
Abstract
Vericiguat is an oral soluble guanylate cyclase stimulator and enhances the cyclic guanosine monophosphate pathway independently of nitric oxide as well as synergistically in normal- and low-nitric oxide conditions. This review describes the pharmacokinetic and pharmacodynamic profile of vericiguat and summarizes the effect of vericiguat on cardiac electrophysiology and population pharmacokinetic/pharmacodynamic relationships. Vericiguat demonstrates virtually complete absorption and increased exposure with food. Vericiguat has high oral bioavailability when taken with food (93.0%) with dose-proportional pharmacokinetics in healthy volunteers. Vericiguat has slightly less than dose-proportional pharmacokinetics with a slight decrease in bioavailability at higher doses in patients with heart failure (HF) with reduced ejection fraction (HFrEF). Vericiguat is a low-clearance drug, with a half-life of approximately 20 h in healthy volunteers and 30 h in patients with HFrEF. Most drug metabolism is achieved by glucuronidation. Vericiguat has pharmacodynamic effects as expected from its pharmacological mechanism of action (i.e., relaxation of the smooth muscles in the vasculature leading to changes in hemodynamics). In the VICTORIA trial (NCT02861534), which enrolled patients with HFrEF, no meaningful exposure–response relationships for the incidence of symptomatic hypotension or syncope were evident. There were no significant imbalances in the incidence of undesirable hemodynamic-related effects (symptomatic hypotension and syncope) in subgroups with HFrEF defined by sex, age, race, and renal impairment. In addition, most patients achieved the 10-mg target dose per the blood pressure-guided titration regimen. No dose adjustments due to body weight, age, sex, race, or hepatic/renal impairment are necessary in adult patients with HFrEF. Observed and predicted changes in vericiguat exposure when co-administered with perpetrator drugs were small and not clinically meaningful. In addition, vericiguat has low potential as a perpetrator to affect exposure and/or pharmacodynamic effects of drugs commonly prescribed in patients with heart failure; therefore, no dose adjustment of these drugs is required in patients taking vericiguat. There is limited experience on the combined use of vericiguat with long-acting nitrates in patients with HFrEF. The ongoing VICTOR trial (NCT05093933), which is investigating vericiguat in patients with HFrEF, permits the co-administration of long-acting nitrates. Combined use of vericiguat and phosphodiesterase type-5 inhibitors has not been studied in patients with HFrEF and is therefore not recommended because of the potential increased risk for symptomatic hypotension. Vericiguat was not associated with electrophysiological abnormalities in preclinical and clinical studies up to the approved dose of 10 mg at steady state. Vericiguat is approved for the treatment of recently decompensated patients with worsening HFrEF. Vericiguat’s safety and efficacy profile in patients with HFrEF will be further characterized by the VICTOR trial (NCT05093933) in adults without recent decompensation and in a pediatric population with HF due to left ventricular systolic dysfunction (VALOR trial, NCT05714085).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Vericiguat is approved as a first-in-class drug for the treatment of recently decompensated patients with worsening heart failure with reduced ejection fraction. |
Vericiguat has a favorable pharmacokinetic and pharmacodynamic profile that was thoroughly characterized in numerous Phase I, II, and III trials; these results informed the initiation and dose titration of vericiguat. |
The recommendations regarding the lack of need for dose adjustments beyond the dose titration are also discussed in the review. |
1 Introduction
More than 60 million people worldwide have heart failure (HF) and approximately 50% of these have HF with reduced left ventricular ejection fraction (HFrEF) [1, 2]. Approximately 6.7 million adults had HF in 2017–2020, representing a prevalence of 2.3% in the USA, compared with approximately 6.0 million (2.1%) in 2015–2018 [3]. The prevalence of HF continues to increase over time due to the aging of the US population [3, 4]. Projections estimate that HF will affect more than 8 million adults in the USA by 2030, corresponding to a 46% increase from 2012 [3]. The European Society of Cardiology (ESC) 2021 guidelines suggest that 1–2% of the global adult population have HF [4].
Guideline-directed medical therapies (GDMT) for HFrEF include angiotensin-converting enzyme (ACE) inhibitors/angiotensin receptor-neprilysin inhibitors, mineralocorticoid receptor antagonists, beta blockers, and sodium–glucose cotransporter 2 inhibitors (SGLT2i) [4, 5]. Nevertheless, there remains substantial residual risk in patients on optimized GDMT, with annualized event rates per 100 patient-years of 13.2–37.8 for the primary endpoint in comparator arms of contemporary HF trials [6].
Vericiguat is a first-in-class soluble guanylate cyclase (sGC) stimulator approved for the treatment of patients with worsening HFrEF in the EU, USA, and other countries [7,8,9]. The ESC 2021 guidelines and the American Heart Association/American College of Cardiology/Heart Failure Society of America 2022 consensus guidelines recommend considering vericiguat to reduce the risk of hospitalization for HF and cardiovascular death in high-risk patients with worsening HFrEF who are already on GDMT [4, 5]. The recommendations are based on the findings of the pivotal Phase III VICTORIA trial (NCT02861534), which assessed vericiguat in adult patients with reduced ejection fraction and recently decompensated chronic HF on a background of standard of care including angiotensin receptor neprilysin inhibitors (n = 731 [15%] at randomization) [4, 5, 10, 11]. Participants who were clinically unstable, defined as having received any intravenous treatment within 24 h prior to randomization, and/or having a systolic blood pressure (SBP) < 100 mmHg or symptomatic hypotension, were excluded from the VICTORIA trial [10]. In the VICTORIA trial, the incidence of the primary composite endpoint comprising death from cardiovascular causes or hospitalization for HF was lower in patients randomized to vericiguat than in those randomized to placebo (35.5% vs 38.5%; hazard ratio [HR], 0.90; 95% confidence interval [CI], 0.82–0.98; p = 0.02) [10].
Multimorbidity and polypharmacy are common in patients with HF [12]. In addition, the prevalence of HF in male and female patients aged ≥ 80 years is high, approximately 7% and 11%, respectively [3]. Therefore, drugs developed for HFrEF must have an appropriate pharmacokinetic and pharmacodynamic profile that makes them suitable for clinical treatment in a vulnerable population. A comprehensive preclinical and clinical study program characterized the pharmacokinetic and pharmacodynamic profile of vericiguat. This review describes the pharmacokinetic and pharmacodynamic profile of vericiguat in animals, healthy volunteers, and patients with HFrEF, including the influence of extrinsic and intrinsic factors. This review also summarizes studies investigating the effect of vericiguat on cardiac electrophysiology and population pharmacokinetic/pharmacodynamic relationships in patients with HFrEF.
2 Physiochemical Properties and Preclinical Pharmacology
2.1 Chemical and Physicochemical Properties
Vericiguat, methyl {4,6-diamino-2-[5-fluoro-1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b] pyridin-3-yl] pyrimidin-5-yl}carbamate (Fig. 1), has a molecular weight of 426.4 g/mol [7, 13]. Vericiguat is a Biopharmaceutics Classification System Class II drug owing to its high permeability (apparent permeability coefficient [Papp] = 111 nm/s for the A-to-B direction and Papp = 464 nm/s for the B-to-A direction across Caco-2 cells) and low solubility [14]. Vericiguat is a lipophilic (logP 2.99) basic compound with a pKa of 4.7 and its solubility is pH-dependent, with lower solubility in neutral conditions compared with acidic conditions [14, 15].

Chemical structure of vericiguat
2.2 Preclinical Pharmacokinetics and Safety Pharmacology
Decreased nitric oxide (NO) availability and sGC functionality in chronic HF lead to impairment of the NO–sGC–cyclic guanosine monophosphate (NO–sGC–cGMP) pathway [16, 17]. Dysfunction of NO–sGC–cGMP signaling leads to cGMP deficiency, endothelial inflammation, and myocardial and vascular dysfunction [16, 17]. Preclinical studies demonstrated the effects of vericiguat on recombinant sGC and sGC-overexpressing cells [13, 18]. In NO-independent conditions, vericiguat (0.01–100 µM) stimulated sGC by 1.7- to 57.6-fold. When combined with an NO donor (diethylamine/NO complex) at the highest tested concentration of vericiguat (100 µM) a much higher stimulation of sGC, more than 340-fold, was observed, which is indicative of a synergistic effect [13, 18,19,20]. The half-maximal effective concentrations of vericiguat in an sGC-overexpressing Chinese hamster ovary cell line with and without an NO donor (S-nitroso-N-acetyl-d,l-penicillamine 100 nM) were 10.6 nM and 1005 nM, respectively [13, 18]. Pharmacodynamic assessments of vericiguat on isolated blood vessels and hearts demonstrated inhibition of pharmacologically induced contractions of saphenous artery rings, aortic rings, and femoral vein rings [13, 18]. Vericiguat also relaxed isolated nitrate-tolerant rabbit saphenous artery rings, with an inhibitory concentration (IC)50 of 5.8 nM [13, 18]. Vericiguat reduced coronary perfusion pressure without affecting heart rate (HR) and contractility, with a maximum reduction of 30% at 10 µM in Langendorff-perfused rodent heart preparations [13, 18]. Finally, vericiguat was associated with significant reductions in mortality, cardiac hypertrophy, atrial natriuretic peptide, and proteinuria in Nω-nitro-l-arginine methyl ester-treated renin transgenic rats [13, 18]. Collectively, the studies demonstrate that vericiguat directly, potently, and selectively stimulates sGC to enhance the cGMP pathway under low-NO conditions, as occur in HF [21]. Vericiguat restores the impaired NO–sGC–cGMP pathway by directly stimulating sGC through a binding site independent of NO and by sensitizing sGC to endogenous NO, thus restoring cGMP production even under low-NO conditions and oxidative stress (Fig. 2) [15, 22].

Adapted from Sandner et al. [16] under the terms of the CC-BY license (https://creativecommons.org/licenses/by/4.0/). cGMP cyclic guanosine monophosphate, Cyb5R3 cytochrome b5 reductase 3, GTP guanosine trisphosphate, sGC soluble guanylyl cyclase
Vericiguat mode of action.
In vitro studies indicate that vericiguat and its N-glucuronide metabolite (M-1) are not inhibitors of major cytochrome P450 (CYP) isoforms (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4) or uridine diphosphate-glucuronosyltransferase (UGT) isoforms (UGT1A1, 1A4, 1A6, 1A9, 2B4, and 2B7), nor are they inducers of CYP1A2, 2B6, and 3A4, at clinically relevant concentrations [13, 15]. Furthermore, vericiguat is not a substrate of organic cation transporter 1 (OCT1), or organic anion transporting polypeptides (OATP1B1 and OATP1B3) [15]. In addition, the M-1 metabolite is not a substrate of permeability glycoprotein (Pgp), breast cancer resistance protein (BCRP), OATP1B1, or OATP1B3 [15]. Vericiguat and M-1 are not inhibitors of drug transporters, including Pgp, BCRP, bile salt export pump, OATP1B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, multidrug and toxin extrusion protein 1 (MATE1), and MATE2K at clinically relevant concentrations [15]. The preclinical studies indicated a low drug–drug interaction potential, later confirmed in dedicated clinical studies, population pharmacokinetic analysis, and in silico modeling simulations at clinically relevant concentrations (Sect. 3.4) [15, 23, 24].
The preclinical pharmacokinetic profile of vericiguat was assessed in rodent and canine models [25]. Overall, vericiguat pharmacokinetics were in line with the data obtained in humans. No significant interspecies differences in vericiguat metabolism were observed; specifically, all metabolites observed in humans were seen in rodents and canines [25].
3 Pharmacokinetic and Pharmacodynamic Properties
3.1 Summary of Vericiguat ADME
3.1.1 Absorption and the Effect of Food
The bioavailability of vericiguat, dose proportionality, and the influence of food was investigated in five Phase I studies for different formulations during drug development (Table 1). Vericiguat is a substrate of active transport proteins (Pgp and BCRP) in vitro [15]. However, in an absolute bioavailability study, the oral bioavailability of vericiguat 10 mg (administered as two 5-mg tablets, fed state) relative to an intravenously administered radiolabeled vericiguat microdose of 20 μg (fed state) was 93.0% (Fig. 3) [14]. The high bioavailability of vericiguat indicates a limited role of Pgp and BCRP during absorption.

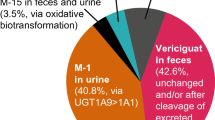
Absorption, metabolism, distribution, and excretion of vericiguat. AUC area under the concentration–time curve, CLsys systemic clearance, Cmax maximum plasma concentration, HFrEF heart failure with reduced ejection fraction, M-1 N-glucuronide metabolite, PK pharmacokinetic, tmax time to reach Cmax, UGT uridine diphosphate-glucuronosyltransferase, Vd/F apparent volume of distribution, Vss volume at steady state
Vericiguat exhibits a positive food effect. In the fed state, the exposure of vericiguat increased by 19% (area under the plasma concentration-time curve; AUC) and 9% (maximum plasma drug concentration; Cmax) after administration of a 5-mg tablet and by approximately 40% (AUC and Cmax) after administration of a 10-mg tablet, compared with the fasted state. The decrease in the rate of absorption is also reflected in a delayed time to Cmax from approximately 1–4 h under fed versus fasted conditions. However, the type of meal (high-fat high-calorie vs low-fat low-calorie) does not meaningfully impact the pharmacokinetics of vericiguat [14]. Vericiguat exposure (AUC and Cmax) was unaffected when administered as a crushed tablet or intact tablet to human volunteers [14].
The dose-proportionality of vericiguat exposure (AUC and Cmax) was demonstrated across the dose range of 1.25–15 mg when given as single oral doses to healthy volunteers. Furthermore, the pharmacokinetics of vericiguat appeared time-independent after multiple dosing up to the highest therapeutically relevant dose of 10 mg [14, 26]. In healthy volunteers, there was no evidence for unexpected accumulation, with accumulation of vericiguat 1.25 mg, 5 mg, and 10 mg in plasma after 7 days of once-daily dosing being between 155% and 171% [27,28,29].
3.1.2 Distribution
Vericiguat exhibits high protein binding at approximately 98%, with serum albumin being the predominant binding component [15, 26]. Therefore, vericiguat is unlikely to be removed by hemodialysis due to high protein binding [7]. The volume of distribution of vericiguat in healthy volunteers was approximately 44 L, indicating low affinity to tissues [14, 30, 31]. In patients with HFrEF, the apparent central volume of distribution was 46.8 L [23].
3.1.3 Metabolism and Elimination
Vericiguat is a low-clearance drug, with a mean plasma clearance of 1.62 L/h following administration to healthy volunteers (Fig. 3) [8, 14, 31]. The half-life of vericiguat is approximately 20 h in healthy volunteers and 30 h in patients with HFrEF [23, 27]. The predominant metabolic pathway of vericiguat is Phase II UGT-mediated conjugation with glucuronic acid to form a pharmacologically inactive N-glucuronide metabolite (M-1) [13, 15]. The primary UGT isoforms contributing to the formation of M-1 are UGT1A9 and UGT1A1, with preferential catalysis by the UGT1A9 isoform [15]. The M-1 metabolite is the only major circulating metabolite in human plasma (72% of total radioactivity AUC) and is the main component identified in urine (41% of the dose) [15]. The CYP-mediated metabolism accounts for less than 5% of vericiguat’s biotransformation [15]. A minor metabolite (M-15), presumably formed by demethylation/decarboxylation/acetylation, was detected as a component in urine and feces (1.9% and 1.6% of the dose, respectively), indicating that oxidative metabolism plays a minor role in vericiguat clearance [15]. Unchanged vericiguat was detected in urine and feces (9.0% and 42.6% of the dose, respectively); however, the amount of unchanged vericiguat recovered in feces is unlikely to be due to non-absorption given its high bioavailability [15]. Ex vivo incubation with human feces under anerobic conditions demonstrated hydrolytic cleavage of M-1 to vericiguat by microbial flora [15]. Therefore, vericiguat observed in feces may result from hydrolysis of M-1 to vericiguat in the gut lumen, as well as possibly from biliary/intestinal secretion of vericiguat [15]. The amount of unchanged vericiguat in urine is consistent with its renal clearance (0.0994–0.149 L/h), indicating that vericiguat is likely excreted through the kidneys via glomerular filtration, with minimal contribution of active secretion or tubular reabsorption [27, 32]. In summary, vericiguat elimination occurs via multiple routes in humans, mainly glucuronidation (to M-1), glomerular filtration of unchanged vericiguat in the kidneys, excretion of unchanged vericiguat via bile/intestinal secretion, and oxidative biotransformation (to M-15).
3.2 Pharmacokinetic Properties in Different Populations
In dedicated Phase I studies, the effects of several intrinsic factors such as age, sex, race, and renal and hepatic impairment on the exposure of vericiguat (AUC and Cmax) were investigated to determine whether dose adjustments due to these characteristics in adult patients with HFrEF were required (Table 1). In addition, an integrated population pharmacokinetics model using data from patients with HFrEF enrolled in the SOCRATES-REDUCED and VICTORIA trials was performed (Table 2) [23].
3.2.1 Age and Sex
No clinically meaningful age-related differences in the pharmacokinetic profile of vericiguat were observed between healthy young volunteers aged 18–45 years and healthy elderly volunteers aged ≥ 65 years [33]. Similarly, age did not affect vericiguat exposure in patients with HFrEF in a clinically relevant manner (Fig. 4) [23]. Cmax and AUC were increased in healthy female participants by 32% and 37%, respectively, compared with healthy male participants [33]. The differences in vericiguat exposure between age groups and sex can be attributed to observed body weight differences [23]. Furthermore, the sex of patients was not a statistically significant covariate in the population pharmacokinetics analysis of patients with HFrEF [23].

Pharmacokinetics of vericiguat in different populations. Figure adapted from vericiguat prescribing information [7]. AUC area under the curve, CI confidence interval, Cmax maximum drug concentration, NT-proBNP N-terminal pro-B-type natriuretic peptide. aDose and body weight-normalized AUC and dose and body weight normalized Cmax after a single-dose administration. bAUC over the dosing interval and Cmax after multiple dose administration. cData from Phase I studies in healthy volunteers. dData from the integrated population pharmacokinetics model of VICTORIA and SOCRATES-REDUCED
3.2.2 Race
Healthy Asian volunteers showed approximately 70% and 80% higher vericiguat AUC and Cmax, respectively, compared to healthy White volunteers [28]. However, when adjusted for body weight, these exposure differences reduced to about 38% (AUC) and 45% (Cmax) [28]. In the population pharmacokinetics analysis, no significant or clinically relevant effect of race, including African American, White, or Asian, was found on any pharmacokinetic parameters after adjusting for body weight in patients with HFrEF [23].
3.2.3 Renal Impairment
In volunteers with mild (creatinine clearance [CLCR] 50–80 mL/min), moderate (CLCR 30 to < 50 mL/min), and severe (CLCR < 30 mL/min) renal impairment and without HFrEF, unbound AUC normalized for body weight was increased by 8%, 73%, and 143%, respectively, compared with healthy control volunteers with normal kidney function (CLCR > 80 mL/min) [8, 23, 34]. There was no correlation between the fraction of unbound vericiguat and serum albumin concentrations [35]. The overall higher exposure did not result in significant effects on blood pressure (BP) and HR [35]. The increases in drug exposure were not accompanied by effects on BP and HR in otherwise healthy volunteers with mild-to-moderate renal impairment [35]. Vericiguat AUC in patients with HFrEF with moderate and severe renal impairment was increased by 13% and 20%, respectively, compared with those with normal kidney function [8, 23]. These differences in vericiguat AUC were primarily driven by body weight and are not considered clinically relevant. In the population pharmacokinetics analysis of patients with HFrEF, estimated glomerular filtration rate (eGFR) had no clinically meaningful effect on vericiguat exposures [23]. Therefore, no dose adjustments are required in non–dialysis-dependent patients with eGFR ≥ 15 mL/min/1.73 m2 [7, 8]. The pharmacokinetics of vericiguat in patients with eGFR < 15 mL/min/1.73 m2 or on dialysis have not been studied [7, 8]. Variations in the magnitude of vericiguat AUC changes between participants with renal impairment in VICTORIA and in the Phase I renal impairment study could be attributed to differences in study designs, food variability, concurrent medications, and potential differences in kidney disease etiology.
3.2.4 Hepatic Impairment
In a dedicated Phase I study, unbound vericiguat AUC (normalized by dose per body weight) in volunteers with mild (Child–Pugh A) and moderate (Child–Pugh B) hepatic impairment was increased by 21% and 47%, respectively, compared with matched healthy control volunteers [30, 36]. These elevations in vericiguat exposure are not considered clinically relevant [37]. Altered levels of bilirubin and albumin, both components of the Child–Pugh score, were assessed as covariates in the population pharmacokinetic evaluation and did not affect vericiguat pharmacokinetics in patients with HFrEF [23]. Therefore, no dose adjustments of vericiguat are required in patients with mild-to-moderate hepatic impairment [7, 8]. Vericiguat has not been investigated in patients with severe hepatic impairment [7, 8].
3.2.5 Population Pharmacokinetics Model
In the integrated population pharmacokinetics model comprising data from SOCRATES-REDUCED and VICTORIA (Table 2), disease-related characteristics that did not influence vericiguat exposure in patients with HFrEF were examined and included baseline N-terminal pro-B-type natriuretic peptide (NT-proBNP), left ventricular ejection fraction, New York Heart Association class, albumin and bilirubin levels, and eGFR [23]. Body weight was the only statistically significant covariate that influenced the exposure of vericiguat at a given dose level [23]. Specifically, baseline and time-varying body weight were predictors of vericiguat apparent clearance and apparent central volume of distribution [23]. However, the effects were generally small and no dose adjustments are required for those with low (< 60 kg) or high (> 90 kg) body weight [23, 38].
3.2.6 Patients with HFrEF
In patients with HFrEF, the estimated steady-state AUC within the dosing interval of 24 h of vericiguat 10 mg, based on the integrated population PK analysis, was 6680 µg×h/L [28]. This is in line with the exposure observed in healthy volunteers after administration of 10-mg vericiguat using the formulation intended for marketing and as recommended in the label, in fed state. Here, AUC was determined as 5600 µg×h/L, indicating that vericiguat pharmacokinetics are generally consistent between healthy volunteers and patients with HFrEF [14].
3.2.7 Summary of Vericiguat Pharmacokinetics in Different Populations
Overall, no dose adjustments due to body weight, age, sex, ethnicity, race, or hepatic/renal impairment are necessary in adult patients with HFrEF.
3.3 Pharmacodynamic Properties in Different Populations
The pharmacodynamic profile of vericiguat, particularly its hemodynamic-related effects, and the impact of intrinsic factors on pharmacodynamic effects is reported from studies in healthy volunteers, patients with HFrEF, and different populations (Table 1).
3.3.1 Healthy Volunteers
In healthy volunteers, vericiguat was associated with increases in cGMP and vasoactive hormones (further details in Sect. 5) [27]. In addition, vericiguat was associated with relevant increases in cardiac parameters (cardiac output and cardiac index) and HR and decreased systemic vascular resistance (further details in Sect. 5) [27]. The observed increase in HR was accompanied by slight decreases in SBP and diastolic BP (DBP) (reductions of approximately 2–3 mmHg with vericiguat 5–15 mg); this association is attributed to a compensatory baroreflex response [27]. There were no relevant differences in hemodynamic changes (HR, SBP, and DBP) with vericiguat in the Phase I studies conducted in healthy Asian volunteers compared with White volunteers [27]. Overall, vericiguat was well tolerated in healthy subjects. The adverse event profile of vericiguat was predominantly associated with its pharmacodynamic mode of action, (i.e., relaxation of smooth muscle in the vasculature leading to hemodynamic changes and gastrointestinal side effects).
3.3.2 Patients with HFrEF
In patients with HFrEF, vericiguat was associated with a small reduction in BP, with BP values returning to baseline after 16 weeks [10]. In the VICTORIA trial, the incidence of symptomatic hypotension in the vericiguat and placebo treatment arms were similar, at 9.1% and 7.9%, respectively (risk difference [95% CI]: 1.2 [− 0.3, 2.8]; p = 0.12) [10]. The incidence of syncope in the vericiguat and placebo treatment arms was also similar, at 4.0% and 3.5% of patients, respectively (risk difference [95% CI]: 0.6 [− 0.5, 1.6]; p = 0.30) [10]. Overall, these small pharmacodynamic differences are not unexpected, given the pharmacological mechanism of action of vericiguat [22]. There were no significant imbalances in the incidence of symptomatic hypotension and syncope in subgroups with HFrEF defined by sex, age, and race [11, 28, 39].
In the VICTORIA trial, initial reductions in SBP were more evident in patients aged > 75 years and > 85 years compared to younger patients, with SBP values returning to baseline thereafter [39]. Furthermore, in subgroups defined by age, symptomatic hypotension and syncope, event rates were similar by randomized treatment (adjusted HR for composite safety outcome [95% CI]: 1.06 [0.78–1.44] for > 75 years and 1.23 [1.01–1.51] for ≤ 75 years, all p for interaction > 0.05) [39]. Although older patients had a slightly lower body weight than younger patients, and thus higher vericiguat exposure (Fig. 4), the proportions of patients achieving the 10-mg target dose per the BP-guided titration regimen in those aged > 75 years and ≤ 75 years were similar (78% and 79%, respectively), indicating that vericiguat is well tolerated in elderly patients [39]. Overall, no differences in safety related to hemodynamic effects of vericiguat were observed between patients aged ≥ 65 years compared to younger patients, but greater sensitivity of some older individuals cannot be ruled out [7].
As previously discussed, body weight is a significant intrinsic factor influencing vericiguat pharmacokinetics [23]. However, in the VICTORIA trial, there was no meaningful exposure–response relationship for symptomatic hypotension and syncope, both prespecified safety outcomes of clinical interest related to hemodynamic effects [28].
In the VICTORIA trial, there was no significant imbalance in the incidence of symptomatic hypotension and syncope in subgroups with renal impairment [28]. In addition, renal impairment did not meaningfully affect the proportion of patients achieving the 10-mg target dose [28].
3.3.3 Summary of Vericiguat Pharmacodynamics in Different Populations
In summary, vericiguat has hemodynamic-related effects as expected from its pharmacological mechanism of action. There was no significant imbalance in the incidence of undesirable hemodynamic-related effects (symptomatic hypotension and syncope) in subgroups with HFrEF defined by sex, age, race, and renal impairment. In addition, most patients achieved the 10-mg target dose per the BP-guided titration regimen; therefore, no dose adjustment of vericiguat is required in geriatric patients, non–dialysis-dependent patients with eGFR ≥ 15 mL/min/1.73 m2, and patients with mild-to-moderate hepatic impairment.
3.4 Pharmacokinetic Drug–Drug Interactions
3.4.1 Pharmacokinetic Drug–Drug Interaction Profile of Vericiguat as a Victim
The effects of co-administered drugs on vericiguat pharmacokinetics, and the effects of vericiguat on the pharmacokinetics of other drugs were investigated in multiple drug–drug interaction studies.
The effects of co-administered drugs on the pharmacokinetics of vericiguat were assessed in dedicated healthy volunteer drug–drug interaction studies with gastric pH-modifying agents (omeprazole and aluminum hydroxide–magnesium hydroxide), a broad-spectrum CYP and transporter inhibitor (ketoconazole), a UGT1A9 inhibitor (mefenamic acid), and a broad-spectrum inducer (rifampicin) (Table 1 and Fig. 5) [15]. In addition, drug–drug interactions with a UGT1A1 inhibitor (atazanavir) were assessed by physiologically based pharmacokinetic modeling (Table 2) [24].

Adapted from Boettcher et al. [15] under the terms of the CC-BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/). AUC area under the concentration–time curve, AUC12,md AUC from time 0 h to 12 h after multiple-dose administration, AUC22 AUC from time 0 h to 22 h, AUC24 AUC from time 0 h to 24 h, AUC24,md AUC24 after multiple-dose administration, AUCτ,md AUC within the dosing interval after multiple dosing, CI confidence interval, Cmax maximum concentration within a dosing interval, Cmax,md Cmax within a dosing interval after multiple-dose administration, Ctrough trough concentration within a dosing interval, LBQ657 sacubitrilat, the active metabolite of sacubitril, PBPK, physiologically based pharmacokinetic
Summary of clinical interactions and pharmacokinetic changes investigating the perpetrator and victim potential of vericiguat.
Pre- and co-administration of omeprazole 40 mg decreased vericiguat mean AUC by 32.2% and mean Cmax by 49.6%; similarly, co-administration with an aluminum hydroxide/magnesium hydroxide combination antacid (10 mL) with vericiguat 5 mg decreased the extent of vericiguat absorption (AUC by 27.1% and mean Cmax by 45.7%) [15]. Notably, this study was conducted in fasting conditions; however, administration of food increases vericiguat exposure and reduces pharmacokinetic variability [14]. As food increases gastric pH, the positive effect of food on wettability mitigates the negative effect of pH on vericiguat solubility [40]. Therefore, vericiguat exposure is less impacted by drugs that increase gastric pH when taken with food. Vericiguat was administered with food in the Phase II and Phase III clinical trials in patients with HFrEF (SOCRATES-REDUCED and VICTORIA) [10]. There was no effect on vericiguat exposure in patients with HFrEF when vericiguat was co-administered with gastric pH-modifying agents in the SOCRATES-REDUCED and VICTORIA trials [23]. Therefore, vericiguat is recommended to be administered with food, with no need for dose adjustments in patients with HFrEF taking medicinal products that increase gastric pH [7, 8].
Pre- and co-administration with ketoconazole 200 mg, administered twice daily, had no clinically relevant effect on the exposure of a single dose of vericiguat 1.25 mg (12.6% increase in AUC and 10.9% increase in Cmax), consistent with the minor contribution of Pgp on vericiguat absorption and CYP enzymes to the overall clearance of vericiguat [15].
Pre- and co-administration of mefenamic acid (total dose of 2500 mg over 48 h) had no clinically relevant effect on the exposure of a single dose of vericiguat 2.5 mg (19.7% increase in AUC and a negligible change in Cmax) [15].
Physiologically based pharmacokinetic modeling indicates that co-administration of atazanavir 400 mg once daily is expected to have no clinically relevant effect on the exposure of vericiguat 10 mg (predicted increase in AUC of 12% and Cmax by 4%) [24]. A single oral dose of vericiguat 10 mg administered after pre-treatment for 6 days and concomitant administration with rifampicin 600 mg once daily was associated with a non-clinically relevant decrease in vericiguat exposure (AUC by 28.7% and Cmax by 8.6%) [15]. Overall, vericiguat has a minimal risk of being a victim of pharmacokinetic drug–drug interactions, as confirmed in studies with healthy volunteers and in patients with HFrEF.
3.4.2 Pharmacokinetic Drug–Drug Interaction Profile of Vericiguat as a Perpetrator
The effects of vericiguat on the pharmacokinetics of index substrates of CYP3A4 (midazolam) and Pgp (digoxin) were assessed in Phase I healthy volunteer studies. The administration of vericiguat 10 mg once daily did not meaningfully impact the exposure of a single administered dose of midazolam 7.5 mg (mean AUC was reduced by 17.8% and Cmax by 23.1%), indicating that vericiguat is not an inhibitor of CYP3A4 [15]. Also, pharmacokinetics of sildenafil, a CYP3A4 substrate, were not meaningfully affected by co-administration with vericiguat [15]. In addition, vericiguat did not affect the exposure of digoxin, indicating that vericiguat is not an inhibitor of Pgp [15]. Overall, vericiguat has a minimal risk of being a clinically relevant perpetrator for common metabolic and excretion pathways such as CYP3A4 and Pgp.
3.4.3 Summary of Vericiguat Pharmacokinetic Drug–Drug Interaction Profile
The observed (and predicted with atazanavir) changes in vericiguat exposure when co-administered with perpetrator drugs were small and not clinically meaningful. The observed changes in exposure of drugs metabolized or excreted by CYP3A4 and Pgp pathways were less than 25% when co-administered with vericiguat.
3.5 Pharmacodynamic Drug–Drug Interactions
Potential pharmacodynamic drug–drug interactions were assessed in several populations with drugs acting on the NO–sGC–cGMP pathway (short-acting nitrates [nitroglycerin], a long-acting nitrate [isosorbide mononitrate], a phosphodiesterase 5 [PDE5] inhibitor [sildenafil]), and other drugs commonly used in patients with HFrEF (sacubitril/valsartan [an angiotensin receptor-neprilysin inhibitor], aspirin [an antithrombotic], and warfarin [an anticoagulant]) (Table 1).
3.5.1 Hemodynamic Effects with Nitrates
Vericiguat was not associated with more than additive hemodynamic effects (HR and BP) in both rodents and canines when co-administered with glycerol trinitrate [30], suggesting a low potential for a pharmacodynamics interaction between vericiguat and nitrates. Based on preclinical results, a drug–drug interaction study in healthy volunteers was conducted, which confirmed that the known effects elicited by a short-acting nitrate (nitroglycerin; decreased BP and increased HR) were unaffected by the co-administration of vericiguat 5 mg in healthy volunteers [41, 42]. In addition, maintenance treatment with vericiguat 2.5–10 mg was well tolerated in patients with chronic coronary syndromes receiving sublingual nitroglycerin spray (0.4 mg) [41]. No statistically significant differences in hemodynamic parameters (SBP, DBP, and HR) were observed between vericiguat and placebo during co-administration with a sublingual nitroglycerin spray (0.4 mg) in patients with chronic coronary syndromes [41]. In a different drug–drug interaction study in patients with chronic coronary syndromes, vericiguat (up-titrated to 10 mg) co-administered with a long-acting nitrate (isosorbide mononitrate 60 mg) was generally well tolerated [43]. Co-administration of isosorbide mononitrate with vericiguat was associated with decreases in SBP and DBP of 2–5 mmHg and increases in HR of 1–2 beats/min [30, 43]. In the Phase III VICTORIA trial, co-administration of short-acting nitrates was allowed per the study protocol; however, long-acting nitrates were prohibited as no clinical data on the combination with vericiguat were available at study initiation [10]. Among the randomized VICTORIA trial participants receiving concomitant nitrates (11.9% of the overall population), 99.8% reported that the combination with either vericiguat or placebo was well tolerated [30].
Overall, co-administration of vericiguat with organic nitrates did not result in a more than additive reduction in BP or increase in HR in dedicated pharmacodynamic studies. Concomitant use with short-acting nitrates was well tolerated in patients with HFrEF in the VICTORIA trial. There is limited experience on the combined use of vericiguat with long-acting nitrates in patients with HFrEF. The European and US labels do not have a contraindication for the concomitant use of nitrates with vericiguat [7, 8]. The ongoing VICTOR trial (NCT05093933), which is investigating vericiguat in patients with HFrEF, permits the co-administration of nitrates, including long-acting nitrates [44]. Results from the VICTOR trial are anticipated to further expand on the efficacy and safety profile of vericiguat when combined with nitrates in a population with stable HFrEF.
3.5.2 Hemodynamic Effects with a PDE5 Inhibitor
Combined treatment of vericiguat with single doses of a PDE5 inhibitor (sildenafil 25 mg, 50 mg, and 100 mg) was generally well tolerated in healthy volunteers [45]. Vericiguat co-administered with sildenafil resulted in an increased frequency of transient nervous system disorder adverse events, most commonly headache and head discomfort (predominantly of mild intensity) [45]. These adverse events are likely attributable to exaggerated smooth muscle relaxation and vasodilatation when two drugs acting on the same NO–sGC–cGMP pathway are combined. Furthermore, the combination was associated with decreases in seated SBP and DBP of ≤ 5.4 mmHg compared with co-administration of placebo with sildenafil [45]. No dose-dependent trend on hemodynamic parameters across the different sildenafil doses was evident [45]. However, concomitant use of vericiguat and PDE5 inhibitors has not been studied in patients with HFrEF [10], and is therefore not recommended because of the potential increased risk for symptomatic hypotension [7, 8, 46].
3.5.3 Pharmacodynamic Effects with Other Drugs Commonly Prescribed in Patients with HF
A study in mice indicated a role of sGC in platelet aggregation [47], and patients with HFrEF often receive antiplatelet and anticoagulants [26]. Administration of vericiguat in healthy volunteers had no effect on bleeding time or platelet aggregation when given alone, and had no additive effect on bleeding time or platelet aggregation when given in combination with aspirin (assessed by arachidonic acid-induced turbidimetry, collagen-induced turbidimetry, arachidonic acid-induced impedance aggregometry, and collagen-induced impedance aggregometry) [26]. Furthermore, vericiguat, when co-administered with warfarin, did not influence coagulation in comparison with warfarin alone (assessed by prothrombin time and activities of factors X, VII, and II) [26].
An interaction study of vericiguat 2.5 mg co-administered with twice-daily treatment of sacubitril/valsartan 97/103 mg in healthy volunteers showed no clinically relevant pharmacodynamic interactions between sacubitril/valsartan and vericiguat (additive BP effects by vericiguat were < 2 mmHg) [26]. In the VICTORIA trial, the observed imbalance in symptomatic hypotension and syncope between the vericiguat and placebo groups was similar in patients with HFrEF taking sacubitril/valsartan versus those not taking sacubitril/valsartan [10, 11].
SGLT2is became indicated therapies in HF only after completion of the VICTORIA trial. They had approved indications as anti-hyperglycemic agents prior to initiation of VICTORIA, and there were no restrictions regarding SGLT2i use in the VICTORIA trial [10]. Overall use of SGLT2i was low in the VICTORIA trial (n = 122 [2.4%]) because at that time SGLT2is were not yet approved for HFrEF [39]. In the VICTORIA trial, the rates of symptomatic hypotension and syncope events were numerically greater in those receiving SGLT2i (HR [95% CI]: 1.32 [0.57–3.06]) compared with those who were not (HR [95% CI]: 1.18 [1.00–1.39]); however, incidences were small (22 overall across randomized treatment arms) and the differences were not statistically significant (p = 0.79) [39]. The proportion of patients titrated to the vericiguat 10-mg dose in those receiving an SGLT2i versus those not receiving an SGLT2i at baseline was similar (74% and 79%, respectively) [39].
3.5.4 Summary of Vericiguat Pharmacodynamic Drug–Drug Interaction Profile
In summary, vericiguat has low potential as a perpetrator to affect exposure and/or pharmacodynamic effects of drugs commonly prescribed in patients with HFrEF, including digoxin, sacubitril/valsartan, aspirin, and warfarin. No dose adjustment of these drugs is required in patients taking vericiguat [7, 8].
4 Cardiac Electrophysiological Profile of Vericiguat
An integrated program assessed the QT/corrected QT interval (QT/QTc) prolongation and proarrhythmic potential of vericiguat, using in vitro, animal, and patient studies [48, 49]. This was complemented with concentration-QTc modeling (Table 2) [50].
Preclinically, vericiguat’s potential for proarrhythmic risk was investigated in mechanistic ion-channel studies in environments simulating normal and diseased physiological conditions [49]. The ion channels investigated were human ether-à-go-go-related gene (hERG), human sodium channel/current isoform 1.5 (hNav1.5), human voltage-gated (L-type) calcium channel/current isoform 1.2 (hCav1.2), co-expression of the human voltage potassium current LQT1 and its accessory subunit, minK (hKvLQT1/mink), and human voltage-gated potassium channel/current isoform 4.3 (hKv4.3) in transfected cell lines [49]. Vericiguat was not associated with inhibition of cardiac ion channels (hERG, hNav1.5, hCav1.2, hKvLQT1/mink, and hKv4.3) at substantial exposure multiples [49]. In animal studies using conscious, telemetered beagle dogs (n = 4 per dose group, vericiguat administered as single oral doses of 0 [vehicle], 0.6, 2.0, and 6.0 mg/kg body weight following a Latin square study design), QTc intervals were not meaningfully prolonged after vericiguat was administered (QTcF change from baseline with the highest administered dose of 1.4% [± 3.6]) [49].
Electrocardiograms (ECGs) were collected by investigators in studies of vericiguat administered to healthy volunteers [27]. No clinically relevant electrophysiological abnormalities were reported in the single- and multiple-dose escalation studies in healthy volunteers receiving vericiguat up to 15 mg once daily (EudraCT: 2011-001627-21 and EudraCT: 2012-000953-30) [27].
Standard 12-lead ECGs in two Phase II studies of vericiguat in patients with HF (SOCRATES-REDUCED [NCT01951625] and SOCRATES-PRESERVED [NCT01951638]) were recorded [51,52,53,54]. The centrally assessed ECG parameters included QT and QTc and were recorded at screening and visits 1–5 [30, 51,52,53,54]. In drug exposure–QTc analyses from both SOCRATES trials, the upper limit of the 90% CI for the change from baseline in QTc in the highest quartile of vericiguat exposure was below 10 msec [30]. In addition, vericiguat trough concentration was negatively correlated with the QT interval frequency-corrected according to Fridericia’s formula (QTcF) [30]. All results were consistent between the two Phase II trials for QT interval frequency-corrected according to Bazett’s formula (QTcB) and QTcF [30].
A dedicated, randomized, placebo-controlled, double-blind, Phase Ib study in patients with chronic coronary syndromes investigated the potential for vericiguat (up-titrated to 10 mg) to prolong QTc interval, using moxifloxacin as a positive control (NCT03504982) [55]. Triplicate ECGs, HR, BP, and samples for vericiguat pharmacokinetics were collected in a synchronized fashion throughout the study. Different evaluation methods including QTcF, QTcB, and the individual method to correct for HR including every time point following administration of vericiguat were tested. All evaluations yielded similar results. Placebo-adjusted changes from baseline in QTcF were < 6 ms with an upper bound of the 90% CI below 10 ms across all investigated time points (up to 7 h post-dose) [55]. Moxifloxacin confirmed the assay sensitivity [55]. Concentration–QTc modeling approaches complemented these results following the principles for model development and evaluation as outlined in the scientific white paper by Garnett et al [56] to estimate individual time-matched, baseline- and placebo-corrected QTcF (ΔΔQTcF) [57]. The modeling results demonstrated a very small positive (not clinically relevant) relationship between ΔΔQTcF (slope estimate of 7.05 ms×L/mg; 95% CI 1.41–12.70 ms×L/mg with the upper bound of the two-sided 90% CI for model-derived QTc < 10 ms), confirming that within the plasma concentration range associated with the recommended target dose of vericiguat 10 mg once daily, there is no clinically relevant QTc effect [50].
In VICTORIA, investigator-read ECGs assessed the impact of vericiguat on QTcF interval in patients with HFrEF [10, 30]. At Week 16, there was no difference in the mean change from baseline in QTcF between treatment groups [30]. In addition, no concerning imbalances in outcomes related to QTc prolongation, such as sudden cardiac death, were identified [58].
Overall, data from comprehensive in vitro and preclinical assessments, as well as Phase I, II, and III clinical trials in healthy volunteers, and patients with chronic coronary syndromes and HFrEF, did not reveal any unwanted effects attributable to vericiguat on cardiac repolarization.
5 Pharmacokinetic/Pharmacodynamic Relationships
The application of model-based strategies to accelerate the drug-development process was one of the objectives of vericiguat’s clinical development. Data from studies of two sGC stimulators, riociguat and nelociguat, in patients with pulmonary hypertension and left ventricular dysfunction or biventricular chronic HF were available, prior to initiation of the Phase II clinical studies with vericiguat. An integrated model incorporating pharmacokinetics and hemodynamics data from riociguat, nelociguat, and vericiguat was developed to justify the appropriate dose range to be tested in the vericiguat dose-finding study SOCRATES-REDUCED in patients with HFrEF (Table 2) [59]. Pharmacokinetics/pharmacodynamics modeling indicated that vericiguat 2.5–10 mg would encompass an efficacious exposure range and that the 1.25-mg dose would be a “non-effective” dose level with respect to hemodynamic parameters [59]. These model-based approaches facilitated the accelerated development of vericiguat by avoiding the need for a proof-of-concept study in patients with HFrEF, thereby enabling immediate progression from Phase I to Phase IIb.
The direct correlations between vericiguat plasma concentrations and various pharmacodynamic measures observed in healthy volunteers across the Phase I program were in line with the expectations based on the mode of action. In particular, vericiguat plasma concentration was correlated with a positive change from baseline in cGMP, renin, adrenaline, and noradrenaline plasma levels [27]. In addition, increases in vericiguat plasma concentration were associated with increases in cardiac output and cardiac index, as well as decreases in systemic vascular resistance, without a clear effect on HR [27, 30]. No overall effect of vericiguat on stroke volume was detected [30].
Based on the results and the experience with other compounds that have a similar mode of action (e.g., riociguat), vericiguat was dosed according to a BP-guided titration regimen in Phase II and Phase III studies enrolling patients with HFrEF.
The vericiguat exposure–response relationship for SBP and NT-proBNP concentration (with NT-proBNP being an indicator of disease severity) in patients with HFrEF was investigated with population pharmacokinetic/pharmacodynamic modeling of data from the Phase II SOCRATES-REDUCED trial (Table 2) [60]. The analysis revealed a significant pharmacokinetic/pharmacodynamic relationship between vericiguat exposure (Cmax) and decreases in SBP on Day 1, but no exposure–response relationships were observed at visit 4 (steady state) [60]. The absence of a relationship between vericiguat exposure at steady state and changes in SBP indicates an adaptation to the effect of vericiguat. This was confirmed by analysis of data from VICTORIA, which showed that SBP reduced slightly over the first 16 weeks (more so in the vericiguat group) and subsequently returned to baseline [10]. The mean reduction in SBP across the course of the study was approximately 1–2 mmHg greater in patients receiving vericiguat than those receiving placebo [7, 8].
Exploratory pharmacokinetic/pharmacodynamic analysis of NT-proBNP, a cardiac biomarker in HFrEF, demonstrated an exposure-dependent reduction of NT-proBNP with vericiguat [60]. This finding is in line with the observed dose-dependent decreases in SOCRATES-REDUCED (p < 0.02), with higher vericiguat doses associated with greater reductions from baseline in NT-proBNP [52]. Exposure-dependent reduction of NT-proBNP with vericiguat was influenced by baseline NT-proBNP [60]. Specifically, a lower exposure-dependent NT-proBNP reduction in patients with HFrEF with higher baseline NT-proBNP was observed [60]. A study utilizing a quantitative model based on real-world data, validated by a model-based meta-analysis of interventional and observational studies, demonstrated that short-term response of NT-proBNP to treatment in patients with HF is predictive of mid- to long-term clinical event rates in clinical trials [61]. Identifying NT-proBNP as a biomarker of disease progression, together with data demonstrating a reduction in NT-proBNP with vericiguat, provided the basis for proceeding with clinical development of vericiguat into Phase III (VICTORIA).
In the VICTORIA trial, vericiguat was associated with a significant and sustained decline in NT-proBNP; this change was associated with an improvement in the primary efficacy outcome [62]. There was an attenuation of benefit on the primary efficacy endpoint observed in patients in the highest NT-proBNP quartile (> 5314 pg/mL) compared with patients in the lower three quartiles [10, 63]. Vericiguat AUCs were similar across the quartiles of NT-proBNP in the integrated population pharmacokinetics analysis, indicating that the observed heterogeneity of treatment effect is not likely to be attributable to differences in drug plasma concentrations in this study population titrated to the maximal dose of vericiguat (10 mg) [23]. Event rates of clinical outcomes were markedly greater in patients with baseline NT-proBNP > 5314 pg/mL compared with those with NT-proBNP ≤ 5314 pg/mL, irrespective of treatment allocation [63]. The high event rate in patients with elevated NT-proBNP is indicative of more severe disease that may be less responsive to pharmacological HF therapies [63].
Data from the VICTORIA trial were used to quantify relationships between vericiguat exposure, hemodynamic parameters (SBP) and prespecified safety events of interest (symptomatic hypotension and syncope). On Day 1, a small but statistically significant correlation between higher vericiguat exposures (Cmax) and decrease from baseline in SBP at 2 h post-dose was noted [28]. However, no such correlation was observed during the remaining days of the titration phase through the duration of the study [28]. Furthermore, no meaningful exposure–response relationships for the incidence of symptomatic hypotension or syncope were evident. These analyses provide further support that vericiguat is well tolerated in patients with HFrEF [30].
6 Discussion and Conclusions
Vericiguat’s pharmacokinetic and drug–drug interaction profile was thoroughly characterized in Phase I studies. Data from patients with HFrEF are consistent with vericiguat’s characterized pharmacokinetic and biopharmaceutical profile in healthy volunteers. Modeling and simulation studies were conducted throughout the vericiguat clinical development program to support investigation of pharmacokinetics, characterize pharmacokinetic-pharmacodynamic relationships, and drug–drug interaction and QT prolongation potential, thereby informing posology and labeling information.
As the UGT isoforms that catalyze the metabolism of vericiguat are primarily expressed in the kidneys and liver, dedicated Phase I studies were performed in patients with renal and hepatic impairment of various degrees [64, 65]. These studies, supported by later analyses in patients with HFrEF, indicate that no dose changes are necessary in people with renal impairment (eGFR ≥ 15 mL/min/1.73 m2) or mild-to-moderate hepatic impairment. The tolerability of vericiguat was further demonstrated by dedicated analyses in vulnerable subgroups enrolled in the VICTORIA trial (patients aged > 75 years, and those with baseline SBP 100–110 mmHg) [39]. Vericiguat was well tolerated in elderly patients (Sect. 3.3). Patients with a baseline SBP < 110 mmHg exhibited a pattern of increasing SBP in both treatment arms [39]. In subgroups defined by SBP (< 110 mmHg and ≥ 110 mmHg), symptomatic hypotension and syncope event rates were similar by randomized treatment (HR [95% CI]: 1.32 [0.99–1.75] for < 110 mmHg and 1.11 [0.90–1.37] for ≥ 110 mmHg, all p > 0.05) [39]. The proportions of patients with SBP < 110 mmHg achieving the 10-mg target dose in the vericiguat and placebo treatment arms were 60% and 62%, respectively [39]. The results indicate that vericiguat can be initiated and up-titrated in patients with a low SBP, in accordance with the BP and well–being-guided dosing titration described in labels [7,8,9].
Vericiguat was not associated with electrophysiological abnormalities in the in vitro, animal, and Phase I studies. No differences in change from baseline in QTcF between treatment groups were evident in the Phase III VICTORIA study and no imbalances in adverse QTc prolongation-related outcomes. Results from these assessments demonstrate a lack of unwanted effect of vericiguat on cardiac repolarization at the recommended target dose of vericiguat 10 mg once daily.
In vitro studies predicted a low pharmacokinetics drug–drug interaction profile for vericiguat, confirmed by dedicated Phase I healthy volunteer studies that observed small variations in vericiguat exposure which were not clinically meaningful. Consequently, no dose adjustments were prespecified in the Phase III VICTORIA study beyond the BP- and wellbeing-guided dosing titration. Treatment with vericiguat was well tolerated in the VICTORIA trial, despite the use of a wide variety of concomitant medications in this vulnerable patient population. The results indicate that vericiguat is suitable for the treatment of a population with multimorbidity and polypharmacy.
Vericiguat has low potential as a perpetrator to affect the pharmacodynamic effects of drugs commonly prescribed in patients with HFrEF. In addition, pharmacodynamic drug–drug interactions were assessed with drugs acting on the NO–sGC–cGMP pathway, including a PDE5 inhibitor (sildenafil). Administration of PDE5 inhibitors for on-demand treatment of erectile dysfunction has been reported in patients with HFrEF. Therefore, a drug–drug interaction study between vericiguat and sildenafil in healthy volunteers was conducted. The aim of the study was to investigate the combined BP effects of vericiguat with the approved doses of sildenafil (25 mg, 50 mg, and 100 mg) for erectile dysfunction. While co-administration of vericiguat with sildenafil was well tolerated in healthy volunteers, the concomitant use of vericiguat and PDE5 inhibitors, such as sildenafil, has not been studied in patients with HFrEF. The co-administration of sildenafil and vericiguat is currently not recommended due to the lack of clinical experience in patients with HF and the potential increased risk for symptomatic hypotension.
The treatment effect of vericiguat on the primary efficacy outcome in VICTORIA was generally consistent across subgroups; however, baseline NT-proBNP was a notable exception. Currently, vericiguat is being investigated in two ongoing studies. In the Phase III VICTOR trial, the efficacy and safety of vericiguat in patients with HFrEF without recent decompensation is being investigated to complement the results of the VICTORIA trial [44]. The aim of the Phase II/III VALOR trial (NCT05714085) is to investigate the efficacy, safety, and pharmacokinetics of vericiguat in pediatric participants with HF due to systemic left ventricular systolic dysfunction using change from baseline in NT-proBNP as the prespecified primary outcome. Dose equivalents for pediatric patients with HF were derived by physiologically based pharmacokinetic modeling to match the target exposure (AUC) observed in adult patients with HFrEF [66].
Vericiguat exposure upon titration based on BP and tolerability at steady state was not correlated with symptomatic hypotension, syncope, or other undesirable hemodynamic effects in the VICTORIA trial. Overall, vericiguat is well tolerated and efficacious in patients with HFrEF.
In summary, the oral sGC stimulator vericiguat has a well-characterized biopharmaceutical, pharmacokinetic, and pharmacodynamic profile that supports once-daily dosing. In addition, vericiguat has a limited drug–drug interaction profile and is well tolerated in patients with HF. Vericiguat is a recommended treatment for worsening HFrEF based on the results from the VICTORIA trial. Vericiguat’s safety and efficacy profile in patients with HFrEF will be further characterized by the VICTOR trial in adults without recent decompensation [44] and in a pediatric population with HF due to left ventricular systolic dysfunction (VALOR trial) [66].
References
GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392(10159):1789–858. https://doi.org/10.1016/S0140-6736(18)32279-7.
Savarese G, Lund LH. Global public health burden of heart failure. Card Fail Rev. 2017;3(1):7–11. https://doi.org/10.15420/cfr.2016:25:2.
Tsao CW, Aday AW, Almarzooq ZI, et al. Heart disease and stroke statistics-2023 update: a report from the American Heart Association. Circulation. 2023;147(8):e93–621. https://doi.org/10.1161/cir.0000000000001123.
McDonagh TA, Metra M, Adamo M, et al. 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2021;42(36):3599–726. https://doi.org/10.1093/eurheartj/ehab368.
Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145(18):e895–1032. https://doi.org/10.1161/cir.0000000000001063.
Butler J, Anstrom K, Armstrong P. Comparing the benefit of novel therapies across clinical trials. Circulation. 2020;142(8):717–9. https://doi.org/10.1161/CIRCULATIONAHA.120.047086.
Food and Drug Administration. VerquvoTM (vericiguat) tablets. Highlights of prescribing information. 2021. www.accessdata.fda.gov/drugsatfda_docs/label/2021/214377s000lbl.pdf. Accessed 17 Oct 2023.
European Medicines Agency. Verquvo summary of product characteristics. 2021. www.ema.europa.eu/en/documents/product-information/verquvo-epar-product-information_en.pdf. Accessed 31 Jul 2023.
Bayer plc. Verquvo 10 mg film-coated tablets summary of product characteristics. 2021. www.medicines.org.uk/emc/product/12775/smpc#gref. Accessed 27 Aug 2021.
Armstrong PW, Pieske B, Anstrom KJ, et al. Vericiguat in patients with heart failure and reduced ejection fraction. N Engl J Med. 2020;382(20):1883–93. https://doi.org/10.1056/NEJMoa1915928.
Senni M, Alemayehu WG, Sim D, et al. Efficacy and safety of vericiguat in patients with heart failure with reduced ejection fraction treated with sacubitril/valsartan: insights from the VICTORIA trial. Eur J Heart Fail. 2022;24(9):1614–22. https://doi.org/10.1002/ejhf.2608.
Mastromarino V, Casenghi M, Testa M, et al. Polypharmacy in heart failure patients. Curr Heart Fail Rep. 2014;11(2):212–9. https://doi.org/10.1007/s11897-014-0186-8.
Follmann M, Ackerstaff J, Redlich G, et al. Discovery of the soluble guanylate cyclase stimulator vericiguat (BAY 1021189) for the treatment of chronic heart failure. J Med Chem. 2017;60(12):5146–61. https://doi.org/10.1021/acs.jmedchem.7b00449.
Becker C, Boettcher M, Muenster U, Loewen S, Lobmeyer M, Mueck W. Results from in vitro and in vivo studies evaluating the bioavailability, effects of food, and administration as crushed tablet suspension on vericiguat pharmacokinetics. AAPS Open. 2022;8(1):16. https://doi.org/10.1186/s41120-022-00063-4.
Boettcher M, Gerisch M, Lobmeyer M, et al. Metabolism and pharmacokinetic drug-drug interaction profile of vericiguat, a soluble guanylate cyclase stimulator: results from preclinical and phase I healthy volunteer studies. Clin Pharmacokinet. 2020;59(11):1407–18. https://doi.org/10.1007/s40262-020-00895-x.
Sandner P, Follmann M, Becker-Pelster E, et al. Soluble GC stimulators and activators: past, present and future. Br J Pharmacol. 2021. https://doi.org/10.1111/bph.15698.
Michalak M, Armstrong PW. Exploring new cardiovascular pathways: are soluble guanylate cyclase stimulators the right direction? Circ Heart Fail. 2018;11(3):e004813. https://doi.org/10.1161/circheartfailure.118.004813.
Follmann M, Becker C, Rossig L, Sandner P, Stasch JP. Discovery and development of the soluble guanylate cyclase stimulator vericiguat for the treatment of chronic heart failure. Contempor Accounts Drug Discov Dev. 2021;27:50.
Becker EM, Alonso-Alija C, Apeler H, et al. NO-independent regulatory site of direct sGC stimulators like YC-1 and BAY 41–2272. BMC Pharmacol. 2001;1:13. https://doi.org/10.1186/1471-2210-1-13.
Hoenicka M, Becker EM, Apeler H, et al. Purified soluble guanylyl cyclase expressed in a baculovirus/Sf9 system: stimulation by YC-1, nitric oxide, and carbon monoxide. J Mol Med (Berl). 1999;77(1):14–23. https://doi.org/10.1007/s001090050292.
Breitenstein S, Roessig L, Sandner P, Lewis KS. Novel sGC stimulators and sGC activators for the treatment of heart failure. Handb Exp Pharmacol. 2017;243:225–47. https://doi.org/10.1007/164_2016_100.
Armstrong PW, Roessig L, Patel MJ, et al. A multicenter, randomized, double-blind, placebo-controlled trial of the efficacy and safety of the oral soluble guanylate cyclase stimulator: the VICTORIA trial. JACC Heart Fail. 2018;6(2):96–104. https://doi.org/10.1016/j.jchf.2017.08.013.
Trujillo ME, Arrington L, Patel Y, et al. Population pharmacokinetics of vericiguat in patients with heart failure with reduced ejection fraction: an integrated analysis. Clin Pharmacol Ther. 2022;112(5):1061–9. https://doi.org/10.1002/cpt.2712.
Frechen S, Ince I, Dallmann A, et al. Applied physiologically-based pharmacokinetic modeling to assess uridine diphosphate-glucuronosyltransferase-mediated drug-drug interactions for vericiguat. CPT Pharmacometrics Syst Pharmacol. 2023. https://doi.org/10.1002/psp4.13059.
Janssen W, Schwarz T, Bütehorn U, et al. Pharmacokinetics and mass balance of vericiguat in rats and dogs and distribution in rats. Xenobiotica. 2022;52(5):453–62. https://doi.org/10.1080/00498254.2022.2082899.
Boettcher M, Loewen S, Gerrits M, Becker C. Pharmacodynamic and pharmacokinetic interaction profile of vericiguat: results from three randomized phase I studies in healthy volunteers. Clin Pharmacokinet. 2021;60(3):337–51. https://doi.org/10.1007/s40262-020-00935-6.
Boettcher M, Thomas D, Mueck W, et al. Safety, pharmacodynamic, and pharmacokinetic characterization of vericiguat: results from six phase I studies in healthy subjects. Eur J Clin Pharmacol. 2021;77:527–37. https://doi.org/10.1007/s00228-020-03023-7.
Food and Drug Administration. Vericiguat (VERQUVO) integrated review—Center for Drug Evaluation and Research. 2020. www.accessdata.fda.gov/drugsatfda_docs/nda/2021/214377Orig1s000IntegratedR.pdf. Accessed 12 Apr 2021.
Bayer Clinical Trials Explorer. Multiple dose escalation phase I study of BAY1021189. https://clinicaltrials.bayer.com/study/15357. Accessed 20 Apr 2023.
European Medicines Agency. Verquvo: EPAR—public assessment report. 2021. www.ema.europa.eu/en/documents/assessment-report/verquvo-epar-public-assessment-report_en.pdf. Accessed 16 Apr 2023.
Bayer Clinical Trials Explorer. Vericiguat absolute bioavailability using microdosing technology. 2023. https://clinicaltrials.bayer.com/study/17114. Accessed 20 Apr 2023.
Bayer Clinical Trials Explorer. Single dose escalation study of BAY1021189. 2023. https://clinicaltrials.bayer.com/study/15355. Accessed 20 Apr 2023.
Bayer Clinical Trials Explorer. Influence of age and gender on the pharmacokinetics of a single oral dose of 5 mg BAY 1021189 as immediate-release tablet in healthy male and female subjects. 2023 https://clinicaltrials.bayer.com/study/15816. Accessed 20 Apr 2023.
ClinicalTrials.gov. A trial to learn how safe vericiguat (BAY1021189) is and the way the body absorbs, distributes and gets rid of vericiguat in participants with kidney disease and in age-, weight- and gender-matched healthy participants. 2021. https://clinicaltrials.gov/ct2/show/NCT04722484. Accessed 16 Apr 2023.
Bayer Clinical Trials Explorer. A trial to learn how safe vericiguat (BAY1021189) is and the way the body absorbs, distributes and gets rid of vericiguat in participants with kidney disease and in age-, weight- and gender-matched healthy participants. 2021. https://clinicaltrials.bayer.com/study/15813/. Accessed 8 May 2023.
Boettcher MA, Lobmeyer M, Meyer M, Mueck W, Trujillo M, Becker C. Vericiguat clinical pharmacology programme: biopharmaceutical properties and potential intrinsic and extrinsic factor effects. Eur Heart J. 2020;41:3328. https://doi.org/10.1093/ehjci/ehaa946.3328.
Bayer Clinical Trials Explorer. A trial to learn how safe vericiguat (BAY1021189) is and the way the body absorbs, distributes and gets rid of vericiguat in participants with liver disease and in age-, weight- and gender-matched healthy participants. 2021. https://clinicaltrials.bayer.com/study/15840. Accessed 8 May 2023.
Department of Health and Aged Care. Australian public assessment report for verquvo. 2022. https://www.tga.gov.au/sites/default/files/2022-07/auspar-verquvo-220707.pdf. Accessed 16 Apr 2023.
Lam CSP, Mulder H, Lopatin Y, et al. Blood pressure and safety events with vericiguat in the VICTORIA trial. J Am Heart Assoc. 2021;10(22):e021094. https://doi.org/10.1161/jaha.121.021094.
University of Washington. NDA 214377—vericiguat—UW drug interaction solutions. 2023. https://didb.druginteractionsolutions.org/citation/214377/. Accessed 8 May 2023.
Boettcher M, Düngen HD, Donath F, et al. Vericiguat in combination with short-acting nitroglycerin in patients with chronic coronary syndromes: the randomized, phase Ib, VENICE study. Clin Pharmacol Ther. 2022;111(6):1239–47. https://doi.org/10.1002/cpt.2574.
Bayer Clinical Trials Explorer. Evaluation of safety, tolerability and the pharmacodynamic effect after treatment of vericiguat and nitroglycerin. 2021. https://clinicaltrials.bayer.com/study/17115. Accessed 8 May 2023.
Boettcher M, Mikus G, Trenk D, et al. Vericiguat in combination with isosorbide mononitrate in patients with chronic coronary syndromes: The randomized, phase Ib, VISOR study. Clin Transl Sci. 2022;15(5):1204–14. https://doi.org/10.1111/cts.13238.
ClinicalTrials.gov. A study of vericiguat (MK-1242) in participants with chronic heart failure with reduced ejection fraction (HFrEF) (MK-1242-035) (VICTOR). 2023. https://clinicaltrials.gov/ct2/show/NCT05093933. Accessed 18 April 2023.
Boettcher M, Nowotny B, Krausche R, Becker C. Evaluation of the influence of sildenafil on the safety, tolerability, pharmacokinetics, and pharmacodynamics of vericiguat in healthy adults. Clin Pharmacokinet. 2023;62(2):321–33. https://doi.org/10.1007/s40262-022-01203-5.
Boettcher M, Becker C. Authors’ reply to Ganijee et al.: Comment on: evaluation of the influence of sildenafil on safety, tolerability, pharmacokinetics, and pharmacodynamics of vericiguat in healthy adults". Clin Pharmacokinet. 2023;62(11):1651–3. https://doi.org/10.1007/s40262-023-01304-9.
Zhang G, Xiang B, Dong A, et al. Biphasic roles for soluble guanylyl cyclase (sGC) in platelet activation. Blood. 2011;118(13):3670–9. https://doi.org/10.1182/blood-2011-03-341107.
Boettcher M-F, Duengen H-D, Corcea V, et al. Vericiguat: a QTc interval study in patients with coronary artery disease. ESC. 2021;2021:2021.
Himmel H, Lagrutta A, Vömel M, et al. Nonclinical cardiovascular assessment of the soluble guanylate cyclase stimulator vericiguat. J Pharmacol Exp Ther. 2023. https://doi.org/10.1124/jpet.122.001368.
Ruehs H, Solms A, Frei M, et al. Assessing QTc effects of vericiguat using two different concentration-QTc modeling approaches. Clin Pharmacokinet. 2023;62(11):1639–48. https://doi.org/10.1007/s40262-023-01282-y.
Bayer Clinical Trials Explorer. Phase IIb safety and efficacy study of four dose regimens of BAY1021189 in patients with heart failure and preserved ejection fraction suffering from worsening chronic heart failure (SOCRATES-PRESERVED). 2021. https://clinicaltrials.bayer.com/study/15829. Accessed 8 May 2023.
Gheorghiade M, Greene SJ, Butler J, et al. Effect of vericiguat, a soluble guanylate cyclase stimulator, on natriuretic peptide levels in patients with worsening chronic heart failure and reduced ejection fraction: The SOCRATES-REDUCED randomized trial. JAMA. 2015;314(21):2251–62. https://doi.org/10.1001/jama.2015.15734.
Pieske B, Butler J, Filippatos G, et al. Rationale and design of the SOluble guanylate Cyclase stimulatoR in heArT failurE Studies (SOCRATES). Eur J Heart Fail. 2014;16:1026–38. https://doi.org/10.1002/ejhf.135.
Pieske B, Maggioni AP, Lam CSP, et al. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: results of the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED EF (SOCRATES-PRESERVED) study. Eur Heart J. 2017;38(15):1119–27. https://doi.org/10.1093/eurheartj/ehw593.
Böttcher M, Dungen HD, Corcea V, et al. Vericiguat: A randomized, phase Ib, placebo-controlled, double-blind, QTc interval study in patients with chronic coronary syndromes. Am J Cardiovasc Drugs. 2023;23(2):145–55. https://doi.org/10.1007/s40256-022-00557-2.
Garnett C, Bonate PL, Dang Q, et al. Scientific white paper on concentration-QTc modeling. J Pharmacokinet Pharmacodyn. 2018;45(3):383–97. https://doi.org/10.1007/s10928-017-9558-5.
Meyer M, Ruehs H, Solms A, et al. A concentration-QTc analysis of vericiguat. Eur Heart J. 2021;42(1):ehab724.0910. https://doi.org/10.1093/eurheartj/ehab724.0910.
ClinicalTrials.gov. A study of vericiguat in participants with heart failure with reduced ejection fraction (HFrEF) (MK-1242-001) (VICTORIA). 2021. https://clinicaltrials.gov/ct2/show/results/NCT02861534. Accessed 17 Apr 2023.
Meyer M, Schneckener S, Loosen R, et al. Leveraging translational approaches for accelerated clinical development of vericiguat. Eur Heart J. 2021;42(1):ehab724.0919. https://doi.org/10.1093/eurheartj/ehab724.0919.
Ruehs H, Klein D, Frei M, et al. Population pharmacokinetics and pharmacodynamics of vericiguat in patients with heart failure and reduced ejection fraction. Clin Pharmacokinet. 2021;60(11):1407–21. https://doi.org/10.1007/s40262-021-01024-y.
Schmitt W, Rühs H, Burghaus R, et al. NT-proBNP qualifies as a surrogate for clinical endpoints in heart failure. Clin Pharmacol Ther. 2021;110(2):498–507. https://doi.org/10.1002/cpt.2222.
Armstrong PW, Zheng Y, Troughton RW, et al. Sequential evaluation of NT-proBNP in heart failure: insights into clinical outcomes and efficacy of vericiguat. JACC Heart Fail. 2022;10(9):677–88. https://doi.org/10.1016/j.jchf.2022.04.015.
Senni M, Lopez-Sendon J, Cohen-Solal A, et al. Vericiguat and NT-proBNP in patients with heart failure with reduced ejection fraction: analyses from the VICTORIA trial. ESC Heart Fail. 2022. https://doi.org/10.1002/ehf2.14050.
Bhatt DK, Mehrotra A, Gaedigk A, et al. Age- and genotype-dependent variability in the protein abundance and activity of six major uridine diphosphate-glucuronosyltransferases in human liver. Clin Pharmacol Ther. 2019;105(1):131–41. https://doi.org/10.1002/cpt.1109.
Margaillan G, Rouleau M, Fallon JK, et al. Quantitative profiling of human renal UDP-glucuronosyltransferases and glucuronidation activity: a comparison of normal and tumoral kidney tissues. Drug Metab Dispos. 2015;43(4):611–9. https://doi.org/10.1124/dmd.114.062877.
ClinicalTrials.gov. Efficacy, safety, and pharmacokinetics of vericiguat in pediatric participants with heart failure due to left ventricular systolic dysfunction (MK-1242-036). 2023. https://clinicaltrials.gov/ct2/show/NCT05714085. Accessed 18 Apr 2023.
Acknowledgements
The authors would like to thank the participants, their families, and all investigators involved in the studies. Editorial support, including fact checking, referencing, figure preparation, formatting, proofreading, and submission was provided by Moamen Hammad, PhD, and Melissa Ward, BA, both of Scion (a division of Prime, London, UK), supported by Bayer AG, Wuppertal, Germany and Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA according to Good Publication Practice guidelines (https://www.acpjournals.org/doi/10.7326/M22-1460).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was funded by Bayer AG, Wuppertal, Germany and Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
Conflict of interest
AF, MM, LR, and CB are employees of Bayer AG and may own stock and/or stock options in Bayer AG. MB was an employee of Bayer AG at the time of the studies. ROB, MET, and EK are employees of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA, and may own stock and/or stock options in Merck & Co., Inc., Rahway, NJ, USA.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Animal experiments described in paragraph one of Sect. 2.2 were conducted in accordance with German animal welfare laws, approved by local authorities, and performed in accordance with the ethical guidelines of Bayer AG. Experiments investigating the clinical pharmacokinetic profile of vericiguat in rodents and canines (paragraph three of Sect. 2.2) were performed in accordance with the respective local legal Animal Protection Law and effective government requirements (approval no. 820/A51). In vivo assessments of QT/QTc (paragraph one of Sect. 4) were conducted with local approval and conformed to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes, or the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Consent to participate
Informed consent was obtained from all individual participants included in the studies.
Consent for publication
Not applicable.
Code availability
Not applicable.
Availability of data and material
Availability of the data underlying this publication will be determined according to Bayer’s commitment to the EFPIA/PhRMA “Principles for responsible clinical trial data sharing”. This pertains to scope, timepoint and process of data access. As such, Bayer commits to sharing upon request from qualified scientific and medical researchers patient-level clinical trial data, study-level clinical trial data, and protocols from clinical trials in patients for medicines and indications approved in the United States (US) and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after January 01, 2014. Interested researchers can use www.vivli.org to request access to anonymized patient-level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information is provided in the member section of the portal. Data access will be granted to anonymized patient-level data, protocols and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that patient privacy is safeguarded.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Fritsch, A., Meyer, M., Blaustein, R.O. et al. Clinical Pharmacokinetic and Pharmacodynamic Profile of Vericiguat. Clin Pharmacokinet (2024). https://doi.org/10.1007/s40262-024-01384-1
Accepted:
Published:
DOI: https://doi.org/10.1007/s40262-024-01384-1