
Abstract
Aim
Randomized pilot study comparing clinical outcomes with bromfenac ophthalmic solution 0.07% versus nepafenac 0.3% ophthalmic suspension administered as identical dosing regimens in patients undergoing uncomplicated phacoemulsification with intraocular lens implantation.
Methods
Forty-nine subjects were treated with bromfenac (n = 25) or nepafenac (n = 24) once daily starting 3 days before cataract surgery, continued on the day of surgery, and for 21 days following surgery, in addition to standard of care. Subjects were followed at 1 day and 7, 21, and 42 days postoperatively. Assessments included best-corrected visual acuity [Early Treatment Diabetic Retinopathy Study (ETDRS)], summed ocular inflammation score (SOIS; anterior chamber cells plus flare grading), macular volume and thickness (spectral domain optical coherence tomography), intraocular pressure, and adverse events.
Results
Treatment groups were similar at baseline. Outcomes for mean letters read (p = 0.20), mean change in macular volume (p = 0.98), and retinal thickness (p = 0.93) were not statistically different between the groups at day 42. Mean SOIS dropped markedly and similarly from post-surgical day 1 to day 7 in both treatment groups and was statistically equivalent to baseline in both groups by day 21. At day 42, 87% of subjects in the bromfenac group and 82% of subjects in the nepafenac group demonstrated stable or improved visual acuity. The proportions of eyes with mean retinal thickness of 10 µm or less at days 7, 21, and 42 were similar for the bromfenac (95.8%, 78.3%, 73.9%, respectively) and nepafenac (91.7%, 87.5%, 66.7%) groups (all p = NS, bromfenac vs. nepafenac).
Conclusion
Both bromfenac 0.07% and nepafenac 0.3% produced positive and similar clinical outcomes with regard to ETDRS visual acuity post-cataract surgery when dosed using identical regimens. Increases in mean retinal thickness and mean macular volume were small and similar between treatments.
Trial Registration number
NCT01847638.
Funding
Bausch & Lomb Incorporated.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cataract, a clouding of the normally clear lens of the eye commonly associated with aging, is the most common cause of blindness worldwide [1]. The standard of care for cataract treatment is sutureless small-incision phacoemulsification with foldable intraocular lens (IOL) implantation [2]. Cataract extraction can dramatically improve vision and vision-related quality of life [3, 4] and has become one of the most common and successful surgical interventions in the USA.
The process of ocular surgery triggers an inflammatory cascade involving cyclooxygenase (COX)-mediated prostaglandin production along with other inflammatory mediators [5]. One consequence of the inflammatory milieu is a breakdown of the blood–retinal barrier with the result being accumulation of intraretinal fluid, macular thickening, or frank edema [6, 7]. Previous research has demonstrated that up to 41% of normal eyes undergoing phacoemulsification experience an increase in macular thickness as measured by spectral domain optical coherence tomography (SDOCT) [6, 8]. Postoperative macular thickening has been linked to reduced visual acuity and contrast sensitivity; transient or permanent impairment of vision has occurred with as little as 10 μm of swelling [9,10,11].
Topical nonsteroidal anti-inflammatory drugs (NSAIDs) are potent inhibitors of COX enzymes and have been established as an effective treatment for the management of peri- and postoperative ocular inflammation [5, 11,12,13]. Two of the latest generation topical NSAIDs approved for the postoperative treatment of pain and inflammation in cataract extraction patients are bromfenac 0.07% ophthalmic solution (Prolensa®; Bausch + Lomb, Rochester, NY) and nepafenac 0.3% ophthalmic suspension (Ilevro®; Alcon Laboratories Inc, Fort Worth, TX). Both compounds are potent inhibitors of the COX-1 and COX-2 enzymes [14,15,16] and both products are the latest iteration of the respective molecules. The newest formulation of bromfenac reflects a lower concentration (0.07% rather than 0.09%) compared with the initial formulation and has a pH slightly closer to that of human tears [17, 18]. The most recent formulation of nepafenac has three times the effective concentration of the molecule relative to older formulations and a 40% smaller particle size for increased absorption, as well as a retention agent that improves bioavailability [19]. Nepafenac is a prodrug, the active metabolite of which is amfenac, a potent COX inhibitor [19].
Both bromfenac and nepafenac have been approved by the US Food and Drug Administration (FDA) for clinical use based on the results of clinical trials confirming efficacy and safety in the treatment of pain and inflammation following cataract surgery [20,21,22,23]. Yet, there are no definitive clinical data to suggest which topical NSAID may result in the best outcomes. Both drugs are approved for once daily (QD) use, starting 1 day prior to surgery, continued on the day of surgery, and for 2 weeks postoperatively. Official nepafenac dosing recommendations include an additional drop administered 30–120 min prior to surgery [24]. This single-site pilot study was designed to provide preliminary comparative clinical data on inflammation-related outcomes with bromfenac 0.07% QD or nepafenac 0.3% QD used according to identical dosing regimens. While older formulations of these medications have been compared in a head-to-head study [25], the author knows of no prior studies directly comparing the current formulations administered with identical dosing schedules.
Methods
Study Design
This was a prospective, single-site, randomized, single-masked, parallel-group pilot study comparing bromfenac ophthalmic solution 0.07% and nepafenac ophthalmic suspension 0.3% in patients undergoing uncomplicated cataract surgery with IOL implantation. The study was initiated at the end of 2013 and completed in March of 2015. The protocol was approved by Sterling Institutional Review Board (Atlanta, GA) on November 2, 2013 and the study was conducted in accordance with the principles of the Declaration of Helsinki approved in 1964 and as revised in 2013. All subjects provided informed consent prior to participation. Participating subjects received study drug, study-related procedures, and study visits at no charge to either themselves or their insurance company.
Subjects
Adults (≥ 18 years of age) with planned unilateral uncomplicated cataract extraction by phacoemulsification with posterior chamber IOL implantation who could postpone second eye surgery for at least 6 weeks were eligible for participation in the study. No other ophthalmic surgical procedures were allowed within 15 days prior to the initiation of study drug or throughout the duration of the study. Screening (baseline) evaluations were conducted within 8 days before the date planned for cataract surgery and included an assessment of medical history and recording of demographic information, complete bilateral ophthalmic examination [best corrected visual acuity (BCVA), SDOCT, biomicroscopic examination, intraocular pressure (IOP) measurement, and dilated funduscopic examination], and urine pregnancy testing for female participants. Eligible patients were required to have a BCVA of 20/200 or better in either eye at screening, and an IOP between 5 and 22 mmHg in the study eye. Exclusion criteria included known hypersensitivity to bromfenac or nepafenac or their components, salicylates, or other NSAIDs; intraocular inflammation (i.e., cells or flare in the anterior chamber as measured on slit lamp examination) in the study eye at screening; superficial punctate keratitis; other active corneal pathology that was considered nonstable, greater than mild, or might compromise assessment of the safety or efficacy of study treatment; any extraocular/intraocular inflammation in the study eye at screening (blepharitis allowed if mild only, and no concurrent conjunctivitis or lid erythema/edema) or ongoing, unresolved uveitis; and history of radial keratotomy, corneal transplant, or corneal refractive surgery in the study eye within the prior 2 years. Patients with type 1 or 2 diabetes were eligible to participate if there were no ocular findings determined to be clinically significant by the principal investigator. Preexisting macular edema, retinal edema, or more than two microaneurysms within the totality of the fundus constituted exclusions from participation. Use of topical, ocular, or inhaled corticosteroids was not permitted for 14 days prior to screening. The following were not allowed for 7 days prior to study drug initiation: ocular, topical, or systemic NSAIDs; ocular, topical, or systemic gentamicin; or cyclosporine ophthalmic emulsion. Ocular prostaglandin use was not allowed for 30 days prior to study entry.
Treatment
Enrolled subjects were randomly assigned, according to a computer-generated list, in a ratio of 1:1 to receive either bromfenac ophthalmic solution 0.07% or nepafenac ophthalmic suspension 0.3%. Study drugs were provided in their original FDA-approved packaging. Subjects were instructed to administer one drop daily of their assigned medication beginning 3 days before surgery, one dose on the day of surgery, then QD for 21 days after surgery. The single dose on the day of surgery was felt to be reflective of common clinical practice and differed from the precisely scheduled dosing on surgery day in the nepafenac 0.3% pivotal trial (exactly 30–120 min prior to surgery).
Uncomplicated phacoemulsification with IOL implantation (Tecnis® 9000series; Abbott Medical Optics Inc, Santa Ana, CA or Alcon SN60WF, Alcon Laboratories, Fort Worth, TX) was performed on one eye per subject by a single surgeon. Standard surgical procedure was followed for all subjects, including preoperative anti-infective besifloxacin 0.6% twice daily (BID) and study NSAID 3 days prior to surgery. Intraoperatively, subjects were administered one drop each of prednisolone acetate 1% and moxifloxacin 0.5%; ketorolac 0.4% was administered immediately postoperatively. Besifloxacin 0.6% was given BID for 10 days postoperatively. With the exception of the single drops of prednisolone acetate and ketorolac on the day of surgery, no other ocular steroids or NSAIDs other than study medication were allowed during the period from 7 days prior to initiation of study medication through the duration of the study.
Assessments
All subjects were required to attend follow-up assessments the day after surgery and then at 7, 21, and 42 days after surgery. For the entire trial, the investigator and any study personnel involved in recording study data were masked to study drug assignment; unmasking was allowed only if necessary for patient safety reasons. The study coordinator reviewed informed consent, inclusion/exclusion criteria with subjects, received the computer-generated list, dispensed study medication, and was responsible for scheduling study visits. Screening evaluations were conducted within 8 days before the date planned for cataract surgery and included a review of medical history and recording of demographic information, complete bilateral ophthalmic examination (BCVA, SDOCT, biomicroscopic examination, IOP measurement, and dilated funduscopic examination), and urine pregnancy test for female participants.
Assessments performed at each study visit included BCVA evaluation [Early Treatment Diabetic Retinopathy Study (ETDRS) letters], biomicroscopic examination, SDOCT, summed ocular inflammation score (SOIS) cell and flare grading, IOP measurement, adverse event (AE) recording, and a review of concomitant medications. At each follow-up visit, BCVA was measured using the ETDRS chart at 4 m in controlled lighting conditions and acuities were recorded using letters read and the logarithm of the minimum angle of resolution system according to the method described by Kaiser [26] (LogMAR VA = 0.1 + LogMAR value of best line read − 0.002 × number of letters read). Bilateral slit lamp biomicroscopy (without pupil dilation) examination was performed to assess intra- and extraocular inflammation. Dilated funduscopic exams were performed at the initial visit and again at the final study visit (day 42 ± 3).
An experienced ophthalmic technician obtained all Stratus optical coherence tomography (OCT) (Carl Zeiss Meditec Inc, Dublin, CA) scans for all study subjects. OCT captures the interference pattern between backscattered light and a reference beam to create three-dimensional images, which can provide structural and quantitative data regarding ocular structures, including the retina. Two scan patterns were used; the fast macular thickness protocol, using six radial line scans through a common central axis (fovea) with a retinal thickness/volume tabular output and a retinal-thickness output report. Central retinal thickness was defined as the distance between the inner limiting membrane of the retina and the inner border of the choriocapillaris measured in the central 1-mm area of the (minimum) 7-mm posterior pole scan. The Stratus software, Version 5.0, (Carl Zeiss Meditec Inc, Dublin, CA) calculated total macular volume within the 7-mm-diameter scanned area, representing a weighted average of the central, inner, and outer subfields multiplied by the area of the grid measured. Macular volume is an objective indicator of macular thickening or swelling and can be used to demonstrate the amount of inflammation after cataract surgery. All scans were reviewed by the principal investigator for quality of centration and signal strength. Ocular inflammation was calculated using SOIS to quantify cells and flare present in the anterior chamber. Anterior chamber cells were assessed using a slit lamp biomicroscope at ×16 magnification with a 0.3 × 1-mm oblique high-intensity beam. Cell counts were measured twice and converted to a grade (Table 1), by which the mean score was calculated. Anterior chamber flare was measured once and graded using the scale included in Table 1. The SOIS was calculated by combining the sum of the cell and flare grades.
Endpoints and Statistical Analysis
Primary efficacy endpoints were changes from baseline to postoperative day 42 (± 7 days) in BCVA (ETDRS letters), OCT measurements of macular volume and retinal thickness, and SOIS. Secondary efficacy endpoints included safety assessments including AEs, both serious and non-serious, and IOP measurements.
As a small pilot study with a planned sample size of 50 subjects, this study was not powered to establish statistical superiority. All analyses of efficacy were conducted on the intent-to-treat population, which included all randomized subjects who received at least one dose of study medication. Subjects were analyzed in the group to which they were randomized. Paired t tests were used for within-subject baseline comparison of results and between-group comparisons. A p value of less than 0.05 was considered statistically significant.
Results
Study Population
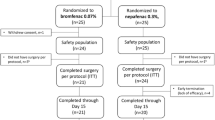
Fifty participants met all inclusion/exclusion criteria and were randomly assigned to receive either bromfenac ophthalmic solution 0.07% QD (n = 25) or nepafenac ophthalmic suspension 0.3% QD (n = 24). Because of a protocol deviation (assessment by non-spectral domain OCT), one subject in the nepafenac group was excluded; thus, the analysis population included 49 subjects (bromfenac, n = 25; nepafenac, n = 24). Demographic and clinical characteristics of subjects and eyes at baseline were similar between the treatment groups (Table 2). In each group, approximately two-thirds of subjects were Caucasian and female. No additional ocular surgery was performed during the study visits. All subjects completed all required study visits and completed their assigned treatment regimens (based on subjective reporting).
Efficacy
In both groups, BCVA changed significantly from baseline to day 42 (p < 0.05) with no significant difference between groups; the mean change in ETDRS acuity was − 0.12 in the bromfenac group and − 0.2 in the nepafenac group (p = 0.2) (Table 3). At day 42, 87% of subjects in the bromfenac group and 82% of subjects in the nepafenac group demonstrated stable or improved visual acuity.
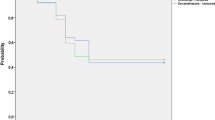
Mean retinal thickness and mean macular volume increased to a small extent at each study visit (Fig. 1) and were significantly greater at day 42 relative to baseline in each group. There were no statistically significant differences between treatments at any visit. The proportions of eyes with mean retinal thickness of 10 µm or less at days 7, 21, and 42 were similar for the bromfenac (95.8%, 78.3%, 73.9%, respectively) and nepafenac (91.7%, 87.5%, 66.7%) groups (p = NS). Small increases from baseline in central subfield thickness were noted at days 7, 21, and 42 in the bromfenac (mean ± SE, 3.2 ± 1.78 µm, 9.1 ± 3.55 µm, 14.7 ± 5.67 µm, respectively) and nepafenac (3.3 ± 2.88 µm, 7.2 ± 2.82 µm, 10.6 ± 3.27 µm) groups (all p = NS, bromfenac vs. nepafenac). At day 42, mean macular volume was 0.35 mm3 in both groups (Fig. 1b). Mean SOIS dropped markedly and similarly from day 1 to day 7 in both treatment groups and was statistically equivalent to baseline in both groups by day 21 (Fig. 2).

Change from baseline in a mean retinal thickness and b mean macular volume at each study timepoint. Study drugs were administered through day 21. Data depict mean ± standard error. No statistically significant differences between treatments at any timepoint

Mean SOIS by study visit (see Table 2 for scoring method). Study drugs were administered through day 21. Data depict mean ± standard error. No statistically significant differences between treatments at any timepoint. NS not significant, SOIS summed ocular inflammation score
Safety
Both study drugs were well tolerated. The most common AE was iritis, reported in 3 (12.0%) patients in the bromfenac group and 7 (29.2%) patients in the nepafenac group. One case of iritis in the bromfenac group required treatment. Other AEs in the bromfenac group included one report each of cystoid macular edema and dizziness; in the nepafenac group, there was one report each of dizziness and ocular pain (both voluntarily reported), and one report each of increased IOP and posterior capsular haze. There were no differences between treatment groups in mean IOP at any study visit and mean IOP did not increase significantly from baseline in either group.
Discussion
This small prospective, randomized pilot study compared clinical outcomes associated with two of the most commonly used and newest formulations of topical ophthalmic NSAIDs, bromfenac 0.07% and nepafenac 0.3%, for the treatment of inflammation and pain following cataract extraction surgery. The safety and efficacy of these two compounds in patients undergoing cataract surgery has been thoroughly examined in independent studies [20,21,22, 27, 28]; however, head-to-head data comparing postoperative outcomes with the two products are limited.
In the current study, patients with similar baseline characteristics used either bromfenac 0.07% or nepafenac 0.3% according to identical dosing regimens that were considered reflective of current standard clinical practice. None of the clinical outcomes evaluated (mean letters read, mean change in macular volume, retinal thickness, SOIS) were statistically different between the treatment groups at any visit. Inflammation, as measured by SOIS, improved markedly in both groups within the first 7 days after surgery, and within 3 weeks, had returned to pre-surgical levels. Measures of retinal thickness and macular volume increased to a small, but significant, degree in both treatment groups. One-quarter of patients in the bromfenac group and one-third of patients in the nepafenac group experienced an increase in retinal thickness of greater than 10 µm.
Speed of visual recovery and extent of visual improvement are common and clinically important factors for all patients having routine cataract surgery. A previous pilot head-to-head study comparing older formulations of bromfenac (0.09%; Bromday™; QD dosing) and nepafenac (0.1%; Nevanac®; three times daily dosing) as part of post-cataract surgery management noted essentially statistical equivalency between the treatments with regard to visual acuity findings, mean change in macular volume, and retinal thickness at each study visit; independently, the change from baseline to week 6 in BCVA was significant in the bromfenac group, but not the nepafenac group [25]. In the same study, mean macular volume and retinal thickening significantly worsened from baseline to week 6 in the nepafenac group only. These findings were not replicated in the current pilot study and may be due to differences in study populations and/or differences inherent to the specific bromfenac and nepafenac formulations used in each study. Additional studies with larger populations will be necessary to further explore differences between the drugs.
A major limitation of this pilot study is the small number of subjects, and the likely lack of power to demonstrate differences between the treatments for the primary outcomes. Study treatment was not masked to patients, but this should not have impacted the assessments which were objective measurements made by masked study personnel.
Conclusions
In this small pilot study, bromfenac 0.07% ophthalmic solution and nepafenac 0.3% ophthalmic suspension, each dosed QD according to the same dosing schedules, were both well tolerated and demonstrated similar efficacy in minimizing post-surgical inflammation and retinal thickening, as well as improving BCVA, following cataract surgery. Further head-to-head comparisons based on larger populations powered to detect differences between treatments will be necessary to more rigorously assess potential differences in safety and effectiveness between these two products.
References
Khairallah M, Kahloun R, Bourne R, et al. Number of people blind or visually impaired by cataract worldwide and in world regions, 1990 to 2010. Invest Ophthalmol Vis Sci. 2015;56(11):6762–9.
Olson RJ, Braga-Mele R, Chen SH, et al. Cataract in the Adult Eye Preferred Practice Pattern®. Ophthalmology. 2017;124(2):P1–119.
Lamoureux EL, Fenwicke E, Pesudovs K, Tan D. The impact of cataract surgery on quality of life. Curr Opin Ophthamlmol. 2011;22(1):19–27.
Javed U, McVeigh K, Scott NW, Azuara-Blanco A. Cataract extraction and patient vision-related quality of life: a cohort study. Eye. 2015;29(7):921–5.
Kim SJ, Flach AJ, Jampol LM. Nonsteroidal anti-inflammatory drugs in ophthalmology. Surv Ophthalmol. 2010;55(2):108–33.
Lobo CL, Faria PM, Soares MA, Bernardes RC, Cunha-Vaz JG. Macular alterations after small-incision cataract surgery. J Cataract Refract Surg. 2004;30(4):752–60.
McColgin AZ, Heier JS. Control of intraocular inflammation associated with cataract surgery. Curr Opin Ophthalmol. 2000;11(1):3–6.
Sourdille P, Santiago PY. Optical coherence tomography of macular thickness after cataract surgery. J Cataract Refract Surg. 1999;25(2):256–61.
Wittpenn JR, Silverstein S, Heier J, et al. A randomized, masked comparison of topical ketorolac 0.4% plus steroid vs steroid alone in low-risk cataract surgery patients. Am J Ophthalmol. 2008;146(4):554–60.
Blumenkranz MS, Haller JA, Kuppermann BD, et al. Correlation of visual acuity and macular thickness measured by optical coherence tomography in patients with persistent macular edema. Retina. 2010;30(7):1090–4.
Flach AJ. The incidence, pathogenesis and treatment of cystoid macular edema following cataract surgery. Trans Am Ophthalmol Soc. 1998;96:557–634.
Juthani VV, Clearfield E, Chuck RS. Non-steroidal anti-inflammatory drugs versus corticosteroids for controlling inflammation after uncomplicated cataract surgery. Cochrane Database Syst Rev. 2017;7:CD010516.
Kessel L, Tendal B, Jørgensen KJ, et al. Post-cataract prevention of inflammation and macular edema by steroid and nonsteroidal anti-inflammatory eye drops: a systematic review. Ophthalmology. 2014;121(10):1915–24.
Kida T, Ogawa T, McNamara TR, Song CK, Gow JA. Evaluation of the human COX-2 inhibition of amfenac, bromfenac, diclofenac, and ketorolac. Paper presented at American Society of Cataract and Refractive Surgery Symposium on Cataract, IOL, and Refractive Surgery; April 27–May 2, 2007. San Diego; 2007.
Waterbury LD, Silliman D, Jolas T. Comparison of cyclooxygenase inhibitory activity and ocular anti-inflammatory effects of ketorolac tromethamine and bromfenac sodium. Curr Med Res Opin. 2006;22(6):1133–40.
Walters T, Raizman M, Ernest P, Gayton J, Lehmann R. In vivo pharmacokinetics and in vitro pharmacodynamics of nepafenac, amfenac, ketorolac, and bromfenac. J Cataract Refract Surg. 2007;33(9):1539–45.
Sheppard JD. Topical bromfenac for prevention and treatment of cystoid macular edema following cataract surgery: a review. Clin Ophthalmol. 2016;10:2099–111.
Prolensa® (bromfenac ophthalmic solution) 0.07% [prescribing information]. Bausch + Lomb, a division of Valeant Pharmaceuticals North America, LLC, Rochester, MN; 2016.
Modi SS, Lehmann RP, Walters TR, et al. Once-daily nepafenac ophthalmic suspension 0.3% to prevent and treat ocular inflammation and pain after cataract surgery: phase 3 study. J Cataract Refract Surg. 2014;40(2):203–11.
Donnenfeld ED, Holland EJ, Stewart RH, Gow JA, Grillone LR, on behalf of the Bromfenac Ophthalmic Solution 0.09% (Xibrom) Study Group. Bromfenac ophthalmic solution 0.09% (Xibrom) for postoperative ocular pain and inflammation. Ophthalmology. 2007;114(9):1653–62.
Silverstein SM, Cable MG, Sadri E, et al. Once daily dosing of bromfenac ophthalmic solution 0.09% for postoperative ocular inflammation and pain. Curr Med Res Opin. 2011;27(9):1693–703.
Nardi M, Lobo C, Bereczki A, et al. Analgesic and anti-inflammatory effectiveness of nepafenac 0.1% for cataract surgery. Clin Ophthalmol. 2007;1(4):527–33.
Lane SS, Modi SS, Lehmann RP, Holland EJ. Nepafenac ophthalmic suspension 0.1% for the prevention and treatment of ocular inflammation associated with cataract surgery. J Cataract Refract Surg. 2007;33(1):53–8.
Ilevro® (nepafenac ophthalmic suspension) 0.3% [prescribing information]. Alcon Laboratories. Fort Worth, TX; 2014.
Cable M. Comparison of bromfenac 0.09% QD to nepafenac 0.1% tid after cataract surgery: pilot evaluation of visual acuity, macular volume, and retinal thickness at a single site. Clin Ophthalmol. 2012;6:997–1004.
Kaiser PK. Prospective evaluation of visual acuity assessment: a comparison of Snellen versus ETDRS charts in clinical practice (an OAS thesis). Trans Am Ophthalmol Soc. 2009;107:311–24.
Miyanaga M, Miyai T, Nejima R, Maruyama Y, Miyata K, Kato S. Effect of bromfenac ophthalmic solution on ocular inflammation following cataract surgery. Acta Ophthalmol. 2009;87(3):300–5.
Miyake K, Ota I, Miyake G, Numaga J. Nepafenac 0.1% verus fluorometholone 0.1% for preventing cystoid macular edema after cataract surgery. J Cataract Refract Surg. 2011;37(9):1581–8.
Acknowledgements
The author wishes to thank all of the patients who participated in this study.
Funding
This study was funded through an independent research grant from Bausch & Lomb Incorporated. Writing and editorial support was provided by Churchill Communications (Maplewood, New Jersey, USA). Article processing fees were funded by Bausch & Lomb Incorporated.
Medical Writing and/or Editorial Assistance
Editorial assistance in the preparation of this article was provided by Sandra Westra, PharmD of Churchill Communications (Maplewood, NJ). Support for this assistance was funded by Bausch & Lomb Incorporated.
Authorship
The author meets the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, takes responsibility for the integrity of the work as a whole, and has given her approval for this version to be published.
Disclosures
Dr. Toyos has participated in clinical trials sponsored by Bausch & Lomb Incorporated and serves on a Bausch & Lomb Incorporated speakers bureau and advisory boards.
Compliance with Ethics Guidelines
The study protocol was approved by Sterling Institutional Review Board (Atlanta, GA) on November 2, 2013 and the study was conducted in accordance with the principles of the Declaration of Helsinki approved in 1964 and as revised in 2013. All subjects provided informed consent prior to participation.
Data Availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.7718657
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Toyos, M.M. Comparison of Once-Daily Bromfenac 0.07% Versus Once-Daily Nepafenac 0.3% in Patients Undergoing Phacoemulsification. Ophthalmol Ther 8, 261–270 (2019). https://doi.org/10.1007/s40123-019-0174-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-019-0174-x