
Abstract
Introduction
This small pilot study is the first direct comparison of the currently marketed formulations of bromfenac (0.07% solution) and nepafenac (0.3% suspension) using identical dosing regimens and including an extra pre-surgical “pulse” dose in patients undergoing cataract surgery.
Methods
Adults scheduled for unilateral phacoemulsification with intraocular lens implantation were randomly assigned to bromfenac 0.07% or nepafenac 0.3%, each given once-daily 1 day prior to surgery, on the day of surgery plus an extra dose 1 h before surgery, and for 14 days after surgery. Assessments included summed ocular inflammation score (SOIS), visual acuity (VA), and retinal thickness measured via optical coherence tomography.
Results
The study population included 49 patients (bromfenac, n = 24; nepafenac, n = 25). The percentage of patients with a SOIS = 0 (no cells or flare) at post-surgical day 15 (primary efficacy endpoint) was statistically similar between the bromfenac (57.1%) and nepafenac (50.0%) treatment groups (intent-to-treat with last observation carried forward) (P = 0.6318). The proportions of patients with an SOIS of 0 at days 3 and 8 were significantly (P < 0.05) higher in the bromfenac group (23.8 and 52.4%, respectively) versus the nepafenac group (0.0 and 20.8%, respectively). Visual acuity was similar between groups at each study visit, as were mean retinal thickness and change from baseline in retinal thickness. Rescue medication (typically difluprednate) was given on or before day 15 to 13 patients in each treatment group (bromfenac, 54.2%; nepafenac, 52.0%). There were no adverse events considered to be related to either treatment.
Conclusions
The results of this small pilot study suggest that once-daily bromfenac 0.07% produces similar benefits with regard to postsurgical inflammation, VA, and retinal thickness as once-daily nepafenac 0.3%, and possibly has a faster onset of anti-inflammatory action, when compared using identical dosing regimens.
Funding
Bausch & Lomb Incorporated.
Trial Registration
NCT03886779.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ocular inflammation is common after cataract surgery and, when untreated, may result in complications such as pain, photophobia, corneal edema, cystoid macular edema (CME), posterior capsule opacification, and increased intraocular pressure (IOP) [1, 2]. Management of post-operative inflammation with nonsteroidal anti-inflammatory drugs (NSAIDs) is considered of value to decrease the risk of long-term complications such as CME and mitigate post-surgical pain [1, 3,4,5]. The administration of topical NSAIDs prior to and after surgery has been shown to be beneficial to facilitate analgesia, CME prevention, and resolution of inflammation [1, 3]. Topical NSAIDs penetrate ocular tissues and elicit anti-inflammatory benefits by inhibiting the formation of cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2), thereby blocking the synthesis of prostaglandins, the most important mediators of inflammation [4, 6, 7].
Two of the most widely prescribed topical NSAIDs for the reduction of pain and inflammation post-cataract surgery are bromfenac and nepafenac. Bromfenac was originally approved by the Food and Drug Administration (FDA) in 1997 and has been marketed as a variety of formulations with varying concentrations and dosing recommendations over the years. It is currently marketed as a once-daily 0.07% ophthalmic solution (Prolensa®; Bausch & Lomb Incorporated, Rochester, NY, USA), indicated for the treatment of post-operative inflammation and reduction of pain after cataract surgery [8]. The approved dosing regimen is once daily, beginning 1 day prior to surgery, continued on the day of surgery and through the first 14 days after surgery [8].
Nepafenac was initially approved by the FDA in 2005 as a 0.1% suspension for administration three times daily. The most recent formulation, a once-daily, 0.3% ophthalmic suspension (Ilevro®, Alcon Laboratories, Inc., Fort Worth, TX, USA), was approved by the FDA in 2012 for the treatment of pain and inflammation associated with cataract surgery [9]. The recommended dosing of nepafenac 0.3% is similar to that for bromfenac 0.07% (once daily, beginning 1 day prior to cataract surgery, continued on the day of surgery and through the first 2 weeks of the post-operative period), but with an additional dose 30–120 min prior to surgery.
Independent clinical trials evaluating bromfenac and nepafenac have demonstrated that each formulation is highly effective and safe for managing post-surgical pain and inflammation [10,11,12,13,14,15,16,17,18,19]. However, there are little data directly comparing these two NSAIDs in head-to-head fashion, particularly with the currently marketed formulations (bromfenac 0.07% and nepafenac 0.3%). This small pilot study is the first direct comparison of bromfenac 0.07% and nepafenac 0.3% using an identical dosing schedule beginning 1 day prior to surgery along with an extra “pulse” dose 1 h before surgery in patients undergoing planned cataract extraction with posterior chamber intraocular lens implantation.
Methods
Study Design
This was a single-center, randomized, investigator-masked, parallel group, active-comparator controlled pilot study (ClinicalTrials.gov Identifier: NCT03886779). The study was performed in accordance with the International Conference on Harmonization Consolidated Good Clinical Practice Guidelines and with the ethical principles of the Helsinki Declaration of 1964 and its later amendments. The study protocol was approved by Quorum Institutional Review Board prior to the screening of subjects. All subjects provided written informed consent before undergoing any study-specific procedures.
Patients
Male or female adult patients (≥ 18 years of age) scheduled for unilateral cataract surgery by phacoemulsification with posterior chamber intraocular lens implantation without other ophthalmic surgical procedures were eligible for inclusion. Other inclusion criteria included a best corrected visual acuity (VA) of 20/200 or better in either eye, IOP in the study eye of between 5 and 22 mmHg, and no other planned ocular surgical procedures in the study or fellow (non-study) eye prior to initiation of study medication use through to the end of the study. Major exclusion criteria included the following: known hypersensitivity to bromfenac, nepafenac, salicylates, or other NSAIDs; evidence of ocular inflammation in the study eye on slit lamp examination at screening (any cell or flare in the anterior chamber [AC]); prior use of tamsulosin (Flomax); use of ocular, topical, or systemic NSAIDs, or cyclosporine ophthalmic emulsion, or other immunomodulators within 7 days; use of any ocular prostaglandin within 30 days prior to initiation of study medication or during the study; use of topical, ocular, inhaled, or systemic corticosteroids within 14 days prior to screening; corneal transplantation or corneal refractive surgery in the study eye within the last 2 years; active (non-stable or greater than mild in severity) corneal pathology in the study eye at screening; and other medical conditions that might confound the study.
Study Treatments and Masking
Eligible patients were assigned sequentially according to a computer-generated randomization list to receive either bromfenac 0.07% or nepafenac 0.3% in a 1:1 ratio. Both medications were to be administered as one drop once daily at the same time each day (preferably in the morning), beginning 1 day prior to surgery (day 1) and continuing on the day of surgery (day 0) and for about 14 days after surgery. On the day of surgery, study personnel instilled 1 additional drop of the assigned study drug into each patient’s study eye 1 h prior to surgery. All surgeries were performed by the lead investigator, and all post-operative assessments were performed by a single sub-investigator, both of whom were masked to the study treatment. Patients were instructed not to instill any other ocular medications within 15 min before or after instilling the study medication dose. They were also advised to use gentle eyelid closure for at least 1 min after instilling the dose into the study eye and then to repeat instillation of a single drop of the study medication if there was uncertainty as to whether successful instillation occurred. Treatment compliance was verbally confirmed with patients and documented at each study visit.
Study medications were provided as commercially available formulations of bromfenac 0.07% (Prolensa) and nepafenac 0.3% (Ilevro), packaged inside of plain white study kit boxes labeled according to the randomization schedule. Patients were to keep the medication inside of the study kit boxes when attending study visits so as not to unmask the investigator. The investigator and sub-investigator were both masked to study treatment for the duration of the study, unless unmasking was required.
In addition to study medication, patients administered besifloxacin 0.6% suspension three times daily (TID) for 2 days prior to surgery, every 2 h while awake on the day of surgery, and TID each day after surgery until the bottle was finished. Pain management, which was not to include NSAIDs or corticosteroids, was to be determined at the discretion of the investigator.
Outcome Measures
Efficacy
The primary efficacy endpoint was the proportion of patients with an absence of AC ocular inflammation on slit-lamp biomicroscopy at the day 15 visit, as defined by a summed ocular inflammation score (SOIS) of grade 0 (no cells or flare). For each assessment, AC cell grade was measured twice and averaged for determination of the visit score; AC flare grade was determined using the grading scale shown in Table 1. The SOIS was calculated by adding the subject’s AC cells and flare grades, with the minimum score of 0 indicating the absence of inflammation and a maximum score of 8 (AC cells > 50 and flare grade 4 [intense]). The SOIS has been used as the primary endpoint variable in numerous studies of bromfenac, including those used for the basis of FDA approval [10, 11, 17, 19,20,21], as well in studies of other ocular NSAIDs, including ketorolac [22] and diclofenac [23].
Secondary efficacy endpoints included the proportion of patients with SOIS of 0 at each study visit; Early Treatment Diabetic Retinopathy Study (ETDRS) Logarithm of the Minimum Angle of Resolution (logMAR) VA at day 15 and change from baseline (screening) to day 15; VA and change from baseline at each study visit; and measurements of retinal thickness and volume at day 42 as measured by optical coherence tomography (OCT) performed using a Zeiss OCT Stratus 3000 scanner (Carl Zeiss Meditec, Inc., Dublin, CA, USA).
Patients with an increase in SOIS compared with the previous post-surgical visit, those with an SOIS that was unchanged for two consecutive visits after surgery, or patients who had any sign or symptom of inflammation sufficiently severe as to require alternative anti-inflammatory treatment could be prematurely discontinued from study treatment at the investigator’s discretion.
Safety
Safety endpoints included the incidence and frequency of treatment-emergent ocular and non-ocular adverse events (AEs), as well as findings from ophthalmological evaluations (including slit-lamp biomicroscopy [each study visit], funduscopic evaluation [days 15 and 42] and IOP measurements [each study visit]).
Statistical Analysis
The study was not powered for statistical superiority since it was considered to be a pilot study of clinical outcomes for a small group of subjects. All efficacy analyses were conducted on the intent-to-treat (ITT) population, defined as all randomized subjects who received at least one dose of study medication and underwent protocol-defined surgery.
The primary efficacy analysis tested the difference between treatment groups in the proportion of subjects with an SOIS of 0 in the study eye at day 15 using the Pearson Chi-squared test with a two-sided alpha = 0.05. For each treatment group, 95% asymptotic normal confidence intervals were computed.
Two analyses of efficacy were performed: one based on data with last observation carried forward (LOCF) and one based on data as observed. Two types of missing values were anticipated in the LOCF data analysis: (1) from subjects who were not responding to test treatment (based on assessment of ocular inflammation) and who may have required alternative medical management (i.e., rescue therapy) and (2) from subjects who missed scheduled evaluations. For the LOCF analysis for the first type of missing data, regarding subjects who received a rescue medication prior to day 15, the efficacy response nearest (on or before) the date of receiving rescue medication was carried forward and used in the determination of the outcome. For the LOCF analysis for the second type of missing data, regarding subjects who missed scheduled evaluations, the efficacy response from the last visit at which it was measured was carried forward and used in the determination of the outcome.
Safety analyses were based on the safety population, which included all patients who received at least one dose of study medication. Safety endpoints were summarized by visit and treatment group and presented separately for data obtained before and after a patient received rescue medication. Treatment-emergent AEs were summarized by system organ class and preferred terms for each treatment group; 95% confidence intervals around the difference between treatment groups in the incidence were constructed using asymptotic normal approximations. Visual acuity, biomicroscopy, and funduscopic findings were summarized using discrete summary statistics.
All statistical testing was two-sided and performed at the 0.05 significance level, unless otherwise noted. All analyses were performed using SAS® Version 9.2 or higher (SAS Institute, Cary, NC, USA).
Results
Patient Characteristics and Disposition
A total of 57 patients were screened, 50 of whom met the eligibility requirements and were randomized to treatment with bromfenac (n = 25) or nepafenac (n = 25) (Fig. 1). One patient randomized to bromfenac withdrew consent, thus the final safety population included 49 patients (bromfenac, n = 24; nepafenac, n = 25). The treatment groups had similar demographic and baseline characteristics with no clinically relevant differences apparent between groups (Table 2). Four patients did not undergo scheduled surgery and were therefore excluded from the efficacy population (bromfenac, n = 3; nepafenac, n = 1). Thus, the ITT population included 21 bromfenac-treated patients and 24 nepafenac-treated patients.

Flow chart of patient disposition. aScreen failures included: epiretinal membranes (n = 2); bleeding due to diabetes (n = 1); macular edema (n = 1); intraocular pressure > 22 mmHg (n = 1); severe uncontrolled anemia (n = 1); best corrected visual acuity worse than 20/200 (n = 1). bPatients received pre-surgical doses of study drug and were included in safety evaluations. ITT Intent-to-treat population
Efficacy
The percentage of patients with an SOIS of 0 at day 15 (primary efficacy endpoint) was statistically similar between the bromfenac (57.1%) and nepafenac (50.0%) treatment groups in the ITT population with LOCF (P = 0.6318) (Fig. 2). Similar findings were noted in the analysis using data as observed (57.1 vs. 60.0%; P = 0.8527).

Percentage of patients with a summed ocular inflammation score (SOIS) of 0 at day 15 (ITT population with last observation carried forward [LOCF] and based on data as observed)
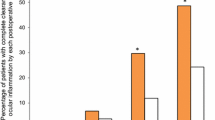
The percentages of patients with an SOIS of 0 at each study visit (ITT with LOCF) are shown in Fig. 3. A statistically greater percentage of patients in the bromfenac group compared to the nepafenac group had an SOIS of 0 at post-surgical days 3 (23.8 vs. 0.0%; P = 0.0112) and 8 (52.4 vs. 20.8%; P = 0.0274); for all other visits, the differences between groups were not statistically significant. Findings were similar in the analysis based on data as observed: differences between groups were significant (P < 0.05) at days 3 (bromfenac, 5/21 [23.8%]; nepafenac, 0/24 [0.0%]) and 8 (bromfenac, 11/20 [55.0%]; nepafenac, 5/24 [20.8%]), but not significant at days 15 (bromfenac, 12/21 [57.1%]; nepafenac, 12/20 [60.0%]) or 42 (bromfenac, 10/10 [100.0%]; nepafenac, 8/8 [100.0%]).

Percentage of patients with a SOIS of 0 at each study visit (ITT population with LOCF). Bars reflect 95% confidence limits. Asterisk indicates P < 0.05 vs. nepafenac
Mean ETDRS logMAR assessments at each study visit and change from baseline are presented in Table 3. At each study visit, mean assessments and changes from baseline were statistically equivalent between treatment groups. Mean change from baseline at day 42 was − 0.13 with bromfenac and − 0.16 with nepafenac.
For the ITT population with LOCF at day 42, there was no difference between the bromfenac (n = 11) and nepafenac (n = 9) groups with regard to the mean (± standard deviation [SD]) retinal thickness (275.8 ± 25.5 vs. 261.0 ± 34.5 µm, respectively; P = 0.2838) or mean change from baseline in retinal thickness (11.0 ± 20.9 vs. 17.3 ± 16.3 µm; P = 0.4669). Findings were similar for the analysis of ITT data as observed (data not shown).
Rescue medication (typically difluprednate) was required on or before day 15 by 13 patients in each treatment group (bromfenac, 54.2%; nepafenac, 52.0%).
Safety
Both study drugs were safe and well tolerated, and there were no AEs that were considered to be related to either bromfenac or nepafenac. A total of 14 patients reported at least one AE, of whom eight were in the bromfenac group (27 AEs) and six were in the nepafenac group (20 AEs). Of the 47 AEs reported, 17 (36.2%) were ocular; these were generally consistent with a typical post-surgical state (e.g., pain, photophobia, mattering/eye discharge), and none were considered related to treatment. There were no reports of punctate keratitis or toxic conjunctivitis. There were two serious events (altered mental state; urinary tract infection), neither of which was considered related to treatment. Overall, 81% of AEs were mild in severity, none were severe, and all were considered to be unrelated to treatment.
Discussion
This is one of the few clinical trials to directly compare bromfenac 0.07% against nepafenac 0.3% used according to identical dosing regimens in patients undergoing cataract surgery, and the only known study to incorporate an extra pulse dose of medication just prior to surgery. The two treatment groups had similar characteristics at baseline. While outcomes were similar for the primary efficacy endpoint (proportions of patients with an SOIS of 0 at day 15), bromfenac was significantly better in terms of the proportion of patients having an SOIS of 0 at earlier timepoints (days 3 and 8), possibly suggesting a more rapid onset of effectiveness with regard to the resolution of inflammation. This was a small pilot study, and the findings would need to be confirmed by larger trials, but the speed of onset is certainly a clinical factor assumed to be of importance to patients. However, it should be noted that VA, an outcome which would have a practical impact on patients, was similar between groups at every study visit. Retinal thickness increased to a small and similar extent in each treatment group.
Rescue medications were given to about half of patients in each treatment group. As is the norm in studies evaluating ophthalmic NSAIDs, the protocol did not permit pre- or post-operative steroid use. Thus, additional anti-inflammatory therapy in some patients was not unexpected and still lessened overall steroid exposure in these patients. Most of the rescues occurred at day 15 (at the end of the study treatment period), if the patient had persistent anterior cell and flare or lingering clinical signs and symptoms. In clinical practice, topical NSAIDs and corticosteroids are often used concomitantly for post-cataract surgery management. The purpose of this study was to compare the two NSAIDS, bromfenac and nepafenac, and not necessarily to establish the monotherapy efficacy of either. The “rescued” patients continued their NSAID—thus “rescue” in this case should be translated as a need for augmentation rather than treatment failure. It remains that about half of patients in each treatment group did not use any rescue medications and were satisfactorily managed with a topical NSAID alone. Further, the criteria for rescue medication use was somewhat subjective and reflects the investigators’ threshold for implementing adjunctive therapy.
Another recent study also compared these two formulations using identical dosing schedules, but ones that were slightly different than the regimen used in the current study [20]. In the prior study, bromfenac 0.07% and nepafenac 0.3% were both dosed once daily for 3 days prior to cataract surgery, once on the day of surgery (no extra “pulse dose” prior to surgery as in the current study), and once daily for 21 days after surgery. Specific study endpoints were slightly different, but the findings corroborated those of the current study, including a similar efficacy between bromfenac 0.07% and nepafenac 0.3% in minimizing post-surgical inflammation and retinal thickening; similar, significant improvements from baseline in VA; and no safety concerns with either drug. Mean SOIS scores were statistically similar between the treatment groups at each study visit.
Pivotal pre-approval clinical studies for bromfenac 0.07% [17] used dosing regimens that were identical to those in the current study, but without the additional “pulse dose” on the day of surgery, which is part of official nepafenac 0.3% dosing recommendations [9]. Interestingly, for the outcome of complete resolution of AC cells and flare by post-operative day 14 or 15, the percentage of patients using bromfenac 0.07% who achieved this outcome was higher in the current study (57.1%) than in a pooled analysis of data from two pivotal clinical studies which did not include the pre-surgical pulse dose (48.6%) [17]. The difference might be attributable, at least in part, to the extra pre-surgical dose employed in the current study. For nepafenac, the percentage of patients achieving complete resolution of AC cells and flare by post-operative day 14 or 15 in the current study (60.0%; data as observed) was slightly lower than that reported in a pivotal phase 3 study which also included a pre-surgical pulse dose (68.4%) [16]. The current study did not include a vehicle control; however, in prior studies, the percentages of vehicle-treated patients achieving complete resolution of AC cells and flare by postoperative day 14 or 15 ranged from 24 to 34% [16, 17].
The strengths of this study included patient randomization and masking of treatment to the investigator. Further, all post-operative evaluations were objective in nature and performed by a single, masked sub-investigator. Patients were not masked to treatment, but this should not have impacted the objective measurement of efficacy outcomes. Subjective measures of pain were not performed, which might be viewed as a limitation of the study, given the clinical relevance of this symptom. The small size of the study population was another limitation. Further, the study was conducted at a single center, thus limiting the generalizability of the findings.
Conclusion
In conclusion, the results of this small pilot study suggest that identically-dosed, once-daily regimens of bromfenac 0.07% and nepafenac 0.3%, including an extra “pulse” dose on the day of surgery, produce similar improvements in post-cataract-surgery measures of VA and retinal thickness, but that bromfenac may have a faster onset of action with regard to resolution of inflammation. Further, larger studies are needed to confirm these preliminary findings.
References
Hoffman RS, Braga-Mele R, Donaldson K, et al. Cataract surgery and nonsteroidal antiinflammatory drugs. J Cataract Refract Surg. 2016;42(9):1368–79.
Walters T, Raizman M, Ernest P, Gayton J, Lehmann R. In vivo pharmacokinetics and in vitro pharmacodynamics of nepafenac, amfenac, ketorolac, and bromfenac. J Cataract Refract Surg. 2007;33(9):1539–45.
O’Brien TP. Emerging guidelines for use of NSAID therapy to optimize cataract surgery patient care. Curr Med Res Opin. 2005;21(7):1131–7.
Lindstrom R. The pharmacologic and pathophysiologic rationale for using NSAIDs in ocular inflammatory disease and ocular surgery. Int Ophthalmol Clin. 2006;46(4):7–11.
Flach AJ. Topical nonsteroidal antiinflammatory drugs in ophthalmology. Int Ophthalmol Clin. 2002;42(1):1–11.
Ahuja M, Dhake AS, Sharma SK, Majumdar DK. Topical ocular delivery of NSAIDs. AAPS J. 2008;10(2):229–41.
Kim SJ, Flach AJ, Jampol LM. Nonsteroidal anti-inflammatory drugs in ophthalmology. Surv Ophthalmol. 2010;55(2):108–33.
Bausch & Lomb Incorporated. Prolensa® (bromfenac ophthalmic solution) 0.07% prescribing information. Bausch + Lomb, a division of Valeant Pharmaceuticals North America LLC, Bridgewater, NJ; 2016.
Alcon Laboratories, Inc. Ilevro™ (nepafenac ophthalmic suspension) 0.3% prescribing Information. Fort Worth, TX; 2019.
Donnenfeld ED, Holland EJ, Stewart RH, Gow JA, Grillone LR, Bromfenac Ophthalmic Solution 0.09% (Xibrom) Study Group. Bromfenac ophthalmic solution 009% (Xibrom) for postoperative ocular pain and inflammation. Ophthalmology. 2007;114(9):1653–62.
Silverstein SM, Cable MG, Sadri E, et al. Once daily dosing of bromfenac ophthalmic solution 0.09% for postoperative ocular inflammation and pain. Curr Med Res Opin. 2011;27(9):1693–703.
Nardi M, Lobo C, Bereczki A, et al. Analgesic and anti-inflammatory effectiveness of nepafenac 0.1% for cataract surgery. Clin Ophthalmol. 2007;1(4):527–33.
Lane SS, Modi SS, Lehmann RP, Holland EJ. Nepafenac ophthalmic suspension 0.1% for the prevention and treatment of ocular inflammation associated with cataract surgery. J Cataract Refract Surg. 2007;33(1):53–8.
Silverstein SM, Jackson MA, Goldberg DF, Muñoz M. The efficacy of bromfenac ophthalmic solution 0.07% dosed once daily in achieving zero-to-trace anterior chamber cell severity following cataract surgery. Clin Ophthalmol. 2014;8:965–72.
Singh RP, Lehmann R, Martel J, et al. Nepafenac 0.3% after cataract surgery in patients with diabetic retinopathy: results of 2 randomized phase 3 studies. Ophthalmology. 2017;124(6):776–85.
Modi SS, Lehmann RP, Walters TR, et al. Once-daily nepafenac ophthalmic suspension 0.3% to prevent and treat ocular inflammation and pain after cataract surgery: phase 3 study. J Cataract Refract Surg. 2014;40(2):203–11.
Walters TR, Goldberg DF, Peace JH, Gow JA, Bromfenac Ophthalmic Solution 0.07% Once Daily Study Group. Bromfenac ophthalmic solution 0.07% dosed once daily for cataract surgery: results of 2 randomized controlled trials. Ophthalmology. 2014;121(1):25–33.
Miyanaga M, Miyai T, Nejima R, Maruyama Y, Miyata K, Kato S. Effect of bromfenac ophthalmic solution on ocular inflammation following cataract surgery. Acta Ophthalmol. 2009;87(3):300–5.
Henderson BA, Gayton JL, Chandler SP, et al. Safety and efficacy of bromfenac ophthalmic solution (Bromday) dosed once daily for postoperative ocular inflammation and pain. Ophthalmology. 2011;118(11):2120–7.
Toyos MM. Comparison of once-daily bromfenac 0.07% versus once-daily nepafenac 0.3% in patients undergoing phacoemulsification. Ophthalmol Ther. 2019;8(2):261–70.
Al-Awadi A, Tokko HA, Chan CC, Somani S. Comparison of 2 regimens of loteprednol etabonate and bromfenac for cataract surgery. Can J Ophthalmol. 2019;54(3):388–94.
Donnenfeld ED, Nichamin LD, Hardten DR, et al. Twice-daily, preservative-free ketorolac 0.45% for treatment of inflammation and pain after cataract surgery. Am J Ophthalmol. 2011;151(3):420–6.e1.
Kraff MC, Martin RG, Neumann AC, Weinstein AJ. Efficacy of diclofenac sodium ophthalmic solution versus placebo in reducing inflammation following cataract extraction and posterior chamber lens implantation. J Cataract Refract Surg. 1994;20(2):138–44.
Acknowledgements
The author expresses his appreciation to the patients who participated in this study.
Funding
This study and publication (i.e., Ophthalmology and Therapy’s “Rapid Service Fee”) were funded by Bausch + Lomb, a division of Bausch Health US, LLC.
Medical Writing and Editorial Assistance
Statistical analyses were performed by Garrick Wallstrom, PhD of SDC (Statistics & Data Corporation), funded by Bausch + Lomb. Medical writing assistance was provided by Sandra Westra, PharmD of Churchill Communications (Maplewood, NJ), funded by Bausch + Lomb.
Authorship
Dr. Silverstein meets the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, takes responsibility for the integrity of the work as a whole, and has given his approval for this version to be published.
Disclosures
Dr. Silverstein is a lecturer and researcher for both Alcon and Bausch + Lomb.
Compliance with Ethics Guidelines
This study was performed in accordance with the International Conference on Harmonization Consolidated Good Clinical Practice Guidelines, and the ethical principles of the Helsinki Declaration of 1964 and its later amendments. The study protocol was approved by Quorum Institutional Review Board prior to screening of subjects. All subjects provided written informed consent before undergoing any study-specific procedures. No identifiable patient information is provided in this manuscript.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.9805418.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License ( http://creativecommons.org/licenses/by-nc/4.0/ ), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Silverstein, S.M. Bromfenac Ophthalmic Solution 0.07% Versus Nepafenac Ophthalmic Suspension 0.3% for Post-Cataract Surgery Inflammation: A Pilot Study of Identical Dosing Regimens with Pre-Surgical “Pulse” Dose. Ophthalmol Ther 8, 577–587 (2019). https://doi.org/10.1007/s40123-019-00215-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-019-00215-y