
Abstract
Introduction
Bromfenac is a well-known topical ophthalmic nonsteroidal anti-inflammatory drug (NSAID) that is commercialized in the USA and other regions of the world. A new formulation, 0.075% bromfenac in DuraSite®, was developed to treat postoperative inflammation and reduce pain in patients who have undergone cataract surgery. We hypothesized that efficacy and safety would be enhanced with twice-daily (BID) dosing compared to once-daily (QD) dosing.
Methods
This was a multicenter, double-masked, comparative study in which 40 and 45 subjects were randomized to groups receiving BID dosing and QD dosing, respectively. Subjects self-instilled the study drug for 14 days postoperative and were followed for an additional 2-week evaluation phase. The primary efficacy endpoint was the proportion of subjects with an anterior chamber cell (ACC) grade of 0 at day 15.
Results
A total of 45 subjects had cleared ACC (grade “0”) at day 15, of whom 21 were in the BID group (52.5%) and 24 were in the QD group (53.5%). A secondary analysis found 7/40 (17.5%) subjects in the BID group and 10/45 (22.2%) subjects in the QD group achieved an ACC grade of 0 at day 8. There were more adverse events in the QD group (n = 16) than in the BID group (n = 12).
Conclusion
Similar outcomes were observed for subjects using Bromfenac 0.075% in DuraSite® in the BID and QD dosing regimens for the treatment of post-cataract surgery inflammation.
Trial registration
ClinicalTrials.gov identifier, NCT01190878.
Funding
InSite Vision (now a division of Sun Pharma).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are a well-accepted treatment to reduce postoperative pain after ocular surgery and to control inflammation [1,2,3,4,5]. There are numerous NSAIDs approved for ophthalmic use in the USA, including bromfenac. Bromfenac is a potent cyclooxygenase inhibitor with a long history of use in various strengths for ophthalmic indications dating back to 2006 [1, 2, 6,7,8,9]. DuraSite® (InSite Vision, Alameda, CA) is a synthetic polymer-based formulation designed to improve solubility, absorption, bioavailability and residence time. Both clinical and nonclinical studies have shown the DuraSite® drug delivery system to be safe and non-toxic [10]. DuraSite® is commercially available in the USA in two antibiotic formulations (one with 1% azithromycin and the other with 0.6% besifloxacin), and DuraSite® technology has also been used in a formulation of loteprednol gel.
In 2016, the Food and Drug Administration approved bromfenac 0.075% administered twice daily (BID) for the treatment of postoperative inflammation and prevention of ocular pain in patients undergoing cataract surgery. The on-label indication recommends that BID dosing begin 1 day before surgery and continue on the day of surgery and for 14 days postsurgery [11].
The purpose of this analysis was to compare two dosing regimens of bromfenac 0.075%: once-daily (QD) and BID in post-cataract surgery patients to assess safety, tolerability and efficacy.
Methods
Study Design
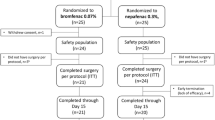
This was a multicenter, randomized, double-masked, parallel-group, comparative subgroup analysis of a larger overall study. Subjects were randomly assigned in a 1:1 ratio to receive 0.075% bromfenac in DuraSite® BID or 0.075% bromfenac in DuraSite® QD according to a validated computer-generated central randomization schedule. There were a total of 85 subjects enrolled: 40 subjects received 0.075% bromfenac BID ("BID group"), 45 subjects received 0.075% bromfenac QD ("QD group"). Subjects were enrolled post-cataract surgery into a 14-day dosing phase, followed by a 2-week evaluation phase. Four visits were required for the study, with two visits taking place during the dosing phase (days 1 and 8) and two during the evaluation phase (days 15 and Day 29). In addition, there was a telephone call on day 3 to obtain visual analog scale (VAS) values for pain/discomfort and photophobia. The subject self-instilled the study medication, not the investigator or his/her study staff.
Key entry criteria included an anterior chamber cell (ACC) grade of ≥2 and anterior chamber flare of ≥2 in the study eye at the baseline examination on the day after surgery (day 1); uneventful phacoemulsification surgery and intraocular lens implantation; avoidance of topical, systemic or inhaled salicylates or NSAIDs within 1 week before cataract surgery, with the exception of oral doses of aspirin at 165 mg/day or lower; avoidance of topical, inhaled or oral corticosteroid within 15 days before cataract surgery and any depot-corticosteroid within 45 days before cataract surgery; no concurrent use of ocular or systemic antihistamines or mast cell stabilizers within 1 week before surgery, a best-corrected visual acuity (BCVA) of at least +1.0 logMAR (Snellen equivalent of 20/200) in the fellow eye (non-study eye) and an intraocular pressure range of >8 and ≤22 mmHg in the study eye.
Key exclusion criteria for the study eye included: a history of severe dry eye, active corneal pathology, Fuchs dystrophy, diabetic retinopathy, previous vitrectomy or epiretinal membrane; any sign of iritis or scleritis; previous glaucoma or refractive surgery in the previous 2 years; chronic or recurrent ocular or systemic disease that may affect wound healing (e.g. diabetes mellitus, systemic connective tissue disease, severe atopic disease); use of any medication that could interfere with normal lacrimation within the week prior to cataract surgery (including, but not limited to, NSAIDs/aspirin, antihistamines or mast cell stabilizers).
Protocol and informed consent forms for this study were reviewed and approved by an Institutional Review Board (IRB) (New England IRB, Needham, MA) and were provided to the contract research organization (ClinOps LLC, San Francisco, CA) before subjects were screened for entry. The study is registered with ClinicalTrials.gov. ID NCT01190878.
Study Drug
The drug 0.075% bromfenac in DuraSite® is preserved with benzalkonium chloride (0.005%). The drug was administered as topical drops in the postoperative eye either QD or BID for 14 days (those in the QD arm were given vehicle drops for the second administration).
DuraSite® is a mucoadhesive material long used to enhance the residence time of a pharmaceutical on the ocular surface, has been evaluated in several other topical ophthalmic formulations, and its efficacy and safety data is well known [12,13,14,15,16].
Subject compliance with instillation frequency was assessed by subject diary.
Primary Efficacy Outcome
The primary efficacy outcome was the proportion of subjects with an ACC grade of 0 at day 15 (see Table 1 for grading). The proportions of subjects with an ACC grade of 0 for the study eye at days 8, 15 and 29, respectively, were summarized using the last observation carried forward (LOCF) method for the intent-to-treat (ITT) population and per protocol (PP) population.
Secondary Outcome: Efficacy
Secondary efficacy endpoints included slit lamp biomicroscopy results at days 8, 15 and 29, respectively, and VAS results (pain or discomfort and photophobia) at days 3, 8, 15 and 29, respectively.
Statistical Analysis
The frequency of subjects with an ACC grade of 0 was compared between the BID and the QD groups at days 8, 15 and 29 using the standard Chi-square test and Fisher’s exact test. To supplement the hypothesis tests, confidence intervals for the difference between the BID and the QD groups at each visit in the proportion of subjects with an ACC grade of 0 were computed using Wald’s (asymptotic) method and the Clopper–Pearson (exact) method.
Mean VAS scores for pain were compared between the BID and the QD groups at days 8, 15 and 29 using an analysis of covariance model with baseline VAS pain score and study site as covariates. Using the same statistical model as the clinical study report, linear contrasts were used to test for equal mean pain scores at each measurement day and to construct confidence intervals (CI) for the difference. A similar analysis was performed to compare mean VAS scores for photophobia between the BID and the QD groups.
Results
The subject disposition for the two study groups is shown in Table 2.
Table 3 shows that there were no major differences between groups at study entry. The majority of subjects were Caucasian in both groups, and the mean age was 71.3 years in the BID group and 70.9 years in the QD group. A similar percentage of subjects were taking at least one medication in addition to the study drug: 39/40 (97.5%) in the BID group and 44/45 (97.8%) in the QD group.
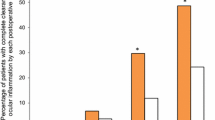
The number of subjects with cleared ACC (grade 0) at day 15 were similar in the two group: 21/40 (52.5%) in the BID group and 24/45 (53.3%) in the QD group (two-sided Fisher’s exact test P = 1.000). The proportion of subjects with an ACC grade of 0 at days 8 and 29 was seven (17.5%) in BID group and ten (22.2%) in the QD group (two-sided Fisher’s exact test P = 0.7866), and 27 (67.5%) in BID group and 27 (60%) in the QD group (two-sided Fisher’s exact test P = 0.5062), respectively. The treatment differences for the proportion of patients with an ACC grade of 0 were computed twice, once with 95% CI (not reported here), and once with 90% CI.
Table 4 shows the VAS results detailing pain or discomfort and photophobia. The difference between the BID and QD groups was not statistically significant.
Safety Evaluation
At least one treatment-emergent adverse event (TEAE) was reported in each group: 11/40 (27.5%) subjects in the BID group and 11/45 (24.4%) subjects in the QD group. Of the 11 subjects with any TEAE, 6/11 (54.5%) in the BID group and 11/11 (100% in the QD group were eye disorders. In the BID group there was one incidence each of the following TEAEs (occurring in 9.1% of subjects): conjunctival cysts, cystoid macular edema (CME), eye inflammation, eye irritation, eye pain, posterior capsule opacification, trichiasis, hernia, cellulitis, foreign body in eye and nephrolithiasis. In the QD group there was one incidence each of the following TEAEs (occurring in 9.1% of subjects): eyelid margin crusting, meibomian gland dysfunction, ocular hypertension, posterior capsule opacification, bronchitis, hordeolum and pulmonary congestion. In the QD group there were two incidences (occurring in 18.2% of subjects) of eye inflammation and eye pruritis. There were four incidences in the QD group (occurring in 36.4%) of iritis. Only one serious TEAE (fecaloma, in the BID group) was reported, but that was deemed not to be related to the study drug.
Only eye inflammation was deemed to be definitely related to the study drug. CME, eye irritation, eyelid margin crusting, eye pruritis and iritis were considered to be possibly related to the study medication. Moderate eye pain in one subject in the BID group led to the subject’s study withdrawal. No other subjects withdrew from the study as a result of AEs.
Best Corrected Visual Acuity
Best corrected visual scuity was assessed in both the study eye and the non-study eye using an Early Treatment Diabetic Retinopathy Study chart (see Table 5). At day 8, one subject in the QD group (2.2%) had a worsening of 4 lines of vision, but that effect had dissipated at day 15. There were no significant changes in intraocular pressure (IOP) between the two groups at day 8, nor were there any statistically significant differences between the study and non-study eye in either group at any time point, with one exception: one subject in the QD group (1/45; 2.2%) reported ocular hypertension, with an increase in IOP from 18 mmHg at day 1 to 35 mmHg at day 29; the issue was resolved with the use of topical medications.
Discussion
This study compared two different dosing regimens (QD and BID) of 0.075% bromfenac in DuraSite® as part of a phase II study and found that the efficacy and safety outcomes were generally similar between the two dosing regimens.
There has been a recent trend to reduce the number of topical medications, especially in an older population likely to be on concomitant medications and in those with chronic diseases where long-term exposure can be deleterious to the ocular surface. However, those concerns are mitigated somewhat by the evidence that non-high risk cataract surgery patients are likely to be on postoperative topical medication for the treatment of inflammation and pain for a shorter amount of time [17].
Noncompliance with dosing regimens is a common complaint in this older patient group [18, 19]. Missed doses may adversely impact a drug’s profile—while the chances for missing a dose with once-daily is smaller than with twice-daily medications, the impact of those missed doses are not equivalent [20]. Comte et al. noted “the pharmacokinetic equivalent of a single missed once-daily dose is 2–3 sequentially omitted twice-daily doses” [20]. Another benefit of twice-daily dosing is that the duration of effect is not diminished as much if/when a patient misses a dose, which may be potentially more relevant when patients are using short-term topical medications. Outside of ophthalmology, twice-daily dosing can have a protective effect against relapse [21] and may provide a greater treatment effect [20, 22,23,24,25] In some cases, QD dosing has shown the greatest fluctuation in pharmacokinetic/pharmacodynamics compared with twice- or even thrice-daily dosing [26].
Anecdotal evidence (later verified through physician interviews, data on file, InSite Vision) determined a challenge deemed difficult to overcome with current topical ophthalmic NSAID preparations—namely, bottle size and volume of medication per prescription. It is not uncommon for anterior segment surgeons to recommend dosing ophthalmic NSAIDs for up to 8 weeks (or longer) in post-cataract patients deemed at higher risk for developing CME (e.g. patients with concurrent diabetes) [17, 27, 28]. However, economic restrictions and/or limitations in coverage by insurance plans pose challenges to many patients in terms of a prescription refill (a second bottle) that would allow longer duration of the therapy, which is much needed for these patients [17]. In this context, Insite Vision designed a patient “use” study to study and track patient’s experience in using ophthalmic eye drops as part of its phase III trial on 0.075% bromfenac. Based on these data and the outcomes from this current subgroup analysis, the compound BromSite was approved for twice-daily dosing in a 5 mL bottle.
Conclusion
In this trial, outcomes from dosing with 0.075% bromfenac BID were equivalent to those from dosing with 0.075% bromfenac QD for the treatment of inflammation and prevention of pain in a uncomplicated postoperative cataract population, and in the ability to achieve ACC grade 0 at day 15.
References
Cho H, Wolf KJ, Wolf EJ. Management of ocular inflammation and pain following cataract surgery: focus on bromfenac ophthalmic solution. Clin Ophthalmol. 2009;3:199–210.
Waterbury LD, Silliman D, Jolas T. Comparison of cyclooxygenase inhibitory activity and ocular anti-inflammatory effects of ketorolac tromethamine and bromfenac sodium. Curr Med Res Opin. 2006;22(6):1133–40.
Donnenfeld ED, Donnenfeld A. Global experience with Xibrom (bromfenac ophthalmic solution) 0.09%: the first twice-daily ophthalmic nonsteroidal anti-inflammatory drug. Int Ophthalmol Clin. 2006;46(4):21–40.
Henderson BA, Gayton JL, Chandler SP, et al. Safety and efficacy of bromfenac ophthalmic solution (Bromday) dosed once daily for postoperative ocular inflammation and pain. Ophthalmology. 2011;118(11):2120–7.
Silverstein SM, Cable MG, Sadri E, et al. Once daily dosing of bromfenac ophthalmic solution 0.09% for postoperative ocular inflammation and pain. Curr Med Res Opin. 2011;27(9):1693–703.
Senju Pharmaceutical Co. Bronuck (package insert). Senju Pharmaceutical Co., Osaka, 2009.
ISTA Pharmaceuticals. Xibrom (package insert). ISTA Pharmaceuticals, Irvine, 2010.
ISTA Pharmaceuticals. Bromday (package insert). ISTA Pharmaceuticals, Irvine, 2011.
Committee for Medicinal Products for Human Use (CHMP). Yellox Assessment Report. Report No.: EMA/431843/2011. European Medicines Agency, London, 2011.
Bowman LM, Si E, Pang J, et al. Development of a topical polymeric mucoadhesive ocular delivery system for azithromycin. J Ocul Pharmacol Ther. 2009;25(2):133–9.
InSite Vision. BromSite (prescribing information). InSite Vision, Alameda, 2016.
Hosseini K, Hutcheson J, Bowman LM. Aqueous humor concentration of Bromfenac 0.09% (Bromday) compared with Bromfenac in DuraSite 0.075% (Bromsite) in cataract patients undergoing phacoemulsification after 3 days dosing. Association for Research in Vision and Ophthalmology. Seattle, 2013.
Luchs J. Azithromycin in DuraSite for the treatment of blepharitis. Clin Ophthalmol. 2010;4:681–8.
Malhotra R, Gira J, Berdy GJ, Brusatti R. Safety of besifloxacin ophthalmic suspension 0.6% as a prophylactic antibiotic following routine cataract surgery results of a prospective parallel-group investigator-masked study. Clin Ophthalmol. 2012;6:855–63.
Protzko E, Bowman L, Abelson M, Shapiro A. Phase 3 safety comparisons for 1.0% azithromycin in polymeric mucoadhesive eye drops versus 0.3% tobramycin eye drops for bacterial conjunctivitis. Invest Ophthalmol Vis Sci. 2007;48(8):3425–9.
Shafiee A, Bowman LM, Hou E, Hosseini K. Ocular pharmacokinetics of bimatoprost formulated in DuraSite compared to bimatoprost 0.03% ophthalmic solution in pigmented rabbit eyes. Clin Ophthalmol. 2013;7:1549–5156.
Singh R, Alpern L, Jaffe GJ, et al. Evaluation of nepafenac in prevention of macular edema following cataract surgery in patients with diabetic retinopathy. Clin Ophthalmol. 2012;6:1259–69.
Robin A, Grover DS. Compliance and adherence in glaucoma management. Indian J Ophthalmol. 2011;59[Suppl]:S93–6.
Tsai T, Robin AL, Smith JP III. An evaluation of how glaucoma patients use topical medications: a pilot study. Trans Am Ophthalmol Soc. 2007;105:29–33 (discussion-5).
Comte L, Vrijens B, Tousset E, et al. Estimation of the comparative therapeutic superiority of QD and BID dosing regimens, based on integrated analysis of dosing history data and pharmacokinetics. J Pharmacokinet Pharmacodyn. 2007;34(4):549–58.
Ruer-Mulard M, Aberer W, Gunstone A, et al. Twice-daily versus once-daily applications of pimecrolimus cream 1% for the prevention of disease relapse in pediatric patients with atopic dermatitis. Pediatr Dermatol. 2009;26(5):551–8.
Tsai T, Kroehl M, Smith S, et al. Efficacy and safety of twice- versus once-daily dosing of lisinopril for the treatment of hypertension. In: 50th Midyear meeting of the American Society of Health-System Pharmacists. New Orleans, LA, 2015.
Eriksson BI, Dahl OE, Buller HR, et al. A new oral direct thrombin inhibitor, dabigatran etexilate, compared with enoxaparin for prevention of thromboembolic events following total hip or knee replacement: the BISTRO II randomized trial. J Thromb Haemost. 2005;3(1):103–11.
Lan AJ, Colford JM, Colford JM Jr. The impact of dosing frequency on the efficacy of 10-day penicillin or amoxicillin therapy for streptococcal tonsillopharyngitis: a meta-analysis. Pediatrics. 2000;105(2):E19.
Zeng C, Wei J, Li H, et al. Comparison between 200 mg QD and 100 mg BID oral celecoxib in the treatment of knee or hip osteoarthritis. Sci Rep. 2015;5:10593.
de la Pena A, Ma X, Reddy S, et al. Application of PK/PD modeling and simulation to dosing regimen optimization of high-dose human regular U-500 insulin. J Diabetes Sci Technol. 2014;8(4):821–9.
Wielders LH, Lambermont VA, Schouten JS, et al. Prevention of cystoid macular edema after cataract surgery in nondiabetic and diabetic patients: a systematic review and meta-analysis. Am J Ophthalmol. 2015;160(5):968–981e33.
Elsawy MF, Badawi N, Khairy HA. Prophylactic postoperative ketorolac improves outcomes in diabetic patients assigned for cataract surgery. Clin Ophthalmol. 2013;7:1245–9.
Acknowledgments
InSite Vision (now a division of Sun Pharma) funded this study and publication charges. Editorial assistance in the preparation of this manuscript was provided by Michelle Dalton, ELS, of Dalton & Associates, Inc. Support for this assistance was funded by Sun Pharma. Statistical analysis was performed by William Krebs, PhD, an independent consultant; support for this assistance was funded by Sun Pharma.
The named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Kamran Hosseini: Sun Pharma: employee. William Trattler: Sun Pharma: consultant.
Compliance with Ethics Guidelines
This study received approval from an institutional review board (New England IRB, Needham, MA) and conformed with the Helsinki Declaration as revised in 2013. Informed consent was obtained from all patients before being included in the study.
Data Availability
The datasets generated during and/or analyzed during the study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/0AF8F060651D636A.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Trattler, W., Hosseini, K. Twice-Daily vs. Once-Daily Dosing with 0.075% Bromfenac in DuraSite: Outcomes from a 14-Day Phase 2 Study. Ophthalmol Ther 6, 277–284 (2017). https://doi.org/10.1007/s40123-017-0102-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-017-0102-x