fuel cells can play a major role in this clean energy transition, but improved performance and durability are needed to enable low-cost PEMFCs with sufficient lifetime. Development of higher-performing electrodes will be critical in enabling accelerated deployment of fuel cells, especially in HDV applications, where the large electrode thickness presents formidable barriers to transport of protons and oxygen. In this review, we have attempted to summarize and analyze the current state of alternative electrode development efforts. While the results of this analysis demonstrate that this field remains at an early stage of maturation, there is nonetheless ample reasons for optimism. Intensification of alternative electrode R&D, coupled with rigorous baselining and attention to MEA best practices, presents one of the most promising paths to enable faster deployment of clean and efficient fuel cell technology. We sincerely hope that our review and analysis can help to guide the PEMFC research community towards realization of a cleaner and better world based on emissions-free fuel cell technology.
References
Shukla, P.R., Skea, J., Slade, R. et al.: Climate change and land: an IPCC special report on climate change, desertification, land degradation, sustainable land management, food security, and greenhouse gas fluxes in terrestrial ecosystems. Foreword Technical and Preface, 35–74 (2019).
U.S. Energy Information Administration. Annual Energy Outlook, 2020.
Sui, P.C., Zhu, X., Djilali, N.: Modeling of PEM fuel cell catalyst layers: status and outlook. Electrochem. Energy Rev. 2, 428–466 (2019). https://doi.org/10.1007/s41918-019-00043-5
Guo, Y.Q., Pan, F.W., Chen, W.M., et al.: The controllable design of catalyst inks to enhance PEMFC performance: a review. Electrochem. Energy Rev. 4, 67–100 (2021). https://doi.org/10.1007/s41918-020-00083-2
Papageorgopoulos, D.: Fuel Cell R&D Overview. 2019 Annual Merit Review and Peer Evaluation Meeting. 33 (2019).
Papageorgopoulos, D.: Fuel Cell Technologies Overview. 2021 Annual Merit Review and Peer Evaluation Meeting. 1–72 (2021).
Martinez, U., Komini Babu, S., Holby, E.F., et al.: Progress in the development of Fe-based PGM-free electrocatalysts for the oxygen reduction reaction. Adv. Mater. 31, 1–20 (2019). https://doi.org/10.1002/adma.201806545
Li, J.K., Sougrati, M.T., Zitolo, A., et al.: Identification of durable and non-durable FeNx sites in Fe-N-C materials for proton exchange membrane fuel cells. Nat. Catal. 4, 10–19 (2020). https://doi.org/10.1038/s41929-020-00545-2
He, Y.H., Tan, Q., Lu, L.L., et al.: Metal-nitrogen-carbon catalysts for oxygen reduction in PEM fuel cells: self-template synthesis approach to enhancing catalytic activity and stability. Electrochem. Energy Rev. 2, 231–251 (2019). https://doi.org/10.1007/s41918-019-00031-9
Xiao, F., Wang, Y.C., Wu, Z.P., et al.: Recent advances in electrocatalysts for proton exchange membrane fuel cells and alkaline membrane fuel cells. Adv. Mater. 33, 202006292 (2021). https://doi.org/10.1002/adma.202006292
Shen, H.J., Thomas, T., Rasaki, S.A., et al.: Oxygen reduction reactions of Fe-N-C catalysts: current status and the way forward. Electrochem. Energy Rev. 2, 252–276 (2019). https://doi.org/10.1007/s41918-019-00030-w
Cheng, L., Khedekar, K., Rezaei Talarposhti, M., et al.: Mapping of heterogeneous catalyst degradation in polymer electrolyte fuel cells. Adv. Energy Mater. 10, 2000623 (2020). https://doi.org/10.1002/aenm.202000623
Khedekar, K., Rezaei Talarposhti, M., Besli, M.M., et al.: Probing heterogeneous degradation of catalyst in PEM fuel cells under realistic automotive conditions with multi-modal techniques. Adv. Energy Mater. 11, 2101794 (2021). https://doi.org/10.1002/aenm.202101794
Shao, Y.Y., Yin, G.P., Gao, Y.Z.: Understanding and approaches for the durability issues of Pt-based catalysts for PEM fuel cell. J. Power Sources 171, 558–566 (2007). https://doi.org/10.1016/j.jpowsour.2007.07.004
Zhang, J.W., Yuan, Y.L., Gao, L., et al.: Stabilizing Pt-based electrocatalysts for oxygen reduction reaction: fundamental understanding and design strategies. Adv. Mater. 33, 2006494 (2021). https://doi.org/10.1002/adma.202006494
Breitwieser, M., Klingele, M., Vierrath, S., et al.: Tailoring the membrane-electrode interface in PEM fuel cells: a review and perspective on novel engineering approaches. Adv. Energy Mater. 8, 1701257 (2018). https://doi.org/10.1002/aenm.201701257
Wang, W., Lv, F., Lei, B., et al.: Tuning nanowires and nanotubes for efficient fuel-cell electrocatalysis. Adv. Mater. 28, 10117–10141 (2016). https://doi.org/10.1002/adma.201601909
Nie, Y., Li, L., Wei, Z.D.: Recent advancements in Pt and Pt-free catalysts for oxygen reduction reaction. Chem. Soc. Rev. 44, 2168–2201 (2015). https://doi.org/10.1039/c4cs00484a
Wang, G.L., Zou, L.L., Huang, Q.H., et al.: Multidimensional nanostructured membrane electrode assemblies for proton exchange membrane fuel cell applications. J. Mater. Chem. A. 7, 9447–9477 (2019). https://doi.org/10.1039/c8ta12382a
Yang, Y.G., Zhou, X.Y., Li, B., et al.: Recent progress of the gas diffusion layer in proton exchange membrane fuel cells: material and structure designs of microporous layer. Int. J. Hydrog. Energy 46, 4259–4282 (2021). https://doi.org/10.1016/j.ijhydene.2020.10.185
Park, S., Lee, J.W., Popov, B.N.: A review of gas diffusion layer in PEM fuel cells: materials and designs. Int. J. Hydrog. Energy 37, 5850–5865 (2012). https://doi.org/10.1016/j.ijhydene.2011.12.148
Weber, A.Z., Borup, R.L., Darling, R.M., et al.: A critical review of modeling transport phenomena in polymer-electrolyte fuel cells. J. Electrochem. Soc. 161, F1254–F1299 (2014). https://doi.org/10.1149/2.0751412jes
Paul, M.T.Y., Kim, D., Saha, M.S., et al.: Patterning catalyst layers with microscale features by soft lithography techniques for proton exchange membrane fuel cells. ACS Appl. Energy Mater. 3, 478–486 (2020). https://doi.org/10.1021/acsaem.9b01754
Shukla, S., Domican, K., Karan, K., et al.: Analysis of low platinum loading thin polymer electrolyte fuel cell electrodes prepared by inkjet printing. Electrochim. Acta 156, 289–300 (2015). https://doi.org/10.1016/j.electacta.2015.01.028
Debe, M.K.: Nanostructured thin film electrocatalysts for PEM fuel cells: a tutorial on the fundamental characteristics and practical properties of NSTF catalysts. ECS Trans. 45, 47–68 (2012). https://doi.org/10.1149/1.3701968
Aizawa, M., Gyoten, H.: Effect of micro-patterned membranes on the cathode performances for PEM fuel cells under low humidity. J. Electrochem. Soc. 160, F417–F428 (2013). https://doi.org/10.1149/2.085304jes
Slack, J.J., Gumeci, C., Dale, N., et al.: Nanofiber fuel cell MEAs with a PtCo/C cathode. J. Electrochem. Soc. 166, F3202–F3209 (2019). https://doi.org/10.1149/2.0151907jes
Hong, S.J., Hou, M., Zhang, H.J., et al.: A high-performance PEM fuel cell with ultralow platinum electrode via electrospinning and underpotential deposition. Electrochim. Acta 245, 403–409 (2017). https://doi.org/10.1016/j.electacta.2017.05.066
Shukla, S., Domican, K., Secanell, M.: Effect of electrode patterning on PEM fuel cell performance using ink-jet printing method. ECS Trans. 64, 341–352 (2014). https://doi.org/10.1149/06403.0341ecst
Choi, S., Jeon, J., Chae, J.S., et al.: Single-step fabrication of a multiscale porous catalyst layer by the emulsion template method for low Pt-loaded proton exchange membrane fuel cells. ACS Appl. Energy Mater. 4, 4012–4020 (2021). https://doi.org/10.1021/acsaem.1c00379
Wilson, M.S., Gottesfeld, S.: Thin-film catalyst layers for polymer electrolyte fuel cell electrodes. J. Appl. Electrochem. 22, 1–7 (1992). https://doi.org/10.1007/BF01093004
M.S. Wilson, Membrane catalyst layer for fuel cells, US5211984A, 18 May 1993. https://image-ppubs.uspto.gov/dirsearch-public/print/downloadPdf/5211984
Raistrick, I.D., Electrode assembly for use in a solid polymer electrolyte fuel cell. US4876115A. 24 Oct 1989. https://image-ppubs.uspto.gov/dirsearch-public/print/downloadPdf/4876115
Litster, S., McLean, G.: PEM fuel cell electrodes. J. Power Sources 130, 61–76 (2004). https://doi.org/10.1016/j.jpowsour.2003.12.055
Owejan, J.P., Owejan, J.E., Gu, W.B.: Impact of platinum loading and catalyst layer structure on PEMFC performance. J. Electrochem. Soc. 160, F824–F833 (2013). https://doi.org/10.1149/2.072308jes
Ahn, C.Y., Ahn, J., Kang, S.Y., et al.: Enhancement of service life of polymer electrolyte fuel cells through application of nanodispersed ionomer. Sci. Adv. 6, eaaw0870 (2020). https://doi.org/10.1126/sciadv.aaw0870
Eguchi, M., Baba, K., Onuma, T., et al.: Influence of ionomer/carbon ratio on the performance of a polymer electrolyte fuel cell. Polymers 4, 1645–1656 (2012). https://doi.org/10.3390/polym4041645
Liu, Y.X., Murphy, M.W., Baker, D.R., et al.: Proton conduction and oxygen reduction kinetics in PEM fuel cell cathodes: effects of ionomer-to-carbon ratio and relative humidity. J. Electrochem. Soc. 156, B970–B980 (2009). https://doi.org/10.1149/1.3143965
Santangelo, P., Cannio, M., Romagnoli, M.: Review of catalyst-deposition techniques for PEMFC electrodes. Tecnica Italiana Italian J. Eng. Sci. 63, 65–72 (2019). https://doi.org/10.18280/ti-ijes.630109
Sassin, M.B., Garsany, Y., Gould, B.D., et al.: Fabrication method for laboratory-scale high-performance membrane electrode assemblies for fuel cells. Anal. Chem. 89, 511–518 (2017). https://doi.org/10.1021/acs.analchem.6b03005
Lee, E., Kim, D.H., Pak, C.: Effects of cathode catalyst layer fabrication parameters on the performance of high-temperature polymer electrolyte membrane fuel cells. Appl. Surf. Sci. 510, 145461 (2020). https://doi.org/10.1016/j.apsusc.2020.145461
Uchida, M., Park, Y.C., Kakinuma, K., et al.: Effect of the state of distribution of supported Pt nanoparticles on effective Pt utilization in polymer electrolyte fuel cells. Phys. Chem. Chem. Phys. 15, 11236 (2013). https://doi.org/10.1039/c3cp51801a
Mauger, S.A., Neyerlin, K.C., Yang-Neyerlin, A.C., et al.: Gravure coating for roll-to-roll manufacturing of proton-exchange-membrane fuel cell catalyst layers. J. Electrochem. Soc. 165, F1012–F1018 (2018). https://doi.org/10.1149/2.0091813jes
Bodner, M., García, H.R., Steenberg, T., et al.: Enabling industrial production of electrodes by use of slot-Die coating for HT-PEM fuel cells. Int. J. Hydrog. Energy 44, 12793–12801 (2019). https://doi.org/10.1016/j.ijhydene.2018.11.091
Shao, M.H., Merzougui, B., Shoemaker, K., et al.: Tungsten carbide modified high surface area carbon as fuel cell catalyst support. J. Power Sources 196, 7426–7434 (2011). https://doi.org/10.1016/j.jpowsour.2011.04.026
Breitwieser, M., Klingele, M., Britton, B., et al.: Improved Pt-utilization efficiency of low Pt-loading PEM fuel cell electrodes using direct membrane deposition. Electrochem. Commun. 60, 168–171 (2015). https://doi.org/10.1016/j.elecom.2015.09.006
Li, D.G., Pan, Y.T., Wang, X.J., et al.: Effect of the catalyst metal content on PEMFC durability. ECS Trans. 92, 589–594 (2019). https://doi.org/10.1149/09208.0589ecst
Kumaraguru, S.: Durable High Power Membrane Electrode Assembly with Low Pt Loading, General Motors, DOE hydrogen program, 2019. https://www.hydrogen.energy.gov/docs/hydrogenprogramlibraries/pdfs/review20/fc156_kumaraguru_2020_p.pdf
Cullen, D.A., Neyerlin, K.C., Ahluwalia, R.K., et al.: New roads and challenges for fuel cells in heavy-duty transportation. Nat. Energy 6, 462–474 (2021). https://doi.org/10.1038/s41560-021-00775-z
Marcinkoski, J.: DOE Advanced Truck Technologies, Subsection of the Electrified Powertrain Roadmap Technical Targets for Hydrogen-Fueled Long-Haul Tractor-Trailer Trucks. Record #: 19006, 31 October 2019. https://www.hydrogen.energy.gov/pdfs
Weber, A.Z., Kusoglu, A.: Unexplained transport resistances for low-loaded fuel-cell catalyst layers. J. Mater. Chem. A. 2, 17207–17211 (2014). https://doi.org/10.1039/c4ta02952f
More, K., Borup, R., Reeves, K.: Identifying contributing degradation phenomena in PEM fuel cell membrane electride assemblies via electron microscopy. ECS Trans. 3, 717–733 (2006). https://doi.org/10.1149/1.2356192
Ramaswamy, N.: Durable Fuel Cell MEA through Immobilization of Catalyst Particle and Membrane Chemical Stabilizer. U.S. Department of Energy Annual Merit Review and Peer Evaluation Meeting (2022).
She, Z.W., Kibsgaard, J., Dickens, C.F., et al.: Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, eaad4998 (2017). https://doi.org/10.1126/science.aad4998
Sun, X.Y., Yu, H.M., Zhou, L., et al.: Influence of platinum dispersity on oxygen transport resistance and performance in PEMFC. Electrochim. Acta 332, 135474 (2020). https://doi.org/10.1016/j.electacta.2019.135474
Chen, J.K., Liu, H.M., Huang, Y.A., et al.: High-rate roll-to-roll stack and lamination of multilayer structured membrane electrode assembly. J. Manuf. Process. 23, 175–182 (2016). https://doi.org/10.1016/j.jmapro.2016.06.022
Bing, Y.H., Liu, H.S., Zhang, L., et al.: Nanostructured Pt-alloy electrocatalysts for PEM fuel cell oxygen reduction reaction. Chem. Soc. Rev. 39, 2184–2202 (2010). https://doi.org/10.1039/b912552c
Park, Y.C., Tokiwa, H., Kakinuma, K., et al.: Effects of carbon supports on Pt distribution, ionomer coverage and cathode performance for polymer electrolyte fuel cells. J. Power Sources 315, 179–191 (2016). https://doi.org/10.1016/j.jpowsour.2016.02.091
Holdcroft, S.: Fuel cell catalyst layers: a polymer science perspective. Chem. Mater. 26, 381–393 (2014). https://doi.org/10.1021/cm401445h
Komini Babu, S., Spernjak, D., Dillet, J., et al.: Spatially resolved degradation during startup and shutdown in polymer electrolyte membrane fuel cell operation. Appl. Energy 254, 113659 (2019). https://doi.org/10.1016/j.apenergy.2019.113659
De Bruijn, F.A., Dam, V.A.T., Janssen, G.J.M.: Review: durability and degradation issues of PEM fuel cell components. Fuel Cells. 8, 3–22 (2008). https://doi.org/10.1002/fuce.200700053
Shao, Y.Y., Liu, J., Wang, Y., et al.: Novel catalyst support materials for PEM fuel cells: current status and future prospects. J. Mater. Chem. 19, 46–59 (2009). https://doi.org/10.1039/b808370c
Ren, X.F., Lv, Q.Y., Liu, L.F., et al.: Current progress of Pt and Pt-based electrocatalysts used for fuel cells. Sustain. Energy Fuels. 4, 15–30 (2020). https://doi.org/10.1039/c9se00460b
Schuler, T., Chowdhury, A., Freiberg, A.T., et al.: Fuel-cell catalyst-layer resistance via hydrogen limiting-current measurements. J. Electrochem. Soc. 166, F3020–F3031 (2019). https://doi.org/10.1149/2.0031907jes
Kongkanand, A., Mathias, M.F.: The priority and challenge of high-power performance of low-platinum proton-exchange membrane fuel cells. J. Phys. Chem. Lett. 7, 1127–1137 (2016). https://doi.org/10.1021/acs.jpclett.6b00216
Fan, J.T., Chen, M., Zhao, Z.L., et al.: Bridging the gap between highly active oxygen reduction reaction catalysts and effective catalyst layers for proton exchange membrane fuel cells. Nat. Energy 6, 475–486 (2021). https://doi.org/10.1038/s41560-021-00824-7
Das, P.K., Li, X.G., Liu, Z.S.: Analysis of liquid water transport in cathode catalyst layer of PEM fuel cells. Int. J. Hydrog. Energy 35, 2403–2416 (2010). https://doi.org/10.1016/j.ijhydene.2009.12.160
Srouji, A.K., Zheng, L.J., Dross, R., et al.: The role of water management on the oxygen transport resistance in polymer electrolyte fuel cell with ultra-low precious metal loading. J. Power Sources 364, 92–100 (2017). https://doi.org/10.1016/j.jpowsour.2017.07.036
Eikerling, M.: Water management in cathode catalyst layers of PEM fuel cells. J. Electrochem. Soc. 153, E58–E70 (2006). https://doi.org/10.1149/1.2160435
Meier, J.C., Galeano, C., Katsounaros, I., et al.: Design criteria for stable Pt/C fuel cell catalysts. Beilstein J. Nanotechnol. 5, 44–67 (2014). https://doi.org/10.3762/bjnano.5.5
Cherevko, S., Kulyk, N., Mayrhofer, K.J.J.: Durability of platinum-based fuel cell electrocatalysts: dissolution of bulk and nanoscale platinum. Nano Energy 29, 275–298 (2016). https://doi.org/10.1016/j.nanoen.2016.03.005
Linse, N., Gubler, L., Scherer, G.G., et al.: The effect of platinum on carbon corrosion behavior in polymer electrolyte fuel cells. Electrochim. Acta 56, 7541–7549 (2011). https://doi.org/10.1016/j.electacta.2011.06.093
Speder, J., Zana, A., Spanos, I., et al.: Comparative degradation study of carbon supported proton exchange membrane fuel cell electrocatalysts: the influence of the platinum to carbon ratio on the degradation rate. J. Power Sources 261, 14–22 (2014). https://doi.org/10.1016/j.jpowsour.2014.03.039
Ahluwalia, R.K., Peng, J.K., Wang, X., et al.: Long-term stability of nanostructured thin film electrodes at operating potentials. J. Electrochem. Soc. 164, F306–F320 (2017). https://doi.org/10.1149/2.0881704jes
Ahluwalia, R.K., Arisetty, S., Peng, J.K., et al.: Dynamics of particle growth and electrochemical surface area loss due to platinum dissolution. J. Electrochem. Soc. 161, F291–F304 (2014). https://doi.org/10.1149/2.051403jes
Towne, S., Viswanathan, V., Holbery, J., et al.: Fabrication of polymer electrolyte membrane fuel cell MEAs utilizing inkjet print technology. J. Power Sources 171, 575–584 (2007). https://doi.org/10.1016/j.jpowsour.2007.07.017
Therdthianwong, A., Saenwiset, P., Therdthianwong, S.: Cathode catalyst layer design for proton exchange membrane fuel cells. Fuel 91, 192–199 (2012). https://doi.org/10.1016/j.fuel.2011.07.003
Song, C.H., Park, J.S.: Membrane–electrode assemblies with patterned electrodes for proton-exchange membrane fuel cells. Chem. Lett. 47, 196–199 (2018). https://doi.org/10.1246/cl.170995
Heinz, O., Aghajani, M., Greenberg, A.R., et al.: Surface-patterning of polymeric membranes: fabrication and performance. Curr. Opin. Chem. Eng. 20, 1–12 (2018). https://doi.org/10.1016/j.coche.2018.01.008
O’Hayre, R., Lee, S.J., Cha, S.W., et al.: A sharp peak in the performance of sputtered platinum fuel cells at ultra-low platinum loading. J. Power Sources 109, 483–493 (2002). https://doi.org/10.1016/S0378-7753(02)00238-0
Zeis, R., Mathur, A., Fritz, G., et al.: Platinum-plated nanoporous gold: an efficient, low Pt loading electrocatalyst for PEM fuel cells. J. Power Sources 165, 65–72 (2007). https://doi.org/10.1016/j.jpowsour.2006.12.007
Choi, W.C., Kim, J.D., Woo, S.I.: Modification of proton conducting membrane for reducing methanol crossover in a direct-methanol fuel cell. J. Power Sources 96, 411–414 (2001). https://doi.org/10.1016/S0378-7753(00)00602-9
Kim, J.H., Sohn, J., Cho, J.H., et al.: Surface modification of Nafion membranes using atmospheric-pressure low-temperature plasmas for electrochemical applications. Plasma Process. Polym. 5, 377–385 (2008). https://doi.org/10.1002/ppap.200700108
Bae, J.W., Cho, Y.H., Sung, Y.E., et al.: Performance enhancement of polymer electrolyte membrane fuel cell by employing line-patterned Nafion membrane. J. Ind. Eng. Chem. 18, 876–879 (2012). https://doi.org/10.1016/j.jiec.2012.01.019
Jang, S., Her, M., Kim, S., et al.: Membrane/electrode interface design for effective water management in alkaline membrane fuel cells. ACS Appl. Mater. Interfaces 11, 34805–34811 (2019). https://doi.org/10.1021/acsami.9b08075
Aizawa, M., Gyoten, H., Salah, A., et al.: Pillar structured membranes for suppressing cathodic concentration overvoltage in PEMFCs at elevated temperature/low relative humidity. J. Electrochem. Soc. 157, B1844–B1851 (2010). https://doi.org/10.1149/1.3502613
Kim, S.M., Kang, Y.S., Ahn, C., et al.: Prism-patterned Nafion membrane for enhanced water transport in polymer electrolyte membrane fuel cell. J. Power Sources 317, 19–24 (2016). https://doi.org/10.1016/j.jpowsour.2016.03.083
Pu, L.J., Jiang, J.J., Yuan, T., et al.: Performance improvement of passive direct methanol fuel cells with surface-patterned Nafion® membranes. Appl. Surf. Sci. 327, 205–212 (2015). https://doi.org/10.1016/j.apsusc.2014.11.087
Maruf, S.H., Wang, L., Greenberg, A.R., et al.: Use of nanoimprinted surface patterns to mitigate colloidal deposition on ultrafiltration membranes. J. Membr. Sci. 428, 598–607 (2013). https://doi.org/10.1016/j.memsci.2012.10.059
Ning, F.D., Bai, C., Qin, J.Q., et al.: Great improvement in the performance and lifetime of a fuel cell using a highly dense, well-ordered, and cone-shaped Nafion array. J. Mater. Chem. A. 8, 5489–5500 (2020). https://doi.org/10.1039/c9ta13666e
Chen, M., Wang, M., Yang, Z.Y., et al.: A novel catalyst layer structure based surface-patterned Nafion® membrane for high-performance direct methanol fuel cell. Electrochim. Acta 263, 201–208 (2018). https://doi.org/10.1016/j.electacta.2018.01.015
Ahn, C.Y., Jang, S., Cho, Y.H., et al.: Guided cracking of electrodes by stretching prism-patterned membrane electrode assemblies for high-performance fuel cells. Sci. Rep. 8, 1–9 (2018). https://doi.org/10.1038/s41598-018-19861-6
Cho, H., Moon Kim, S., Sik Kang, Y., et al.: Multiplex lithography for multilevel multiscale architectures and its application to polymer electrolyte membrane fuel cell. Nat. Commun. 6, 1–8 (2015). https://doi.org/10.1038/ncomms9484
Jang, S., Kim, M., Kang, Y.S., et al.: Facile multiscale patterning by creep-assisted sequential imprinting and fuel cell application. ACS Appl. Mater. Interfaces 8, 11459–11465 (2016). https://doi.org/10.1021/acsami.6b01555
Paul, M.T.Y., Saha, M.S., Qi, W.L., et al.: Microstructured membranes for improving transport resistances in proton exchange membrane fuel cells. Int. J. Hydrog. Energy 45, 1304–1312 (2020). https://doi.org/10.1016/j.ijhydene.2019.05.186
Kang, S., Bae, G., Kim, S.K., et al.: Performance of a MEA using patterned membrane with a directly coated electrode by the bar-coating method in a direct methanol fuel cell. Int. J. Hydrog. Energy 43, 11386–11396 (2018). https://doi.org/10.1016/j.ijhydene.2018.04.086
Barambu, N.U., Bilad, M.R., Wibisono, Y., et al.: Membrane surface patterning as a fouling mitigation strategy in liquid filtration: a review. Polymers 11, 1687 (2019). https://doi.org/10.3390/polym11101687
Omosebi, A., Besser, R.S.: Patterning the cathode catalyst layer-membrane interface of a PEMFC for elevated power density. ECS Trans. 41, 883–889 (2011). https://doi.org/10.1149/1.3635622
Chi, W.S., Jeon, Y., Park, S.J., et al.: Fabrication of surface-patterned membranes by means of a ZnO nanorod templating method for polymer electrolyte membrane fuel-cell applications. ChemPlusChem 79, 1109–1115 (2014). https://doi.org/10.1002/cplu.201402083
Koh, J.K., Jeon, Y., Cho, Y.I., et al.: A facile preparation method of surface patterned polymer electrolyte membranes for fuel cell applications. J. Mater. Chem. A. 2, 8652–8659 (2014). https://doi.org/10.1039/c4ta00674g
Jeon, Y., Kim, D.J., Koh, J.K., et al.: Interface-designed membranes with shape-controlled patterns for high-performance polymer electrolyte membrane fuel cells. Sci. Rep. 5, 16394 (2015). https://doi.org/10.1038/srep16394
Khan, A., Nath, B.K., Chutia, J.: Conical nano-structure arrays of platinum cathode catalyst for enhanced cell performance in PEMFC (proton exchange membrane fuel cell). Energy 90, 1769–1774 (2015). https://doi.org/10.1016/j.energy.2015.07.002
Omosebi, A., Besser, R.S.: Ultra-low mass sputtered and conventional catalyst layers on plasma-etched nafion for PEMFC applications. Fuel Cells. 17, 762–769 (2017). https://doi.org/10.1002/fuce.201600183
Yakovlev, Y.V., Nováková, J., Kúš, P., et al.: Highly developed nanostructuring of polymer-electrolyte membrane supported catalysts for hydrogen fuel cell application. J. Power Sources 439, 227084 (2019). https://doi.org/10.1016/j.jpowsour.2019.227084
Seol, C., Jang, S., Lee, J., et al.: High-performance fuel cells with a plasma-etched polymer electrolyte membrane with microhole arrays. ACS Sustain. Chem. Eng. 9, 5884–5894 (2021). https://doi.org/10.1021/acssuschemeng.1c00059
Omosebi, A., Besser, R.S.: Electron beam assisted patterning and dry etching of nafion membranes. J. Electrochem. Soc. 158, D603–D610 (2011). https://doi.org/10.1149/1.3615938
Lee, D.H., Jo, W., Yuk, S., et al.: In-plane channel-structured catalyst layer for polymer electrolyte membrane fuel cells. ACS Appl. Mater. Interfaces 10, 4682–4688 (2018). https://doi.org/10.1021/acsami.7b16433
Spernjak, D., Mukundan, R., Borup, R.L., et al.: Enhanced water management of polymer electrolyte fuel cells with additive-containing microporous layers. ACS Appl. Energy Mater. 1, 6006–6017 (2018). https://doi.org/10.1021/acsaem.8b01059
Murata, S., Imanishi, M., Hasegawa, S., et al.: Vertically aligned carbon nanotube electrodes for high current density operating proton exchange membrane fuel cells. J. Power Sources 253, 104–113 (2014). https://doi.org/10.1016/j.jpowsour.2013.11.073
Zhang, C.K., Yu, H.M., Li, Y.K., et al.: Supported noble metals on hydrogen-treated TiO2 nanotube arrays as highly ordered electrodes for fuel cells. Chemsuschem 6, 659–666 (2013). https://doi.org/10.1002/cssc.201200828
Wang, C., Waje, M., Wang, X., et al.: Proton exchange membrane fuel cells with carbon nanotube based electrodes. Nano Lett. 4, 345–348 (2004). https://doi.org/10.1021/nl034952p
Wu, H.J., Yuan, T., Huang, Q.H., et al.: Polypyrrole nanowire networks as anodic micro-porous layer for passive direct methanol fuel cells. Electrochim. Acta 141, 1–5 (2014). https://doi.org/10.1016/j.electacta.2014.06.149
Hezarjaribi, M., Jahanshahi, M., Rahimpour, A., et al.: Gas diffusion electrode based on electrospun Pani/CNF nanofibers hybrid for proton exchange membrane fuel cells (PEMFC) applications. Appl. Surf. Sci. 295, 144–149 (2014). https://doi.org/10.1016/j.apsusc.2014.01.018
Sun, R.L., Xia, Z.X., Shang, L., et al.: Hierarchically ordered arrays with platinum coated PANI nanowires for highly efficient fuel cell electrodes. J. Mater. Chem. A. 5, 15260–15265 (2017). https://doi.org/10.1039/c7ta02500a
Xia, Z.X., Wang, S.L., Jiang, L.H., et al.: Bio-inspired construction of advanced fuel cell cathode with Pt anchored in ordered hybrid polymer matrix. Sci. Rep. 5, 16100 (2015). https://doi.org/10.1038/srep16100
Fu, X.D., Wang, S.L., Xia, Z.X., et al.: Aligned polyaniline nanorods in situ grown on gas diffusion layer and their application in polymer electrolyte membrane fuel cells. Int. J. Hydrog. Energy 41, 3655–3663 (2016). https://doi.org/10.1016/j.ijhydene.2015.12.193
Chen, M., Wang, M., Yang, Z.Y., et al.: High performance and durability of order-structured cathode catalyst layer based on TiO2@PANI core-shell nanowire arrays. Appl. Surf. Sci. 406, 69–76 (2017). https://doi.org/10.1016/j.apsusc.2017.01.296
Choi, J., Wycisk, R., Zhang, W.J., et al.: High conductivity perfluorosulfonic acid nanofiber composite fuel-cell membranes. Chemsuschem 3, 1245–1248 (2010). https://doi.org/10.1002/cssc.201000220
Ballengee, J.B., Pintauro, P.N.: Composite fuel cell membranes from dual-nanofiber electrospun mats. Macromolecules 44, 7307–7314 (2011). https://doi.org/10.1021/ma201684j
Park, J.H., Ju, Y.W., Park, S.H., et al.: Effects of electrospun polyacrylonitrile-based carbon nanofibers as catalyst support in PEMFC. J. Appl. Electrochem. 39, 1229–1236 (2009). https://doi.org/10.1007/s10800-009-9787-4
Zhang, W.J., Pintauro, P.N.: High-performance nanofiber fuel cell electrodes. Chemsuschem 4, 1753–1757 (2011). https://doi.org/10.1002/cssc.201100245
Waldrop, K., Wycisk, R., Pintauro, P.N.: Application of electrospinning for the fabrication of proton-exchange membrane fuel cell electrodes. Curr. Opin. Electrochem. 21, 257–264 (2020). https://doi.org/10.1016/j.coelec.2020.03.007
Chan, S., Jankovic, J., Susac, D., et al.: Electrospun carbon nanofiber catalyst layers for polymer electrolyte membrane fuel cells: fabrication and optimization. J. Mater. Sci. 53, 11633–11647 (2018). https://doi.org/10.1007/s10853-018-2411-4
Álvarez, G., Alcaide, F., Cabot, P.L., et al.: Electrochemical performance of low temperature PEMFC with surface tailored carbon nanofibers as catalyst support. Int. J. Hydrog. Energy 37, 393–404 (2012). https://doi.org/10.1016/j.ijhydene.2011.09.055
Chung, S., Shin, D., Choun, M., et al.: Improved water management of Pt/C cathode modified by graphitized carbon nanofiber in proton exchange membrane fuel cell. J. Power Sources 399, 350–356 (2018). https://doi.org/10.1016/j.jpowsour.2018.07.126
Mooste, M., Kibena-Põldsepp, E., Vassiljeva, V., et al.: Electrocatalysts for oxygen reduction reaction based on electrospun polyacrylonitrile, styrene–acrylonitrile copolymer and carbon nanotube composite fibres. J. Mater. Sci. 54, 11618–11634 (2019). https://doi.org/10.1007/s10853-019-03725-z
Wang, Y., Li, G., Jin, J.H., et al.: Hollow porous carbon nanofibers as novel support for platinum-based oxygen reduction reaction electrocatalysts. Int. J. Hydrog. Energy 42, 5938–5947 (2017). https://doi.org/10.1016/j.ijhydene.2017.02.012
Cavaliere, S., Subianto, S., Savych, I., et al.: Dopant-driven nanostructured loose-tube SnO2 architectures: alternative electrocatalyst supports for proton exchange membrane fuel cells. J. Phys. Chem. C 117, 18298–18307 (2013). https://doi.org/10.1021/jp404570d
Chen, S.X., Du, H.D., Wei, Y.P., et al.: Fine-tuning the cross-sectional architecture of antimony-doped tin oxide nanofibers as Pt catalyst support for enhanced oxygen reduction activity. Int. J. Electrochem. Sci. 12, 6221–6231 (2017). https://doi.org/10.20964/2017.07.42
Ji, Y., Cho, Y.I., Jeon, Y., et al.: Design of active Pt on TiO2 based nanofibrous cathode for superior PEMFC performance and durability at high temperature. Appl. Catal. B Environ. 204, 421–429 (2017). https://doi.org/10.1016/j.apcatb.2016.11.053
Kayarkatte, M.K., Delikaya, Ö., Roth, C.: Polyacrylic acid-Nafion composites as stable catalyst support in PEM fuel cell electrodes. Mater. Today Commun. 16, 8–13 (2018). https://doi.org/10.1016/j.mtcomm.2018.02.003
Simotwo, S.K., Kalra, V.: Study of Co-electrospun nafion and polyaniline nanofibers as potential catalyst support for fuel cell electrodes. Electrochim. Acta 198, 156–164 (2016). https://doi.org/10.1016/j.electacta.2016.02.209
Nabil, Y., Cavaliere, S., Harkness, I.A., et al.: Novel niobium carbide/carbon porous nanotube electrocatalyst supports for proton exchange membrane fuel cell cathodes. J. Power Sources 363, 20–26 (2017). https://doi.org/10.1016/j.jpowsour.2017.07.058
Kim, M., Kwon, C., Eom, K., et al.: Electrospun Nb-doped TiO2 nanofiber support for Pt nanoparticles with high electrocatalytic activity and durability. Sci. Rep. 7, 44411 (2017). https://doi.org/10.1038/srep44411
Sun, Y.Y., Cui, L.R., Gong, J., et al.: Design of a catalytic layer with hierarchical proton transport structure: the role of nafion nanofiber. ACS Sustain. Chem. Eng. 7, 2955–2963 (2019). https://doi.org/10.1021/acssuschemeng.8b03910
Hwang, M., Elabd, Y.A.: Impact of ionomer resistance in nanofiber-nanoparticle electrodes for ultra-low platinum fuel cells. Int. J. Hydrog. Energy 44, 6245–6256 (2019). https://doi.org/10.1016/j.ijhydene.2019.01.083
Wang, X.H., Richey, F.W., Wujcik, K.H., et al.: Effect of polytetrafluoroethylene on ultra-low platinum loaded electrospun/electrosprayed electrodes in proton exchange membrane fuel cells. Electrochim. Acta 139, 217–224 (2014). https://doi.org/10.1016/j.electacta.2014.06.139
Wei, M., Jiang, M., Liu, X.B., et al.: Graphene-doped electrospun nanofiber membrane electrodes and proton exchange membrane fuel cell performance. J. Power Sources 327, 384–393 (2016). https://doi.org/10.1016/j.jpowsour.2016.07.083
Choi, S., Yuk, S., Lee, D.H., et al.: Rugged catalyst layer supported on a Nafion fiber mat for enhancing mass transport of polymer electrolyte membrane fuel cells. Electrochim. Acta 268, 469–475 (2018). https://doi.org/10.1016/j.electacta.2018.02.097
Gao, Y.Y., Hou, M., He, L., et al.: A new insight into the stability of nanofiber electrodes used in proton exchange membrane fuel cells. J. Energy Chem. 42, 126–132 (2020). https://doi.org/10.1016/j.jechem.2019.06.017
Brodt, M., Wycisk, R., Pintauro, P.N.: Nanofiber electrodes with low platinum loading for high power hydrogen/air PEM fuel cells. J. Electrochem. Soc. 160, F744–F749 (2013). https://doi.org/10.1149/2.008308jes
Brodt, M., Han, T., Dale, N., et al.: Fabrication, in-situ performance, and durability of nanofiber fuel cell electrodes. J. Electrochem. Soc. 162, F84–F91 (2014). https://doi.org/10.1149/2.0651501jes
Brodt, M., Wycisk, R., Dale, N., et al.: Power output and durability of electrospun fuel cell fiber cathodes with PVDF and nafion/PVDF binders. J. Electrochem. Soc. 163, F401–F410 (2016). https://doi.org/10.1149/2.0711605jes
Chintam, K., Waldrop, K., Baker, A.M., et al.: Improved water management of electrospun nanofiber membrane electrode assemblies at high current densities measured in operando using neutron radiography. ECS Trans. 92, 125–134 (2019). https://doi.org/10.1149/09208.0125ecst
Slack, J.J., Brodt, M., Cullen, D.A., et al.: Impact of polyvinylidene fluoride on nanofiber cathode structure and durability in proton exchange membrane fuel cells. J. Electrochem. Soc. 167, 054517 (2020). https://doi.org/10.1149/1945-7111/ab77fb
Chevalier, S., Lavielle, N., Hatton, B.D., et al.: Novel electrospun gas diffusion layers for polymer electrolyte membrane fuel cells: Part I. Fabrication, morphological characterization, and in situ performance. J. Power Sources 352, 272–280 (2017). https://doi.org/10.1016/j.jpowsour.2017.03.098
Chan, S., Jankovic, J., Susac, D., et al.: Electrospun carbon nanofiber catalyst layers for polymer electrolyte membrane fuel cells: structure and performance. J. Power Sources 392, 239–250 (2018). https://doi.org/10.1016/j.jpowsour.2018.02.001
Zeng, Y.C., Zhang, H.J., Wang, Z.Q., et al.: Nano-engineering of a 3D-ordered membrane electrode assembly with ultrathin Pt skin on open-walled PdCo nanotube arrays for fuel cells. J. Mater. Chem. A. 6, 6521–6533 (2018). https://doi.org/10.1039/c7ta10901f
Qi, M.M., Zeng, Y.C., Hou, M., et al.: Free-standing and ionomer-free 3D platinum nanotrough fiber network electrode for proton exchange membrane fuel cells. Appl. Catal. B Environ. 298, 120504 (2021). https://doi.org/10.1016/j.apcatb.2021.120504
Chen, H., Snyder, J.D., Elabd, Y.A.: Electrospinning and solution properties of nafion and poly(acrylic acid). Macromolecules 41, 128–135 (2008). https://doi.org/10.1021/ma070893g
Kabir, S., Van Cleve, T., Khandavalli, S., et al.: Toward optimizing electrospun nanofiber fuel cell catalyst layers: microstructure and Pt accessibility. ACS Appl. Energy Mater. 4, 3341–3351 (2021). https://doi.org/10.1021/acsaem.0c03073
Khandavalli, S., Sharma-Nene, N., Kabir, S., et al.: Toward optimizing electrospun nanofiber fuel cell catalyst layers: polymer–particle interactions and spinnability. ACS Appl. Polym. Mater. 3, 2374–2384 (2021). https://doi.org/10.1021/acsapm.0c01354
Dong, B., Gwee, L., Salas-de la Cruz, D., et al.: Super proton conductive high-purity nafion nanofibers. Nano Lett. 10, 3785–3790 (2010). https://doi.org/10.1021/nl102581w
Ramaswamy, N., Gu, W.B., Ziegelbauer, J.M., et al.: Carbon support microstructure impact on high current density transport resistances in PEMFC cathode. J. Electrochem. Soc. 167, 064515 (2020). https://doi.org/10.1149/1945-7111/ab819c
Mohseninia, A., Kartouzian, D., Eppler, M., et al.: Influence of structural modification of micro-porous layer and catalyst layer on performance and water management of PEM fuel cells: hydrophobicity and porosity. Fuel Cells. 20, 469–476 (2020). https://doi.org/10.1002/fuce.201900203
Mohseninia, A., Kartouzian, D., Schlumberger, R., et al.: Enhanced water management in PEMFCs: perforated catalyst layer and microporous layers. Chemsuschem 13, 2931–2934 (2020). https://doi.org/10.1002/cssc.202000542
Zlotorowicz, A., Jayasayee, K., Dahl, P.I., et al.: Tailored porosities of the cathode layer for improved polymer electrolyte fuel cell performance. J. Power Sources 287, 472–477 (2015). https://doi.org/10.1016/j.jpowsour.2015.04.079
Cho, Y.H., Jung, N., Kang, Y.S., et al.: Improved mass transfer using a pore former in cathode catalyst layer in the direct methanol fuel cell. Int. J. Hydrog. Energy 37, 11969–11974 (2012). https://doi.org/10.1016/j.ijhydene.2012.05.031
Kim, O.H., Cho, Y.H., Kang, S.H., et al.: Ordered macroporous platinum electrode and enhanced mass transfer in fuel cells using inverse opal structure. Nat. Commun. 4, 2473 (2013). https://doi.org/10.1038/ncomms3473
Zhao, J.S., He, X.M., Wang, L., et al.: Addition of NH4HCO3 as pore-former in membrane electrode assembly for PEMFC. Int. J. Hydrog. Energy 32, 380–384 (2007). https://doi.org/10.1016/j.ijhydene.2006.06.057
Fischer, A., Jindra, J., Wendt, H.: Porosity and catalyst utilization of thin layer cathodes in air operated PEM-fuel cells. J. Appl. Electrochem. 28, 277–282 (1998). https://doi.org/10.1023/A:1003259531775
Henkensmeier, D., Dang, Q.K., Nambi Krishnan, N., et al.: Ortho-Dichlorobenzene as a pore modifier for PEMFC catalyst electrodes and dense Nafion membranes with one porous surface. J. Mater. Chem. 22, 14602–14607 (2012). https://doi.org/10.1039/c2jm32146g
Yu, Z., Carter, R.N., Zhang, J.: Measurements of pore size distribution, porosity, effective oxygen diffusivity, and tortuosity of PEM fuel cell electrodes. Fuel Cells. 12, 557–565 (2012). https://doi.org/10.1002/fuce.201200017
Lee, M.C., Tan, C., Ravanfar, R., et al.: Ultrastable water-in-oil high internal phase emulsions featuring interfacial and biphasic network stabilization. ACS Appl. Mater. Interfaces 11, 26433–26441 (2019). https://doi.org/10.1021/acsami.9b05089
Kim, S.M., Ahn, C.Y., Cho, Y.H., et al.: High-performance fuel cell with stretched catalyst-coated membrane: one-step formation of cracked electrode. Sci. Rep. 6, 26503 (2016). https://doi.org/10.1038/srep26503
Yoon, Y.: Effect of pore structure of catalyst layer in a PEMFC on its performance. Int. J. Hydrog. Energy 28, 657–662 (2003). https://doi.org/10.1016/s0360-3199(02)00156-8
White, R.T., Wu, A., Najm, M., et al.: 4D in situ visualization of electrode morphology changes during accelerated degradation in fuel cells by X-ray computed tomography. J. Power Sources 350, 94–102 (2017). https://doi.org/10.1016/j.jpowsour.2017.03.058
Hwang, G.S., Kim, H., Lujan, R., et al.: Phase-change-related degradation of catalyst layers in proton-exchange-membrane fuel cells. Electrochim. Acta 95, 29–37 (2013). https://doi.org/10.1016/j.electacta.2013.02.017
Guo, Q.H., Qi, Z.G.: Effect of freeze-thaw cycles on the properties and performance of membrane-electrode assemblies. J. Power Sources 160, 1269–1274 (2006). https://doi.org/10.1016/j.jpowsour.2006.02.093
Kumano, N., Kudo, K., Suda, A., et al.: Controlling cracking formation in fuel cell catalyst layers. J. Power Sources 419, 219–228 (2019). https://doi.org/10.1016/j.jpowsour.2019.02.058
Huang, D.C., Yu, P.J., Liu, F.J., et al.: Effect of dispersion solvent in catalyst ink on proton exchange membrane fuel cell performance. Int. J. Electrochem. Sci. 6, 2551–2565 (2011). https://doi.org/10.1016/S1452-3981(23)18202-2
Tranter, T.G., Tam, M., Gostick, J.T.: The effect of cracks on the in-plane electrical conductivity of PEFC catalyst layers. Electroanalysis 31, 619–623 (2019). https://doi.org/10.1002/elan.201800553
Hizir, F.E., Ural, S.O., Kumbur, E.C., et al.: Characterization of interfacial morphology in polymer electrolyte fuel cells: micro-porous layer and catalyst layer surfaces. J. Power Sources 195, 3463–3471 (2010). https://doi.org/10.1016/j.jpowsour.2009.11.032
Yin, Y., Li, R.T., Bai, F.Q., et al.: Ionomer migration within PEMFC catalyst layers induced by humidity changes. Electrochem. Commun. 109, 106590 (2019). https://doi.org/10.1016/j.elecom.2019.106590
Ramani, D., Singh, Y., Orfino, F.P., et al.: Characterization of membrane degradation growth in fuel cells using X-ray computed tomography. J. Electrochem. Soc. 165, F3200–F3208 (2018). https://doi.org/10.1149/2.0251806jes
Babu, S.K., Atkinson, R.W., III., Papandrew, A.B., et al.: Vertically oriented polymer electrolyte nanofiber catalyst support for thin film proton-exchange membrane fuel cell electrodes. ChemElectroChem 2, 1752–1759 (2015). https://doi.org/10.1002/celc.201500232
Wang, Y., Ruiz Diaz, D.F., Chen, K.S., et al.: Materials, technological status, and fundamentals of PEM fuel cells: a review. Mater. Today 32, 178–203 (2020). https://doi.org/10.1016/j.mattod.2019.06.005
Debe, M.K.: Electrocatalyst approaches and challenges for automotive fuel cells. Nature 486, 43–51 (2012). https://doi.org/10.1038/nature11115
Çögenli, M.S., Mukerjee, S., Yurtcan, A.B.: Membrane electrode assembly with ultra low platinum loading for cathode electrode of PEM fuel cell by using sputter deposition. Fuel Cells. 15, 288–297 (2015). https://doi.org/10.1002/fuce.201400062
Yang, G.Q., Yu, S.L., Mo, J.K., et al.: Impacts of catalyst nanolayers on water permeation and swelling of polymer electrolyte membranes. J. Power Sources 448, 227582 (2020). https://doi.org/10.1016/j.jpowsour.2019.227582
Ergul-Yilmaz, B., Yang, Z.W., Perry, M.L., et al.: Microstructural evolution and ORR activity of nanocolumnar platinum thin films with different mass loadings grown by high pressure sputtering. J. Electrochem. Soc. 167, 134514 (2020). https://doi.org/10.1149/1945-7111/abba90
Lai, Y.C., Huang, K.L., Tsai, C.H., et al.: Sputtered Pt loadings of membrane electrode assemblies in proton exchange membrane fuel cells. Int. J. Energy Res. 36, 918–927 (2012). https://doi.org/10.1002/er.1883
Gruber, D., Ponath, N., Müller, J., et al.: Sputter-deposited ultra-low catalyst loadings for PEM fuel cells. J. Power Sources 150, 67–72 (2005). https://doi.org/10.1016/j.jpowsour.2005.02.076
Ostroverkh, A., Johanek, V., Dubau, M., et al.: Novel fuel cell MEA based on Pt-C deposited by magnetron sputtering. ECS Trans. 80, 225–230 (2017). https://doi.org/10.1149/08008.0225ecst
Cullen, D.A., Lopez-Haro, M., Bayle-Guillemaud, P., et al.: Linking morphology with activity through the lifetime of pretreated PtNi nanostructured thin film catalysts. J. Mater. Chem. A. 3, 11660–11667 (2015). https://doi.org/10.1039/c5ta01854d
Steinbach, A.J., Allen, J.S., Borup, R.L., et al.: Anode-design strategies for improved performance of polymer-electrolyte fuel cells with ultra-thin electrodes. Joule 2, 1297–1312 (2018). https://doi.org/10.1016/j.joule.2018.03.022
Jiang, S.F., Yi, B.L., Zhang, C.K., et al.: Vertically aligned carbon-coated titanium dioxide nanorod arrays on carbon paper with low platinum for proton exchange membrane fuel cells. J. Power Sources 276, 80–88 (2015). https://doi.org/10.1016/j.jpowsour.2014.11.093
Jiang, S.F., Yi, B.L., Zhang, H.J., et al.: Vertically aligned titanium nitride nanorod arrays as supports of platinum–palladium–cobalt catalysts for thin-film proton exchange membrane fuel cell electrodes. ChemElectroChem 3, 734–740 (2016). https://doi.org/10.1002/celc.201500571
Inaba, M., Suzuki, T., Hatanaka, T., et al.: Fabrication and cell analysis of a Pt/SiO2Platinum thin film electrode. J. Electrochem. Soc. 162, F634–F638 (2015). https://doi.org/10.1149/2.0201507jes
Jiang, S.F., Yi, B.L., Cao, L.S., et al.: Development of advanced catalytic layer based on vertically aligned conductive polymer arrays for thin-film fuel cell electrodes. J. Power Sources 329, 347–354 (2016). https://doi.org/10.1016/j.jpowsour.2016.08.098
Mardle, P., Ji, X.C., Wu, J., et al.: Thin film electrodes from Pt nanorods supported on aligned N-CNTs for proton exchange membrane fuel cells. Appl. Catal. B Environ. 260, 118031 (2020). https://doi.org/10.1016/j.apcatb.2019.118031
Tian, Z., Lim, S.H., Poh, C.K., et al.: A highly order-structured membrane electrode assembly with vertically aligned carbon nanotubes for ultra-low Pt loading PEM fuel cells. Adv. Energy Mater. 1, 1205–1214 (2011). https://doi.org/10.1002/aenm.201100371
Hu, L.M., Zhang, M.X., Komini Babu, S., et al.: Ionic conductivity over metal/water interfaces in ionomer-free fuel cell electrodes. ChemElectroChem 6, 2659–2666 (2019). https://doi.org/10.1002/celc.201900124
Zenyuk, I.V., Litster, S.: Modeling ion conduction and electrochemical reactions in water films on thin-film metal electrodes with application to low temperature fuel cells. Electrochim. Acta 146, 194–206 (2014). https://doi.org/10.1016/j.electacta.2014.08.070
Galbiati, S., Morin, A., Pauc, N.: Supportless platinum nanotubes array by atomic layer deposition as PEM fuel cell electrode. Electrochim. Acta 125, 107–116 (2014). https://doi.org/10.1016/j.electacta.2014.01.061
Kim, J.M., Jo, A., Lee, K.A., et al.: Conformation-modulated three-dimensional electrocatalysts for high-performance fuel cell electrodes. Sci. Adv. 7, eabe9083 (2021). https://doi.org/10.1126/sciadv.abe9083
Fidiani, E., AlKahfi, A.Z., Absor, M.A.U., et al.: Au-doped PtAg nanorod array electrodes for proton-exchange membrane fuel cells. ACS Appl. Energy Mater. 5, 14979–14989 (2022). https://doi.org/10.1021/acsaem.2c02528
Wei, Z.X., Su, K.H., Sui, S., et al.: High performance polymer electrolyte membrane fuel cells (PEMFCs) with gradient Pt nanowire cathodes prepared by decal transfer method. Int. J. Hydrog. Energy 40, 3068–3074 (2015). https://doi.org/10.1016/j.ijhydene.2015.01.009
Galbiati, S., Morin, A., Pauc, N.: Nanotubes array electrodes by Pt evaporation: Half-cell characterization and PEM fuel cell demonstration. Appl. Catal. B Environ. 165, 149–157 (2015). https://doi.org/10.1016/j.apcatb.2014.09.075
Marconot, O., Pauc, N., Buttard, D., et al.: Vertically aligned platinum copper nanotubes as PEM fuel cell cathode: elaboration and fuel cell test. Fuel Cells. 18, 723–730 (2018). https://doi.org/10.1002/fuce.201700242
Zeng, Y.C., Shao, Z.G., Zhang, H.J., et al.: Nanostructured ultrathin catalyst layer based on open-walled PtCo bimetallic nanotube arrays for proton exchange membrane fuel cells. Nano Energy 34, 344–355 (2017). https://doi.org/10.1016/j.nanoen.2017.02.038
Paulus, U.A., Veziridis, Z., Schnyder, B., et al.: Fundamental investigation of catalyst utilization at the electrode/solid polymer electrolyte interface. J. Electroanal. Chem. 541, 77–91 (2003). https://doi.org/10.1016/s0022-0728(02)01416-x
An, S.J., Litster, S.: In situ, ionic conductivity measurement of ionomer/binder-free Pt catalyst under fuel cell operating condition. ECS Trans. 58, 831–839 (2013). https://doi.org/10.1149/05801.0831ecst
Pan, S.F., Qin, J.Q., Ning, F.D., et al.: Well-dispersed nafion array prepared by the freeze-drying method to effectively improve the performance of proton exchange membrane fuel cells. ACS Sustain. Chem. Eng. 9, 16770–16777 (2021). https://doi.org/10.1021/acssuschemeng.1c06304
Babu, S.K., Spendelow, J.S., Mukundan, R.: Coaxial nanowire electrode. US20200321626A1, 8 Oct 2020. https://image-ppubs.uspto.gov/dirsearch-public/print/downloadPdf/20200321626
Borup, R., Weber, A.: FC-PAD: Fuel Cell Performance and Durability Consortium, DOE Fuel Cell Technologies Office Annual Merit Review. (2019). https://www.energy.gov/eere/fuelcells/fc-pad-fuel-cell-consortium-performance-and-durability
Cuynet, S., Caillard, A., Kaya-Boussougou, S., et al.: Membrane patterned by pulsed laser micromachining for proton exchange membrane fuel cell with sputtered ultra-low catalyst loadings. J. Power Sources 298, 299–308 (2015). https://doi.org/10.1016/j.jpowsour.2015.08.019
Jiao, K., Xuan, J., Du, Q., et al.: Designing the next generation of proton-exchange membrane fuel cells. Nature 595, 361–369 (2021). https://doi.org/10.1038/s41586-021-03482-7
Acknowledgements
This work was supported in part by the Hydrogen and Fuel Cell Technologies Office (HFTO), Office of Energy Efficiency and Renewable Energy, US Department of Energy (DOE) through the Million Mile Fuel Cell Truck (M2FCT) consortium, technology managers G. Kleen and D. Papageorgopoulos. Financial support for this work from the Laboratory Directed Research and Development (LDRD) program at Los Alamos National Laboratory (LANL) is gratefully acknowledged (Projects 20200200DR, 20210915PRD2, 20200777PRD4). ChungHyuk Lee acknowledges the support of the Natural Sciences and Engineering Research Council of Canada (NSERC). Gaoqiang Yang acknowledges the support of National Natural Science Foundation of China (Grant No. 751202400064), Hunan University under Research Start-up Fund (Grant No. 521118010179), and “100 Talents Project” of Hunan Province.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have not disclosed any competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, G., Lee, C., Qiao, X. et al. Advanced Electrode Structures for Proton Exchange Membrane Fuel Cells: Current Status and Path Forward. Electrochem. Energy Rev. 7, 9 (2024). https://doi.org/10.1007/s41918-023-00208-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41918-023-00208-3