
Abstract
The knowledge about the level of genetic diversity and population structure in natural populations of Artemisia annua L. is a primary step in breeding programs for development of new cultivars with higher artemisinin level and better quality of secondary metabolites composition. We used PCR-based “retrotransposon-microsatellite amplified polymorphisms” (REMAPs) to study insertional polymorphism in A. annua genome to assess genetic variability and population structure in a collection of 118 accessions collected from north and northwest of Iran. Twenty-five primer combinations of 10 retrotransposon and seven ISSR primers amplified a total of 693 clear and unambiguous fragments in the studied accessions. The average number of bands, polymorphic bands, polymorphism, effective number of alleles, Shannon’s information index and expected heterozygosity were 27.72, 24.76, 88.14%, 1.47, 0.42 and 0.28, respectively. The analysis of molecular variance revealed high genetic variation present within sampled geographical regions. Distance-based cluster analysis assigned the studied accessions into four clusters according to their geographical origin, which were also confirmed by principal coordinate analysis. In model-based Bayesian clustering, the maximum value of Δk was obtained when the collection of 118 assayed A. annua accessions assigned into two subgroups (K = 2). The results showed the high genetic variation in the collection of Iranian sweet wormwood which revealed by REMAP markers indicating the reliability and efficiency of this marker system for analysis of genetic diversity and population structure of A. annua.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Sweet wormwood (Artemisia annua L., 2n = 18) is a fragrant bushy wind-pollinated annual herb (Daviesa et al. 2011), widely used in the therapy of fever and malaria (Tzeng et al. 2007). Cultivated sweet wormwood varieties contain from 0.01 to 2% of artemisinin (the most important secondary metabolite) in dry matter (Muangphrom et al. 2016). However, some transgenic lines, e.g., transgenic line HFD82 had higher artemisinin content up to 3.2% in dry matter (Shen et al. 2018). Additionally, this medicinal plant represents a rich source of essential oils, coumarins, polyacetylenes (Bhakuni et al. 2001), and is well-known as a natural herbicide and its flowers have antiperiodic, antiseptic, digestive, anti-tumor, antiprotozoal and antibacterial properties (Cavar et al. 2012). Essential oils are used also in the cosmetics, health care and drug industry (Gupta et al. 2002). Artemisinin and related compounds isolated from A. annua have been shown to be effective against SARS-CoV-2/coronavirus disease 2019 (COVID-19) and has saved millions of lives (Soni et al. 2022).
The knowledge about genetic diversity and population structure is essential for breeding program of plant species (Yildiz and Arbizu 2022). Various DNA marker systems, such as Random Amplified Polymorphic DNA (RAPD) (Nguyen et al. 2017), Amplified Fragment Length Polymorphism (AFLP) (Bovard et al. 2016), Inter-Simple Sequence Repeat (ISSR) (Gaafar et al. 2017; Nguyen et al. 2017), Simple Sequence Repeat (SSR) (Kumar et al. 2014) and Expressed Sequence Tag–Simple Sequence Repeat (EST–SSR) (Kumar et al. 2014) have been used to analyze genetic diversity and relationships among different Artemisia species. Compared to other DNA marker systems (i.e., SSR, AFLP), retrotransposon-based markers are easy to use and more effective in detecting genetic polymorphism (Kalendar et al. 1999, 2011). To the best of our knowledge, this is the first report on characterization of A. annua accessions using insertional polymorphism. However, in some medicinal plants retrotransposon-based markers have been used for analysis of genetic diversity and population structure (Doungous et al. 2015). Due to their dynamic nature and universality, retrotransposons contribute to the variation of genome size and genomic architecture in medicinal plants (Sorkheh et al. 2017).
The dispersion, ubiquity, prevalence, high heterogeneity and genomic dynamism of RTN-like elements in plant genomes made them a suitable DNA-fingerprinting tool. Retrotransposon-Microsatellite Amplified Polymorphism (REMAP) and Inter-Retrotransposon Amplified Polymorphism (IRAP) have been widely used to assess the insertional polymorphism of LTR-retrotransposons and genetic diversity of various plant species (Kalendar et al. 2011). REMAP markers have been successfully applied to the analysis of genetic relationships, phylogenetic evolution and genetic diversity in plant species. Sorkheh et al. (2017) studied the organization and distribution of Ty1-copia and Ty3-gypsy elements in almond genome using DNA fragments corresponding to reverse transcriptase domain of copia-like and gypsy-like retrotransposons. Abbasi Holasou et al. (2016) studied insertional polymorphism in Iranian populations of flax (Linum usitatissimum L.) and reported higher genetic diversity in Zagros populations compared with the populations originated from Alborz.
Although, the northern part of Iran is a major sweet wormwood growing region in Asia (Naghavi et al. 2019), few works, especially at the molecular level have been done to characterize Iranian A. annua collection. Therefore, molecular characterization of this collection may be the first effective step in the further breeding programs. The aims of the present study were: (i) to evaluate cross-species transferability of retrotransposon markers in A. annua, and (ii) to assess genetic diversity and structure of A. annua collection based on insertional polymorphism.
2 Materials and methods
Plant materials and DNA isolation –
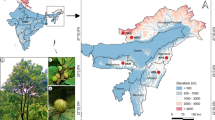
A total of 118 native sweet wormwood (A. annua) accessions collected in East Azerbaijan, Gilan, Mazandaran and Golestan provinces of Iran were used in the present study (Supplementary Table 1, Fig. 1). The seeds were planted in pots containing a mixture of peat and vermiculite and were kept in greenhouse with an ambient temperature of 25 °C. Genomic DNA was isolated from young leaves of 60 days old seedlings using CTAB protocol (Ausubel et al. 1995). The quality and quantity of DNA samples was assessed using 0.8% agarose gel electrophoresis and spectrophotometer (Bio- Photometer 6131, Eppendorf, Germany).

Geographical distribution of the A. annua accessions used in this study
REMAP analysis –
Seventy-seven REMAP primer combinations, derived from 11 RTN primers designed based on retrotransposon families Nikita, Sukkula, Bare 1, LTR1, LTR6149, LTR6150, Tnt-1, ToRTL1, Tnt1.OL16, 3′LTR and Ty1-Copia and 32 ISSR primers were tested (Supplementary Table 2 and Table 1). The PCR reaction mixture was prepared in a final volume of 10 μL containing 2 μL of genomic DNA, 4 μL of Master Mix, 3 μL of dd H2O, and 1 μL of each primer. The amplification was performed at 94 °C for 5 min, followed by 40 cycles of 94 °C for 60 s, 60 s for annealing temperature (for annealing temperature of particular primer combination, see Table 1), and 72 °C for 2 min, with a final extension of 7 min at 72 °C. The amplicons were resolved by 4% ultrathin (0.2 mm) non-denature polyacrylamide gel and detected stained by ethidium bromide in Gel-Scan 3000 electrophoresis system (Corbett, Sydney, Australia). To estimate the size of the amplified fragments, a 100 bp Plus DNA Ladder 100 to 3000 bp was used.
Data analysis –
The presence or absence of clear and reproducible bands were scored as 1 or 0, respectively. For each primer combination, the number of bands, percentage of polymorphic bands (PPB), number of bands with a frequency higher than or equal to 5%, number of private bands, number of less common bands with frequency lower than or equal to 25% and 50%, expect heterozygosity (He), number of effective alleles (Ne), Shannonʼs information index (I), number of migrants between groups (Nm) and genetic differentiation (PhiPT) were calculated for each geographical region using GenAlEx 6.4 (Peakall and Smouse 2006). Analysis of molecular variance (AMOVA) was performed to estimate distribution of genetic diversity. Distance-based cluster analysis using Minimum Evolution (ME) algorithm and number of differences evolutionary distance coefficient implemented in MEGA 4.0 (Tamura et al. 2007) was used to group the studied accessions. In addition, population structure was inferred using model-based cluster analysis implemented in Structure 2.3.1 software (Pritchard et al. 2000). The analysis was performed based on admixture model using 50,000 generations of burn in period, 100,000 MCMC iterations with K = 1–10 (K: number of sub-populations). The most likely K value was determined by the log likelihood of the data [LnP(D)] and an ad hoc statistic, ΔK, using web-based software STRUCTURE HARVESTER version 0.6.92 (Earl and Von Holdt 2012). To better understand and describe the genetic structure, principal coordinate analysis (PCoA) was performed using GenAlEx 6.4 (Peakall and Smouse 2006).
3 Results
Polymorphism of REMAP markers –
Twenty-five out of 77 tested primer combinations were successful in amplifying fragments and produced a total of 693 fragments (with the size from 100 to 3000 bp) in the investigated 118 individuals. The overall polymorphism was 88% (619 fragments were polymorphic). All the RTN primers, except LTR6149 in combination with at least one ISSR primer generated distinguishable and polymorphic banding pattern (Supplementary Table 2 and Table 1). The average number of polymorphic fragments per primer combination was 24.76 and Tnt-1/ISSR11 combination produced the highest number of bands (51) and polymorphic bands (45). The banding patterns generated by ToRTL1/ISSR6, ToRTL1/ISSR26 and ToRTL1/ISSR42 primer combinations were 100% polymorphic. The mean Ne, I, and He values were 1.47, 0.42 and 0.28, respectively, and the highest value of these parameters was recorded for Sukkula/ISSR11 primer combination. Figure 2 shows the banding pattern of ToRTL1/ISSR35 and Sukkula/ISSR11 primer combinations in some sweet wormwood accessions.

Typical representation of REMAP banding patterns of different Sweet wormwood (A. annua) accessions generated by a ToRTL1/ISSR35 and b Sukkula/ISSR11
Genetic diversity and population structure –
The ME algorithm clustered the investigated accessions based on their geographical origin. The first and third clusters consisted mostly of accessions from Mazandaran and Golestan provinces, respectively. The accessions from Gilan province were grouped in the second cluster along with two accessions from Mazandaran, i.e., Mazandaran-Niasteh and Mazandaran-Eiman Abad. Whereas, the fourth cluster grouped accessions of mixed origin (Fig. 3).

Unrooted dendrogram of showing grouping of 118 Sweet wormwood accessions based on REMAP data using Minimum evolution clustering algorithm (based on number of differences)
Principal component analysis (PCoA) data for all 115 accessions are shown in Fig. 4. The first three coordinates explained 31.00, 19.82 and 15.31% of the total molecular variability, respectively. The PCoA revealed genetic differentiation of the sweet wormwood accessions also into four clusters clearly divided according to their geographical origin (Fig. 4). The grouping pattern obtained by PCoA was to some extent similar to that of cluster analysis.

Principal coordinate analysis (PCoA) of 118 Sweet wormwood accessions based on REMAP markers. Each individual is represented by one symbol (symbol color correspond to sampled region)
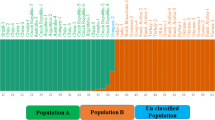
Analysis of genetic relationships and population structure using Bayesian model-based clustering revealed slow increase in the log-likelihoods from K = 2 to 6 (Fig. 5b). However, using the method of Evanno et al. (2005), maximal ΔK occurred at K = 2 (Fig. 5a), followed by K = 4 and 6, indicating the whole population can be divided into two sub-populations which could be further subdivided into four and six groups. Figure 5c shows bar plots for K = 2, 4 and 6. Based on K = 2, sub-population I contained of 14 accessions of admixture samples and sub-population II consisted of 104 accessions from Gilan, Golestan, and Mazandaran provinces. But grouping based on K = 4 could distinguish accessions from Gilan, Mazandaran and Golestan provinces. Azerbaijan accessions with few accessions from Gilan, Golestan, and Mazandaran were grouped in a separate cluster. Considering K = 6, the sup-population 1 included 46 accessions from Mazandaran. The sub-population 2 was composed of accessions from Azerbaijan (2) and Gilan (4). Moreover, the sub-population 3 for K = 6 was mainly composed of Golestan (3) and Mazandaran (3). The Golestan accessions (20) were mostly included in sub-population 4. The sub-population 5 as admixture group consisted of 2, 2 and 1 accessions from Golestan, Gilan, and Mazandaran provinces. The sub-population 6 in the K = 6 model was mainly comprised of Gilan (33) accessions along one accession of Mazandaran (2).

The pattern of population structure of the 118 Sweet wormwood (A. annua) accessions inferred using REMAP data. a Estimated LnP(D) of possible clusters (K) from 1 to 10; b ΔK based on the rate of change of LnP(D) between successive K and c Population structure based on K = 2, 4 and 6
The characteristics of REMAP markers for the geographical populations are presented in Table 2. The percentage of polymorphic loci ranged from 81.10 (Golestan) to 87.01 (Mazandaran), with an average of 84.08. Three accession-specific REMAP markers were identified in Gilan, Golestan and Mazandaran populations. Mean expected heterozygosity varied from 0.250 (Mazandaran) to 0.258 (Gilan) with an average of 0.253. The amount of gene flow among geographical populations calculated indirectly from PhiPT (Nm = 0.25[(1/PhiPT)-1]) ranged from 4.03 (Golestan and Mazandaran) to 4.81 (Golestan and Gilan) with an average of 4.36. Nei genetic distance between populations ranged from 0.050 (Gilan and Mazandaran) to 0.055 (Gilan and Golestan) with an average of 0.052. The minimum and maximum genetic differentiation were observed between Golestan and Gilan populations (PhiPT = 0.094) Golestan and Mazandaran populations (PhiPT = 0.11), respectively (Table 3). Analysis of molecular variance (AMOVA) revealed higher genetic variability within populations (90%) compared with among populations (10%). Azerbaijan population was not included in this analysis due to insufficient sample size.
4 Discussion
Retrotransposon insertional polymorphism –
The insertional polymorphism of A. annua genome was assessed by mean of REMAP technique, which detects polymorphism in the amplified regions between retrotransposon insertion and microsatellite loci (Kalendar et al. 1999). Since transposable elements represent 60% of the A. annua genome, of which more than 22.69% are LTR-retrotransposons (Shen et al. 2018), retrotransposon-based DNA-fingerprinting could be an ideal technique to study the genome wide diversity of accessions or breeding lines. This is the first report of REMAP-based assessment of retrotransposon insertional polymorphism in A. annua populations. Twenty-five RTN and ISSR primers combinations amplified distinguishable and scorable banding pattern which used for analysis of genetic diversity and population structure in 118 Sweet wormwood accessions collected from north and northwest of Iran. All RTN primers (Sukkula, ToRTL1, Tnt-1, LTR1, Nikita, Tnt1.OL16, Ty1-Copia, LTR6150, Bare1 and 3′LTR) except LTR6149 amplified DNA fragments for A. annua genome in combination with at least one ISSR primer indicating the insertion of retrotransposons near to microsatellite loci in Sweet wormwood genome. The Sukkula and ToRTL1 derived from Gramineae and Solanaceae, respectively, were the most frequent retrotransposon families in Sweet wormwood genome and primers designed based on their LTR regions could amplify higher number of fragments in combination with several ISSR primers. It may also suggest possible role of these retrotransposon families in the, organization, and evolution of the A. annua genome. Du et al. (2013) and Asadkhani Mamaghani et al. (2015) mentioned that grasses share transcriptionally, translationally, and integrationally active RTN families.
Sorkheh et al. (2017) in analysis of genetic diversity in wild almond reported that the LTR primers designed based of retrotransposon sequences from one species can be used in related Rosaceae genera, but the polymorphism of non-native retrotransposons from Prunus spp. L. was low compared with that of non-native retrotransposons from Lotus japonicas L. In the present study, Tnt-1 primer derived from Rosaceae in combination with ISSR11 amplified the highest number of fragments (45) in A. annua genome (from which 88.2% were polymorphic) followed by Ty1-Copia/ISSR16 (43 bands and 91.5% polymorphism), Tnt-1/ISSR26 (38 bands and 97.4% polymorphism) and Sukkula/ISSR11 (37 bands and 94.9% polymorphism). This is comparable to report of Sharma and Nandineni (2014), who amplified the lowest (12) and highest (40) number of fragments using Sukkula/UBC808 and LTR6149/UBC812 primers combinations. Banding pattern derived from Tnt-1 RTN primer in sweet wormwood were characterized by large number of clear bands and high polymorphism indicating that Tnt-1, Ty1-Copia and Sukkula insertion pattern in near SSR motifs is closer than the insertion pattern of other mentioned retroelements and these RTNs families were the most active retrotransposon in Iranian Sweet wormwood genome. Also mentioned RTNs families play an important role in the structure, organization, and evolution of Sweet wormwood genome that may be of use in certain applications. The insertion of Sukkula and other RTN families in vicinity of microsatellite motifs has been previously reported in the genome of Indian potato cultivars (Sharma and Nandineni 2014).
In our study, the high percentage of polymorphism detected by REMAP (88%) markers suggested the activities of the RTN families used in the Iranian Sweet wormwood genome. Much polymorphism was detected by REMAPs, showing the number of LTRs present in the genome (Kalendar et al. 1999), explaining the high average percentage of REMAP polymorphism. Karimi et al. (2014) in a study of genetic diversity in a cultivated tarragon (Artemisia dracunculus L.) Iranian germplasm including 29 genotypes reported that, SRAP markers (83.14%) were more efficient than the ISSR (69.97%) assay for detection of polymorphism. Bovard et al. (2016) reported 64.5% polymorphism among Iranian A. annua populations using AFLP markers.
The transferability of RTN primers across different genera and species and even sometimes plant families has been examined in several studies. Teo et al. (2005) used the IRAP technique to assess transferability of RTN primers designed based on barley RTN families for identification and characterization of banana cultivars and classification of Musa genome constitutions. Alavi-Kia et al. (2008) used seven LTR-retrotransposon primers derived from barley genome for analysis of genetic diversity and phylogenetic relationships in various species of the Crocus genus. Rahmani et al. (2015) used LTR-retrotransposons primers derived from barley and rice to assess genetic diversity in Crataegus. Asadkhani Mamaghani et al. (2015) reported transferability of barley RTN primers for studying Hypericum perforatum L. The barley LTR primers were also successfully used in Iranian populations of Leonurus cardiaca L. based on IRAP analysis (Soorani et al. 2013). Due to high level of polymorphism revealed by REMAP markers in the current and previous studies, this marker system could efficient tool for the evaluation of the A. annua germplasm at DNA level for productivity enhancement.
Genetic relationships and population structure of sweet wormwood (A. annua) genotypes –
The genetic relationship of the investigated sweet wormwood (A. annua) accessions from four geographical regions was assessed based on REMAP data using distance and model-based cluster analyses. Distance-based cluster analysis (Fig. 3) assigned the accessions into four major groups mainly in concordance with their geographical origins. Some accessions from Mazandaran, Gilan and Golestan provinces with similar semiarid and cool climate were clustered in the same groups. Admixed genetic structure close to Mazandaran province is characteristic for accessions Mazandaran-Niasteh and Mazandaran-Eiman Abad. Accessions in the admixture class may have an intricate history involving intercrossing or feasibly resulting from the gene flown between populations. A putative reason to this might be linked to seed dispersal. The high genetic distance between some accessions from the same population could be due to geographical barriers within the region Migration of accessions among regions, followed by mixing and introgression with preexisting germplasm, could be another reason. The genetically diverse group identified in this study can be used to derive parental lines for sweet wormwood breeding. Gaafar et al. (2017) analyzed Judean wormwood collected in Egypt using ISSR markers and they reported concordance between molecular and geographical grouping. Model-based cluster analysis revealed maximum ΔK at K = 2, however, there was increase in log-likelihoods from K = 2 to 6 with maximum value at K = 6. The population structure revealed by this method was to some extent consistent with the grouping obtained by distance-based clustering (Fig. 5a–c). Biplot of the 118 sweet wormwood accessions based on two first coordinate in PCoA assigned them into four groups. It was evident from distance and model-based cluster analyses and PCoA that grouping of sweet wormwood accessions based on molecular data was according to their geographical origins and accessions from same province were closely associated. Classification of accessions from different populations in the same cluster may be is due to their ecological characteristics of collection sites. Gaafar et al. (2017), Huang et al. (2011) also reported similar findings for sweet wormwood genotypes with the aid of ISSR and ITS markers.
The moderate to high level of genetic variation (I = 0.31, He = 0.20, Ne = 1.34) detected in the studied populations might be ascribed to nature of floral biology of A. annua, creation of bottleneck effect during A. annua selection and evaluation, and moderate insertional activity of the used RTNs. Floral biology and pollination method are the driving force behind the genetic diversity. It has been demonstrated that long-lived and outcrossing species are capable of maintaining higher levels of genetic diversity compared with annuals or short lived perennials (Salameen et al. 2020).
Doungous et al. (2015) stated that the genetic variation in medicinal plants is strongly dependent on plant life form, geographical range, pollen dispersal mechanisms, and natural selection. In this study, genetic diversity (I, He, Ne) at the population level was relatively high in population Gilan than in other populations, related to the complex and diverse mountain environment. Gaafar et al. (2017) in analysis of Egyptian A. judaica reported low level of genetic diversity (measured by Nei gene diversity) ranging from 0.00 to 0.19 with an average of 0.14 and Shannon’s index ranging from 0.00 to 0.28 with an average of 0.20. Huang et al. (2014) studying genetic diversity of 290 Chinese A. halodendron accessions using ISSR markers reported moderate level of genetic variation (I = 0.32, Ne = 1.39, and He = 0.22). Nybom and Bartish (2000) reported that levels of genetic variation are strongly dependent on plant life form, geographical range, pollen dispersal mechanisms, genetic equilibrium, and natural selection. A reduction in genetic variation might suggest a decline in adaptation to a changing environment, leading to an increased danger of extinction and increased inbreeding. Low PhiPT values may be attributed to the close distance, convenient transportation, and similar environmental conditions between populations.
Based on REMAP data, we identified high level of polymorphism and moderate genetic variability within the investigated collection of A. annua. It indicates that the three studied geographical populations are genetically different and are valuable sources for hybridization program and even each of the studied populations could be utilized as independent genetic resource for improvement of Iranian A. anuua germplasm. Our results showed that with decreasing the geographical distance between the populations, gene flow was increased. Therefore, geographical and ecological factors may played a significant role in genetic variation of different populations. Diversity parameters revealed the presence of higher genetic variability in the studied materials suggesting the studied sweet wormwood accessions can provide useful building blocks for future breeding programs to enhance sweet wormwood productivity. Similar observations have also been reported for other Artemisia species such as A. judaica (Gaafar et al. 2017) and A. halodendron (Huang et al. 2011, 2014).
It is concluded that REMAP markers provide powerful tools to study genetic relationships among sweet wormwood accessions. Moderate to high genetic diversity of Iranian Sweet wormwood indicates the need for hybridization program to extend the genetic base of Iranian sweet wormwood germplasm. Introduction of useful traits from the ancient primitive forms of wild species could increase the polymorphism of modern sweet wormwood varieties. Besides, the use of the strong and complementary statistical methods such as distance and model-based cluster analyses and PCoA proved to be useful for the determination of genetic relationships among sweet wormwood accessions and determination of population structure. This information might be very useful in planning sweet wormwood breeding programs and defining strategies for germplasm conservation. Knowledge of the population structure has great importance for studies focusing on association mapping as well, which can detect association between phenotypes and linked markers on the basis of linkage disequilibrium.
Data availability
The data are available on request.
References
Abbasi Holasou H, Abdollahi Mandoulakani B, Jafari M, Bernousi I (2016) Use of IRAP and REMAP markers to interpret the population structure of Linum usitatissimum from Iran. Biol Sect Bot. https://doi.org/10.1515/biolog-2016-0042
Alavi-Kia SS, Mohammadi SA, Aharizad S, Moghaddam M (2008) Analysis of genetic diversity and phylogenetic relationships in Crocus genus of Iran using inter-retrotransposon amplified polymorphism. Biotechnol Biotech Equip 22:795–800
Asadkhani Mamaghani R, Mohammadi SA, Aharizad S (2015) Transferability of barley retrotransposon primers to analyze genetic structure in Iranian Hypericum perforatum L. populations. Turk J Bot 39:664–672
Ausubel FM, Brent R, Kingston RE, Moore DD, Struhl K, Albright LM, Coen DM, Varki A (1995) Current protocols in molecular biology. John Wiley, New York
Bhakuni RS, Jain DC, Sharma RP, Kumar S (2001) Secondary metabolites of Artemisia annua and their biological activity. Curr Sci 80:35–48
Bovard R, Naghavi MR, Omidi M, Torabi S, Parvaneh S, Safakish Kashani MB (2016) The study of genetic diversity in different plant populations of Artemisia anuua L. native to northern Iran by molecular marker AFLP. J Biodivers Environ Sci 9:51–59
Cavar S, Maksimovic M, Vidic D, Paric A (2012) Chemical composition and antioxidant and antimicrobial activity of essential oil of Artemisia annua L. from Bosnia. Ind Crops Prod 37:479–485
Daviesa MJ, Atkinson CJ, Burns C, Arroo R, Woolley J (2011) Increases in leaf artemisinin concentration in Artemisia annua in response to the application of phosphorus and boron. Ind Crops Prod 34:1465–1473
Doungous O, Kalendar R, Adiobo A, Schulman AH (2015) Retrotransposon molecular markers resolve cocoyam (Xanthosoma sagittifolium) and taro (Colocasia esculenta) by type and variety. Euphytica. https://doi.org/10.1007/s10681-015-1537-6
Du XY, Hu QN, Zhang QL, Wang YB, Luo ZR (2013) Transferability of retrotransposon primers derived from Persimmon (Diospyros kaki Thunb.) across other plant species. Genet Mol Res 12:1781–1795
Earl DA, Von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing structure output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Gaafar RM, Hamouda MM, Sayed Ahmed HI, El-Shazlyb HH, Badr A (2017) Genetic differentiation in the medicinal plant Artemisia judaica L. populations in Saint-Catherine area, South Sinai, Egypt. Plant Gene 12:80–87
Gupta SK, Singh P, Bajpai P, Ram G, Singh D, Gupta MM, Jain DC, Khanuja SP, Kumar S (2002) Morphogenetic variation artemisinin and volatile oil in Artemisia annua. Ind Crops Prod 16:217–224
Huang W, Zhao X, Zhao X, Zhao H, Wang S, Lian J (2011) A combined approach using ISSR and ITS analysis for the characterization of Artemisia halodendron from Horqin sandy land, northern China. Biochem Syst Ecol 39:346–351
Huang W, Zhao X, Zhao X, Li Y, Lian J, Yun J (2014) Relationship between the genetic diversity of Artemisia halodendron and climatic factors. Acta Oecol 55:97–103
Kalendar R, Grab T, Regina M, Souniemi A, Schulman AH (1999) IRAP and REMAP: two new retrotransposon-based DNA fingerprinting techniques. Theor Appl Genet 98:704–711
Kalendar R, Flavell AJ, Ellis THN, Moisy C, Schulman AH (2011) Analysis of plant diversity with retrotransposon-based molecular markers. Heredity 106:520–530
Karimi A, Hadian J, Farzaneh M, Khadivi-Khub A (2014) Evaluation of genetic variability, rust resistance and marker-detection in cultivated Artemisia dracunculus from Iran. Gene 554:224–232
Kumar J, Bajaj P, Mishra GP, Bala Singh Sh, Singh H, Naik PK (2014) Utilization of EST-derived SSRs in the genetic characterization of Artemisia annua L. genotypes from Ladakh, India. Indian J Biotechnol 13:464–472
Muangphrom P, Seki H, Muranaka T (2016) Artemisinin-based antimalarial research: application of biotechnology to the production of artemisinin, its mode of action, and the mechanism of resistance of Plasmodium parasites. J Nat Med 70:318–334
Naghavi MR, Aryakia E, Hadi S, Ghafoori H, Mousavi H, Shahzadeh Fazeli SA (2019) Characterization of morphological, phytochemical and molecular diversity of Artemisia annua accessions in Hyrcanian area of Iran. J Agri Sci Technol 21:1265–1276
Nguyen HT, Inotai K, Radacsi P, Ladanyi M, Eva Zamborine-Nemeth E (2017) Morphological, phytochemical and molecular characterization of intraspecific variability of wormwood (Artemisia absinthium L.). J Appl Bot Food Qual 90:238–245
Nybom H, Bartish IV (2000) Effects of life history traits and sampling strategies on genetic diversity estimates obtained with RAPD markers in plants. Perspect Plant Ecol Evol Syst 3:93–114
Peakall R, Smouse PE (2006) GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rahmani M-S, Shabanian N, Khadivi-Khub A, Woeste KE, Badakhshan H, Alikhani L (2015) Population structure and genotypic variation of Crataegus pontica inferred by molecular markers. Gene 572:123–129
Salameen FA, Habibi N, Al Ahmad S, Kumar V, Dashti J, Talebi L, Al Doaij B (2020) Genetic diversity analysis of Rhanterium eppaposum Oliv. by ISSRs reveals a weak population structure. Curr Plant Biol 21:100138
Sharma V, Nandineni MR (2014) Assessment of genetic diversity among Indian potato (Solanum tuberosum L.) collection using microsatellite and retrotransposon based marker systems. Mol Phylogenet Evol 73:10–17
Shen Q, Zhang L, Liao Z, Wang Sh, Shi P, Liu M, Pan Q, Wang Y, Lv Z, Lu X, Zhang F, Ma Y, Chen M, Hao X, Li L, Lv G, Zhou Y, Sun X, Brodelius PE, Rose JKC (2018) The genome of Artemisia annua provides insight into the evolution of Asteraceae family and artemisinin biosynthesis. Mol Plant 11:776–788
Soni R, Shankar G, Mukhopadhyay P, Gupta V (2022) A concise review on Artemisia annua L.: a major source of diverse medicinal compounds. Ind Crops Prod. https://doi.org/10.1016/j.indcrop.2022.115072
Soorani A, Nazeri V, Fatthi R, Khadivi A (2013) DNA fingerprinting of Leonurus cardiaca L. germplasm in Iran using amplified fragment length polymorphism and interretrotransposon amplified polymorphism. Biochem Syst Ecol 50:438–447
Sorkheh K, Koohi Dehkordi M, Ercisli S, Hegedus A, Halasz J (2017) Comparison of traditional and new generation DNA markers declares high genetic diversity and differentiated population structure of wild almond species. Sci Rep 7:5966
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Teo CH, Tan SH, Ho CL, Faridah QZ, Othman YR, Heslop-Harrison JS, Kalendar R, Schulman AH (2005) Genome constitution and classification using retrotransposon-based markers in the orphan crop banana. J Plant Biol 48:96–105
Tzeng TC, Lin YL, Jong TT, Chang CMJ (2007) Ethanol modified supercritical fluids extraction of scopoletin and artemisinin from Artemisia annua L. Sep Purif Technol 56:18–24
Yildiz M, Arbizu CI (2022) Inter-primer binding site (iPBS) retrotransposon markers provide insights into the genetic diversity and population structure of carrots (Daucus, Apiacae). Turk J Agric For 46:214–223
Acknowledgements
The authors gratefully acknowledge the laboratory facilities provided by Genomics & Molecular Breeding Lab, Center of Excellence in Cereal Molecular Breeding, University of Tabriz, Iran. The research was financially supported by the University of Tabriz incorporation with the Iran National Elites Foundation, under vote NO. 102-1565.
Funding
This research was financially supported by the University of Tabriz incorporation with the Iran National Elites Foundation, under vote NO. 102-1565.
Author information
Authors and Affiliations
Contributions
HAH, SAM, and NV conceptualized and designed the experiment; HAH and SAM analyzed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Abbasi Holasou, H., Valizadeh, N. & Mohammadi, S.A. Molecular insights into the genetic diversity and population structure of Artemisia annua L. as revealed by insertional polymorphisms. Braz. J. Bot 46, 51–60 (2023). https://doi.org/10.1007/s40415-022-00860-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40415-022-00860-x