
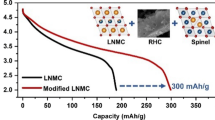
Abstract
Lithium-rich materials (LRMs) are among the most promising cathode materials toward next-generation Li-ion batteries due to their extraordinary specific capacity of over 250 mAh g−1 and high energy density of over 1 000 Wh kg−1. The superior capacity of LRMs originates from the activation process of the key active component Li2MnO3. This process can trigger reversible oxygen redox, providing extra charge for more Li-ion extraction. However, such an activation process is kinetically slow with complex phase transformations. To address these issues, tremendous effort has been made to explore the mechanism and origin of activation, yet there are still many controversies. Despite considerable strategies that have been proposed to improve the performance of LRMs, in-depth understanding of the relationship between the LRMs’ preparation and their activation process is limited. To inspire further research on LRMs, this article firstly systematically reviews the progress in mechanism studies and performance improving attempts. Then, guidelines for activation controlling strategies, including composition adjustment, elemental substitution and chemical treatment, are provided for the future design of Li-rich cathode materials. Based on these investigations, recommendations on Li-rich materials with precisely controlled Mn/Ni/Co composition, multi-elemental substitution and oxygen vacancy engineering are proposed for designing high-performance Li-rich cathode materials with fast and stable activation processes.
Graphical abstract
The “Troika” of composition adjustment, elemental substitution, and chemical treatment can drive the Li-rich cathode towards stabilized and accelerated activation.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In recent decades, concerns over the energy crisis and the related environmental issues have dramatically increased the demand on electric vehicles (EVs) and hybrid electric vehicles (HEVs). The performance of EVs and HEVs is mainly determined by the high-energy-density Li-ion batteries (LIBs) [1]. The energy density of a LIB depends on its specific capacity and operational voltage, which are currently mainly limited by the cathode materials. However, the state-of-the-art commercialized LIBs cathodes such as LiMn2O4 (140 mAh g−1) [2], LiFePO4 (170 mAh g−1) [3], LiCoO2 (150 mAh g− 1) [4] and LiNixMnyCozO2 (NMC, where N, M, C represent Ni, Mn and Co, respectively, and x + y + z = 1) (around 200 mAh g− 1 depending on components) [2] working in the 3.0–4.0 V (vs. Li + /Li) potential range can hardly satisfy the demand. Although researchers are developing alternative new LIB systems, they are still far from practical applications [5,6,7,8,9,10,11,12]. Among LIB cathode materials, lithium-rich materials (LRMs) are promising candidates for EVs due to their superior specific capacity (over 280 mAh g− 1) [13], low cost, and excellent energy density (over 1 000 Wh kg− 1) [14]. As a unique phenomenon of LRMs during the initial charge of over 4.5 V [15], the activation process provides extra capacity compared to conventional layered cathode materials. Activation of the LRMs involves an oxygen anion redox reaction and Li extraction from the Li2MnO3 phase. These reactions determine the electrochemical performance such as specific capacity, cycling stability and rate capability of LRMs. However, the activation process is thermodynamically slow with severe structural degradation, which drastically hinders the commercialization of LRMs. To understand the process of activation, great efforts have been made to reveal the structure and electrochemical change during activation, but there are still many debates [16,17,18]. At the same time, various methods are proposed to eliminate the major obstacles against practical application of the materials, namely low Coulombic efficiency, poor cycling stability, voltage decay, and voltage hysteresis. These strategies can also influence the rate, extent, and stability of activation; unfortunately, they are rarely noticed. There are excellent review articles focusing on the various aspects of LRMs including the activation mechanism, the methods to suppress the irreversible oxygen loss, or the strategies to improve the stability [19,20,21,22,23,24]. However, to our knowledge, there is a lack of reviews that specifically highlight the strategies to control the activation process of LRMs, which plays an important role in determining the specific capacity, Coulombic efficiency, cycling performance, voltage retention, and voltage hysteresis.
This review aims to provide new insights on the understanding of the activation process and discuss the strategies that can effectively accelerate and stabilize the activation, in terms of compositional control, elemental substitution, and chemical treatment. We further propose guidelines of activation-tuning strategies, future perspectives, and research directions for Li-rich cathode materials. Rational selection/combination of chemical composition control, multi-elemental substitution and oxygen vacancy engineering are promising strategies toward LRMs with high capacity, fast activation, and stable cycling performance.
2 Structures, Activation Mechanism and Challenges of LRMs
2.1 Crystal Structure of LRMs
Lithium-rich cathodes, which are usually notated as xLi2MnO3·(1 − x)LiMnO2 or Li1+xM1−x O2 (M = Mn, Ni or Co), were first developed from the compound Li2MnO3 by Thackeray and co-workers in 1991 [25]. Similar to other intercalation cathode materials, the structure significantly influences the performance of LRMs. As the notation of LRMs xLi2MnO3·(1 − x)LiMO2 implies, this kind of material is a combination of two phases [15, 26, 27]. The first phase is Li2MnO3 (sometimes denoted as Li[Li1/3Mn2/3]O2) with a monoclinic structure (space group C2/m), in which 1/3 of the Mn sites are occupied by Li. The second phase is LiMO2 (M = Mn, Ni or Co) with a rhombohedral structure (space group R\(\overline{3}\)m). As shown in Fig. 1a, these two phases have similar structures, a close-packed cubic oxygen array, and all the octahedral sites are occupied by lithium (Li), transition metals (TMs) or Li/TM. Therefore, the two phases are highly compatible with each other [27]. Figure 1b demonstrates the combination of LiMO2 and Li2MnO3 phases. These two different systems have a similar layered structure, but the latter can provide extra Li source [26]. Figure 1c shows the different arrangements of oxygen arrays in conventional layered oxide and Li-rich Li2MnO3 oxide [15].

Copyright © 2013, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. b Combination of LiMO2 and Li2MnO3 phases in LRMs. Reprinted with permission from Ref. [26]. Copyright © 2015 Elsevier B.V. c Oxygen array arrangement in LiMO2 and Li2MnO3. Reprinted with permission from Ref. [15]. Copyright © 2013, American Chemical Society. d Typical XRD pattern of LRMs. Reprinted with permission from Ref. [28]. Copyright © 2015, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. e HAADF and ABF STEM images of the R\(\overline{3}\)m LiTMO2 structure and C2/m Li2MnO3 structure projected along the [100]mon direction. Insets: simulated local atomic arrangement. Reprinted with permission from Ref. [27]. Copyright © 2013, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim
a Schematic crystal models of the R\(\overline{3}\)m LiTMO2 and the C2/m Li2MnO3 phases. Reprinted with permission from Ref. [27].
To date, it is still debatable whether the crystal structure of LRMs is two phases co-existing as a solid solution or separate phases in different local areas. Earlier, X-ray diffraction was applied to characterize the structure of LRMs. From the typical X-ray diffraction (XRD) pattern shown as Fig. 1d, the main peaks can fit well with the R\(\overline{3 }\)m space group, while the broad and weak diffraction peaks in the 20°–25° range can indicate the existence of the space group C2/m [28]. However, there are debates on the source of these small peaks. Some researchers believe that these peaks indicate LiMn6 cation ordering in the Li2MnO3 phase, but the others claim that this indexes lithium ordering in the transition metal layer. As XRD can only show the overall crystal structure but cannot distinguish a solid solution from separated phases, more advanced characterization techniques are needed to analyze the local atomic arrangement of LRMs.
In recent years, a series of advanced techniques such as high-resolution transmission electron microscopy (HRTEM), high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM), annular bright-field (ABF) STEM, diffraction scanning transmission electron microscopy (D-STEM) and electron energy-loss spectroscopy (EELS) were applied, attempting to directly visualize the local structure of LRMs. Although considerable research effort has been made in this field, there is still no clear conclusion on the local structure. Different research groups had contradictory explanations for the results. Some researchers gave evidence supporting the solid solution model [17, 29,30,31]. For example, Ferreira et al. only observed the monoclinic C2/m phase in their Li1.2Ni0.2Mn0.6O2 sample, and confirmed the absence of an R\(\overline{3 }\)m phase with D-STEM, indicating a single phase and a solid solution in this material. They believed that the broad weak peaks in the 20°–35° range in the XRD pattern are the result of thin planar defects in the (001) plane [31]. On the contrary, some researchers observed separated phases with HAADF-STEM or ABF-STEM [27, 30, 32]. For instance, Zhou et al. found individual domains of both monoclinic and rhombohedral symmetry within the same grain in their Li1.2Mn0.567Ni0.166Co0.067O2 sample, as shown in Fig. 1e, suggesting the co-existence of two separated phases [27]. Furthermore, many researchers tried to explain the reason for the different results mentioned above. It has been found that the structure of LRMs can be affected by multiple factors, such as composition, lithium ion content, synthesis temperature and cooling rates [33,34,35,36,37,38,39,40,41]. The widely accepted theory is that the composition, especially the content of lithium ions, is one of the most important factors controlling the local structure. It is believed that a higher ratio of lithium and transition metals, around 2:1, can make the structure of the LiMO2 phase shift to that of Li2MnO3. In this case, the overall structure will tend to form a homogeneous solid solution. However, a lower ratio of Li and TM cannot maintain periodic ordering of Li and TM, resulting in local separation of phases [33, 38]. Currently, the consensus tends to the solid solution model, but due to the complexity of LRMs, the study on their local structure needs to be continued in the future.
2.2 Activation Mechanism of LRMs
The LRMs family commonly has capacity larger than 250 mAh g−1 with a high-voltage plateau of 4.5 V. This is contributed by the unique activation process during the first charge–discharge cycle. Many researchers have attempted to determine the mechanism of this phenomenon. As shown in Fig. 2a, the initial charge cycle can be divided into two regions: the first region is 2.0–4.5 V, while the second is 4.5–4.8 V [42]. Within the first region, the TMs in the LiMO2 phase are oxidized to the M4+ valence state, along with the extraction of lithium ions. However, the Li2MnO3 phase cannot be oxidized, as the manganese is already in the Mn4+ valence state. Due to the difference in TM content, materials with variable composition can deliver various amounts of capacity at this stage. Meanwhile, in the second higher voltage stage, the oxygen in the lattice is oxidized. Simultaneously, Li ions in the Li2MnO3 phase are extracted [43]. Same as the first charge process, the mechanism of first discharge is also complex. During the first discharge, the Li ions re-insert into the cathode, and the TMs are reduced and revert to their initial state. However, not all the oxidized oxygen can be reduced in this process. Instead, some of the Mn4+ ions are reduced to Mn3+. During the first discharge, Li ions cannot fully return to the lattice, which also explain the high irreversible capacity in the first cycle. Operando neutron diffraction clearly demonstrated the Li+ movement during the initial electrochemical cycle of LRMs. During the initial charge, the capacity of the slope area is mainly contributed by the extraction of Li ions from the Li layer, while the capacity of the plateau region is provided by Li extraction from TM layers. Later in the first discharge, the Li can easily reinsert into the Li layers, but is difficult to intercalate back to TM layers (Fig. 2b) [44].

Copyright © 2018, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. b Electrochemical charge/discharge profile with corresponding lithium occupancy at different states of delithiation/lithiation for high Li-rich (left) and low Li-rich (right). Reprinted with permission from Ref. [44]. Copyright © 2016, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. c In situ Raman spectra directly demonstrating oxygen redox. Reprinted with permission from Ref. [46]. Copyright © 2018, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. d Schematic mechanism of activation reaction for Li[NixLi(1−2x)/3Mn(2−x)/3]O2 during the initial charge and discharge cycle (top) and charge–discharge curve (bottom). Reprinted with permission from Ref. [64]. Copyright © 2014, American Chemical Society. e Illustration of different proposed activation mechanisms. Reprinted with permission from Ref. [58]. Copyright © 2019, Springer Nature Limited. f Schematic of the structural, electron donation and Mn–O electron exchange evolutions during the charging of Li2MnO3. Reprinted with permission from Ref. [57]. Copyright © 2020, Elsevier
a Typical charge/discharge curve of LRM. Reprinted with permission from Ref. [42].
Since the first study on LRMs, researchers have made tremendous effort to explain the origin of activation process. However, the mechanism is still debated. There are various explanations for the activation of LRMs, which can be classified as six types of theories: oxygen dimer theory [45,46,47,48], oxygen ejection theory [16, 17, 49,50,51], trapped O2 gas redox theory [52,53,54,55], electron hole theory [13, 56], Mn high valence redox theory [57,58,59,60] and side reaction theory [61,62,63,64,65].
In the early reports, researchers believed the formation of oxygen dimers such as O2−/O22− or O2−/O2n− and their reversible redox originates the activation of LRM [45, 46]. As the essence of activation, many researchers have found evidence that the oxygen redox involves the formation of oxygen dimers such as O2−/O22− or O2−/O2n− [46,47,48]. For example, Fig. 2c clearly visualizes the oxygen redox reaction during the cycling [46]. Similarly, X-ray photoemission spectroscopy (XPS) and electron paramagnetic resonance (EPR) can directly identify oxygen dimers (O2)n− species (O–O distance 2.42 Å) [47]. Although these oxygen dimers are highly unstable and tend to be further oxidized or disproportioned into oxygen gas, researchers believe most of these dimers can reversibly return to the LRM lattice.
However, not all the researchers observed the existence of oxygen dimers, and thus proposed the oxygen ejection mechanism. Some studies stated that the initial capacity loss is caused by the simultaneous removal of O from the structure during the first charge [49]. Earlier studies confirmed that the O2 originates from the oxygen near the surface of the lattice during first charge [17]. The removal of oxygen led to oxygen vacancies, which in turn, decreased the number of available Li+ ion sites. Therefore, not all the Li+ ions can reversibly reinsert into the lattice. Furthermore, with these oxygen vacancies, the TMs tend to migrate into the Li layer, initiating the transformation to a spinel structure, which finally results in the breakdown of the crystal lattices [16]. This is a self-accelerated process, as the oxygen vacancies can reduce the TMs’ migration free energy, while the TMs’ migration can in turn promote the oxygen vacancy formation [50]. However, this theory has difficulty in explaining the lattice evolution by activation, as not all the TM migration is related to the activation. TM migration can occur in un-activatable cathode materials without the activation process, but the activation of LRM can accelerate the TM migration [51]. More importantly, this oxygen ejection cannot fully explain the high capacity of LRMs, as the charge is far beyond the amount that the released O2 gas can provide [53, 55]. There is no doubt that the extra charge capacity is provided by oxygen oxidation during charging, but if all the oxidized oxygen is irreversibly lost from the lattice, the reduction of TMs on discharging cannot fully compensate the charge during discharge.
To further explain the reversible high capacity of LRMs, the trapped O2 gas redox mechanism was proposed. This mechanism partially affirmed the oxygen ejection mechanism, but revealed further details. In this theory, although the formation of O2 gas is a fact, not all the O2 gas is irreversibly released during the activation. Instead, the majority of O2 is reserved in the bulk, and can participate in the subsequent reduction reaction. Thus, only the oxygen near the material surface will be released, whereas most oxygen in the lattice is reserved and can participate in the subsequent redox [53, 55]. High-resolution resonant inelastic X-ray scattering (HR-RIXS) spectroscopy, soft X-ray absorption spectroscopy (SXAS) and solid-state 17O magic angle spinning (MAS) NMR spectroscopy can directly prove the existence of molecular O2 gas after initial charging of LRMs, and 13% of the total lattice O participates in O2 formation. However, only 3% is detected in released O2 gas. Therefore, the majority of O2 must be trapped in the lattice. During the charge, the O2 in the bulk contributes to most of the capacity (~ 75%) when charged over 4.6 V. At the end of charge, the free O2 is directly observed instead of peroxo O22−, while the O2 is lost after discharge, indicating that this process is reversible. On the one hand, with the O extracted from the lattice at the > 4.6 V region, there is an irreversible rearrangement of TM honeycomb ordering. In this process, TMs migrate to Li sites, while the Li tends to cluster during the discharge. Such lattice rearrangement will leave the space to accommodate O2. These O2 are close to the TM redox center, and therefore can be reversibly reduced to O2− during the discharge. On the other hand, the O released from the surface will lead to layered-to-spinel phase transformation, which prevents the further loss of lattice oxygen [54]. However, such oxygen behavior is not the same in pure Li2MnO3, which is more representative to the activation study, as the capacity of this material is almost solely related to the oxygen redox. All the O in Li2MnO3 is coordinated with 4 Li ions and 2 TM ions. During the charge of Li2MnO3, the Li ions are extracted, accompanied by oxygen oxidization. These oxidized oxygen ions are undercoordinated and unstable, and can be easily released from the lattice. The soft X-ray spectroscopy, NMR and scanning transmission electron microscopy (STEM) reveal that O2 gas formation is the only way of charge compensation. Similar to other LRMs, the oxygen loss from the surface of Li2MnO3 is also not reversible. But in Li2MnO3, the oxygen loss will result in a highly disordered phase on the surface, rather than a spinel phase [52]. Although the trapped O2 gas redox mechanism can explain the reversible capacity, it cannot clarify the origin and location of oxygen redox. For example, some researchers believe that the TMs’ migration into Li layers lead to oxygen displacement, which is partially irreversible. Such displacement is the origin of oxygen redox, but it is only an oxygen disordering, but not a loss from the lattice [60]. More interestingly, recent theoretical calculations indicate that bulk oxygen redox occurs in the LiMO2 phase rather than the Li2MnO3 phase, while the Li2MnO3 is merely a source of extra Li. However, as this mechanism proposal is only based on the computational study, experimental verification is needed in the future research [59]. In addition, it is obvious that the activation behavior in Co/Ni containing LRM and pure Li2MnO3 is different. This is likely related to the O–Co/O–Ni bonding. The change in oxygen surrounding environment and electron configuration will change the way by which the electron extracted from the oxygen, as well as the way of oxygen movement.
With the support of advanced characterization techniques, researchers can see more details of the oxygen behavior during the activation of LRMs. Based on the new findings, the electron hole mechanism is proposed. This theory not only explains the reversible high capacity of LRMs, but also points out the origin of electrons involved in the activation. For example, during the initial charge of the material, when the voltage is \(\leqslant\) 4.5 V, oxygen is released from the lattice, although it is mostly in the form of CO2, rather than expected O2 gas. The source of such CO2 is not the electrolyte. Oppositely, the O atom in the CO2 is released from the lattice. However, the amount of released oxygen can only provide 0.05 e− per formula unit, while the actual electron involved in the activation is 0.55 e−. Thus, the other 0.5 e− must be provided by an alternative electrochemical process. Firstly, the Raman spectra did not show any evidence of O–O bonds with ∼1.45 Å length, which excluded the formation of O22− peroxide species. Secondly, K-edge XANES spectra of Mn, Ni, Co and O proved that there is no further oxidation on TMs beyond 4 + valence, but the MnO6 octahedrons are distorted. Furthermore, RIXS reveal that electronic environment around the oxygen changed significantly when the material is charged over 4.5 V. Therefore, the extra 0.5 e− is provided by electron holes on the oxygen, and the electron holes are located in the oxygen of Mn4+–O bonds. These electron holes on oxygen are reversible, whereas the oxygen released from the lattice is not reversible and will cause low initial Coulombic efficiency [13]. The DFT calculation also supports the electron hole theory. However, the results indicate that the electron originates from the Li–O–Li bond rather than the TM–O bond. In the simulation model, the excess Li in the LRMs will lead to a specific oxygen environment, in which one oxygen coordinates with four Li ions and two TM ions. In this environment, the unit has a linear orphaned Li–O–Li configuration, and the electron in this configuration is labile. When the material is deeply delithiated, the labile electron will be extracted from the Li–O–Li configuration, and leave electron holes [56].
In addition to the commonly accepted oxygen redox mechanism, there is also a highly debated Mn high valence redox mechanism. Recently, first-principle calculations suggested that Mn may experience oxidation beyond 4 + during activation of LRMs, as shown in Fig. 2e. In these calculations, Mn ions can redox between 4 + and 7 + , accompanied by simultaneous Mn migration from octahedral sites to tetrahedral sites or formation of oxygen dimers [58]. Similar theoretical calculations (Fig. 2f) suggest that Mn high valence redox can also occur in pure Li2MnO3. During charging, the ability of oxygen redox decreased with the reduced Li content in Li2MnO3, while the probability of Mn4+/Mn6+ or Mn4+/Mn7+ redox is increased at the same time. Then, the high valence Mn6+ or Mn7+ oxidizes their surrounding O ions and results in molecular O2 and Mn4+ [57]. However, practical experiments ruled out the Mn4+/Mn7+ redox mechanism in Li2MnO3 by quantitatively analyzing the capacity contributed by the activation process, as the charge change in Mn4+/Mn7+ redox cannot match with the actual capacity observed during cycling [59]. In addition, the most recent work reveals the change of O and Mn valences using operando XANES. This work provides further evidence against the hypothesis that Mn in LRMs can be oxidized beyond 4 + valence. It is obvious that Mn high valence redox theory has many conflicting reports. The reason of such variation may relate to the difference in LRM composition that researchers used, thus this theory needs to be further explored.
The electrolyte side-reaction mechanism is also a theory to explain the activation of LRMs. As a common phenomenon, LRMs can react with the electrolyte during the cycling [63]. In particular, the charging of LRMs will result in oxygen containing species forming on the material surface [62]. These byproducts, such as Li2CO3, can be reversibly formed and decomposed during the cycling, thus significantly contributing to the capacity [61]. In addition to the electrolyte oxidation, it is also reported that the activation of Li2MnO3 is caused by the reaction between cathodes and electrolytes, which finally leads to exchange of Li+ and H+ [65]. Both electrodes can react with the electrolyte during the cycling, forming a series of intermediate products such as H+, Li2CO3 and LiOH, and finally increase the capacity of LRMs. Such reactions are shown in Fig. 2d [64]. As the side reaction between an electrode and an electrolyte is difficult to be characterized, this theory still needs to be further studied.
It should be noted that various studies usually observed different phenomena in the experiments or theoretical calculations, which result in direct conflicting conclusions. However, in the above-mentioned works, most of the LRM objects are in varying chemical composition. As a complex type of materials, different ways of TM and O bonding will lead to significant changes in oxygen redox behavior. Therefore, the studies based on the same template materials are more meaningful for a reliable comparison. Despite the controversy regarding the mechanisms, oxygen redox is the determining process of the LRM activation because it decides the capacity and reversibility of the LRMs. Therefore, the adjustment of the activation extent and reversibility, in principle, relies on the tuning on oxygen redox behavior.
2.3 Challenges on the Practical Application of LRMs
Although LRMs have capacities far more than the traditional cathode materials for LIBs, there are still challenges hindering their practical application, such as lower capacity than the theoretical values, poor initial Coulombic efficiency, capacity/voltage decay and voltage hysteresis.
Low practical capacity. Depending on the composites, the LRMs have theoretical capacities of ~ 400 mAh g−1. However, only ~ 60%–75% (250–300 mAh g−1) of the theoretical capacities are practically achieved, whereas traditional cathode materials, such as LiMn2O4 [2] and LiFePO4 [3], already reached their theoretical capacity. This is partially because the capacity of layered cathode materials is limited by their structures. Taking typical layered LiCoO2 as an example, it has alternating Li layers and TM layers in its lattice. When delithiating, the spacing between TM layers will increase due to electrostatic repulsion. Eventually, its lattice structure will collapse under deep delithiation. Thus, this material can only deliver 150 mAh g−1 reversible capacity out of 274 mAh g−1 theoretical capacity, which means 55% of the Li+ ions are released [4, 66]. In contrast, LRMs are not a typically perfect layered material, as there is extra Li in the TM layers as shown in Fig. 1a [27]. This structure is more resistant to the electrostatic repulsion during delithiation, especially when the Li from the TM layers is extracted [67]. Thus, theoretically, LRMs are capable of releasing more Li than LiCoO2. However, the actual capacity of LRMs is far behind its theoretical value. For example, the most commonly studied LRM material, Li1.2Mn0.6Ni0.2O2, has a theoretical capacity of 378 mAh g−1, but the reported practical capacity is usually 200–300 mAh g−1, meaning only ~ 50%–75% of the Li is extracted [68, 69]. This can be attributed to the oxygen evolution during the activation, as the oxygen loss will induce TM diffusion, which blocks the Li extraction path [70].
Low initial Coulombic efficiency. During activation of LRMs, there are two irreversible processes: the oxygen ions are partially lost from the lattice, and some TMs migrate to the Li layer. Both facts can hinder the Li re-insertion into the lattice, causing low initial Coulombic efficiency [44, 71]. Although part of the initial capacity loss is attributed to the formation of a solid-electrolyte interface (SEI), the total capacity loss is far beyond that of the traditional layered LIB cathode materials, indicating the majority of the capacity is still sacrificed by the oxygen loss [72, 73]. Thus, stabilized activation with suppressed oxygen loss and TM migration can effectively improve the Coulombic efficiency.
Capacity/voltage decay. In addition to capacity and efficiency issues, the oxygen loss and TM migration during activation of LRMs can also cause continuous capacity/voltage decay with cycling [74]. Within these two, TM migration is the main factor triggering the capacity/voltage decay, while oxygen evolution is the accelerator [51]. Specifically, TM migration can occur in the absence of oxygen redox. Such migration will cause a phase transition from layered to spinel or quasi-spinel structure, which have a lower charge/discharge voltage [15, 16, 75]. At the same time, the oxygen loss and TM reduction in the lattice will weaken the TM–O bonds, especially the Ni–O bond, which leads to the TM migration to Li sites [76, 77], while Co can accelerate this process [78]. Moreover, after irreversible oxygen loss from the lattice, Mn4+ will be reduced to Mn3+ to compensate the charge during discharge. As Mn4+/Mn3+ redox has lower potential than that of O−/O2− or O2/O2−, the overall voltage of LRMs is decreased. Simultaneously, the loss of oxygen reduces the covalency of TM–O bonds, and suppresses the oxygen redox. As a consequence, the capacity of LRMs is decreased [79, 80].
Voltage hysteresis. LRMs suffer from massive voltage hysteresis between charge and discharge, which dramatically reduces their energy density. Although the mechanism of such a problematic phenomenon is still unclear, most researchers believe it is related to the TM and oxygen activity during the activation [54, 81,82,83,84,85,86,87,88,89]. Early research attributed the hysteresis to the TM migration and lattice restructuring [88, 89], but more recent studies demonstrate that oxygen redox is the key reason for hysteresis. Some researchers considered that voltage hysteresis originates from the energy difference between the formation and breaking of O–O or O–TM–O bonds [82, 86]. Others, however, believe the oxygen evolution during activation will cause irreversible lattice rearrangement and re-coordination of the oxygen environment, which in turn results in a decrease of voltage during discharge [54]. The most recent research decoupled the correlation of TM migration and hysteresis, but confirmed that hysteresis is highly related to the charge transfer in TM–O bonds. The voltage hysteresis will decrease with the increased TM–O covalence [81].
From the current challenges of LRMs, it is obvious that all these problems with LRMs are related to their activation process, especially the oxygen evolution and TM migration. Therefore, the strategies to optimize the activation process are important to design better LRMs.
3 Strategies to Optimize the Activation Process
Despite the fact that Li-rich cathode materials have excellent specific capacity, the Coulombic efficiency, capacity/voltage cycling stability, voltage hysteresis and activation kinetics hinder the commercialization of this kind of material. Many strategies are focused on mitigating the problematic poor rate performance, low Coulombic efficiency, severe voltage/capacity drop and voltage hysteresis of LRMs during cycling [24, 90,91,92,93,94]. However, it is rarely noticed that these methods can also change the activation behavior of LRMs. In the following section of this review, the influences of modification strategies, including composition adjustment, elemental substitution, and chemical treatment on the activation of LRMs, will be extracted from the literature and discussed in detail. Accordingly, a guideline of strategy selection towards accelerated and stabilized activation for high-performance LRMs will be proposed.
3.1 Compositional Adjustment
The performance of LRMs is related to their chemical composition. The commonest LRMs consist of manganese (Mn), nickel (Ni) or cobalt (Co), and each transition metal has different effects on the lattice structure and electrochemical behavior of the cathodes.
As one of the major transition metals in Li-rich NMC materials, the content of Mn not only determines the ratio of the Li-rich phase Li2MnO3 in the material but also determines the pathway of activation. The activation pathway can be analyzed by the in situ electrochemical quartz crystal microbalance (EQCM) technique. By comparing pure hexagonal LiNi0.3Co0.3Mn0.4O2, pure monoclinic Li2MnO3 and the Li-rich composite 0.5Li2MnO3·0.5LiNi0.3Co0.3Mn0.4O2, the importance of Mn can be demonstrated. In the Li-rich composite material, the hexagonal and monoclinic phases exhibit synergistic effects and therefore yield a significantly higher capacity than either parent phase. Such synergistic effects can also alter the activation process. During activation, the Li2MnO3 phase in the LRM decomposed into Li+, O22− and MnO2, rather than Li2O and MnO2 in pure Li2MnO3. As shown in Fig. 3a–c, this different activation pathway provides an additional source of Li+ that contributes to the extra capacity [95]. In addition to the synergistic effect, Mn also shows an unexpected electrochemical behavior during the activation of LRMs. As shown in Fig. 3d–h, in the initial charging process, Mn4+ in the Li[Li0.2Ni0.16Mn0.56Co0.08]O2 Li-rich materials partially reduced to Mn3+ at voltages > 4.5 V, instead of keeping its valence as the previous studies had suggested. This unique Mn valence evolution is in opposition to the general electrochemical reaction, where the valence increases as voltage increases [96]. The first-principles calculations demonstrate that during activation the oxygen O2− in the lattice is oxidized to an O2δ− dimer to compensate the charge increase due to delithiation. The Mn4+ remains unchanged at the early stage, acting as an electrocatalyst. However, with the increased extent of O2δ− dimer oxidization, the dimer releases electrons back to the Mn ions, which ultimately causes the reduction of Mn4+ to Mn3+. The electron loss from Mn and electron gain of O are illustrated in Fig. 3i and j [97]. All the above reports indicate that Mn is an essential element in common Mn-based LRM, as it triggers the anionic redox reaction of oxygen.

Copyright © 2019, American Chemical Society. d Initial cycling curve and voltage points for XANES measurement; e–h XANES spectra for O and Mn pre-edge at different voltages. Reprinted with permission from Ref. [96]. Copyright © 2019, American Chemical Society. i Average electron loss from the Mn and gain of oxygen at different stages of delithiation; j average magnetic moment values at different stages of delithiation. Reprinted with permission from Ref. [97]. Copyright © 2019, American Chemical Society
a Proposed electrochemical reactions in initial charge; b proposed electrochemical reactions in initial discharge; c illustration of proposed electrochemical behavior at high voltage above 4.5 V. Reprinted with permission from Ref. [95].
Ni is another key component in LRMs. In 2014, Wang et al. systematically studied the role of Ni by controlling its content in their Co-free LRMs. With increase in Ni content in their Li4/3–2x/3Mn2/3–x/3NixO2 (0.09 \(\leqslant\) x \(\leqslant\) 0.2), the materials show distinctly faster activation in the initial cycle. This is shown by the extension of the high-voltage activation plateau with an increase in the Ni to Mn ratio and faster reaching to the highest capacity (Fig. 4a and b). Through computational simulation, it is found that Ni in the Li layers can significantly reduce the energy barrier of oxygen evolution, while the oxygen removal can in turn cause Ni to diffuse into the Li layers. The whole process is like a chain reaction. Therefore, a little increase in Ni content can result in a significant increase in the activation rate. But with an appropriate ratio of Ni and Mn, the activation rate, capacity and structural stability can be balanced, as illustrated in Fig. 4c [28, 41]. Recent research investigated the role of Ni in LRMs within the model lattices Li1.15Ni0.47Sb0.38O2 (L1.15NSO), Li1.10Ni0.53Sb0.37O2 (L1.10NSO), and Li4NiTeO6 (LNTO), which eliminated the influence from other transition metals such as Mn and Co. As is consistent with previous reports, Ni2+ to Ni4+ is the main active redox cation during lower voltage charging before activation. However, it is found that not all the Ni can be oxidized at this stage. Ni ions on the particle surface are reduced in the activation region (where oxygen redox occurs), but not fully oxidized to Ni4+, because the evolution of oxygen can partially reduce the Ni. The process is shown in Fig. 4d. Although this reaction can significantly suppress the generation of oxygen gas, such a low oxidation state of Ni on the particles surface can hinder the Ni4+ in the bulk being fully reduced back to Ni2+ during discharge. This inevitably results in decreased reversibility. Therefore, the surface of Ni-containing LRMs must be protected [98]. Due to the higher electronegativity of Ni, the Ni2+/Ni4+ redox couple can deliver a higher voltage than that of Mn3+/Mn4+. Higher Ni content can lead to a higher activation voltage plateau and lower capacity than that of lower Ni materials (Fig. 4e and f). This indicates that too much Ni may have a negative effect on activation. Although the explanation for this phenomenon is not the focus of this work, our conjecture lies in excessive amount of Ni inducing over-activation [99]. Despite the fact that increased Ni content can improve the Coulombic efficiency and operating voltage and cycling stability of the LRM, the capacity, especially the part contributed by activation, was significantly sacrificed as shown in Fig. 4g. They further pointed out (in Fig. 4h) that Ni has a higher and stabler redox potential, as it has a much lower energy gap than that of Mn and Co when oxidized to lose the outermost electron. Ni can also decrease the bond energy of TM3d–O2p and non-bonding O2p. This can promote the formation of O1 but suppress the O2 coordination environment, and increase the valence state of Mn and Co [100]. Interestingly, some of these studies used Co-containing LRMs, and their capacities are much higher than that observed in Co-free LRMs, indicating the important role of Co in increasing the capacity of LRMs.

a Initial charge/discharge curves of LRMs with different Mn/Ni ratios; b prolonged cycling performance of these materials; c influence of Mn/Ni ratios on structure evolution and electrochemical behavior of LRMs. Reprinted with permission from Ref. [41]. Copyright © 2014, Tsinghua University Press and Springer-Verlag Berlin Heidelberg. d Schematic diagram of Ni and O behavior on the surface of particles. Reprinted with permission from Ref. [98]. Copyright © 2020, American Chemical Society. e The impact of Ni content on capacity and f specific energy. Reprinted with permission from Ref. [99]. Copyright © 2018, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. g The change of electrochemical behavior with different Ni contents; h 3d orbitals electronic state of Mn/Co/Ni ions in LRMs at different voltages and corresponding band structures. Reprinted with permission from Ref. [100]. Copyright © 2019, Elsevier Ltd. All rights reserved
Co is another key transition metal determining the activation process of LRMs. In the earlier study of LRMs, Co was found be able to trigger a capacity-increase-upon-cycling (CIUC) phenomenon in their Li[CoxLi1/3−x/3Mn2/3−2x/3]O2 (x = 0.087, 0.100, and 0.118) LRMs. With higher Co content, the capacity increase was faster, indicating faster activation, as shown in Fig. 5a–d [101]. With theoretical calculations, it is revealed that Co can shorten the non-bonding oxygen bond, significantly promoting the oxygen redox reaction. Also, similar to Ni, such promotion in oxygen evolution can in turn facilitate the Co diffusion and eventually become a chain reaction. This is explained by theoretical calculations (shown in Fig. 5e) which demonstrate that the removal and oxygen evolution of oxygen significantly reduces the Mn and Co migration energy barrier. Thus, the activation of the Li2MnO3 phase can be remarkably accelerated with a small amount of Co doping by exhibiting much larger capacity (shown in Fig. 5f) [102]. In addition, Co can induce extra hybrid trigonal lattice arrangement, thus promoting the activation process. The materials with Co can achieve a 256 mAh g−1 discharge capacity in the initial cycle, while the materials without Co can only deliver ~ 100 mAh g−1 [103]. Later, Wang and co-workers systematically studied a series of LRMs with various Co contents. With increase in Co content, the voltage plateau of activation shifted to a lower voltage, and the capacity contributed by activation was increased. This result clearly demonstrates that Co can accelerate the electrochemical activation of LRMs. However, with Co, the TMs will rapidly migrate from octahedral sites to cubic sites in the lithium layer during the initial 30 cycles, resulting in voltage decay and spinel-like phase formation. Then, most of the TMs have moved from cubic sites to their original octahedral sites with the mechanism illustrated in Fig. 5g, and the voltage is stabilized [104]. The acceleration in activation is related to the fact that the overlapping area between O2− 2p and Co3+ 3d6 bands is much larger than that with Ni3+ 3d7 and Mn3+ 3d4, as shown in Fig. 5h. This eases the oxidization of O in the lattice, hence accelerating the activation. However, the poor stability of the Co4+/Co3+ redox couple can eventually cause rapid voltage fading [105]. In general, LRMs with higher Co content (Fig. 5i) have an accelerated activation rate while materials with higher Ni (Fig. 5j) have a higher cycling stability. In addition to this unique function, the ratio of transition metals also has a profound influence on the activation process of LRMs. Changing the Ni/Co ratio can alter the lattice structure and cation arrangement in the LRMs, as shown in the XRD patterns in Fig. 5k. Materials with higher Ni content have a wider Li+ diffusion path due to the larger ionic radii of Ni than Co, which can lead to higher rate performance [106]. Notably, in all the Co containing LRMs discussed above, the activation rate increased with increase in Co content. Materials with higher Co content can be fully activated to their maximum capacity in fewer cycles than those with lower Co content. However, the cycling stability of those with a higher Co ratio show a deteriorative trend.

Copyright © 2014, Elsevier Ltd. g Schematic of TM ions migration. Reprinted with permission from Ref. [104]. Copyright © 2018, Elsevier Ltd. h Comparison of the band diagrams of Co/Ni/Mn-based layered oxides. Reprinted with permission from Ref. [105]. Copyright © 2006, Elsevier B.V. i Charge/discharge curves of LRMs with higher Co content; and j with lower Co content; k corresponding XRD patterns of these two materials. Reprinted with permission from Ref. [106]. Open access, Copyright © 2018, Electrochemical Society
a–d The electrochemical of LRMs with different Co contents. Reprinted with permission from Ref. [101]. Copyright ©2014, The Royal Society of Chemistry. e Change of energy barriers of TM ions with oxygen removal in Co containing LRMs; f cycling performance of Co containing LRMs. Reprinted with permission from Ref. [102].
During recent years, with the increase in concern regarding environmental damage, people are trying to avoid using Co in battery materials. Therefore, attempts to replace expensive and toxic Co with iron (Fe) have become a hot topic in the LRMs area due to the low-cost and nontoxicity of Fe. This family of materials usually contain Mn, Ni and Fe, so they are also known as MNF LRMs [107, 108]. The Fe3+/Fe4+ redox couple proved to be involved in the electrochemical reaction below 4.5 V. During the initial cycling, as shown in Fig. 6a and b, it is clear that Co containing Li1.2Ni0.13Co0.13Mn0.54O2 has a much higher capacity than that of Fe containing Li1.2Ni0.13Fe0.13Mn0.54O2, especially regarding the charge capacity contributed by activation (250 mAh g−1 of Co-based Li-rich and 180 mAh g−1 of Fe-based Li-rich). Thanks to the lower activation extent, as proven in Fig. 6c, Fe-based LRMs show better capacity retention during prolonged cycling. Moreover, during the activation, the conversion of the Li2MnO3 phase to the spinel phase is significantly retarded in the Fe-based LRM. The mechanism is still not clear, but it is speculated that the larger ionic radius of Fe3+ compared to that of Co3+ can impede the ion’s migration during activation, hence preventing the formation of the spinel structure [109]. In Fe containing LRM Li1.2Mn0.5Ni0.2Fe0.1O2, although the initial discharge capacity can achieve 226 mAh g−1, similar to that of common Li1.2Mn0.55Ni0.15Co0.10O2 (232 mAh g−1), the majority of the capacity is contributed by the Ni2+/Ni3+ and Fe3+/Fe4+ redox couples. The activation can only provide ~ 60 mAh g−1. XRD, XAFS, XANES and EDS analysis proved that the activation reaction, and the oxygen evolution, only occur in the Li2MnO3 phase as the number of Mn–O neighbors decreased upon the cycling, while the number of Ni–O and Fe–O neighbors remains unchanged (Fig. 6d). However, the activation of the Li2MnO3 phase did not lead to oxygen gas generation as NMC LRMs do. Instead, it converted to LiMnO2−x and Li2O2/Li2O. During the activation, only the Ni–O bond is expanded, while the Mn–O and Fe–O environment has a negligible variation (Fig. 6e). Such changes caused the lattice expansion, which in turn, resulted in a voltage change, as the expansion of the lattice can reduce the electrochemical potential of Li [110].

Copyright © 2016, Elsevier B.V. d Interatomic distances for Mn–O, Ni–O and Fe–O at different cycles; e number of near neighbors for Mn–O, Ni–O and Fe–O at different cycles. Reprinted with permission from Ref. [110]. Copyright ©2018, Electrochemical Society. f Voltage profile for the Li2Ru1−ySnyO3 with different Li extraction contents and y values; g the voltage profile upon cycling, capacity retention (the lower left inset) and rate capability (the top right inset) for a Li2Ru0.75Sn0.25O3; h the reversible (blue) and irreversible (green) Li-ions with different y values. Reprinted with permission from Ref. [47]. Copyright © 2013, Nature Publishing Group. i In-situ Raman spectra of LRMs; j cycling profile of Ru-based LRMs; k rate capability of the material. Reprinted with permission from Ref. [111]. Copyright © 2019, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim
a Initial cycles for Co containing LRMs; b initial cycles for Fe containing LRMs; c CV curves for Co and Fe containing LRMs. Reprinted with permission from Ref. [109].
In addition to Fe, some sporadic research on other transition ion replacements such as tin (Sn), ruthenium (Ru) and titanium (Ti) has also been tried in place of Co. These materials can still be electrochemically activated and exhibit O2−/O2n− anion redox at voltage above 4.5 V. Ru-based layered LRM was first reported by Tarascon et al. In this work, the TMs in the LRM are replaced by Sn and Ru, finally forming Li2Ru1−ySnyO3. As Sn is electrochemically inactive, the investigation on the single redox cation Ru4+/Ru5+ became feasible. In this composition, more Ru led to a higher reversible capacity (up to 250mAh g−1), while more Sn resulted in more stable cycling (Fig. 6f–h). Most importantly, the authors discovered that the reaction Ru6+ + O2− → Ru5+ + O− prevented the generation of O2 gas during the activation [47]. Ru-based LRMs can also form a rock-salt structure, in which Ru is in a 5+ valence state and cannot be further oxidized. Thus, the charge compensation below activation voltage is accomplished by the Ni2+/Ni4+ redox couple, while the O2−/O2n− redox can compensate the charge during activation. Notably, the lattice structure of this material is different from typical rock-salt LRMs. All the Ni, Ru and part of the Li occupy the 16d sites, while the 16c sites are fully filled by Li. Such a structure can facilitate the ordering of Li, leading to unobstructed Li diffusion paths and improving the kinetics. Also, the Li–O–Li arrangements are found in both the Li and TM layers in the lattice, which promotes oxygen evolution during activation. Additionally, Ru has a strong bond with O, only allowing O2− to be partially oxidized to O−, preventing it from being over oxidized and generating O2 gas during activation, proven by the highly reversible peak evolution of peroxo-oxygen in Fig. 6i. Therefore, this material exhibits high capacity (260 mAh g−1 at 10 mA g−1 current density), unnoticeable capacity/voltage decay over 150 cycles (94.2% capacity and nearly 100% voltage retention) and good rate performance, as shown in Fig. 6j and 6k [111].
Similarly, in Ti-based rock-salt LRMs, though the initial capacity is comparable to that of common NMC-based LRM, the capacity fading seems severer than the latter. Unlike common NMC-based LRMs, Mn ions in this Ti-based LRM are in the 3+ valence state, and the Mn3+/Mn4+ redox couple can compensate the charge difference at voltages < 4.5 V. In contrast, Ti is in the 4+ valence state and remains unchanged during the whole charge/discharge cycle. However, similar to NMC-based LRM, long cycling tests with different cutoff voltages reveal that the activation process is the main factor responsible for the capacity decay [111].
In summary, each composition of transition metals in LRMs has its unique function. Mn is the fundamental element of common LRMs. Firstly, it can trigger the activation of LRMs as the redox center at high voltage above 4.5 V. Secondly, Mn can introduce the synergistic effect between the Li2MnO3 phase and the LiTMO2 phase, altering the activation process by regulating the decomposition of the Li2MnO3 phase. This phase will decompose into Li+, O22− and MnO2, providing more Li which contributes to the extra capacity. Finally, Mn can absorb some of the electrons released by O during activation, increasing the reversibility during cycling. Ni has both positive and negative influences on LRMs. The major function of Ni is to elevate the operational voltage, stabilize the lattice structure, improve the Coulombic efficiency and promote the activation process when Ni content is low. Additionally, it has a similar effect to Mn in that it can absorb some electrons during activation and suppress the generation of O2 gas. However, if the Ni content is increased over a certain level, it will encumber the performance. Firstly, high Ni content will have the opposite effect to low Ni content and inhibit the activation. Secondly, as a result of the similar radius of Ni2+ and Li+, Ni2+ can easily occupy the Li+ sites. In fact, thermodynamically, the occupancy of Ni2+ in the Li 2b sites is preferable to the occupation of TM 4g sites in Li layers. The replacement of Ni with Li in TM layers is also possible. These Ni ions can block the diffusion pathways of Li, hindering the reaction. Co is widely believed to accelerate the activation of LRMs, as Co has a suitable outermost electron arrangement to ease the oxidization of O in the lattice. However, the Co4+/Co3+ redox couple has a low voltage and is unstable. This results in low operational voltages and rapid voltage decay of LRMs. Therefore, the balance between Mn, Ni and Co is critical to adjusting the performance toward a higher activation rate without sacrificing too much of the capacity and voltage of LRMs. Table 1 and Fig. 7 summarize the electrochemical performance of LRMs with variable Mn/Ni/Co composition. It is conspicuous that the LRMs with lager Mn/Ni content and appropriate amount of Co have higher overall capacity and activation contribution. In addition, Co composites can clearly improve the initial capacity. This is because the overlapping area between O2− 2p and Co3+ 3d6 bands is much larger than that with Ni3+ 3d7 and Mn3+ 3d4, and eases the oxidization of O in the lattice. Therefore, other elements with this property may also accelerate the activation of LRMs.

a The composition of LRMs with different Mn/Ni/Co ratios reported in the literature; b corresponding charge capacity and corresponding contribution of activation of such composition in the initial cycle
In addition to the commonest Mn, Ni and Co in LRMs, Fe, Ru, Sn and Ti have been studied by researchers to replace expensive and toxic Co. Fe has the advantage of low-cost and nontoxicity, and the capacity retention can be significantly improved in prolonged cycling. However, Fe can retard the activation of the Li2MnO3 phase, and result in lower capacity than NMC-based LRM. Thus, the future research of MNF-based Li-rich cathodes needs to be focused on enhancing the activation. Ru is a superior element for LRMs in both the layered and rock-salt structure. The most important advantage of Ru is to significantly promote activation while simultaneously eliminating the problematic O2 gas generation due to the strong bond between Ru and O. However, the high price of Ru can hamper the practical application of Ru-based LRMs. Sn can obviously enhance the stability of LRMs, but it can also damage the capacity as it is electrochemically inactive. The practical value of Sn may therefore be limited. Ti is also an abundant and nontoxic element. Ti-based LRMs can deliver comparable initial capacity as NMC-based LRMs, but the former suffers from rapid capacity decay with long cycling. Therefore, the priority of this kind of material is to improve the capacity retention. The influences of Fe, Ru, Sn and Ti components are also summarized and listed in Tables 1 and 2.
3.2 Elemental Substitution
Elemental substitution is also a promising strategy to enhance activation kinetics. The composition of LRMs consists of three essential parts: lithium, transition metals and oxygen. Thus, elemental substitution can be divided into three categories: cation doping that replaces Li, cation doping that replaces the TM ions, and anion doping that replaces the O in LRMs. Cation doping, such as sodium (Na), potassium (K), magnesium (Mg), aluminum (Al), iron (Fe), cobalt (Co), and ruthenium (Ru) cations, can increase the electronic conductivity and enlarge the size of the lattice. These changes can accelerate the migration of Li ions [18, 33, 112,113,114,115,116]. Anion doping is mainly focused on the selection of fluorine (F), chlorine (Cl), and sulfur (S). Using F doping as an example, the F-ions can form strong M–F bonds with TMs, which decrease the lattice size and stabilize the spinel structure during cycling. F− is also found to reduce the impedance of the cathode. On balance, however, F− doping can improve the rate capability by stabilizing the lattice structure and reducing the resistance, but at the expense of losing capacity by reducing the amount of diffusible Li+.
In the category of cations, the dopant can be further classified into alkali metals, alkaline-earth metals, transition metals and other metals. Alkali metals are mostly used to substitute for Li in LRMs. Among them, sodium (Na) and potassium (K) are the most commonly used alkali metals to dope LRMs due to their abundance and low cost, while the research on expensive rubidium (Rb) and cesium (Cs) doping is limited. All the alkali metals can stay in the lattice during Li diffusion and act as pillars of the lattice, which can enlarge the Li slab and improve the rate performance, as shown in Fig. 8a, b and c [114, 117,118,119]. In addition, these elements can largely increase the electronic conductivity of LRMs. Due to having the lowest ionic radii of the above-mentioned alkali metals, Na preferably occupies Li sites in the doping process. In addition to improving the rate performance, most Na ions stay in the Li layer during extraction of Li, supporting the structure and retarding the volume change and lattice collapse [118]. Under such an effect, Na changes the local environment of both Ni and O and lengthens the Ni–O bond. Thus, the layered to spinel conversion is suppressed because the Li/Ni disordering is thermodynamically restrained [120]. In addition, during activation, some of the Na is exchanged with Li from the electrolyte. This induces Ni migration to the Na vacancies and inhibits the formation of the spinel phase. This in turn significantly improves the cycling stability and Coulombic efficiency [121]. The ion exchange may also result in a thinner SEI film and reduce the charge transfer resistance during cycling [122]. Most importantly, although Na does not have a direct effect on activation capacity, the Na doped LRM only needs a single activation cycle (Fig. 8d) while the pristine material needs about a dozen cycles (Fig. 8e) to be fully activated. Qing and co-workers attribute this improvement in activation rates to the easier diffusion of Li ions caused by the pinning effect of Na, which also significantly improves the cycling reversibility [122]. However, as the Na replaces active Li in the lattice, Na doping will sacrifice some capacity in exchange for improving stability.

Copyright © 2013, the Electrochemical Society. d Initial cycling curves of pristine LRMs; e initial cycling curves of Na-doped LRMs. Reprinted with permission from Ref. [122]. Copyright © 2015, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. f Schematic diagram of Mn migration during charging in pristine LRMs; g schematic diagram of Mn migration during charging in K-doped LRMs. Reprinted with permission from Ref. [115]. Copyright © 2014, American Chemical Society
a Unit cell of Li2MnO3; b unit cell of LiMO2, c unit cell of NaMO2. Reprinted with permission from Ref. [118].
As an alkali metal, K can also replace some Li in the Li layers and enlarge the Li diffusion paths [123]. However, with a much larger ionic radius than that of the Na dopant, the K ion was reported to cause slight lattice distortion and introduce strain on the surrounding area. Such distortion and strain can result in a thicker LiO2 inter-slab layer in the lattice, which reduces the energy gap of Li migration and eventually accelerates the activation. In addition to the faster activation, as shown in Fig. 8f and g, K stays in the lattice as a pillar and blocks the adjacent octahedral sites. Thus, the migration of Mn ions and formation of the spinel phase is prevented as this process needs three empty adjacent octahedral sites to be thermodynamically favored. The large K ions can also reinforce the oxygen framework, which stabilizes the lattice during activation [115]. Additionally, K doping may alter the local orientation of the lattice in a direction which reduces the Li/Mn/Ni disordering, which eventually leads to a high capacity and rate capability [124]. Moreover, K doping is also speculated to promote Mn to participate in the electrochemical reaction and contribute to the capacity, although the mechanism for this is still unclear. Due to the large radius of K, the mobility of K is limited. The ion exchange of K and Li in the electrolyte is therefore difficult [124].
Apart from Na and K ions, only limited research has been conducted on the large alkali metals like Rb and Cs. Rb doping has a similar effect on LRMs as that of Na and K. However, though the mechanism is still unclear, Rb is also speculated to cause Mn participation in the activation [114]. While Cs proved to promote activation as the Li extraction from the Li2MnO3 phase is found to be accelerated. It is also notable that excessive doping can cause undesirable spinel phase formation [117].
It can be concluded that all the alkali metals including Na, K, Rb and Cs can replace Li in LRMs. These alkali metal ions can enlarge the lattice to facilitate the extraction/insertion of Li to enhance the Coulombic efficiency and rate performance. Also, they can stay in the lattice to act like pillars, stabilizing the lattice and blocking the migration paths of transition metals, hence improving the cycle stability. The main differences between these alkali metals are: (1) Na doping can lower the activation voltage, despite none of the papers mentioning the mechanism, and (2) K, Rb and Cs are speculated to be able to accelerate the activation of the Li2MnO3 phase, but the mechanism is still unknown. In addition, as summarized in Table 3, no evidence shows that one alkali metal is significantly more effective than others, so Na and K doping are more practically applicable in industry due to their abundance and low cost. Future research should therefore focus on the mechanism of the change of activation voltage and activation rates induced by Na/K doping.
The research on alkaline-earth metal doping in LRMs is mostly conducted with magnesium (Mg), calcium (Ca) and barium (Ba) in the past few years. All these metals have the same type of pillar effect, but each of them has some unique features. For instance, as the smallest alkaline-earth metal, Mg can either substitute for Li or TMs (preferably Ni) in LRMs [125,126,127]. When replacing Li, Mg only exhibits the pillar effect, but has negligible influence on the activation process [125]. When Mg occupies both Li and Ni sites, in addition to the pillar effect, the activation voltage near the end of the charge is also reduced to some extent. This indicates that Mg can promote oxygen redox at higher voltages. This phenomenon was not explained in this paper, and it is worth more research towards better understanding [127].
Unlike Mg, Ca can only substitute for Li in the LRMs. Early theoretical calculations predicted that Ca would preferentially occupy Ni sites in the LRMs, and the lattice of Ca doped LRM was unstable, especially during the charging process. This result rules out the possibility of practical TM substitution by Ca [128]. Then, experimental work proves that Ca indeed can only be doped in Li layers [129]. After Ca doping, the activation voltage of doped LRMs is higher than that of pristine and the overall capacity is therefore increased. This indicates that oxygen redox still occurs during activation, but is more difficult. This phenomenon can be attributed to the high electronegativity of Ca2+. The Ca–O bond is stronger and more ionic than the Mg–O bond, and so the oxygen gas generation is thermodynamically more difficult. Therefore, the oxygen loss during the activation is suppressed without sacrificing capacity.
Ba is found to substitute for Ni rather than Li, possibly because it has a much larger ionic radius compared with Mg and Ca. Ba-doped LRMs show a significantly shortened but elevated activation plateau compared to that of pristine samples. Meanwhile, the discharge capacity is not reduced. This indicates that oxygen loss during activation is suppressed. Although there is a lack of direct evidence, it is hypothesized that Ba2+ can stabilize the generation of oxygen radical intermediates formed during activation. Thus, in the presence of Ba2+, which has low positive charge density, the oxygen dimer intermediates cannot be further oxidized to O2 gas [130].
In the above literature on alkaline-earth metals (Table 4), it can be concluded that alkaline-earth metals can stabilize the lattice of Li-materials, and most importantly, suppress oxygen loss during activation. Although there is no evidence that shows that these metals can accelerate the activation, they also have no clear negative impact on the activation process. In future, it may be possible to dope alkaline-earth metals in LRMs as an activation stabilizer while simultaneously introducing other methods to accelerate the activation. This strategy is likely to simultaneously achieve fast activation and stable cycling.
In the past few years, there was significant research into the substitution of transition metals in LRMs. Doping of transition metals was extensively studied. According to the first principal calculations on the reaction energy of oxygen evolution during activation in LRMs consisting of 3d, 4d and 5d metals, metals with the 3d electron arrangement, or 4d and 5d metals with the d0 electronic configuration, are predicted to have irreversible oxygen evolution. Heavier 4d metals with a d2−4 electronic configuration or 5d metals with a d2−6 electronic configuration are predicted to reduce or even suppress the irreversible oxygen redox during activation [131].
Titanium (Ti) is a typical 3d metal with a d0 electronic configuration. In the experimental work, as predicted, Ti-doped LRMs exhibit significant irreversible oxygen evolution during the initial cycle in the 2.0–4.8 V voltage window [132]. However, when the voltage window is reduced to 2.00–4.45 V, the irreversibility is prevented without loss of discharge capacity (Fig. 9a and b). The mechanism is that Ti4+ has completely empty d orbitals, and can therefore only coordinate with adjacent O2− by ionic bonding and not through covalent bonding. As the O2p-Ti3d hybridization state is near the Fermi level, the Ti in the lattice can ease the oxygen evolution and activate the Li2MnO3 phase. Thus, Ti doped LRM can be activated at 4.4 V, about 0.1 V lower than that of Ti-free LRMs. When the voltage window is reduced to 2.00–4.45 V, the irreversibility is prevented without loss of discharge capacity. Furthermore, such O2p-Ti3d hybridization can weaken the O2p-TM3d hybridization of other TM ions, especially Ni and Co, which suppresses the O2 gas generation during activation. It is noteworthy that Ti can be doped in both the TM and Li layers to provide pillar effect (Fig. 9c) [132]. In addition, Ti doping is believed to alleviate lattice deformation during activation [133]. Thus, Ti doping can also improve the lattice’s structural stability in the activation process.

Copyright © 2019, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. d Phase evolution paths of pristine (P-type) and Cr-doped (C-type) LRMs. Reprinted with permission from Ref. [138]. Copyright © 2016, The Royal Society of Chemistry. e Change of initial cycling with different contents of Cr doping. Published by Elsevier Ltd. Reprinted with permission from Ref. [139]. Copyright © 2018, Hydrogen Energy Publications LLC. f Initial cycling of LRMs with different levels of Fe-doping and g corresponding dQ/dV curves. Reprinted with permission from Ref. [142]. Copyright © 2014, Elsevier Ltd. h Initial cycling of LRMs with different levels of Cu-doping. Reprinted with permission from Ref. [143]. Copyright © 2018, Elsevier Ltd. and Techna Group S.r.l. i Initial discharge curves of LRM with different levels of Zn-doping. Reprinted with permission from Ref. [144]. Copyright © 2015, Elsevier Ltd. and Techna Group S.r.l
a Initial cycle of Ti-doped LRM at different voltage windows; b CV profile of pristine and Ti-doped LRM; c schematic of Ti-doped LRM. Reprinted with permission from Ref. [132].
Zirconium (Zr) with a 4d0 orbital has also been used to dope LRMs, showing similar pillar effect. However, Zr doping has no obvious positive impact on activation, giving a similar activation capacity to undoped pristine materials. In fact, Zr can diminish the redox of other transition metals, resulting in a much lower capacity for both charging and discharging [134].
Cr is an electrochemically active metal with 3d electrons, which can provide extra Cr3+/Cr6+ redox couples. Thus, Cr is active and doping with Cr in LRMs will not sacrifice the overall capacity by introducing inactive materials. Earlier reports pointed out that Cr can suppress oxygen loss during activation [135, 136]. Some studies even found that Cr can accelerate the activation of the Li2MnO3 phase as a catalyst, because the initial capacity increases with Cr content [137]. However, the lower voltage slope before activation and the high-voltage activation plateau were not clearly distinguished. Thus, this improvement of capacity may have been contributed by the extra Cr3+/Cr6+ redox couple rather than an extended Li2MnO3 phase activation. The later reports supported this speculation by clearly showing the prolonged TM redox slope and shortened activation plateau [136, 138, 139]. For example, a reduced capacity contribution from activation is observed in the Cr doped LRMs [138]. With the help of in-situ synchrotron XRD, researchers confirmed that Cr can suppress the activation of the Li2MnO3 phase (Fig. 9d). Although such suppression can improve the lattice structure’s stability, the capacity contributed by oxygen redox is sacrificed. However, this loss of capacity can be compensated by the extra Cr3+/Cr6+ redox couple. Thus, the overall capacity of Cr-doped LRM is still larger than that of pristine [138]. As a result, increasing the Cr doping content shortens the activation plateau and gradually shifts it to a lower voltage (Fig. 9e). This change can be attributed to the existence of a stronger Cr–O bond (427 kJ mol−1) than TM–O bond (402 kJ mol−1 for Mn–O, 391.6 kJ mol−1 for Ni–O and 368 kJ mol−1 for Co–O), which retards the O22− oxidation during the activation [139].
Iron (Fe) is another commonly used dopant metal with the 3d electronic configuration, and is not as toxic as Cr. Unfortunately, the shortening of the capacity plateau and the prolonging of the TM redox slope are still observed by researchers [140, 141]. Fe can promote oxygen redox during activation because high valence Fe3+δ ions from the oxidation of Fe3+ during low voltage charging (below 4.5 V), can later capture the electron released by oxygen during high-voltage charging above 4.5 V. This reaction can accelerate the oxygen evolution from the lattice. However, such a reaction does not involve any Li+ extraction because the electrons released from the oxygen ions are trapped in the cathode by the Fe3+δ ions. The capacity is therefore not increased (Fig. 9f). The observed improvement of capacity is from the extra Fe3+/Fe3+δ redox couple, as proven by the dQ/dV curve shown in Fig. 9g [142]. All these results are in good agreement with Fe-based LRMs as mentioned above, suggesting that Fe is mainly a structural stabilizer rather than an activation accelerator.
Besides the commonly used metals, the research on other 3d transition metals such as copper (Cu) and zinc (Zn) is limited. Sorboni and co-workers examined the influence of Cu doping in their Li1.25Mn0.50Ni0.125Co0.125O2 cathode, and found that the initial capacity, and especially the portion contributed by activation decay, decreased with increased Cu content (Fig. 9h). This result suggests that Cu doping can suppress the activation of LRMs. The authors explain the decay in overall capacity by speculating that (1) the Cu2+ ions occupy Li+ sites and inhibit the diffusion of Li+, and (2) the existence of Cu2+ can cause oxidation of Ni2+, which decreases the number of Ni2+/Ni4+ redox couples. However, the electrochemically inactive Cu can stabilize the structure of LRM, resulting in better cycling stability [143]. Zn has pillar effect, helping to improve the rate performance, but the doped LRMs show clear capacity decay with increased Zn content compared to that of pristine materials (Fig. 9i). There are two reasons for this change. Firstly, Zn2+ may occupy Li+ sites due to having a similar size to Li+, which decreases the extractable Li content. Secondly, the strong Zn–O bond can impede oxygen redox during activation [144, 145].
In all the above-mentioned transition metals with 3d electronic orbitals (Table 5), only Ti facilitates the activation process, while Cr, Fe, Cu and Zn can suppress the activation but improve the cycling stability. Among them, electrochemically active Cr and Fe can compensate for the capacity loss from activation by providing extra redox couples. However, Cu and Zn sacrifice capacity contributions from both TM redox and oxygen redox, suggesting that they may not be suitable dopants for achieving a high capacity.
The options for transition metals with 4d2−4 or 5d2−6 configuration are Nb, Mo, Tc, Ru, Rh, Pd, Ag, Cd, W, Re, Os, Ir, Pt, Au and Hg. Among them, only niobium (Nb), molybdenum (Mo), ruthenium (Ru), tungsten (W), and rhenium (Re) substitutions were experimentally or theoretically studied for LRMs. First principal calculations predicted that Nb can provide extra free electrons to compensate for the charge during activation and suppress the oxygen gas generation, evidenced by more Li removal before O2 generation energy reaches to zero than that of other dopant (Fig. 10a). The strong binding between Nb and O also reduced the oxygen evolution (Fig. 10b). The experiment also supports this calculation’s result [146]. Nb thermodynamically occupied Li sites near the particle surface (Fig. 10c), and it remained in a 5+ valence state during electrochemical cycling. Although Nb did not change the TM’s valence state and bulk oxygen environment, it can reduce the local TM3d-O2p hybridization. The weakened TM3d-O2p hybridization and strong Nb–O bond synergistically stabilized the local oxygen framework and suppressed the oxygen oxidation during activation. However, this only occurs near the Nb-containing particle surface, while the bulk oxygen ions are still active during cycling. Therefore, the initial charge/discharge capacities are not reduced, and are even slightly increased as shown in Fig. 10d. The increase in capacity is speculated to be caused by Li vacancies induced by Nb doping in Li layers that can lead to more Li insertion upon discharge [147]. In the other experimental work on Nb doping, in which Nb was homogenously doped into the bulk, the initial discharge capacity, cycling stability and rate performance were increased after Nb doping. This is because Nb5+ (0.64 Å) is larger than Mn4+ (0.53 Å) and Co3+ (0.54 Å) and can enlarge the interlayer spacing, facilitating Li+ diffusion. However, the charge capacity, including the part contributed by activation, shows no obvious change [148]. However, some researchers also observed an increased overall capacity but reduced activation contribution with Nb doping. They proved the suppression in activation through theoretical calculations and reported the formation energy of oxygen vacancies in LRMs is significantly increased from 2.63 to 3.66 eV after Nb doping (Fig. 10e and f). In addition, the existence of Nb in Li sites can reduce the energy barrier of Li+ diffusion, which results in improved cycling stability and rate performance. The easier transport of Li+ also leads to the TM’s redox slope gradually shifting towards lower voltages with increased Nb content (Fig. 10g) [149].

a Calculated O2 release energy with different metal dopings at different delithiation states; b binding energy between different metals and oxygen. Reprinted with permission from Ref. [146]. Copyright © 2015, American Chemical Society. c Schematic of Nb-doped structure; d initial cycling of LRM before and after Nb-doping. Reprinted with permission from Ref. [147]. Copyright © 2018, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. Schematic of structure and Nb–O binding energy when Nb is in e TM layers and f Li layers; g initial cycling profile with different levels of Nb-doping. Reprinted with permission from Ref. [149]. Copyright © 2019, American Chemical Society. h Initial cycling profile before and after Mo-doping; corresponding dQ/dV curves i before and j after Mo-doping. Reprinted with permission from Ref. [151]. Copyright © 2019, Elsevier B.V. k Initial cycling profile with different levels of W-doping. Reprinted with permission from Ref. [153]. Copyright © 2019, Springer-Verlag GmbH Germany, part of Springer Nature. l Change of bond lengths/distances in angstrom before and after Re-doping. Reprinted with permission from Ref. [154]. Copyright © 2019, American Chemical Society
Mo is also predicted to have a similar size to Mn/Ni/Co and strong bonding with oxygen according to first principle calculations. But the Li migration in Mo doped LRM is predicted to be more difficult due to a higher energy barrier and narrower Li diffusion paths. The operational voltage is also predicted to be lower than that of pristine [26]. However, the experimental work of two other groups proved that Li diffusion is actually not hindered after Mo doping, as they observed higher rate performance and lower electrochemical impedance [150, 151]. In Guo and co-workers’ report, both the initial charge and discharge capacity is lower in Mo doped LRMs compared to that of pristine materials, because electrochemically inactive Mo6+ reduces the number of redox couples. However, the activation shows no obvious change with or without Mo [150]. While Chu et al. point out that the doping of Mo6+ can reduce the valence state of Ni, and so the charge plateau voltage is lower than that of pristine. Extra Ni2+ can provide more electrons to boost the capacity. Moreover, the activation plateau is less distinguishable after Mo doping (Fig. 10h), possibly due to the decrease in Mn content. However, the differential plots of capacity versus voltage (dQ/dV) curves and ex-situ XPS indicate stabler oxygen redox in the Mo-doped sample (Fig. 10i and j) [151]. It is notable that the capacity change in the above two reports are in conflict. The contrasting results may originate from the different Mo substitution sites.
As discussed above, in the composition adjustment section, Ru can accelerate the activation and prevent oxygen gas generation if used in large fractions in LRMs. Due to Ru being an expensive metal, such a high Ru content is not practical for industrial applications. To investigate the effect of low Ru content, Song and co-workers reported on a Ru trace doped LRM. In this work, Ru substitutes for Co and enlarges the interlayer space of the lattice, which increases Li mobility in the material. The charging voltage plateau above 4.5 V is apparently prolonged after Ru doping, indicating deeper activation [39]. However, the improvement of cycling stability is not as significant as the Ru-based LRM, possibly because the Ru content is not high enough.
W is a 5d transition metal with strong covalent bonding with oxygen, so it is expected to stabilize the lattice structure of LRMs during cycling. In the past 3 years, the influence of W doping on electrochemical performance both on the surface and in the bulk were studied [152, 153]. In both reports, the activation plateau during initial charging is prolonged after W doping (Fig. 10k), indicating a larger activation extent [153]. However, the underlying mechanism is still not clear. At the same time, the initial discharge capacities also increased, suggesting suppressed irreversible oxygen evolution. In addition, W doped LRMs show better capacity and voltage retention in long cycling. This may be related to the strong W–O bonding, which can restrain the migration of transition metal ions and the subsequent formation of a spinel phase.
Re is the last metal doping studied in the category of 5d transition metal. Re7+ has a similar size as Mn4+, so it can preferably substitute the Mn sites in TM layers and therefore will not block the diffusion paths of Li. Although the influence of Re on activation is not given directly, it is speculated to promote the activation due to the resulting higher discharge capacity. From the theoretical simulations, Re has much stronger bonding with oxygen (627 kJ mol−1) than that of Mn (362 kJ mol−1), Sn (528 kJ mol−1) and Ru (528 kJ mol−1). Therefore, it can significantly lengthen the surrounding O–O bond from 1.235 to 2.72 Å (Fig. 10l). This suppresses the oxygen gas generation during deep delithiation [154].
In all the reports on 4d and 5d transition metals substitution in LRMs, as summarized in Table 6, these elements proved to improve the rate performance due to the reduced Li diffusion barrier. In addition, they can stabilize the lattice during cycling because of their strong bonding with oxygen. Among them, Nb has no obvious influence on the depth of activation. Mo also shows no apparent impact on activation. However, depending on the substitution sites, Mo can either lessen the overall capacity by reducing the number of TM redox couples or increase the capacity by reducing Ni3+ to Ni2+ therefore providing an extra electron during cycling. Ru doped LRMs show deeper activation, but the stability is not as good as the Ru-based LRMs. This may suggest that the Ru content needs to be increased. W doping can increase the activation extent in the cases of surface doping or bulk doping, but the mechanism is still unknown. For Re, although the results of pure Re doping are not given directly, it is hypothesized to promote the activation.
In addition to alkali metals, alkaline-earth metals and transition metals, the research on other metals is mainly focused on Al, Ga and Sn. As the Al–O (512 kJ mol−1) bond is stronger than Mg–O bond (368 kJ mol−1), the lattice structure of Al doped LRMs is more stable, alleviating the layered to spinel transformation [155]. Additionally, Al has a tiny ionic radius (0.51 Å), even smaller than that of Li+ (0.68 Å), and so the doping of Al can substitute for Li, Ni or Co under different synthesis conditions. Regardless of the method, Al doping can suppress the cation disorder in Li-layers [155,156,157,158,159]. In the Co-free LRMs, electrochemically inactive Al replaces active Ni, so the discharge voltage plateau shifts to a lower voltage (Fig. 11a). However, Al doping can improve the rate performance due to having higher electronic conductivity than that of Mn and Ni. More interestingly, the activation voltage near the end of charging is also decreased slightly, but the reason is still not clear [156]. In Co composite LRMs, Al preferentially replaces Co. In some Co-substitution cases, the lower shift of the oxygen redox voltage plateau is more distinct than that of Ni-substitution cases, indicating easier activation (Fig. 11b) [157, 158]. In addition to lowering the activation voltage, the increase of capacity contributed by oxygen redox is observed in some trace Al-doped LRMs. Thus, Al doping should have the capability to accelerate activation to some extent [159]. Apart from occupying TM sites, Al can also substitute for the Li site in LRMs (Fig. 11c and d). When Al occupies Li sites, the lattice is expanded under the force of strong electrostatic repulsion. Such repulsion also causes the rearrangement of local atoms. With Al introduced into the structure, some Mn irreversibly migrates into the Li layer and forms a spinel phase, some transition metals reduce to a lower valence state, and a portion of Li in the Li layers is pushed into the TM layers. These can cause the increase of activation capacity, because there are more Li+ ions to extract at higher voltages. Additionally, the activation voltage also shows a significant downward shift, but the mechanism has not been investigated yet [158].

a Initial cycling profiles of Li[Li0.09Mn0.65*(0.91 − x)Ni0.35*(0.91 − x)Alx]O2 with different x values, when Al replaces Ni. Reprinted with permission from Ref. [156]. Copyright © 2013, Springer-Verlag Berlin Heidelberg. b Initial cycling profile of Al-doped LRMs with different doping contents replacing Co, where (i) is pristine, and Al content increase from (ii) to (v). Reprinted with permission from Ref. [157]. Copyright © 2016, Science China Press and Springer-Verlag Berlin Heidelberg. Atomic arrangement and structures of Al-doped c LiMO2 and d Li2MnO3 phase. Reprinted with permission from Ref. [158]. Copyright © 2018, American Chemical Society. e Energy barrier for TM migration before and after Ga-doping. Reprinted with permission from Ref. [160]. Copyright © 2016, Elsevier Ltd. f Initial cycling profiles of LRMs before and after surface Sn-doping. Reprinted with permission from Ref. [163]. Copyright © 2019, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. g Initial cycling profiles of LRMs with different levels of bulk Sn-doping. Reprinted with permission from Ref. [165]. Copyright © 2017, Springer-Verlag GmbH Germany
As an XIII main-group element, Ga has some similar characteristics to Al. Doped Ga can either occupy Li or TM sites in LRMs by different synthesis methods. When Ga substitutes for Li, it can occupy the tetrahedral site in Li-layers. This process can create three Li vacancies and cause the rearrangement of local atoms, finally leading to a shortening of the Li–O bond and a lengthening of the TM–O bond. Although the rearrangement of atoms has a negligible effect on the activation extent, it raises the energy barrier of TM ions migrating into tetrahedral sites and therefore suppresses the formation of the spinel phase during activation (Fig. 11e) [160]. When Ga substitutes for Ni, it acts like a structural stabilizer by enhancing the TM–O bond, which is beneficial for reducing oxygen loss and improving cycling stability. However, the capacity and rate performance decay slightly, and the activation voltage is shifted to a higher region. All of these phenomena indicate that oxygen redox is inhibited, which has a negative impact on the activation of the Li2MnO3 phase [161].
Sn is also an extensively studied dopant for LRMs. Sn4+ has a larger ionic radius (0.71 Å) than Mn4+ (0.53 Å) and Ni2+ (0.69 Å). Doped Sn4+ ions therefore occupy Mn or Ni sites and can result in an expansion of the lattice. This can facilitate the diffusion of Li and better the rate capability. Moreover, when doped in the bulk, the electrochemically inactive Sn does not contribute to the capacity, but acts as a pillar to ensure a robust crystal structure during charge/discharge. Notably, although the initial capacity, including activation capacity, is not changed after Sn doping, the activation voltage was somehow increased [162]. A similar phenomenon is found in surface coating induced Sn doped LRMs, in which the activation voltage is elevated with a significantly reduced initial charge capacity (Fig. 11f). However, both the charge and discharge capacity increased remarkably in the following few cycles. These facts suggest that the activation is slowed down [163]. In contrast to the suppressed oxygen redox mentioned above, some researchers observed a pronounced trend of increased activation rate with increased Sn doping content. They believe that the enhanced activation is a result of improved electronic conductivity caused by Sn doping, as Sn is a commonly used electron donor [164]. Similarly, it is also reported that an appropriate amount of Sn doping can lead to improved activation capacity and discharge capacity. However, these reports attribute the improvement to stronger Sn–O bonds (relative to TM–O bonds) stabilizing the structure rather than any conductivity improvements. In addition, LRMs doped with small amounts of Sn were observed to have lower activation voltages, while excess Sn doping increased the activation voltage (Fig. 11g). This indicates that the doping content is critical to the battery performance [165].
In all of the above-mentioned reports, Al, Ga and Sn doping in LRMs can influence the activation process but in a different manner and to a different extent. Al can be doped into either Li or TM sites and stabilize the lattice structure during cycling regardless of the occupying sites. More importantly, it can reduce the activation voltage and promote activation. However, the related mechanism is still unrevealed. Similar to Al, Ga can also substitute for Li or TMs. However, the different occupancy of Ga leads to a different influence on the activation process. Specifically, Ga doped in Li sites has a negligible impact on activation, while Ga in TM sites inhibits the activation. In all of the above discussed literature, Sn is found to ease and stabilize the activation of the Li2MnO3 phase in LRMs. But it is notable that the Sn content used is critical to the performance. According to the reports, a low level of Sn doping can improve the activation rate, while a high level of Sn doping can deteriorate the performance.
Anion doping is the broad alternative to cation doping. Instead of replacing TM ions, O2− ions are replaced with, most commonly, F−, Cl−, S2−, or various polyanions. Unlike cation doping, where it can be difficult to determine precisely which TM ions are being replaced, the anion dopant always replaces O2− due to it being the only anion in the system. The changing bond is associated with better rate capability, rate stability and reduced voltage decay [166]. Most pertinently for this paper, a key benefit of anion doping is the effect it has on the activation of Li-rich cathodes. Anion doping changes the average oxidation state of the TM ions [167], and the strength of the TM–O bond [168], both of which have a significant effect on activation. In general, anion doping tends to reduce the lattice decay from activation by increasing the capacity contribution < 4.5 V and reducing the capacity contribution from 4.5 to 4.8 V [169] (Table 7).
The first research to be conducted into Li-rich anion doping was by Kang and Amine in 2005 [170]. They synthesized Li[Li0.2Ni0.15+0.5zCo0.10Mn0.55−0.5z]O2−zFz (0 \(\leqslant\) z \(\leqslant\) 0.10). This report established several important results. Firstly, it established that F-doping improved the cycling stability at the cost of some initial capacity [170]. The pristine sample had a very slightly larger initial capacity (5–10 mAh g−1), but this capacity faded to \(\sim\) 70% after 40 cycles. At z = 0.02 and z = 0.05, meanwhile, it retained \(\sim\) 83% and 93% capacity respectively. Additionally, F-doping caused a shift in the secondary peaks during the charge/discharge process, during both the initial and subsequent cycles. These voltage peaks are associated with the redox peaks of the transition metals, and tend to shift to lower voltages as the number of cycles increases. However, as the F content increases, the voltages shifts are smaller in magnitude [170, 171]. This indicates that F is stabilising the TM ions during cycling, although this paper offers no explanation of the cause. It is also worth noting the significant capacity drop from z = 0.05 to z = 0.10, along with the unusual increase in capacity over the first few cycles. LRMs are normally associated with large irreversible capacity loss after the first cycle, and so this result is in stark contrast both to lower amounts of F-doping and to the pristine sample. Importantly, however, this paper only charged the cathode to 4.6 V during electrochemical testing, meaning that the full activation process was not studied in this report. Despite this, this report is important because it established that F-doping was a viable method for improving the stability of Li-rich cathodes.
The next stage of research was an attempt to determine the mechanism through which fluorine was eliciting the improved capacity retention, and especially its effect on the activation stage. In 2007, Wu and Manthiram demonstrated that doping with F decreased the capacity peak in the first charge cycle associated with irreversible capacity loss [172]. This was good evidence that F-doping made it more difficult for oxygen to leave the lattice during charging. It explained why F-doping decreased the initial capacity (less O2− leaving means less anionic redox capacity contribution) and why it improved cycling capacity (less O2− leaving means less lattice breakdown). Li and Fan confirmed this result in 2013, and also found that the suppression of O2− oxidation increased with increase in F content [173]. However, the reason for the greater oxygen retention was still unclear (Fig. 12).

Influence of different elemental substitutions on charge, discharge, and activation capacities
There were competing theories to explain F’s effect on oxidation. Most of the initial evidence simply pointed to a decrease in the initial capacity. It had been established, through studies on non-Li-rich cathodes, that F-doping impeded the Li de/intercalation process [174]. In this paper, Kim et al. suggest that the strong covalent bond may disturb the intercalation of Li+ ions. Despite offering no evidence or explanation for why this occurs, it became a commonly accepted explanation for the initial drop in capacity [173, 175, 176]. However, the addition of fluorine is likely to have the opposite effect and aid Li+ de/intercalation. The activation barrier for Li+ diffusion is largely based on the c-parameter and the valence state of the TM ions. The Li+ diffusion activation barrier monotonically decreases with increasing inter-slab distance [177], as is intuitively expected. F-doping generally increases the initial lattice spacing [175, 178, 179]. Part of this is because, although F− is smaller than O2− (1.33 Å vs. 1.40 Å), Ni2+ is larger than Ni3+ (0.69 Å vs. 0.56 Å) and so the lattice was expanded [178, 180]. Additionally, the stronger Li–F bonds create a repulsive force in the oxide layer. In an undoped material, Ni mostly exists as Ni4+ with full t2g orbitals, i.e. Ni4+ with (t2g↑)3(t2g↓)3 configuration. With F-doping, the Ni4+ transitions to Ni3+ with (t2g↑)3(t2g↓)3(eg↑)1. This (eg↑)1 orbital repels the p orbital electrons in O2−, causing a lengthening of the Ni–O bond and an additional increase in the c-parameter [168]. Decreasing the TM valence also has a second beneficial effect on Li+ diffusion. The positive charge of the TM ion acts an electrostatic barrier to Li+ diffusion. Due to charge compensation, anion doping lowers the valence state of the TM ion to which it is attached [169, 178, 181]. This lowers the charge of the TM ion, which in turn lowers the barrier for Li+ to diffuse through. The reduced activation is therefore unlikely to be explained by the strong Li–F bond reducing Li+ diffusion.
An alternative explanation is that the strong bond, instead of preventing Li+ diffusion in the Li+ layer, prevents Li+ migration from the TM layer to the Li layer. There is evidence that Cl and S doping increases the migration energy barrier for TM’s being adjacent to the dopant [182]. However, F-doping lowers the energy barrier for adjacent TM. This can be explained in terms of the ionic size sequence S > Cl > O > F, where the larger S and Cl ions sterically hinder migration while the smaller F ion widens the diffusion channel [182]. As the capacity loss is observed in both Cl and F doping, the inhibition of Li+ migration cannot explain the inhibited activation.
The inhibited activation must therefore be explained in terms of the oxygen removal. This explanation was developed concurrently with researchers gaining a greater understanding of the reaction mechanism itself. The pristine activation mechanism has already been discussed, and Wang et al. successfully related this mechanism to anion doping [183]. They observed that F-doping increased the capacity contribution from the Li deintercalation phase (< 4.4 V) while decreasing the contribution from activation (> 4.4 V). This effect is observed regardless of if F-doping is confined to the surface or spread throughout the bulk [183]. It is very difficult to oxidize Mn4+ to Mn5+, and oxygen will oxidize preferentially. With the addition of fluorine, however, some of the Mn4+ is converted to Mn3+ (the same process occurs for the other TMs). This increases both the redox potential and the Li+ extraction from TM oxidation, which accounts for the increased capacity from the pre-activation phase. Additionally, oxygen vacancies facilitate the layered-to-spinel transition by lowering the barrier for cation mixing and by causing additional strain on the lattice [184, 185]. Suppressing the activation therefore also explains the improved stability. Despite this, the reasons for the activation suppression are still unclear. It has been observed consistently; however there is as yet no satisfactory explanation. F-doping appears to increase the ratio of Li2MnO3 to LiTMO2, which leads one to predict an increase in activation [169]. It is also known that F-doping weakens the TM–O bond on average, which should make extraction easier. It is possible that F-doping simply decreases the oxygen available and as a result lowers the activation. Uncovering the mechanism behind the suppression of anion doping would allow for a more rational design of anion doped materials, and would particularly increase the opportunities for synergistic co-doping with cations [186].
In summary of all the above-mentioned element substitution, alkali metals can replace Li, alkaline-earth metals replace either Li or Mn/Ni/Co, transition metals replace Mn/Ni/Co, other metals replace either Li or Mn/Ni/Co, and anions replace oxygen in LRMs. Each element has different effects on the lattice structure and electrochemical performance of LRMs. Specifically, alkali metals can enlarge the lattice to facilitate the extraction/insertion of Li and stay in the lattice as pillars to stabilize the structure. Among them, Na can lower the activation voltage, while K, Rb and Cs are speculated to accelerate the activation of the Li2MnO3 phase. Alkaline-earth metals including Mg, Ca and Ba, can stabilize the lattice of Li-materials, and suppress the oxygen loss during activation. They have no positive or negative impact on the activation process. Thus, these elements are possible to act as activation stabilizers, while simultaneously doping with other methods to accelerate the activation. Transition metals, a big family of elements, can be divided according to their valence electronic configurations as 3d, 4d and 4d transition metals. In 3d transitions metals, only Ti can accelerate the activation, while Cr, Fe, Cu and Zn can suppress the activation. Among them, electrochemically active Cr and Fe can provide extra redox couples to compensate for the capacity loss from activation, but inactive Cu and Zn cannot. Thus, Cu and Zn may not be suitable dopants towards high capacity. In all the reports on 4d and 5d transition metals they can improve the rate performance due to a reduced Li diffusion barrier and stabilization of the lattice during cycling because of strong bonding with oxygen. Among them, Nb and Mo has no obvious influence on the depth of activation. Ru doping can lead to significantly deeper activation, but the stability is not as good as the Ru-based LRMs. W doping can increase the activation extent, but the mechanism is still unknown. Re is also conjectured to be able to promote the activation, but still needs more evidence. Within other metals including Al, Ga and Sn doping in LRMs, Al can reduce the activation voltage, and slightly promote the activation. For Ga, varying occupancy of Ga leads to different influences on the activation process. Specifically, Ga doped in Li sites has negligible impact on activation, while Ga in TM sites inhibits the activation. Sn is able to ease and stabilize the activation of Li2MnO3 phase in LRMs, but content is critical. Low levels of Sn doping improve the activation rate, while excess Sn doping deteriorates the performance. The research on anion substitution is limited, while F is the most commonly used anion. F can reduce the oxygen activity and suppress the O2 gas generation. More study on anion substitution in LRMs is recommended for future research.
It is clear that doping of K, Rb, Cs, Ti, Ru, W and Re can accelerate the activation, Cr, Fe, Cu, Zn and F can suppress the activation, while Na, Mg, Ca, Ba, Nb and Mo can stabilize the oxygen redox but have no significant influence on the activation extent. It is hard to achieve both fast activation and stable cycling at the same time with single element doping. Therefore, in the future research, it is recommended to simultaneously dope more than one element, adding both activation accelerators and activation stabilizers in the material. In addition, to date, most of the research on element substitution in LRMs is related to the cycling stability. Although the activation behavior after doping is clearly changed, the phenomenon is usually ignored, so the mechanism is also not discussed. As one of the most important processes determining the electrochemical performance, the influence of elemental doping on activation of the Li2MnO3 phase is worthy to be studied in the future research. From the fundamental perspective, the influence of dopants may involve the rearrangement of local lattices and change of ion migration during the activation.
3.3 Chemical Treatment
The activation process of Li-rich cathodes usually leads to complex and significant structural changes, which results in severe capacity and voltage decay. In addition, the kinetics of activation also usually limits the activation rate and extent. In recent years, various chemical treatments are applied to LRMs to improve the electrochemical performance. Most of these strategies share a similar concept, namely, to chemically pre-activate the Li2MnO3 phase and create Li/O vacancies to stabilize the lattice upon cycling. With chemical treatment, those materials also show faster and deeper activation. By the properties of the reported chemicals, they can be classified as reductive agents, oxidative agents, and acids.
Reductive agents are the most commonly used chemicals to treat LRMs. Among them, NH3 is one of the most studied reductive agents. The first direct NH3 gas treatment on LRM was reported in 2012 [187], which can introduce N into the surface of LRMs and partially reduce the valence state of Ni and Mn. These factors can weaken the binding energy of Ni–O and Mn–O, improve the activity of Ni/Mn ions and facilitate the extraction of Li+. Therefore, the treated material shows a much higher capacity and faster oxygen redox [187]. In a later work, Mn and Co are found to be partially reduced by NH3 gas (Fig. 13a and b), along with lithium and oxygen loss from the surface lattice [188]. With appropriate treatment time, the initial activation extent and discharge capacity increased slightly. The cycling stability in terms of capacity and voltage is also improved.

Copyright © 2017, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. c Schematic of structure change during initial charging before (upper) and after (lower) CO2 treatment. Reprinted with permission from Ref. [189]. Copyright © 2016, Springer Nature. d Schematic of the surface and the lattice of LRMs after urea treatment; e initial cycling profile before and after urea treatment, and f corresponding dQ/dV curves; g corresponding prolonged cycling performance, the inset is the voltage decay profile. Reprinted with permission from Ref. [67]. Copyright © 2020, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim
Average oxidation state of a Co and b Mn before and after NH3 treatment. Reprinted with permission from Ref. [188].
Meng and co-workers used CO2 gas produced from pyrolysis of NH4HCO3 as a reductive agent to treat the LRMs. After treatment, lattice oxygen is extracted from the particle surface at thickness of 10–20 nm. Specifically, about 4% oxygen is removed, mostly from the Li2MnO3 phase. To compensate for charge difference, Mn4+ is partially reduced to Mn3+ along with the removal of Li+, which can reduce the electrochemical impedance of electron and Li+ migration. Although the amount of lattice oxygen is reduced with the treatment, the initial charge curve shows negligible change, indicating that activation is not hindered. The authors explain that the oxygen vacancies can decrease the oxygen partial pressure during the evolution of oxygen, which promotes oxygen redox in the bulk, but in the form of O2−/O− or O22− rather than O2−/O2. Thus, the structural stability of treated LRM is significantly improved without sacrificing the capacity contributed by activation. The schematic diagram of the structure before and after the treatment, and the change of lattices during initial charging are shown in Fig. 13c [189]. It is worth mentioning that the authors reported the NH4HCO3 decomposition product CO2 gas plays the key role in treatment reaction. However, NH4HCO3 is an ammonium containing chemical, which can produce both NH3 and CO2 during high temperature pyrolysis, while the former can also react with LRMs to create oxygen vacancies. Further investigation on pure CO2 treatment is worth doing in better understanding of the atmospheric impact on the LRMs.
Inspired by the previous research on NH3 and NH4HCO3 treatment, in 2019, Wang and co-workers designed a bifunctional urea treatment on LRM. This method utilizes the pyrolysis products of urea, i.e. C3N4 and NH3, to simultaneously introduce oxygen vacancies and a surface protective layer (Fig. 13d). After treatment, in addition to the oxygen vacancies introduced by reaction between LRM and NH3, C3N4 was coated on the surface of the LRM particles, preventing the direct contact between the electrode material and the electrolyte, hence suppressing the side reaction. At the same time, Mn4+ ions near the surface are partially reduced by NH3, along with lattice oxygen extraction. With the generated oxygen vacancies, the Co-free LRM Li1.84Mn0.94Ni0.19O3 shows dramatic improvement in activation capacity, which is increased from 126.2 to 176.6 mAh g−1 (Fig. 13e). The corresponding dQ/dV reveals significantly enhanced oxygen redox activity (Fig. 13f). In addition, the slow stepwise activation as a nature of Co-free and Ni-poor LRM is also remarkably accelerated. Specifically, the treated material can be fully activated to its maximum discharge capacity (270 mAh g–1) within 45 cycles, which is over 100 cycles earlier than that of pristine material (Fig. 13g). The in-situ synchrotron XRD reveals that, in urea treated material, more Li+ ions are extracted from the transition layers upon high voltage charging above 4.5 V, further evidence for the faster activation [67].
Glucose is also a reductive chemical used to pre-activate the LRMs, but has different mechanisms of reaction with LRM. During the hydrothermal treatment, glucose did not directly react with the material. Instead, glucose decomposed into carbon species, while the generated carbon later reduced Mn in the material. During the treatment, Li and O are also extracted to compensate for the charge. Similarly, such changes can result in a surface spinel phase and improve the stability and rate performance. The initial activation is supressed because of the consumption of the Li2MnO3 phase during treatment, but the discharge capacity is not reduced [190]. This may be related to the Li vacancies generated in the treatment. Dopamine has a similar multi-step reaction mechanism with LRM. During heat treatment, dopamine was first coated on the surface of particles, and then, it was decomposed to carbon species, which finally reduced the Mn4+ and induced a surface spinel phase. After treatment, the activation plateau was also slightly shortened, but not as significantly as that of glucose treated material. In the following cycles, the charge capacity is higher than that of pristine material, indicating higher oxygen redox activity and reversibility [191]. Both glucose and dopamine treatment can essentially induce carbon reduction reaction on LRMs. However, dopamine treated materials show a higher activation extent than that of glucose treated material, possibly because dopamine treatment consumes less of the Li2MnO3 phase.
In addition to reductive chemicals, oxidative chemicals are also commonly used to treat LRMs. For example, Na2S2O8 can oxidize the lattice oxygen near the surface of LRMs, from O2− to O22−. To compensate for the charge, Li+ can be extracted from the lattice. Then during the following annealing process, unstable O22− species can be disproportionated to O2− and O2. The generation of the latter can leave oxygen vacancies, resulting in a LiMn2O4-type spinel phase. Such a reaction is speculated to occur in the Li2MnO3 phase, as the oxidization states of Mn, Ni and Co are not changed during the treatment. Notably, the treatment slightly prolongs the charge plateau, indicating promoted activation [192]. This may be related to the stable transition metal valence state in the treatment. The transition metals stay in their highest oxidation states, which force more oxygens to participate in the charge compensation during electrochemical activation.
The other oxidative chemical (NH4)2S2O8 can oxidize the Ni2+ to Ni3+ on the surface of LRMs. To compensate for the charge, Li will be extracted during the treatment. With these changes, the surface of layered LRM will be converted to a spinel phase (Fig. 14a). After treatment, the discharge capacity and rate performance increased significantly, owing to the Li vacancies and spinel-phase-induced fast Li transportation. For charging, the voltage vs. the capacity slope below 4.5 V shifts to a higher range due to increased Ni3+/Ni4+ redox couples. However, the activation plateau shows negligible change (Fig. 14b), indicating no important change to the activation process [193].

© 2015, The Royal Society of Chemistry. c Schematic diagram of lattice change during L-ascorbic acid treatment. Reprinted with permission from Ref. [195]. Copyright © 2017, the Electrochemical Society. d Change of the surface during H3BO3 treatment and corresponding schematic of lattice change. Reprinted with permission from Ref. [197]. Copyright © 2018, Elsevier B.V. e Initial cycling profile before and after H2SO4 treatment; f O2 and g CO2 gas generation amount during initial cycling before and after H2SO4 treatment. Reprinted with permission from Ref. [198]. Copyright © 2020, American Chemical Society
a Schematic of the change during (NH4)2S2O8 (APS) treatment; b initial cycling profile before and after (NH4)2S2O8 treatment. Reprinted with permission from Ref. [193]. Open Access, Copyright
Besides reductive and oxidative chemicals, acids are also effective at pre-activating LRMs. NH4H2PO4 is a moderate acid, and it can leach the Li out of the surface of LRMs as well as reduce Mn4+ to Mn3+ to compensate for the charge difference. Then, with further calcination, the Li-deficient surface will convert into a spinel phase. This phase improves the Li+ diffusion and stabilizes the lattice structure [194]. Although it is not discussed in this report, the activation plateau is slightly prolonged in both 0.1 C and 1 C current densities, indicating the enhancement of Li2MnO3 phase activation.
L-ascorbic acid is a mild acid with strong reducibility, and has been investigated in both Co-free and Co-containing LRMs. In Co-free LRMs, L-ascorbic acid extracts Li+ from the surface of LRMs due to its acidity. At the same time, Mn4+ is reduced to Mn2+ and dissolves from the surface owing to the reducibility of L-ascorbic acid. After the following calcination process, a surface spinel phase layer will be generated (Fig. 14c). However, the thickness of such a spinel phase is only about 2 nm, because Ni2+ will segregate near the surface of the particle, and prevent the further leaching of L-ascorbic acid [195]. In Co-containing LRMs, Li, Mn and O are extracted during the treatment. The Mn valence state is partially reduced, possibly because Co can increase the oxygen activity during the treatment. The formed spinel phase is smaller than that in Co-free LRMs. Ni and Co enrich on the surface [196]. In both cases, the treated materials show improved discharge capacity, rate performance and cycling stability, due to the surface formed spinel phase. However, after treatment, the Co-free material shows lower activation capacity compared to the pristine, while the Co-containing sample shows higher activation capacity. Although the mechanism is still unknown, the speculation lies into change of structure and ion arrangement caused by Co.
H3BO3 can leach Li and O from the surface of LRMs, and leave Li/O vacancies. At the same time, a small part of Mn is also extracted while the remaining Mn4+ ions near the surface are partially reduced. Ni and Co can migrate into the Li vacancies in Li-layers, and can result in the transformation of a Li-rich layered phase to a Ni/Co-rich rock salt phase (Fig. 14d). Such a new phase can improve the structural stability and kinetics of Li+ and electron transfer, beneficial to the cycle stability and rate performance. The Li vacancies can accommodate more Li+ during intercalation, leading to a higher discharge capacity. Although the whole process will consume the effected Li2MnO3 phase and reduce the activation capacity, the generated oxygen vacancies can prevent the oxygen gas evolution during activation by reducing the oxygen partial pressure [197].
According to the report from McCloskey and co-workers, dilute H2SO4 can also pre-activate LRMs by extracting Li/O from the surface of the particle and partially reduce the oxidation state of Mn4+. Similar to other acid treatment, H2SO4 treatment also results in shortening the activation plateau due to loss of the Li2MnO3 phase during treatment. But the discharge capacity is not sacrificed because Li vacancies allow more Li+ to insert into the material (Fig. 14e). Interestingly, after treatment, differential electrochemical mass spectrometry (DEMS) reveals that the oxygen gas generation is completely suppressed (Fig. 14f), and the release of CO2 is also significantly reduced (Fig. 14g). However, the ex-situ titrations of electrodes and soft X-ray mapping of resonant inelastic X-ray scattering (mRIXS mapping) prove the oxygen redox in the bulk is not affected. Further with isotopically labeled C and O near the surface, the authors found oxygen redox occurs on the surface of LRMs at about 4.2 V, then gradually extends to the bulk during the further charging. But it is worth noting that oxygen gas generation only occurs on the surface. All of these results suggest H2SO4 treatment can fully suppress the oxygen gas evolution at the expense of a minor surface Li2MnO3 phase, but without sacrificing the bulk oxygen redox activity [198].
As the core component of LRMs, research on bare Li2MnO3 is the most direct and convincing way to investigate the influence of chemical treatment. For example, upon treatment with stearic acid, Li2MnO3 is partially converted to m-LiMnO2. The removal of Li/O and reduction of Mn4+ to Mn3+ during the treatment are very similar to that of initial electrochemical activation of Li2MnO3. At the same time, Mn–O bonds are lengthened, which ease the Li extraction and oxygen evolution. Therefore, with this treatment, the initial capacity of Li2MnO3 increases significantly, and the activation voltage plateau shifts lower. Notably, the capacity contributed by activation increases from about 130 to 166 mAh g−1 (Fig. 15a) [199]. Moreover, the initial capacity increased to more than double compared to that of pristine samples. Using XPS and Electron paramagnetic resonance (EPR), it is confirmed that the improvement of electrochemical performance is attributed to the oxygen vacancies introduced during treatment (Fig. 15b) [200]. Firstly, oxygen vacancies located at 8j sites in the lattice prolong the Mn–O bond and enlarge the lattice. This can facilitate Li+ diffusion. Secondly, Mn4+ is partially reduced to Mn3+, which provides more electrons to compensate for the charge during Li+ extraction (Fig. 15c). The reduction of Mn also results in a lower shift of redox voltage. Thirdly, any existing oxygen vacancies increase the formation enthalpies of new oxygen vacancies created during activation, hence improving the structural stability (Fig. 15d). Finally, oxygen vacancies thermodynamically increase the Mn migration energy barrier during activation, which also improves the lattice stability upon cycling (Fig. 15e) [201]. All these factors ensure stabilized cycling with suppressed oxygen evolution. Although the oxygen gas generation is suppressed with pre-introduced oxygen vacancies, the oxygen redox upon cycling still takes part in the electrochemical reaction. Therefore, Li2MnO3 shows stepwise activation in the initial cycles in some cases [201, 202]. However, oxygen-vacancy containing Li2MnO3 doesn’t always demonstrate the apparent activation effect [200, 203]. Such a difference is very likely related to the amount of oxygen vacancies in the lattice, as excess vacancies may profoundly change the lattice structure and impede the oxygen redox. Furthermore, the researchers used different battery testing conditions, specifically the voltage window. This may also have a significant influence on electrochemical behavior, especially the activation process (Fig. 16).

Copyright © 2013, Elsevier Ltd. b EPR spectra of Li2MnO3 with different levels of stearic acid treatment (1 Gauss = 1×10−4 T). Reprinted with permission from Ref. [200]. Copyright © 2019, Elsevier B.V. c Change of the Mn valence state, d enthalpy of additional oxygen vacancy generation and e Mn migration energy barrier of Li2MnO3 with (red) and without (black) oxygen vacancies. Reprinted with permission from Ref. [201]. Copyright © 2016, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim
a Initial cycling profiles of Li2MnO3 before and after stearic acid treatment. Reprinted with permission from Ref. [199].

Influence of different chemical treatments on charge, discharge, and activation capacities
In summary, all the chemicals including reduction agents, oxidation agents and acid treatments, are essentially related to oxygen vacancies on the particle’s surface. Oxygen vacancies can suppress the O2 gas generation by preventing the formation of oxygen radicals during the activation. According to the above literature, oxygen vacancies can also influence the activation behavior of LRMs. Interestingly, in the Co-containing materials, the chemical treatments usually show negligible influence on the extent of activation, but only stabilize the oxygen redox upon cycling. On contrary, in the Co-free LRMs or even bare Li2MnO3, the activation is not only stabilized, but also significantly accelerated. This phenomenon is highly likely related to the impact of Co. As mentioned earlier in the composition adjustment section, Co is an activation accelerator, so the improvement of activation induced by oxygen vacancies is inconspicuous. Therefore, the chemical treatment strategy is more suitable for Co-free LRMs, which have the nature of slow activation (Table 8).
4 Summary and Outlook
Activation is the dominant process of Li-rich cathode materials determining their electrochemical performance. Due to this unique phenomenon, LRMs can deliver much higher specific capacity (> 250 mAh g−1) than that of conventional layered metal oxides. However, the activation also involves complex charge compensation, ion migration, and phase transformation, originated from the oxygen redox reaction at a voltage above 4.5 V vs. Li/Li+. To date, the mechanism of activation and the origin of oxygen redox are still ongoing debates. This review provided insightful understandings on this important research topic. Currently, The O22−/On− redox involving oxygen dimers is the most commonly accepted theory, but other mechanisms are also proposed based on first-principal calculations and advanced in/ex-situ characterizations. The research on further in-depth understanding of the activation mechanism is still important to better guide the design of LRMs with faster and more stable activation.
In this review, the design strategies for controlling activation including composition adjustment, elemental substitution and chemical treatment are discussed. As the fundamental component of LRMs, their chemical composition plays a critical role in the electrochemical performance. Mn plays a critical role in LRMs, triggering and regulating the activation process. Small amounts of Ni can promote the activation, but too much Ni will inhibit the activation and limit the Li+ diffusion. Meanwhile, Co can significantly enlarge the overall capacity and accelerate the activation. Thus, the materials with high Mn/Ni ratios and appropriate Co content can deliver much higher capacity than that of their counterparts. However, due to the toxicity and cost of Co, efforts have been put into replacing the Co with other metals. In the reported attempts, Fe and Sn can stabilize the cycling but will sacrifice the capacity. Ti is able to help retain the capacity but deteriorate the stability. Ru can dramatically stabilize the oxygen redox with high capacity, but it is too expensive to be commercialized. Therefore, exploring alternative elements to replace the Co without sacrificing the performance and cost is still a trend of the future research.
Elemental substitution is also an effective way to adjust the activation of LRMs. In the reported attempts, K, Rb, Cs, Ti, Ru, W, and Re can accelerate the activation, Cr, Fe, Cu, Zn, and F can suppress the activation, while Na, Mg, Ca, Ba, Nb, and Mo can stabilize the oxygen redox but has no significant influence on the activation extent. In the future research, it is worth applying more than one doping element, including both an activation accelerator and a stabilizer at the same time, to achieve faster activation and higher capacity without loss of cycling stability. In addition, the current research mainly focuses on cation doping, but lacks investigation into anion doping which is also of interest and importance to be studied.
The chemical treatment can be divided into reduction agent treatments, oxidation agent treatments, and acid treatments. However, all these reported treatments are essentially related to creating oxygen vacancies on the particle surface. In general, oxygen vacancies can suppress the O2 gas generation because they reduce the oxygen partial pressure, but the improvement also depends on the composition of LRMs. Specifically, oxygen vacancies can significantly accelerate the activation in Co-free LRMs, but result in negligible improvement in Co-containing LRMs. Therefore, the induction of oxygen vacancies is more suitable for Co-free LRMs. Moreover, in the future, chemical treatment with functions other than creating oxygen vacancies is worth exploring.
According to the above-mentioned investigations, different strategies can provide LRMs with high-capacity, accelerated activation, and stabilized cycling. It is recommended to apply multiple strategies at the same time to achieve high-performance LRMs. In addition, during the literature review, we noticed that researchers are using different testing conditions to perform the electrochemical cycling test, especially the voltage window and current density. These conditions have a profound influence on the electrochemical performance and activation behavior of LRMs. This results in difficulties in evaluating the influence of each design strategy. Thus, standardizing testing parameters and procedures is highly demanded in the future research of battery systems. Furthermore, to better understand the activation of LRMs, in-situ technologies are recommended for the future research. For example, in-situ XRD can reflect the crystal structure transformation, in-situ XAS can reveal the chemical bond and element environment change, in-situ Raman spectra can uncover the evolution of oxygen, and in-situ TEM can directly demonstrate the lattice deformation. In recent years, computer simulations and machine learning techniques have been developing rapidly, which are powerful tools to better understand the activation process and predict promising strategies for faster and more stable activation of LRMs. In addition to the insightful understanding of the activation process, the studies on other key challenges of LRMs in terms of the voltage decay and voltage hysteresis issues are also of significant importance towards the commercial application of LRMs.
Data Availability
The authors declare the data and materials used in this paper support their published claims and comply with field standards.
Code availability
Not applicable.
References
Mwasilu, F., Justo, J.J., Kim, E.K., et al.: Electric vehicles and smart grid interaction: a review on vehicle to grid and renewable energy sources integration. Renew. Sustain. Energy Rev. 34, 501–516 (2014). https://doi.org/10.1016/j.rser.2014.03.031
Rozier, P., Tarascon, J.M.: Review: Li-rich layered oxide cathodes for next-generation Li-ion batteries: chances and challenges. J. Electrochem. Soc. 162, A2490–A2499 (2015). https://doi.org/10.1149/2.0111514jes
Goodenough, J.B., Park, K.S.: The Li-ion rechargeable battery: a perspective. J. Am. Chem. Soc. 135, 1167–1176 (2013). https://doi.org/10.1021/ja3091438
Whittingham, M.S.: Lithium batteries and cathode materials. Chem. Rev. 104, 4271–4302 (2004). https://doi.org/10.1021/cr020731c
Chen, K.F., Yin, S., Xue, D.F.: Active La–Nb–O compounds for fast lithium-ion energy storage. Tungsten 1, 287–296 (2019). https://doi.org/10.1007/s42864-019-00029-2
Zuo, J.H., Gong, Y.J.: Applications of transition-metal sulfides in the cathodes of lithium–sulfur batteries. Tungsten 2, 134–146 (2020). https://doi.org/10.1007/s42864-020-00046-6
Cao, Z.J., Zhang, Y.Z., Cui, Y.L.S., et al.: Harnessing the unique features of MXenes for sulfur cathodes. Tungsten 2, 162–175 (2020). https://doi.org/10.1007/s42864-020-00047-5
Teng, X.L., Sun, X.T., Guan, L., et al.: Self-supported transition metal oxide electrodes for electrochemical energy storage. Tungsten 2, 337–361 (2020). https://doi.org/10.1007/s42864-020-00068-0
Sun, C.B., Zhong, Y.W., Fu, W.J., et al.: Tungsten disulfide-based nanomaterials for energy conversion and storage. Tungsten 2, 109–133 (2020). https://doi.org/10.1007/s42864-020-00038-6
Wang, W.-P., Zhang, J., Li, X.-T., et al.: Stabilizing the electrochemistry of lithium-selenium battery via in situ gelated polymer electrolyte: a look from anode. Chem. Res. Chin. Univ. 37, 298–303 (2021). https://doi.org/10.1007/s40242-021-0448-4
Liu, F., Cui, Y.: Zeolite-based electrolyte accelerating the realization of solid-state Li-air battery. Chem. Res. Chin. Univ. 37, 801–802 (2021). https://doi.org/10.1007/s40242-021-1224-1
Zhang, C., Liu, D.H., Geng, C.N., et al.: Solution-based preparation of high sulfur content sulfur/graphene cathode material for Li-S battery. Chem. Res. Chin. Univ. 37, 323–327 (2021). https://doi.org/10.1007/s40242-021-0036-7
Luo, K., Roberts, M.R., Hao, R., et al.: Charge-compensation in 3d-transition-metal-oxide intercalation cathodes through the generation of localized electron holes on oxygen. Nat. Chem. 8, 684–691 (2016). https://doi.org/10.1038/nchem.2471
Chen, L., Fan, X.L., Hu, E.Y., et al.: Achieving high energy density through increasing the output voltage: a highly reversible 5.3 V battery. Chem 5, 896–912 (2019). https://doi.org/10.1016/j.chempr.2019.02.003
Boulineau, A., Simonin, L., Colin, J.F., et al.: First evidence of manganese–nickel segregation and densification upon cycling in Li-rich layered oxides for lithium batteries. Nano Lett. 13, 3857–3863 (2013). https://doi.org/10.1021/nl4019275
Gu, M., Belharouak, I., Zheng, J.M., et al.: Formation of the spinel phase in the layered composite cathode used in Li-ion batteries. ACS Nano 7, 760–767 (2013). https://doi.org/10.1021/nn305065u
Armstrong, A.R., Holzapfel, M., Novák, P., et al.: Demonstrating oxygen loss and associated structural reorganization in the lithium battery cathode Li[Ni0.2Li0.2Mn0.6]O2. J. Am. Chem. Soc. 128, 8694–8698 (2006). https://doi.org/10.1021/ja062027+
Boulineau, A., Simonin, L., Colin, J.F., et al.: Evolutions of Li1.2Mn0.61Ni0.18Mg0.01O2 during the initial charge/discharge cycle studied by advanced electron microscopy. Chem. Mater. 24, 3558–3566 (2012). https://doi.org/10.1021/cm301140g
de Biasi, L., Schwarz, B., Brezesinski, T., et al.: Chemical, structural, and electronic aspects of formation and degradation behavior on different length scales of Ni-rich NCM and Li-rich HE-NCM cathode materials in Li-ion batteries. Adv. Mater. 31, 1900985 (2019). https://doi.org/10.1002/adma.201900985
Ko, M., Oh, P., Chae, S., et al.: Considering critical factors of Li-rich cathode and Si anode materials for practical Li-ion cell applications. Small Weinheim Der Bergstrasse Ger. 11, 4058–4073 (2015). https://doi.org/10.1002/smll.201500474
Zhang, K., Li, B., Zuo, Y.X., et al.: Voltage decay in layered Li-rich Mn-based cathode materials. Electrochem. Energy Rev. 2, 606–623 (2019). https://doi.org/10.1007/s41918-019-00049-z
Yang, J.G., Niu, Y.B., Wang, X., et al.: A review on the electrochemical reaction of Li-rich layered oxide materials. Inorg. Chem. Front. 8, 4300–4312 (2021). https://doi.org/10.1039/d1qi00687h
Hu, S.J., Pillai, A.S., Liang, G.M., et al.: Li-rich layered oxides and their practical challenges: recent progress and perspectives. Electrochem. Energy Rev. 2, 277–311 (2019). https://doi.org/10.1007/s41918-019-00032-8
Nayak, P.K., Erickson, E.M., Schipper, F., et al.: Review on challenges and recent advances in the electrochemical performance of high capacity Li- and Mn-rich cathode materials for Li-ion batteries. Adv. Energy Mater. 8, 1702397 (2018). https://doi.org/10.1002/aenm.201702397
Rossouw, M., Thackeray, M.: Lithium manganese oxides from Li2MnO3 for rechargeable lithium battery applications. Mater. Res. Bull. 26, 463–473 (1991). https://doi.org/10.1016/0025-5408(91)90186-P
Lee, S.H., Moon, J.S., Lee, M.S., et al.: Enhancing phase stability and kinetics of lithium-rich layered oxide for an ultra-high performing cathode in Li-ion batteries. J. Power Sources 281, 77–84 (2015). https://doi.org/10.1016/j.jpowsour.2015.01.158
Yu, H.J., Ishikawa, R., So, Y.G., et al.: Direct atomic-resolution observation of two phases in the Li1.2Mn0.567Ni0.166Co0.067O2 cathode material for lithium-ion batteries. Angew. Chem. Int. Ed. 52, 5969–5973 (2013). https://doi.org/10.1002/anie.201301236
Ye, D.L., Zeng, G., Nogita, K., et al.: Understanding the origin of Li2MnO3 activation in Li-rich cathode materials for lithium-ion batteries. Adv. Funct. Mater. 25, 7488–7496 (2015). https://doi.org/10.1002/adfm.201503276
Lei, C.H., Bareño, J., Wen, J.G., et al.: Local structure and composition studies of Li1.2Ni0.2Mn0.6O2 by analytical electron microscopy. J. Power Sources 178, 422–433 (2008). https://doi.org/10.1016/j.jpowsour.2007.11.077
Bareño, J., Lei, C.H., Wen, J.G., et al.: Local structure of layered oxide electrode materials for lithium-ion batteries. Adv. Mater. 22, 1122–1127 (2010). https://doi.org/10.1002/adma.200904247
Jarvis, K.A., Deng, Z.Q., Allard, L.F., et al.: Atomic structure of a lithium-rich layered oxide material for lithium-ion batteries: evidence of a solid solution. Chem. Mater. 23, 3614–3621 (2011). https://doi.org/10.1021/cm200831c
Gu, M., Belharouak, I., Genc, A., et al.: Conflicting roles of nickel in controlling cathode performance in lithium ion batteries. Nano Lett. 12, 5186–5191 (2012). https://doi.org/10.1021/nl302249v
Thackeray, M.M., Johnson, C.S., Vaughey, J.T., et al.: Advances in manganese-oxide “composite” electrodes for lithium-ion batteries. J. Mater. Chem. 15, 2257 (2005). https://doi.org/10.1039/b417616m
Wu, F., Li, N., Su, Y.F., et al.: Can surface modification be more effective to enhance the electrochemical performance of lithium rich materials? J. Mater. Chem. 22, 1489–1497 (2012). https://doi.org/10.1039/c1jm14459f
Shi, S.J., Tu, J.P., Tang, Y.Y., et al.: Combustion synthesis and electrochemical performance of Li[Li0.2Mn0.54Ni0.13Co0.13]O2 with improved rate capability. J. Power Sources 228, 14–23 (2013). https://doi.org/10.1016/j.jpowsour.2012.11.091
Zheng, J.M., Wu, X.B., Yang, Y.: A comparison of preparation method on the electrochemical performance of cathode material Li[Li0.2Mn0.54Ni0.13Co0.13]O2 for lithium ion battery. Electrochim. Acta 56, 3071–3078 (2011). https://doi.org/10.1016/j.electacta.2010.12.049
Wang, C.C., Jarvis, K.A., Ferreira, P.J., et al.: Effect of synthesis conditions on the first charge and reversible capacities of lithium-rich layered oxide cathodes. Chem. Mater. 25, 3267–3275 (2013). https://doi.org/10.1021/cm402181f
Liu, Y.J., Gao, Y.Y., Dou, A.C.: Influence of Li content on the structure and electrochemical performance of Li1+xNi0.25Mn0.75O2.25+x/2 cathode for Li-ion battery. J. Power Sources 248, 679–684 (2014). https://doi.org/10.1016/j.jpowsour.2013.10.006
Song, B.H., Lai, M.O., Lu, L.: Influence of Ru substitution on Li-rich 0.55Li2MnO3·0.45LiNi1/3Co1/3Mn1/3O2 cathode for Li-ion batteries. Electrochim. Acta 80, 187–195 (2012). https://doi.org/10.1016/j.electacta.2012.06.118
McCalla, E., Rowe, A.W., Shunmugasundaram, R., et al.: Structural study of the Li–Mn–Ni oxide pseudoternary system of interest for positive electrodes of Li-ion batteries. Chem. Mater. 25, 989–999 (2013). https://doi.org/10.1021/cm4001619
Ye, D.L., Sun, C.H., Chen, Y., et al.: Ni-induced stepwise capacity increase in Ni-poor Li-rich cathode materials for high performance lithium ion batteries. Nano Res. 8, 808–820 (2015). https://doi.org/10.1007/s12274-014-0563-3
Zuo, Y.X., Li, B., Jiang, N., et al.: A high-capacity O2− type Li-rich cathode material with a single-layer Li2 MnO3 superstructure. Adv. Mater. 30, 1707255 (2018). https://doi.org/10.1002/adma.201707255
Koyama, Y., Tanaka, I., Nagao, M., et al.: First-principles study on lithium removal from Li2MnO3. J. Power Sources 189, 798–801 (2009). https://doi.org/10.1016/j.jpowsour.2008.07.073
Liu, H.D., Chen, Y., Hy, S., et al.: Operando lithium dynamics in the Li-rich layered oxide cathode material via neutron diffraction. Meet. Abstr. (2016). https://doi.org/10.1149/ma2016-01/2/260
Oishi, M., Yamanaka, K., Watanabe, I., et al.: Direct observation of reversible oxygen anion redox reaction in Li-rich manganese oxide, Li2MnO3, studied by soft X-ray absorption spectroscopy. J. Mater. Chem. A 4, 9293–9302 (2016). https://doi.org/10.1039/c6ta00174b
Li, X., Qiao, Y., Guo, S.H., et al.: Direct visualization of the reversible O2−/O− redox process in Li-rich cathode materials. Adv. Mater. 30, 1705197 (2018). https://doi.org/10.1002/adma.201705197
Sathiya, M., Rousse, G., Ramesha, K., et al.: Reversible anionic redox chemistry in high-capacity layered-oxide electrodes. Nat. Mater. 12, 827–835 (2013). https://doi.org/10.1038/nmat3699
McCalla, E., Abakumov, A.M., Saubanère, M., et al.: Visualization of O–O peroxo-like dimers in high-capacity layered oxides for Li-ion batteries. Science 350, 1516–1521 (2015). https://doi.org/10.1126/science.aac8260
Lu, Z.H., Beaulieu, L.Y., Donaberger, R.A., et al.: Synthesis, structure, and electrochemical behavior of Li[NixLi1/3–2x/3Mn2/3–x/3]O2. J. Electrochem. Soc. 149, A778 (2002). https://doi.org/10.1149/1.1471541
Qian, D.N., Xu, B., Chi, M.F., et al.: Uncovering the roles of oxygen vacancies in cation migration in lithium excess layered oxides. Phys. Chem. Chem. Phys. 16, 14665–14668 (2014). https://doi.org/10.1039/c4cp01799d
Li, N., Hwang, S., Sun, M.L., et al.: Unraveling the voltage decay phenomenon in Li-rich layered oxide cathode of no oxygen activity. Adv. Energy Mater. 9, 1902258 (2019). https://doi.org/10.1002/aenm.201902258
Guerrini, N., Jin, L.Y., Lozano, J.G., et al.: Charging mechanism of Li2MnO3. Chem. Mater. 32, 3733–3740 (2020). https://doi.org/10.1021/acs.chemmater.9b04459
Koga, H., Croguennec, L., Ménétrier, M., et al.: Different oxygen redox participation for bulk and surface: a possible global explanation for the cycling mechanism of Li1.20Mn0.54Co0.13Ni0.13O2. J. Power Sources 236, 250–258 (2013). https://doi.org/10.1016/j.jpowsour.2013.02.075
House, R.A., Rees, G.J., Pérez-Osorio, M.A., et al.: First-cycle voltage hysteresis in Li-rich 3d cathodes associated with molecular O2 trapped in the bulk. Nat. Energy 5, 777–785 (2020). https://doi.org/10.1038/s41560-020-00697-2
Koga, H., Croguennec, L., Ménétrier, M., et al.: Operando X-ray absorption study of the redox processes involved upon cycling of the Li-rich layered oxide Li1.20Mn0.54Co0.13Ni0.13O2 in Li ion batteries. J. Phys. Chem. C 118, 5700–5709 (2014). https://doi.org/10.1021/jp412197z
Seo, D.H., Lee, J., Urban, A., et al.: The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials. Nat. Chem. 8, 692–697 (2016). https://doi.org/10.1038/nchem.2524
Zhang, Z.H., Zhao, S., Wang, B.Y., et al.: Local redox reaction of high valence manganese in Li2MnO3-based lithium battery cathodes. Cell Rep. Phys. Sci. 1, 100061 (2020). https://doi.org/10.1016/j.xcrp.2020.100061
Radin, M.D., Vinckeviciute, J., Seshadri, R., et al.: Manganese oxidation as the origin of the anomalous capacity of Mn-containing Li-excess cathode materials. Nat. Energy 4, 639–646 (2019). https://doi.org/10.1038/s41560-019-0439-6
Rana, J., Papp, J.K., Lebens-Higgins, Z., et al.: Quantifying the capacity contributions during activation of Li2MnO3. ACS Energy Lett. 5, 634–641 (2020). https://doi.org/10.1021/acsenergylett.9b02799
Zuba, M.J., Grenier, A., Lebens-Higgins, Z., et al.: Whither Mn oxidation in Mn-rich alkali-excess cathodes? ACS Energy Lett. 6, 1055–1064 (2021). https://doi.org/10.1021/acsenergylett.0c02418
Hong, J., Lim, H.D., Lee, M., et al.: Critical role of oxygen evolved from layered Li–excess metal oxides in lithium rechargeable batteries. Chem. Mater. 24, 2692–2697 (2012). https://doi.org/10.1021/cm3005634
Yabuuchi, N., Yoshii, K., Myung, S.T., et al.: Detailed studies of a high-capacity electrode material for rechargeable batteries, Li2MnO3–LiCo1/3Ni1/3Mn1/3O2. J. Am. Chem. Soc. 133, 4404–4419 (2011). https://doi.org/10.1021/ja108588y
Castel, E., Berg, E.J., El Kazzi, M., et al.: Differential electrochemical mass spectrometry study of the interface of xLi2MnO3·(1–x)LiMO2 (M = Ni, Co, and Mn) material as a positive electrode in Li-ion batteries. Chem. Mater. 26, 5051–5057 (2014). https://doi.org/10.1021/cm502201z
Hy, S., Felix, F., Rick, J., et al.: Direct in situ observation of Li2O evolution on Li-rich high-capacity cathode material, Li[NixLi(1–2x)/3Mn(2–x)/3]O2 (0 \(\leqslant\) x \(\leqslant\) 0.5). J. Am. Chem. Soc. 136, 999–1007 (2014). https://doi.org/10.1021/ja410137s
Robertson, A.D., Bruce, P.G.: The origin of electrochemical activity in Li2MnO3. Chem. Commun. Camb. Engl. 23, 2790–2791 (2002). https://doi.org/10.1039/b207945c
Liu, Q., Su, X., Lei, D., et al.: Approaching the capacity limit of lithium cobalt oxide in lithium ion batteries via lanthanum and aluminium doping. Nat. Energy 3, 936–943 (2018). https://doi.org/10.1038/s41560-018-0180-6
Lin, T.E., Schulli, T., Hu, Y.X., et al.: Faster activation and slower capacity/voltage fading: a bifunctional urea treatment on lithium-rich cathode materials. Adv. Funct. Mater. 30, 1909192 (2020)
Luo, D., Ding, X.K., Fan, J.M., et al.: Accurate control of initial coulombic efficiency for lithium-rich manganese-based layered oxides by surface multicomponent integration. Angew. Chem. Int. Ed. 59, 23061–23066 (2020). https://doi.org/10.1002/anie.202010531
Zhu, Z., Gao, R., Waluyo, I., et al.: Stabilized co-free Li-rich oxide cathode particles with an artificial surface prereconstruction. Adv. Energy Mater. 10, 2001120 (2020). https://doi.org/10.1002/aenm.202001120
Ku, K., Kim, B., Jung, S.K., et al.: A new lithium diffusion model in layered oxides based on asymmetric but reversible transition metal migration. Energy Environ. Sci. 13, 1269–1278 (2020). https://doi.org/10.1039/c9ee04123k
Fell, C.R., Qian, D.N., Carroll, K.J., et al.: Correlation between oxygen vacancy, microstrain, and cation distribution in lithium-excess layered oxides during the first electrochemical cycle. Chem. Mater. 25, 1621–1629 (2013). https://doi.org/10.1021/cm4000119
Kim, S.P., van Duin, A.C.T., Shenoy, V.B.: Effect of electrolytes on the structure and evolution of the solid electrolyte interphase (SEI) in Li-ion batteries: a molecular dynamics study. J. Power Sources 196, 8590–8597 (2011). https://doi.org/10.1016/j.jpowsour.2011.05.061
Kim, J., Lee, J., Ma, H., et al.: Battery cathodes: controllable solid electrolyte interphase in nickel-rich cathodes by an electrochemical rearrangement for stable lithium-ion batteries. Adv. Mater. 30, 1870029 (2018). https://doi.org/10.1002/adma.201870029
Wu, Y., Ma, C., Yang, J.H., et al.: Probing the initiation of voltage decay in Li-rich layered cathode materials at the atomic scale. J. Mater. Chem. A 3, 5385–5391 (2015). https://doi.org/10.1039/c4ta06856d
Zheng, J.M., Xu, P.H., Gu, M., et al.: Structural and chemical evolution of Li- and Mn-rich layered cathode material. Chem. Mater. 27, 1381–1390 (2015). https://doi.org/10.1021/cm5045978
Gent, W.E., Lim, K., Liang, Y.F., et al.: Coupling between oxygen redox and cation migration explains unusual electrochemistry in lithium-rich layered oxides. Nat. Commun. 8, 2091 (2017). https://doi.org/10.1038/s41467-017-02041-x
Xu, B., Fell, C.R., Chi, M.F., et al.: Identifying surface structural changes in layered Li-excess nickel manganese oxides in high voltage lithium ion batteries: a joint experimental and theoretical study. Energy Environ. Sci. 4, 2223 (2011). https://doi.org/10.1039/c1ee01131f
Shen, S., Hong, Y., Zhu, F., et al.: Tuning electrochemical properties of Li-rich layered oxide cathodes by adjusting Co/Ni ratios and mechanism investigation using in situ X-ray diffraction and online continuous flow differential electrochemical mass spectrometry. ACS Appl. Mater. Interfaces 10, 12666–12677 (2018). https://doi.org/10.1021/acsami.8b00919
Hu, E.Y., Yu, X.Q., Lin, R.Q., et al.: Evolution of redox couples in Li- and Mn-rich cathode materials and mitigation of voltage fade by reducing oxygen release. Nat. Energy 3, 690–698 (2018). https://doi.org/10.1038/s41560-018-0207-z
Ku, K., Hong, J., Kim, H., et al.: Suppression of voltage decay through manganese deactivation and nickel redox buffering in high-energy layered lithium-rich electrodes. Adv. Energy Mater. 8, 1800606 (2018). https://doi.org/10.1002/aenm.201800606
Li, B., Sougrati, M.T., Rousse, G., et al.: Correlating ligand-to-metal charge transfer with voltage hysteresis in a Li-rich rock-salt compound exhibiting anionic redox. Nat. Chem. 13, 1070–1080 (2021). https://doi.org/10.1038/s41557-021-00775-2
Sun, G., Yu, F.D., Zhao, C.T., et al.: Decoupling the voltage hysteresis of Li-rich cathodes: electrochemical monitoring, modulation anionic redox chemistry and theoretical verifying. Adv. Funct. Mater. 31, 2002643 (2021). https://doi.org/10.1002/adfm.202002643
Han, M., Jiao, J.Y., Liu, Z.P., et al.: Eliminating transition metal migration and anionic redox to understand voltage hysteresis of lithium-rich layered oxides. Adv. Energy Mater. 10, 1903634 (2020). https://doi.org/10.1002/aenm.201903634
Assat, G., Glazier, S.L., Delacourt, C., et al.: Probing the thermal effects of voltage hysteresis in anionic redox-based lithium-rich cathodes using isothermal calorimetry. Nat. Energy 4, 647–656 (2019). https://doi.org/10.1038/s41560-019-0410-6
Liu, H., Zhao, C., Qiu, Q., et al.: What triggers the voltage hysteresis variation beyond the first cycle in Li-rich 3d layered oxides with reversible cation migration? J. Phys. Chem. Lett. 12, 8740–8748 (2021). https://doi.org/10.1021/acs.jpclett.1c02185
Taylor, Z.N., Perez, A.J., Coca-Clemente, J.A., et al.: Stabilization of O–O Bonds by d0 Cations in Li4+xNi1–xWO6 (0 \(\leqslant\) x \(\leqslant\) 0.25) Rock Salt oxides as the origin of large voltage hysteresis. J. Am. Chem. Soci. 141, 7333–7346 (2019). https://doi.org/10.1021/jacs.8b13633
Assat, G., Delacourt, C., Corte, D.A.D., et al.: Editors’ choice—practical assessment of anionic redox in Li-rich layered oxide cathodes: a mixed blessing for high energy Li-ion batteries. J. Electrochem. Soc. 163, A2965–A2976 (2016). https://doi.org/10.1149/2.0531614jes
Croy, J.R., Gallagher, K.G., Balasubramanian, M., et al.: Examining hysteresis in composite xLi2MnO3·(1–x)LiMO2 cathode structures. J. Phys. Chem. C 117, 6525–6536 (2013). https://doi.org/10.1021/jp312658q
Assat, G., Foix, D., Delacourt, C., et al.: Fundamental interplay between anionic/cationic redox governing the kinetics and thermodynamics of lithium-rich cathodes. Nat. Commun. 8, 2219 (2017). https://doi.org/10.1038/s41467-017-02291-9
Wang, Z.Y., Liu, E.Z., He, C.N., et al.: Effect of amorphous FePO4 coating on structure and electrochemical performance of Li1.2Ni0.13Co0.13Mn0.54O2 as cathode material for Li-ion batteries. J. Power Sources 236, 25–32 (2013). https://doi.org/10.1016/j.jpowsour.2013.02.022
Shi, S.J., Tu, J.P., Mai, Y.J., et al.: Effect of carbon coating on electrochemical performance of Li1.048Mn0.381Ni0.286Co0.286O2 cathode material for lithium-ion batteries. Electrochim. Acta 63, 112–117 (2012). https://doi.org/10.1016/j.electacta.2011.12.082
Zheng, J.M., Gu, M., Xiao, J., et al.: Functioning mechanism of AlF3 coating on the Li- and Mn-rich cathode materials. Chem. Mater. 26, 6320–6327 (2014). https://doi.org/10.1021/cm502071h
Gao, J., Kim, J., Manthiram, A.: High capacity Li[Li0.2Mn0.54Ni0.13Co0.13]O2-V2O5 composite cathodes with low irreversible capacity loss for lithium ion batteries. Electrochem. Commun. 11, 84–86 (2009). https://doi.org/10.1016/j.elecom.2008.10.036
Gallagher, K.G., Kang, S.H., Park, S.U., et al.: xLi2MnO3·(1–x)LiMO2 blended with LiFePO4 to achieve high energy density and pulse power capability. J. Power Sources 196, 9702–9707 (2011). https://doi.org/10.1016/j.jpowsour.2011.07.054
Yin, Z.W., Peng, X.X., Li, J.T., et al.: Revealing of the activation pathway and cathode electrolyte interphase evolution of Li-rich 0.5Li2MnO3·0.5LiNi0.3Co0.3Mn0.4O2 cathode by in situ electrochemical quartz crystal microbalance. ACS Appl. Mater. Interfaces 11, 16214–16222 (2019). https://doi.org/10.1021/acsami.9b02236
Simonelli, L., Sorrentino, A., Marini, C., et al.: Role of manganese in lithium- and manganese-rich layered oxides cathodes. J. Phys. Chem. Lett. 10, 3359–3368 (2019). https://doi.org/10.1021/acs.jpclett.9b01174
Chen, Z.L., Li, J., Zeng, X.C.: Unraveling oxygen evolution in Li-rich oxides: a unified modeling of the intermediate peroxo/superoxo-like dimers. J. Am. Chem. Soc. 141, 10751–10759 (2019). https://doi.org/10.1021/jacs.9b03710
Ting, M., Burigana, M., Zhang, L.T., et al.: Impact of nickel substitution into model Li-rich oxide cathode materials for Li-ion batteries. Chem. Mater. 32, 849–857 (2020). https://doi.org/10.1021/acs.chemmater.9b04446
Shi, J.L., Xiao, D.D., Ge, M.Y., et al.: High-capacity cathode material with high voltage for Li-ion batteries. Adv. Mater. 30, 1705575 (2018). https://doi.org/10.1002/adma.201705575
Sun, G., Yu, F.D., Que, L.F., et al.: Local electronic structure modulation enhances operating voltage in Li-rich cathodes. Nano Energy 66, 104102 (2019). https://doi.org/10.1016/j.nanoen.2019.104102
Ye, D.L., Ozawa, K., Wang, B., et al.: Capacity-controllable Li-rich cathode materials for lithium-ion batteries. Nano Energy 6, 92–102 (2014). https://doi.org/10.1016/j.nanoen.2014.03.013
Ye, D.L., Wang, B., Chen, Y., et al.: Understanding the stepwise capacity increase of high energy low-Co Li-rich cathode materials for lithium ion batteries. J. Mater. Chem. A 2, 18767–18774 (2014). https://doi.org/10.1039/c4ta03692a
Lanina, E.V., Zhuravlev, V.D., Ermakova, L.V., et al.: Electrochemical performances of composite cathode materials Li1.2Ni0.17Co0.10Mn0.53O2 and Li1.2Ni0.2Mn0.6O2. Electrochim. Acta 212, 810–821 (2016). https://doi.org/10.1016/j.electacta.2016.07.010
Wu, Y.Q., Xie, L.Q., He, X.M., et al.: Electrochemical activation, voltage decay and hysteresis of Li-rich layered cathode probed by various cobalt content. Electrochim. Acta 265, 115–120 (2018). https://doi.org/10.1016/j.electacta.2018.01.181
Manthiram, A., Choi, J.: Chemical and structural instabilities of lithium ion battery cathodes. J. Power Sources 159, 249–253 (2006). https://doi.org/10.1016/j.jpowsour.2006.04.028
Hamad, K.I., Xing, Y.C.: Effect of cobalt and nickel contents on the performance of lithium rich materials synthesized in glycerol solvent. J. Electrochem. Soc. 165, A2470–A2475 (2018). https://doi.org/10.1149/2.0311811jes
Zhao, T.L., Zhang, Y.X., Li, Y.T., et al.: Electrochemical activation of novel Fe-based Li-rich cathode material for lithium-ion batteries. J. Alloys Compd. 741, 597–603 (2018). https://doi.org/10.1016/j.jallcom.2018.01.161
Zhao, T.L., Ji, R.X., Yang, H.D., et al.: Distinctive electrochemical performance of novel Fe-based Li-rich cathode material prepared by molten salt method for lithium-ion batteries. J. Energy Chem. 33, 37–45 (2019). https://doi.org/10.1016/j.jechem.2018.08.005
Laisa, C.P., Nanda Kumar, A.K., Chandrasekaran, S.S., et al.: A comparative study on electrochemical cycling stability of lithium rich layered cathode materials Li1.2Ni0.13M0.13Mn0.54O2 where M = Fe or Co. J. Power Sources 324, 462–474 (2016). https://doi.org/10.1016/j.jpowsour.2016.05.107
Aryal, S., Timofeeva, E.V., Segre, C.U.: Structural studies of capacity activation and reduced voltage fading in Li-rich, Mn-Ni-Fe composite oxide cathode. J. Electrochem. Soc. 165, A71–A78 (2018). https://doi.org/10.1149/2.0031802jes
Li, X., Qiao, Y., Guo, S.H., et al.: A new type of Li-rich rock-salt oxide Li2Ni1/3Ru2/3O3 with reversible anionic redox chemistry. Adv. Mater. 31, 1807825 (2019). https://doi.org/10.1002/adma.201807825
Dianat, A., Seriani, N., Bobeth, M., et al.: Effects of Al-doping on the properties of Li–Mn–Ni–O cathode materials for Li-ion batteries: an ab initio study. J. Mater. Chem. A 1, 9273 (2013). https://doi.org/10.1039/c3ta11598d
Qing, R.P., Shi, J.L., Xiao, D.D., et al.: Cathode materials: enhancing the kinetics of Li-rich cathode materials through the pinning effects of gradient surface Na+ doping. Adv. Energy Mater. 6, 1501914 (2016). https://doi.org/10.1002/aenm.201670035
Li, N., He, Y.S., Wang, X.P., et al.: Incorporation of rubidium cations into Li1.2Mn0.54Co0.13Ni0.13O2 layered oxide cathodes for improved cycling stability. Electrochim. Acta 231, 363–370 (2017). https://doi.org/10.1016/j.electacta.2017.01.137
Li, Q., Li, G.S., Fu, C.C., et al.: K+-doped Li1.2Mn0.54Co0.13Ni0.13O2: a novel cathode material with an enhanced cycling stability for lithium-ion batteries. ACS Appl. Mater. Interfaces 6, 10330–10341 (2014). https://doi.org/10.1021/am5017649
Tabuchi, M., Nabeshima, Y., Takeuchi, T., et al.: Synthesis of high-capacity Ti- and/or Fe-substituted Li2MnO3 positive electrode materials with high initial cycle efficiency by application of the carbothermal reduction method. J. Power Sources 221, 427–434 (2013). https://doi.org/10.1016/j.jpowsour.2012.08.055
Ding, X., Li, Y.X., Deng, M.M., et al.: Cesium doping to improve the electrochemical performance of layered Li1.2Ni0.13Co0.13Mn0.54O2 cathode material. J. Alloys Compd. 791, 100–108 (2019). https://doi.org/10.1016/j.jallcom.2019.03.297
Lim, S.N., Seo, J.Y., Jung, D.S., et al.: Rate capability for Na-doped Li1.167Ni0.18Mn0.548Co0.105O2 cathode material and characterization of Li-ion diffusion using galvanostatic intermittent titration technique. J. Alloys Compd. 623, 55–61 (2015). https://doi.org/10.1016/j.jallcom.2014.09.203
Liu, Z.Z., Zhang, Z., Liu, Y.M., et al.: Facile and scalable fabrication of K+-doped Li1.2Ni0.2Co0.08Mn0.52O2 cathode with ultra high capacity and enhanced cycling stability for lithium ion batteries. Solid State Ion. 332, 47–54 (2019). https://doi.org/10.1016/j.ssi.2018.12.021
Hu, Y.Y., Qin, Z.Z., Pei, J., et al.: Reduced lithium/nickel disorder degree of sodium-doped lithium-rich layered oxides for cathode materials: experiments and calculations. ChemElectroChem 7, 246–251 (2020). https://doi.org/10.1002/celc.201901846
Ates, M.N., Jia, Q.Y., Shah, A., et al.: Mitigation of layered to spinel conversion of a Li-rich layered metal oxide cathode material for Li-ion batteries. J. Electrochem. Soc. 161, A290–A301 (2013). https://doi.org/10.1149/2.040403jes
Wang, D., Liu, M.H., Wang, X.Y., et al.: Facile synthesis and performance of Na-doped porous lithium-rich cathodes for lithium ion batteries. RSC Adv. 6, 57310–57319 (2016). https://doi.org/10.1039/c6ra09042g
Ding, X., Li, Y.X., He, X.D., et al.: Surface Li+/K+ exchange toward double-gradient modification of layered Li-rich cathode materials. ACS Appl. Mater. Interfaces 11, 31477–31483 (2019). https://doi.org/10.1021/acsami.9b07659
Zheng, Z., Guo, X.D., Zhong, Y.J., et al.: Host structural stabilization of Li1.232Mn0.615Ni0.154O2 through K-doping attempt: toward superior electrochemical performances. Electrochim. Acta 188, 336–343 (2016). https://doi.org/10.1016/j.electacta.2015.12.021
Xu, H.J., Deng, S.N., Chen, G.H.: Improved electrochemical performance of Li1.2Mn0.54Ni0.13Co0.13O2 by Mg doping for lithium ion battery cathode material. J. Mater. Chem. A 2, 15015–15021 (2014). https://doi.org/10.1039/c4ta01790k
Yi, T.F., Li, Y.M., Yang, S.Y., et al.: Improved cycling stability and fast charge–discharge performance of cobalt-free lithium-rich oxides by magnesium-doping. ACS Appl. Mater. Interfaces 8, 32349–32359 (2016). https://doi.org/10.1021/acsami.6b11724
Hoang, K.: Doping Li-rich cathode material Li2MnO3: interplay between lattice site preference, electronic structure, and delithiation mechanism. Phys. Rev. Materials 1, 075404 (2017). https://doi.org/10.1103/physrevmaterials.1.075404
Lo, W.T., Yu, C., Leggesse, E.G., et al.: Understanding the role of dopant metal atoms on the structural and electronic properties of lithium-rich Li1.2Ni0.2Mn0.6O2 cathode material for lithium-ion batteries. J. Phys. Chem. Lett. 10, 4842–4850 (2019). https://doi.org/10.1021/acs.jpclett.9b01516
Laisa, C.P., Ramesha, R.N., Ramesha, K.: Enhanced electrochemical performance of lithium rich layered cathode materials by Ca2+ substitution. Electrochim. Acta 256, 10–18 (2017). https://doi.org/10.1016/j.electacta.2017.10.029
Li, J.F., Zhan, C., Lu, J., et al.: Improve first-cycle efficiency and rate performance of layered-layered Li1.2Mn0.6Ni0.2O2 using oxygen stabilizing dopant. ACS Appl. Mater. Interfaces 7, 16040–16045 (2015). https://doi.org/10.1021/acsami.5b04343
Xie, Y., Saubanère, M., Doublet, M.L.: Requirements for reversible extra-capacity in Li-rich layered oxides for Li-ion batteries. Energy Environ. Sci. 10, 266–274 (2017). https://doi.org/10.1039/c6ee02328b
Liu, S., Liu, Z.P., Shen, X., et al.: Li–Ti cation mixing enhanced structural and performance stability of Li-rich layered oxide. Adv. Energy Mater. 10, 1903065 (2020). https://doi.org/10.1002/aenm.201901530
Urban, A., Abdellahi, A., Dacek, S., et al.: Electronic-structure origin of cation disorder in transition-metal oxides. Phys. Rev. Lett. 119, 176402 (2017). https://doi.org/10.1103/physrevlett.119.176402
Dahiya, P.P., Ghanty, C., Sahoo, K., et al.: Suppression of voltage decay and improvement in electrochemical performance by zirconium doping in Li-rich cathode materials for Li-ion batteries. J. Electrochem. Soc. 165, A3114–A3124 (2018). https://doi.org/10.1149/2.0751813jes
Yabuuchi, N., Yamamoto, K., Yoshii, K., et al.: Structural and electrochemical characterizations on Li2MnO3-LiCoO2-LiCrO2 system as positive electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 160, A39–A45 (2012). https://doi.org/10.1149/2.045301jes
Song, B.H., Zhou, C.F., Wang, H.L., et al.: Advances in sustain stable voltage of Cr-doped Li-rich layered cathodes for lithium ion batteries. J. Electrochem. Soc. 161, A1723–A1730 (2014). https://doi.org/10.1149/2.0461410jes
Singh, G., Thomas, R., Kumar, A., et al.: Electrochemical behavior of Cr-doped composite Li2MnO3-LiMn0.5Ni0.5O2 cathode materials. J. Electrochem. Soc. 159, A410–A420 (2012). https://doi.org/10.1149/2.059204jes
Song, B.H., Day, S.J., Sui, T., et al.: Mitigated phase transition during first cycle of a Li-rich layered cathode studied by in operando synchrotron X-ray powder diffraction. Phys. Chem. Chem. Phys. 18, 4745–4752 (2016). https://doi.org/10.1039/c5cp04801j
Li, H.M., Guo, H.J., Wang, Z.X., et al.: Improving rate capability and decelerating voltage decay of Li-rich layered oxide cathodes by chromium doping. Int. J. Hydrog. Energy 43, 11109–11119 (2018). https://doi.org/10.1016/j.ijhydene.2018.04.203
Yi, T.F., Li, Y.M., Cai, X.D., et al.: Fe-stabilized Li-rich layered Li1.2Mn0.56Ni0.16Co0.08O2 oxide as a high performance cathode for advanced lithium-ion batteries. Mater. Today Energy 4, 25–33 (2017). https://doi.org/10.1016/j.mtener.2017.03.005
Billaud, J., Sheptyakov, D., Sallard, S., et al.: Li/Fe substitution in Li-rich Ni, Co, Mn oxides for enhanced electrochemical performance as cathode materials. J. Mater. Chem. A 7, 15215–15224 (2019). https://doi.org/10.1039/c9ta00399a
Liu, X.Y., Huang, T., Yu, A.S.: Fe doped Li1.2Mn0.6−x/2Ni0.2−x/2FexO2 (x\(\leqslant\)0.1) as cathode materials for lithium-ion batteries. Electrochim. Acta 133, 555–563 (2014). https://doi.org/10.1016/j.electacta.2014.04.085
Sorboni, Y.G., Arabi, H., Kompany, A.: Effect of Cu doping on the structural and electrochemical properties of lithium-rich Li1.25Mn–Ni0.125Co0.125O2 nanopowders as a cathode material. Ceram. Int. 45, 2139–2145 (2019). https://doi.org/10.1016/j.ceramint.2018.10.122
Zhao, J.K., Wang, Z.X., Guo, H.J., et al.: Synthesis and electrochemical characterization of Zn-doped Li-rich layered Li[Li0.2Mn0.54Ni0.13Co0.13]O2 cathode material. Ceram. Int. 41, 11396–11401 (2015). https://doi.org/10.1016/j.ceramint.2015.05.102
Wei, X., Yang, P.H., Li, H.L., et al.: Synthesis and properties of mesoporous Zn-doped Li1.2Mn0.54Co0.13Ni0.13O2 as cathode materials by a MOFs-assisted solvothermal method. RSC Adv. 7, 35055–35059 (2017). https://doi.org/10.1039/c7ra05106a
Gao, Y.R., Wang, X.F., Ma, J., et al.: Selecting substituent elements for Li-rich Mn-based cathode materials by density functional theory (DFT) calculations. Chem. Mater. 27, 3456–3461 (2015). https://doi.org/10.1021/acs.chemmater.5b00875
Liu, S., Liu, Z.P., Shen, X., et al.: Surface doping to enhance structural integrity and performance of Li-rich layered oxide. Adv. Energy Mater. 8, 1802105 (2018). https://doi.org/10.1002/aenm.201802105
Hu, X., Guo, H.J., Peng, W.J., et al.: Effects of Nb doping on the performance of 0.5Li2MnO3·0.5LiNi1/3Co1/3Mn1/3O2 cathode material for lithium-ion batteries. J. Electroanal. Chem. 822, 57–65 (2018). https://doi.org/10.1016/j.jelechem.2018.05.015
Zubair, M., Li, G.Y., Wang, B.Y., et al.: Electrochemical kinetics and cycle stability improvement with Nb doping for lithium-rich layered oxides. ACS Appl. Energy Mater. 2, 503–512 (2019). https://doi.org/10.1021/acsaem.8b01534
Pan, W., Peng, W.J., Guo, H.J., et al.: Effect of molybdenum substitution on electrochemical performance of Li[Li0.2Mn0.54Co0.13Ni0.13]O2 cathode material. Ceram. Int. 43, 14836–14841 (2017). https://doi.org/10.1016/j.ceramint.2017.07.232
Guo, L.M., Tan, X.H., Liu, S.N., et al.: Considerable capacity increase of high-nickel lithium-rich cathode materials by effectively reducing oxygen loss and activating Mn4+/3+ redox couples via Mo doping. J. Alloys Compd. 790, 170–178 (2019). https://doi.org/10.1016/j.jallcom.2019.03.172
Huang, J.J., Liu, H.D., Hu, T., et al.: Enhancing the electrochemical performance of Li-rich layered oxide Li1.13Ni0.3Mn0.57O2 via WO3 doping and accompanying spontaneous surface phase formation. J. Power Sources 375, 21–28 (2018). https://doi.org/10.1016/j.jpowsour.2017.11.048
Zhang, N., Sun, Y.Y., Zhao, L., et al.: Improving the electrochemical performance of lithium-rich cathode materials Li1.2Mn0.54Ni0.13Co0.13O2 by a method of tungsten doping. Ionics 25, 5239–5247 (2019). https://doi.org/10.1007/s11581-019-03103-4
Yu, Z., Ning, F.H., Li, B., et al.: Mitigating voltage decay of Li-rich layered oxide by incorporation of 5d metal rhenium. J. Phys. Chem. C 123, 18870–18876 (2019). https://doi.org/10.1021/acs.jpcc.9b05683
Guo, H.C., Xia, Y.G., Zhao, H., et al.: Stabilization effects of Al doping for enhanced cycling performances of Li-rich layered oxides. Ceram. Int. 43, 13845–13852 (2017). https://doi.org/10.1016/j.ceramint.2017.07.107
Tang, Z.H., Li, X.H., Wang, Z.X.: Effects of Al doping for Li[Li0.09Mn0.65*0.91Ni0.35*0.91]O2 cathode material. Ionics 19, 1495–1501 (2013). https://doi.org/10.1007/s11581-013-0881-6
Yi, T.F., Han, X., Yang, S.Y., et al.: Enhanced electrochemical performance of Li-rich low-Co Li1.2Mn0.56Ni0.16Co0.08−xAlxO2 (0\(\leqslant\)x\(\leqslant\)0.08) as cathode materials. Sci. China Mater. 59, 618–628 (2016). https://doi.org/10.1007/s40843-016-5097-7
Yan, W.C., Xie, Y., Jiang, J.C., et al.: Enhanced rate performance of Al-doped Li-rich layered cathode material via nucleation and post-solvothermal method. ACS Sustainable Chem. Eng. 6, 4625–4632 (2018). https://doi.org/10.1021/acssuschemeng.7b03634
Luo, M., Zhang, R., Gong, Y.Q., et al.: Effects of doping Al on the structure and electrochemical performances of Li[Li0.2Mn0.54Ni0.13Co0.13]O2 cathode materials. Ionics 24, 967–976 (2018). https://doi.org/10.1007/s11581-017-2269-5
Song, J.H., Shim, J.H., Kapylou, A., et al.: Suppression of voltage depression in Li-rich layered oxide by introducing GaO4 structural units in the Li2MnO3-like nano-domain. Nano Energy 30, 717–727 (2016). https://doi.org/10.1016/j.nanoen.2016.09.028
Yu, T.H., Li, J.L., Xu, G.F., et al.: Improved cycle performance of Li[Li0.2Mn0.54Co0.13Ni0.13]O2 by Ga doping for lithium ion battery cathode material. Solid State Ion. 301, 64–71 (2017). https://doi.org/10.1016/j.ssi.2017.01.008
Zhao, Y.J., Xia, M.H., Hu, X.S., et al.: Effects of Sn doping on the structural and electrochemical properties of Li1.2Ni0.2Mn0.8O2 Li-rich cathode materials. Electrochim. Acta 174, 1167–1174 (2015). https://doi.org/10.1016/j.electacta.2015.05.068
Li, Q.Y., Zhou, D., Zhang, L.J., et al.: Lithium-ion batteries: tuning anionic redox activity and reversibility for a high-capacity Li-rich Mn-based oxide cathode via an integrated strategy. Adv. Funct. Mater. 29, 1970064 (2019). https://doi.org/10.1002/adfm.201970064
Chen, H., Hu, Q.Y., Peng, W.J., et al.: New insight into the modification of Li-rich cathode material by stannum treatment. Ceram. Int. 43, 10919–10926 (2017). https://doi.org/10.1016/j.ceramint.2017.05.129
Zhou, L., Liu, J., Huang, L.S., et al.: Sn-doped Li1.2Mn0.54Ni0.13Co0.13O2 cathode materials for lithium-ion batteries with enhanced electrochemical performance. J. Solid State Electrochem. 21, 3467–3477 (2017). https://doi.org/10.1007/s10008-017-3688-y
Xie, D.Z., Zhou, W.S., Lin, K.S., et al.: Doping effect of fluoride anion on microstructural and electrochemical properties of lithium-rich cathode materials. Mater. Lett. 253, 82–85 (2019). https://doi.org/10.1016/j.matlet.2019.06.047
Kageyama, M., Li, D.C., Kobayakawa, K., et al.: Structural and electrochemical properties of LiNi1/3Mn1/3Co1/3O2−xFx prepared by solid state reaction. J. Power Sources 157, 494–500 (2006). https://doi.org/10.1016/j.jpowsour.2005.08.002
Yan, H.J., Li, B., Yu, Z., et al.: First-principles study: tuning the redox behavior of lithium-rich layered oxides by chlorine doping. J. Phys. Chem. C 121, 7155–7163 (2017). https://doi.org/10.1021/acs.jpcc.7b01168
Wang, Y., Gu, H.T., Song, J.H., et al.: Suppressing Mn reduction of Li-rich Mn-based cathodes by F-doping for advanced lithium-ion batteries. J. Phys. Chem. C 122, 27836–27842 (2018). https://doi.org/10.1021/acs.jpcc.8b08669
Kang, S.H., Amine, K.: Layered Li(Li0.2Ni0.15+0.5zCo0.10Mn0.55−0.5z)O2−zFz cathode materials for Li-ion secondary batteries. J. Power Sources 146, 654–657 (2005). https://doi.org/10.1016/j.jpowsour.2005.03.152
Li, B., Yan, H.J., Ma, J., et al.: Manipulating the electronic structure of Li-rich manganese-based oxide using polyanions: towards better electrochemical performance. Adv. Funct. Mater. 24, 5112–5118 (2014). https://doi.org/10.1002/adfm.201400436
Wu, Y., Manthiram, A.: Effect of Al3+ and F− doping on the irreversible oxygen loss from layered Li[Li0.17Mn0.58Ni0.25]O2 cathodes. Electrochem. Solid-State Lett. 10, A151 (2007). https://doi.org/10.1149/1.2720637
Li, H.X., Fan, L.Z.: Effects of fluorine substitution on the electrochemical performance of layered Li-excess nickel manganese oxides cathode materials for lithium-ion batteries. Electrochim. Acta 113, 407–411 (2013). https://doi.org/10.1016/j.electacta.2013.09.135
Kim, G.H., Kim, M.H., Myung, S.T., et al.: Effect of fluorine on Li[Ni1/3Co1/3Mn1/3]O2−zFz as lithium intercalation material. J. Power Sources 146, 602–605 (2005). https://doi.org/10.1016/j.jpowsour.2005.03.045
Zhang, H.L., Song, T.F.: Synthesis and performance of fluorine substituted Li1.05(Ni0.5Mn0.5)0.95O2−xFx cathode materials modified by surface coating with FePO4. Electrochim. Acta 114, 116–124 (2013). https://doi.org/10.1016/j.electacta.2013.10.030
Thackeray, M.M., Kang, S.H., Johnson, C.S., et al.: Li2MnO3-stabilized LiMO2 (M = Mn, Ni, Co) electrodes for lithium-ion batteries. J. Mater. Chem. 17, 3112 (2007). https://doi.org/10.1039/b702425h
Kang, K., Ceder, G.: Factors that affect Li mobility in layered lithium transition metal oxides. Phys. Rev. B 74, 094105 (2006). https://doi.org/10.1103/physrevb.74.094105
Liu, T., Zhao, S.X., Gou, L.L., et al.: Electrochemical performance of Li-rich cathode material, 0.3Li2MnO3–0.7LiMn1/3Ni1/3Co1/3O2 microspheres with F-doping. Rare Met. 38, 189–198 (2019). https://doi.org/10.1007/s12598-018-1168-x
Zheng, J.M., Wu, X.B., Yang, Y.: Improved electrochemical performance of Li[Li0.2Mn0.54Ni0.13Co0.13]O2 cathode material by fluorine incorporation. Electrochim. Acta 105, 200–208 (2013). https://doi.org/10.1016/j.electacta.2013.04.150
Liao, L., Wang, X.Y., Luo, X.F., et al.: Synthesis and electrochemical properties of layered Li[Ni0.333Co0.333Mn0.293Al0.04]O2−zFz cathode materials prepared by the Sol-gel method. J. Power Sources 160, 657–661 (2006). https://doi.org/10.1016/j.jpowsour.2005.12.095
Li, X.L., Kang, F.Y., Shen, W.C., et al.: Improvement of structural stability and electrochemical activity of a cathode material LiNi0.7Co0.3O2 by chlorine doping. Electrochim. Acta 53, 1761–1765 (2007). https://doi.org/10.1016/j.electacta.2007.08.025
Kong, F.T., Liang, C.P., Longo, R.C., et al.: Conflicting roles of anion doping on the electrochemical performance of Li-ion battery cathode materials. Chem. Mater. 28, 6942–6952 (2016). https://doi.org/10.1021/acs.chemmater.6b02627
Breddemann, U., Erickson, E.M., Davis, V., et al.: Fluorination of Li-rich lithium-ion-battery cathode materials by fluorine gas: chemistry, characterization, and electrochemical performance in half cells. ChemElectroChem 6, 3337–3349 (2019). https://doi.org/10.1002/celc.201900733
Wang, J.Y., Yang, M.C., Zhao, C., et al.: Unveiling the benefits of potassium doping on the structural integrity of Li-Mn-rich layered oxides during prolonged cycling by dual-mode EPR spectroscopy. Phys. Chem. Chem. Phys. 21, 24017–24025 (2019). https://doi.org/10.1039/c9cp04204k
Li, L., Song, B.H., Chang, Y.L., et al.: Retarded phase transition by fluorine doping in Li-rich layered Li12Mn0.54Ni0.13Co0.13O2 cathode material. J. Power Sources 283, 162–170 (2015). https://doi.org/10.1016/j.jpowsour.2015.02.085
Guo, B., Zhao, J.H., Fan, X.M., et al.: Aluminum and fluorine co-doping for promotion of stability and safety of lithium-rich layered cathode material. Electrochim. Acta 236, 171–179 (2017). https://doi.org/10.1016/j.electacta.2017.03.133
Zhang, H.Z., Qiao, Q.Q., Li, G.R., et al.: Surface nitridation of Li-rich layered Li(Li0.17Ni0.25Mn0.58)O2 oxide as cathode material for lithium-ion battery. J. Mater. Chem. 22, 13104 (2012). https://doi.org/10.1039/c2jm30989k
Erickson, E.M., Sclar, H., Schipper, F., et al.: High-temperature treatment of Li-rich cathode materials with ammonia: improved capacity and mean voltage stability during cycling. Adv. Energy Mater. 7, 1700708 (2017). https://doi.org/10.1002/aenm.201700708
Qiu, B., Zhang, M.H., Wu, L.J., et al.: Gas–solid interfacial modification of oxygen activity in layered oxide cathodes for lithium-ion batteries. Nat. Commun. 7, 12108 (2016). https://doi.org/10.1038/ncomms12108
Fan, J.M., Li, G.S., Li, B.Y., et al.: Reconstructing the surface structure of Li-rich cathodes for high-energy lithium-ion batteries. ACS Appl. Mater. Interfaces 11, 19950–19958 (2019). https://doi.org/10.1021/acsami.9b02827
Ku, L., Cai, Y.X., Ma, Y.T., et al.: Enhanced electrochemical performances of layered-spinel heterostructured lithium-rich Li1.2Ni0.13Co0.13Mn0.54O2 cathode materials. Chem. Eng. J. 370, 499–507 (2019). https://doi.org/10.1016/j.cej.2019.03.247
Han, S.J., Qiu, B., Wei, Z., et al.: Surface structural conversion and electrochemical enhancement by heat treatment of chemical pre-delithiation processed lithium-rich layered cathode material. J. Power Sources 268, 683–691 (2014). https://doi.org/10.1016/j.jpowsour.2014.06.106
Yang, C., Zhang, Q., Ding, W.X., et al.: Improving the electrochemical performance of layered lithium-rich cathode materials by fabricating a spinel outer layer with Ni3+. J. Mater. Chem. A 3, 7554–7559 (2015). https://doi.org/10.1039/c5ta00009b
Zheng, Y., Chen, L., Su, Y.F., et al.: An interfacial framework for breaking through the Li-ion transport barrier of Li-rich layered cathode materials. J. Mater. Chem. A 5, 24292–24298 (2017). https://doi.org/10.1039/c7ta08735g
Xu, K.J., Pang, S.L., Wang, Y.G., et al.: Role of L-ascorbic acid-based treatment toward improving the electrochemical performance of Li-rich layered oxide. J. Electrochem. Soc. 164, A2348–A2354 (2017). https://doi.org/10.1149/2.0731712jes
Pang, S.L., Zhu, M., Xu, K.J., et al.: Enhanced electrochemical performance of Li1.2Mn0.54Ni0.13Co0.13O2 via L-ascorbic acid-based treatment as cathode material for Li-ion batteries. J. Electrochem. Soc. 165, A1897–A1902 (2018). https://doi.org/10.1149/2.1481809jes
Ding, Z.P., Zhang, C.X., Xu, S., et al.: Stable heteroepitaxial interface of Li-rich layered oxide cathodes with enhanced lithium storage. Energy Storage Mater. 21, 69–76 (2019). https://doi.org/10.1016/j.ensm.2018.12.004
Ramakrishnan, S., Park, B., Wu, J., et al.: Extended interfacial stability through simple acid rinsing in a Li-rich oxide cathode material. J. Am. Chem. Soc. 142, 8522–8531 (2020). https://doi.org/10.1021/jacs.0c02859
Zhang, Q.G., Hu, X.H., Zhan, D., et al.: Pyrolysis of in situ formed lithium stearate: an effective strategy to activate Li2MnO3. Electrochim. Acta 113, 424–432 (2013). https://doi.org/10.1016/j.electacta.2013.09.127
Sun, Y., Zan, L., Zhang, Y.X.: Enhanced electrochemical performances of Li2MnO3 cathode materials via adjusting oxygen vacancies content for lithium-ion batteries. Appl. Surf. Sci. 483, 270–277 (2019). https://doi.org/10.1016/j.apsusc.2019.03.210
Lim, J.M., Kim, D., Lim, Y.G., et al.: Mechanism of oxygen vacancy on impeded phase transformation and electrochemical activation in inactive Li2MnO3. Chem. Electro. Chem. 3, 943–949 (2016). https://doi.org/10.1002/celc.201600067
Tan, X., Liu, R., Xie, C.X., et al.: Modified structural characteristics and enhanced electrochemical properties of oxygen-deficient Li2MnO3−δ obtained from pristine Li2MnO3. J. Power Sources 374, 134–141 (2018). https://doi.org/10.1016/j.jpowsour.2017.11.004
Kubota, K., Kaneko, T., Hirayama, M., et al.: Direct synthesis of oxygen-deficient Li2MnO3−x for high capacity lithium battery electrodes. J. Power Sources 216, 249–255 (2012). https://doi.org/10.1016/j.jpowsour.2012.05.061
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. The authors declare the financial support from Australian Research Council through its Discovery and Linkage Programs.
Author information
Authors and Affiliations
Contributions
The concept of this review was designed by Tongen Lin, Yuxiang Hu and Lianzhou Wang. The literature searching and data analysis on the main part of the review were performed by Tongen Lin and Trent Seaby. The figures were prepared by Tongen Lin, Shanshan Ding and Ying Liu. The copyright permissions were requested by Shanshan Ding. The first draft of the manuscript was written by Tongen Lin and Trent Seaby, and all authors commented on previous versions of the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare no conflicts of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, T., Seaby, T., Hu, Y. et al. Understanding and Control of Activation Process of Lithium-Rich Cathode Materials. Electrochem. Energy Rev. 5 (Suppl 2), 27 (2022). https://doi.org/10.1007/s41918-022-00172-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41918-022-00172-4