
Abstract
Attention-deficit/hyperactivity disorder (ADHD) is a common childhood-onset neurodevelopmental disorder characterised by persistent inattention, hyperactivity and impulsivity. Moreover, ADHD is commonly associated with other comorbid diseases (depression, anxiety, bipolar disorder, etc.). The ADHD symptomatology interferes with subject function and development. The treatment of ADHD requires a multidisciplinary approach based on a combination of non-pharmacological and pharmacological treatments with the aim of ameliorating the symptomatology; among first-line pharmacological treatments are stimulants [such as methylphenidate (MPH) and lisdexamfetamine dimesylate (LDX)]. In this review we explored recent ADHD- and stimulants-related literature, with the aim of compiling available descriptions of molecular pathways altered in ADHD, and molecular mechanisms of current first-line stimulants MPH and LDX. While conducting the narrative review, we applied structured search strategies covering PubMed/MEDLINE database and performed handsearching of reference lists on the results of those searches. The aetiology and pathophysiology of ADHD are incompletely understood; both genetic and environmental factors have been associated with the disorder and its grade of burden, and also the relationship between the molecular mechanisms of pharmacological treatments and their clinical implications. The lack of comprehensive understanding of the underlying molecular pathology makes both the diagnosis and treatment difficult. Few published studies evaluating molecular data on the mechanism of action (MoA) of MPH and LDX on ADHD are available and most of them are based on animal models. Further studies are necessary to improve the knowledge of ADHD pathophysiology and how the MoAs of MPH and LDX differentially modulate ADHD pathophysiology and control ADHD symptomatology.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Several lines of evidence suggest that the neurotransmitter imbalance, neuroinflammation and defective immunoregulation, circadian system dysfunction and altered neural viability and neurodegeneration are pathophysiological processes related to attention-deficit/hyperactivity disorder (ADHD) disorder and their comorbidities. |
We reviewed the literature for evidence that explains how the mechanisms of action (MoAs) of methylphenidate (MPH) and lisdexamfetamine dimesylate (LDX) act on these pathophysiological processes and control ADHD symptomatology. |
More evidence has been found on the effect of MPH and LDX on neurotransmitter imbalance and neural viability and neurodegeneration. Regarding their role in neuroinflammation and defective immunoregulation as well as circadian system, there are few data available. |
Despite the published studies on MPH and LDX, the few published molecular data available are based on animal models. Further studies are necessary to improve the knowledge of ADHD pathophysiology and how the MoAs of the MPH and LDX differentially modulate ADHD pathophysiology and control its symptomatology. |
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a common childhood-onset neurodevelopmental disorder characterised by a symptomatology based on persistent inattention, hyperactivity, and impulsivity that has been considered a progressively ceaseless condition [1]. The ADHD symptomatology interferes with subject function and development according to the Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-5) diagnostic criteria [2] impairing daily function [3, 4]. Moreover, ADHD is commonly associated with other comorbid diseases (depression, anxiety, bipolar disorder, etc.) [5], and in adult age it is a risk factor for various mental disorders [6, 7]. Therefore, ADHD is a significant burden for the affected youngsters, adults, their families and society everywhere.
The worldwide prevalence of ADHD is around 4–12% in children and 2.5–5% in adults [8,9,10,11]. It is estimated that 15–50% of children diagnosed with ADHD carry its manifestations into adulthood, but ADHD symptoms are not as easily defined in adulthood as in childhood. Generally speaking, symptoms are likely to adapt when growing into adulthood. Hyperactivity and impulsivity in adults seem to be reduced, as hyperactivity is expressed as an inner tension, and impulsivity becomes more verbal than physical, while inattentiveness can be retained [12, 13].
ADHD, as with other psychiatric disorders, is not easy to diagnose. In this sense, their diagnosis has been criticized because it is not based on a biological testing, and it has been considered subjective [14]. There is no medical test that determines the presence of the condition. However, there are some manuals that establish its diagnosis criteria, the DSM-5 and the International Classification of Diseases 11th revision (ICD-11) being the most usual ones, which observe behaviour defects and study symptoms such as hyperactivity, inattentiveness and impulsivity [15]. One of the reasons that hinder the diagnosis and treatment of this disorder is the lack of complete understanding of the underlying molecular pathology [16]. However, the main features of the diagnosis are the presentation of developmentally inappropriate levels of hyperactive-impulsive and/or inattentive symptoms, or both combined, for at least 6 months and at different settings, with possible impairment of life tasks. The disorder can affect highly intelligent people, often co-occurring with other psychiatric disorders [14].
Following a primary diagnosis of ADHD in a child, adolescent or adult, clinicians have at their disposal a wide range of non-pharmacological and pharmacological treatment options [17] which are administered in combination with a multidisciplinary approach [17, 18], as recommended by current treatment guidelines [19].
Non-pharmacological therapy includes several procedures as behavioural interventions, such as training of parent behaviour and/or social skills [20,21,22]; cognitive training, focused in reducing ADHD symptoms by improving performance in specific neuropsychological functions using electronic interfaces (computers, tablets, smartphones) or noncomputerized methods that allow performance reassessment so that training is adaptive [23,24,25]; neurofeedback based on improving self-control over brain activity patterns (real-time electroencephalogram (EEG) data monitoring of, for example, theta (vigilance) and beta (concentration and neuronal excitability waves) [26,27,28]; or coaching programs, focused on improving executive functions [29,30,31,32].
Pharmacologic treatment is based on stimulants [amphetamines, such as the prodrug lisdexamfetamine dimesylate (LDX), and methylphenidate (MPH)] and nonstimulants (atomoxetine, guanfacine, clonidine, bupropion, modafinil) [17]. Stimulants have generally been used as first-line pharmacologic treatment owing to a higher efficacy in symptomatology reduction compared to nonstimulant medications in all groups of age (children, adolescents and adults) [17, 33, 34]. In this sense, a systematic review and network meta-analysis carried out in 2018 using 133 double-blind randomised controlled trials positioned MPH in children and adolescents, and amphetamines in adults, as preferred first-choice medications for the short-term treatment of ADHD [34]. Nonstimulants are considered as second-line medication and are administered when stimulants are contraindicated or because of lack of response or intolerance [35].
Despite stimulant medications for ADHD being among the most effective drugs in psychiatry [36], there is a substantial placebo effect in subjects with ADHD [37], which could be explained by a synergy between placebo effects that influence the parent of the patient with ADHD and those acting on the clinician when interviewing the parent. Moreover, a nocebo effect affecting patient tolerability is reported, and it is mainly translated into dropouts due to adverse events or any other reason, and weight loss [37], indicating that explanation of potential adverse events due to medication must be expressed better to minimise nocebo effects.
Although the effects of stimulant medications are similar, they show different specific mechanisms of action (MoAs). Both MPH and amphetamines act by blocking presynaptic dopamine (DA) and norepinephrine (NE) transporters, thus increasing catecholamine transmission; however, amphetamines additionally increase the presynaptic efflux of DA [38]. In the case of LDX, the exact MoA in ADHD is not fully understood [39, 40]. It is presumed that is likely related to a blockage of the reuptake of NE and DA into the presynaptic neuron and an increase in the release of these monoamines into the extraneuronal space [39]. Despite this, the molecular mechanisms of stimulant medications are not fully understood. These mechanisms, or how patients respond to these drugs, can be further complicated by their interaction with patients’ genotypes; in this sense, pharmacogenomics research seeks to explain how some drugs are more effective and or better tolerated for specific genotypes in improving patient outcomes [41]. Most of the pharmacogenomic markers for psychiatric drugs compiled by regulatory agencies to date are related to drug metabolism rather than to the mechanism of action [42]. Some stimulants, such as amphetamines, and non-stimulants, such as atomoxetine, have been reported to be substrates of cytochrome P450s, and their performance has been linked to CYP2D6 genotype [41, 42]. While MPH’s US Food and Drug Administration (FDA) label does not contain pharmacogenomic biomarkers, a lot of research has been performed on the impact of genotype on MPH efficacy, and several biological pathways have been suggested to potentially affect MPH mechanisms through patient genotype, mostly concerning monoamine pathways [43,44,45,46,47,48]. Although the research in this field has been increasing in recent years, and has the potential to affect prescription and possibly improve the outcomes of patients with ADHD [49], no clear guidelines have been developed and there is still much to understand in this regard [50]. Our review will not cover this issue further, as this aspect has been extensively reviewed by other authors [41, 51,52,53].
The purpose of this review is to summarize the molecular evidence around the mechanisms of stimulant drugs on the molecular pathophysiology of ADHD in children and adults.
Methodology
While it is not a formal systematic review, we applied structured search strategies covering PubMed/MEDLINE database. In addition, we performed handsearching of reference lists in the articles identified through the structured searches.
To describe ADHD at the molecular level, we first explored the landscape of available molecular information on ADHD to obtain a picture of the pathophysiological processes involved. We reviewed indexed literature reviews in PubMed database, using the following search string (January 2020): (“Attention deficit hyperactivity disorder” [Title] OR “ADHD” [Title] OR “Attention-Deficit/Hyperactivity Disorder” [Title]) AND (“pathogenesis” [[Title/Abstract] OR “pathophysiology” [Title/Abstract] OR “molecular” [Title/Abstract]) AND Review [ptyp]; we explored these results full-length, and reviewed their list of references as well as PubMed “related articles” to completely cover all published molecular pathophysiological knowledge.
Then, we explored the molecular information around first-line stimulant drugs, to identify described direct protein targets and drug-induced molecular changes. Thus, we performed a literature search in PubMed database considering the drugs’ generic and commercial names and the following search string (April 2020): (“molecular” [Title/Abstract] OR “mechanism” [Title/Abstract] OR “pathophysiology” [Title/Abstract] OR “pathogenesis” [Title/Abstract] OR “mode” [Title/Abstract] OR “action” [Title/Abstract] OR “signalling” [Title/Abstract] OR “signalling ”[Title/Abstract] OR “expression” [Title/Abstract] OR “activation” [Title/Abstract] OR “inhibition” [Title/Abstract] OR “activity” [Title/Abstract]). Again, we explored these results full-length, and reviewed their list of references as well as PubMed “related articles” to identify relevant leads.
Finally, we used keywords from the ADHD pathophysiology description to identify published data relating these processes to the drugs and drug-related molecular changes. Equivalent searches and evaluation of the results were performed for both drugs. All the results were contextualised considering the potential clinical implications of these mechanisms as per the known role of the molecular mechanisms in behaviour regulation.
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
ADHD Aetiology and Pathophysiology
ADHD Aetiology
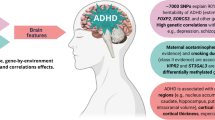
The ADHD aetiology is complex with a multiple pathophysiological entity involving multiple genetic and environmental factors, acting together, and creating a spectrum of neurobiological responsibility [54] (Fig. 1). In this sense, gender may play a role in ADHD aetiology with an incidence rate of 2:1 in boys versus girls [55].

ADHD aetiology, pathophysiological processes and symptoms scheme
Genetic Factors
Genetics has a great weight in ADHD, with a heritability ranging from 60% to 90% according to several studies carried out in twins [54, 56]. However, other studies using single-nucleotide polymorphism (SNP) analysis have estimated that SNP-based heritability ranged between 10% and 28% [57]. The variability in these percentages could explain the modulation in the ADHD manifestation, indicating a mixture of both dominant and recessive genes that work with complex patterns of polygenic transmission [54]. Another factor that supports an ADHD genetic background is the probability of developing ADHD being around 2–8 times more likely in the close relatives of patients with ADHD than relatives of unaffected individuals [58]. Several risk genes have been found in the human genome [59], but only a limited number of small impact size ADHD genes have been identified [60]. Genome-wide association studies (GWAS) have indicated genes playing a role in ADHD that could be linked to neuronal plasticity mechanisms (neuronal migration, cell adhesion and neuron proliferation) [61] or to deficiencies in the synthesis or release of neurotransmitters (NTs), mainly DA, but also serotonin and NE [56, 62]. However, ADHD-associated mutations have been reported to include a wide range of different physiological processes including circadian rhythm mechanisms [63].
Environmental Factors
The estimated involvement of environmental factors in ADHD pathogenesis is around 20–30% [64], highlighting prenatal, perinatal and postnatal difficulties, psychological adversities, exposure to chemical contaminants, or iron deficiency as factors that increase the probability of developing ADHD [54]; predisposing environmental factors increase the risk of developing ADHD based on a genetically or biologically determined vulnerability pattern.
Reduced maternal thyroid capacity, poor maternal wellness, maternal age, post-maturity of the foetus, long working hours, maternal stress, foetal stress, prenatal exposure to alcohol and tobacco, and antepartum haemorrhage are prenatal factors that could influence ADHD development [54, 58, 65]. Obesity and nutritional deficiencies (deficiency in the intake of essential fatty acids), lower serum levels of ferritin, and iron deficiency, as postnatal factors, have been suggested to promote abnormalities in the brain and thereby influence the onset of ADHD [66,67,68,69].
Regarding the influence of psychosocial adversity in ADHD disorder development, extreme parental discord, poor socioeconomic status, paternal violence, inconsistent parent–child relationships, maternal psychiatric disability, parental disapproval, family anxiety and foster placement have been reported as family random factors associated with behavioural disorders in childhood [58, 70].
Environmental factors such as chemical contaminants (pesticides and harmful agricultural chemicals) may promote the neural structure impairments involved in ADHD [58].
Comorbid Conditions
ADHD disorder is usually accompanied by other comorbid conditions. In fact, patients with ADHD are frequently diagnosed with another neuropsychiatric conditions, which can mask ADHD [71].
A significant correlation has been demonstrated between intellectual disability and ADHD onset [72]. Then, intellectual development that is conditioned by cognitive and behavioural problems associated with ADHD [72] precedes the onset of ADHD. In this sense, a specific polymorphism of the brain-derived neurotrophic factor (BDNF) is significantly associated with both ADHD and intellectual disability [73]. In fact, intellectual disability appears before ADHD and it might condition the presence of ADHD.
A wide variety of other neurodevelopmental disorders and cognitive and behavioural problems have been found in ADHD: dyslexia, reasoning disorder, dyscalculia, written language expression disorder, language difficulties, motor control difficulties and also autism spectrum disorders [72].
Furthermore, ADHD has demonstrated a high degree of comorbidity of other psychological and mental conditions such as insomnia, anxiety, conduct disorder, sleep disturbance, oppositional defiant disorder, binge eating disorder, tic disorder, bipolar disorder, personality disorder, drug and substance dependence and mood disturbances, among others [72, 74]. Most of these conditions/disorders usually appear later in development and can manifest as a result of ADHD, but this is not always the case with early stage behavioural problems directly related to ADHD [58].
Pathophysiological Processes and Specific Molecules Involved in ADHD
ADHD pathophysiology is largely unknown. There are several causative cellular and molecular processes related to the ADHD development, among which NT imbalances or alterations in neuronal plasticity and neuronal survival are very well described. Nevertheless, other less studied processes are gaining relevance as regards their involvement in ADHD, such as the role of neuroinflammation and alteration of circadian rhythms.
Neurotransmitter Imbalance
The prefrontal cortex (PFC), caudate and cerebellum are the main brain regions responsible for ADHD development [75]. These regions are involved in controlling consciousness, feelings, impulses, attitudes and behaviour [76]. Slower maturation or decreased size of the PFC and also diminished PFC, caudate or cerebellum activation have been found in patients with ADHD [77, 78]. The pathway function within such regions is highly responsive to the neurochemical setting and is regulated by NTs such as DA, NE, serotonin (5-HT), glutamate and glutamate/gamma-aminobutyric acid (GABA) [75]. Deficiencies in catecholamines such as NE and DA [75, 79, 80] or dysregulation of such NTs, which are essential for normal brain function, including executive and attentional functions [33], are among the causing physiological alterations that have been reported to explain ADHD development.
DA Deficiency
DA plays an essential role in mediating regulation of the cortical system, memory, mood, vascular structure, anticipation of events, motivation, behaviour inhibition, decision-making and problem-solving [16, 81, 82]. Dysregulation of the dopaminergic system is related to the ADHD symptoms and signs [54]. This system is involved in motivation and reward/avoidance behaviours; in this sense, lower DA levels in the PFC could lead to less motivation, resulting in inattention. This might also be the reason for the promiscuous reward-seeking behaviours found in hyperactive-impulsive patients with ADHD [83].
DA interacts with five different receptors localized in postsynaptic neurons [75], which are dysregulated in several brain regions of patients with ADHD. In fact, the dysfunction of DA receptors (DRD1–5) and DA transporter (DAT-1) is the main reason for the altered activity in the dopaminergic system, playing a significant role in the pathogenesis of ADHD [84]. Moreover, DAT-1 is a DA transporter whose function is the reuptake of extracellular DA in the synaptic space [83]. Increased density of DAT-1 in patients with ADHD has been reported, resulting in inadequate extracellular DA levels in these brain regions. MPH and amphetamine-derived compounds (AMPH), the two most widely used drugs for ADHD, act on DAT-1 by inhibiting its transporter function and thereby increasing the levels of extracellular DA [75].
NE Deficiency
NE, also known as noradrenaline (NA), is an important NT in behaviour control, playing a key role in high-level cognitive processes such as working memory and inhibitory response, which appear to be disrupted in ADHD [79], producing irritability and anxiety [85]. NE is also involved in regulating attention [86], in particular those related to emotional and neutral emotions [87], and this signalling network has been found altered in different brain areas in patients with ADHD [88]. NE-mediated signal transduction speed and duration are affected by NE transporter (NET) protein, as it is involved in NE reuptake [79]. Abnormalities in NET function contribute to ADHD development by decreasing the extracellular NE levels. Therefore, treatments for ADHD, such as MPH and AMPH, increase the reservoir of NE and thereby attenuate the hyperactivity and impulsivity experienced in ADHD [75].
Serotonin (5-HT) Imbalance
The imbalance of excitatory serotonin function has also been related to ADHD development [89, 90]. Serotonin dysfunction plays a mediator role in ADHD-related hyperactive and impulsive behaviours [91] inducing impaired control of impulses, violent behaviour, concentration and appetite [89]. Moreover, serotonin regulates DA activity through its receptors 5-hydroxytryptamine receptor 1B (5-HTR1B) or 5-hydroxytryptamine receptor 2A (5-HTR2A). The dysfunction of these receptors can lead to problems of serotonin–DA dynamics, resulting in ADHD symptoms [89, 90]. In addition, other proteins related to 5-HT metabolism, such as tryptophan 5-hydroxylase 2 (TPH2) and nitrous oxide synthase 1 (NOS-1), which reduces the serotonin transporter (SERT or SLC6A4) cell surface localization increasing the levels of extracellular serotonin, might be involved in the pathophysiology of ADHD [90, 92].
Glutamate Imbalance
Glutamate signalling is the most common excitatory synapse NT [93] and deficiencies in this signalling has been associated with ADHD symptoms [94], although it is unknown whether ADHD is associated with differences in glutamate levels [95,96,97].
GABA Imbalance
Alteration of glutamate/GABA equilibrium has been related to a diminished capability to focus on demanding tasks [98] and to result in DA dysregulation, causing inhibition of defective pyramidal neurons and a decrease shift in excitation towards tasks regulated in the PFC [99, 100]. In this sense, higher glutamate levels and reduced GABA levels have been observed in children with ADHD, which indicates a bias towards neuron excitation that might be implicated in hyperactivity induction, although without a firm conclusion [101].
Neuroinflammation and Defective Immunoregulation
Neuroinflammation is hypothesized to affect brain growth and thereby raises the risk of neurodevelopmental disorders through pathways such as glial activation, increased oxidative stress, problems related to neuronal growth and impaired NT activity [60]. In ADHD, low levels of cortisol, an important hormone in the hypothalamus–pituitary–adrenal (HpA) axis, have been observed in children [102,103,104,105]. The HpA axis is a hormonal system that controls stress responses [106] and affects several hormonal behaviours [107, 108]. Furthermore, HpA axis involvement in typical ADHD comorbidities has been reported [109]. This promotes an imbalance in pro-inflammatory T helper 1 cell (Th1)/anti-inflammatory T helper 2 cell (Th2) cytokine profile making a more pro-inflammatory environment in ADHD [60, 102], which can be related to a defective regulatory T cell (Treg) regulation. Pro-inflammatory cytokines such as tumour necrosis factor beta (TNFβ), interferon alpha (IFNα), interleukin-1 (IL-1), IL-5, and IL-16 are found upregulated, whereas the anti-inflammatory cytokines IL-4 and IL-13 are downregulated, resulting in an inflammatory process that causes neuronal damage and contributes to ADHD symptomatology [60]. This dysfunction of the immunoregulation by Treg cells and imbalance in the Th1/Th2 response might be a crucial factor in the development of allergies in children with ADHD [110], or a factor relating Th2-mediated hypersensitivity to ADHD development [111, 112].
Circadian System Dysfunction
Circadian rhythm regulates some biological activities such as sleep and mood [113] through various mechanisms such as the transcription-translation feedback loop (TTFL), which includes two transcription activators: circadian locomotor output cycles kaput (CLOCK) and brain and muscle ARNT-like protein 1 (BMAL1). Both activators create a heterodimer, which inhibits the transcription of repressor proteins Period 2 (PER2) and cryptochrome (CRY) that regulates the daily rhythms of locomotor activity, metabolism and behaviour [114]. Mutations in CLOCK gene have been associated with ADHD, indicating that the disrupted circadian pathways could exacerbate the symptoms of this disease [113], including ADHD-associated aggressiveness symptoms [115]. Moreover, the circadian system regulates dopaminergic system and therefore DA production is subjected to circadian rhythm variations [113]. In this sense, the dopaminergic DA receptor 4 (DRD4) gene is strongly involved in translating light into electrical signals in the retina, and there is a strong circadian pattern in its transcription [63].
ADHD also involves a delayed sleep-phase symptom (sleep disturbances) typical of sleep onset insomnia [116] and other behavioural symptoms related to ADHD [63]. In children with ADHD, irregular sleep cycles, troubled sleep initiation, reduced sleep effectiveness with greater wakefulness periods and increased nocturnal activity have been observed, among others [117]. Although delayed sleep-phase is not enough to developed ADHD symptoms, it has been reported that treatments for sleep problems have promoted a reduction of ADHD symptoms [63]. Moreover, a deficit in melatonin levels has been found in adults and children with ADHD [117], and melatonin has been used as a treatment for children with sleep problems diagnosed with ADHD [118].
Sleep disturbances and deficit of alertness have also been found to be linked to abnormal eating behaviours, including binge eating, which is a common ADHD comorbidity; this link is hypothesized to involve the hypocretin/orexin system through hypoactivation of hypocretin/orexin neurons from perifornical and dorsomedial hypothalamic neurons (involved in arousal) and overactivation of hypocretin/orexin neurons from lateral hypothalamus (involved in reward seeking, including feeding) [119]. Understanding these pathways can lead to the development of therapies for patients with ADHD that improve their wakefulness and reduce abnormal reward-seeking behaviours (such as binge eating).
Altered Neural Viability and Neurodegeneration
Neuronal network morphogenesis is based on an effective equilibrium between neuroplasticity and neurodegeneration, as well as the formation of new cells through proliferation/survival signalling and cytoskeletal structures [120]. ADHD seems to present a morphogenesis dysregulation [92, 121], with a high number of neurophysiological difficulties caused by neuronal network dysfunctions in the brain. It has been observed that the inability to modulate neural connections can promote a reduction of brain volumes and inefficient neural networks [92]. These impairments might be the result of neurodegeneration, neurogenesis problems, altered proliferation–differentiation balance, deficient neurotrophic factors and, thereby, loss of neuronal viability [92]. Besides, oxidative stress results in a notable problem in terms of neuronal damage which can lead to all of the defects mentioned above [92].
There are several proteins and molecular processes involved in neuronal survival, including trophic factors, NTs or neuronal function regulators. One such protein involved is BDNF, which stimulates neuronal development and maintenance, regulates NT function and contributes in neuronal plasticity processes, such as long-term potentiation and learning [122]. BDNF dysregulation is associated with ADHD. Neurotrophin-3 (NTF-3) has been also related to neuron survival [92]. Furthermore, it has been reported that NTF-3 promotes bone marrow neural stem cell proliferation and differentiation into cholinergic neurons [123]. Glutamate and some of its receptors are also involved in neurogenesis and proliferation [92]. Alterations in glutamate receptors (GRM-1, GRM-5, GRM-7 and GRM-8) have been associated with ADHD [124]. Other proteins related to proliferation and neurodevelopment that are altered in ADHD are cyclic AMP response element-binding protein (CREB), cyclin D1 and NOS-1, cadherin-13 (CADH13) and E3 ubiquitin-protein ligase parkin (PARK2) [92].
Among the molecular processes involved, it has been shown that patients with ADHD present an ineffective oxidative stress response [125]. In this sense, paraoxonase-1 (PON-1) (antioxidant agent) has been reported to be downregulated in patients with ADHD [126]. Also, it has been shown that ephrin stimulates excitatory synapses development [127,128,129] and is closely related to neurogenesis, plasticity and learning [93]. Moreover, it has been suggested that ephrin family members have a role in ADHD-related synaptogenesis alterations [92, 130].
Molecular Mechanism of Stimulant Treatments
In order to obtain a full picture of the MoA of stimulants (MPH and LDX), one must consider not only direct targets but also other molecular mediators known to be modulated by the drugs. In the following sections, known and well-established drug targets are reviewed, along with other molecular changes induced by either MPH or LDX, which might be valuable information to help fully understand the molecular consequences of stimulant treatment and how these mechanisms impact clinical observations.
Methylphenidate (MPH)
MPH is a NE–DA reuptake inhibitor; therefore, it causes an increase of catecholamines in the synaptic cleft [shown by positron emission tomography (PET) imaging] [131]. It is the first-line pharmacological treatment recommended in current guidelines [132, 133] for children of at least 6 years of age and adults with ADHD. MPH is the most commonly prescribed medication to treat ADHD [134, 135].
MPH has several formulations, including novel modified release systems to extend its effect. The main slow release systems are Concerta®, an osmotic release system (OROS technology) with 22% immediate and 78% extended release; Medikinet®, modified release (also known as Medikinet XL® or Medikinet Retard®), which is based in a multiarticular beads release, that combines 50% immediate and 50% extended release [136]; Equasym XL® which is based on capsules containing 30% immediate and 70% extended release beads [137]; and Rubicrono®, an extended release formulation based on MPH hydrochloride [138].
Drug Molecular Targets and Drug-Induced Molecular Changes
MPH has a psychostimulant effect through different direct target proteins, and several drug-induced molecular changes have also been described for MPH.
MPH has been reported to modulate with pharmacological action three human protein targets (Table 1): it acts as an inhibitor of DAT (SLC6A3) [139] and NET (SLC6A2) [139] (both transporters of DA and NE, respectively), and as an agonist for the 5-hydroxytryptamine (serotonin) receptor 1A (HTR1A) [140, 141]. All these targets are involved in the presynaptic signalling; therefore, MPH has a role in regulating three main NT pathways: DA, NE and serotonin (Fig. 2).

Mechanism of LDX/MPH in the synapse: 1. Patient with ADHD without treatment. 2. Patient with ADHD treated with MPH 3. Patient with ADHD treated with LDX
Besides these direct targets, the indirect modulation of several other proteins has been reported for MPH. First, other NT pathways have been reported to be stimulated by MPH, including adrenergic, DA and glutamate receptors (e.g. alpha-2A adrenergic receptor [ADRA2A] and alpha-2B adrenergic receptor [ADRA2B], beta-1 adrenergic receptor [ADRB1], DA receptor 1A [DRD1], DA receptor 1B [DRD5], DA receptor 2 [DRD2]) and glutamate receptor 1 (GRIA1) [142,143,144]. Furthermore, MPH has been reported to modulate neurotrophic factors involved in neuronal survival and plasticity, such as BDNF and its BDNF/NT-3 growth factor receptor (NTRK2); the effect of MPH over these factors is unclear, since some experiments report inhibition [145], whereas others report activation, improving BDNF plasma concentrations in children with ADHD [146].
MPH is also able to modulate other more general cellular processes, such as apoptosis, which might also impact neuronal survival. The apoptosis-related proteins reported to be modulated by MPH include apoptosis regulator Bcl-2 (BCL2), apoptosis regulator BAX (BAX), caspase-3 (CASP3) and cytochrome c1, haem protein, mitochondrial precursor (CYC1) [147]. Interestingly, canonical inflammation mediators, such as interleukin-1 beta (IL-1B) and TNF are induced in MPH treatment, suggesting a potential immunomodulatory role for the drug, although the evidence does not readily support it. CREB1, involved in synchronization of circadian rhythmicity, has also been shown to be modulated by MPH treatment [148]. Finally, MPH has been reported to modulate intracellular mediators and transcription factors involved in neuronal signalling, including adenylate kinase isoenzyme 1 (AK1), mitogen-activated protein kinase 3 (MAPK3), proto-oncogene c-Fos (FOS), calcium/calmodulin-dependent protein kinase type IV (CAMK4) and Ras-related C3 botulinum toxin substrate 1 (RAC1) [149].
Lisdexamfetamine Dimesylate (LDX)
LDX is the first stimulant prodrug indicated and recommended by guidelines in children at least 6 years of age when the MPH treatment is not optimal [132, 150], and also as first-line treatment in adults [133, 151] and for binge eating disorder [40] in the USA. Elvanse® (Vyvanse® in the USA) or LDX is a long-term release prodrug of dextroamphetamine (d-amphetamine) [152]. LDX is itself pharmacologically inactive, but following oral administration it is converted by rate-limited enzymatic hydrolysis to l-lysine and d-amphetamine [152]. The active form of the drug has a central nervous system stimulating activity by the primary inhibition of DAT, NET, trace amine-associated receptor 1 (TAAR1) and vesicular monoamine transporter 2 (SLC18A2), among other targets, therefore regulating the reuptake and release of catecholamines (primarily NE and DA) on the synaptic cleft [153].
Once administrated, LDX is converted to the active drug d-amphetamine, through cleavage of the l-lysine (an essential amino acid) in the bloodstream carried out by the erythrocytes. All the subsequent analysis was carried out taking into account the active compound, since the parental prodrug is biologically inactive [154].
Drug Molecular Targets and Drug-Induced Molecular Changes
LDX has been reported to modulate with pharmacological action of seven human protein targets (Table 2). On the one hand, it acts as activator of TAAR1. On the other hand, six protein targets are inhibited by LDX: synaptic vesicular amine transporter (SLC18A2), DAT (SLC6A3), NET (SLC6A2), SERT (SLC6A4) and amine oxidase [flavin-containing] (MAO) A and B [153,154,155,156,157]. All of these targets are involved in the presynaptic signalling, and LDX therefore has a role in regulating three main NT pathways: DA, NE and serotonin. Moreover, LDX has a role as a neuromaintenance agent since it inhibits MAOA and MAOB oxidative enzymes (Fig. 2).
LDX has been reported to indirectly modulate several proteins. First, LDX induces activation of the known dopaminergic and adrenergic receptors DA receptor 1A (DRD1), DA receptor 1B (DRD5), DRD2, ADRA2A, ADRA2B and alpha-2C adrenergic receptor (ADRA2C) [155, 158, 159], contributing to neurotransmission signalling. Furthermore, LDX has been reported to modulate several inflammation mediators such as TNF, IL-4, IL-6 and IL-10 [160]. As it occurs with MPH, TNF is induced upon LDX treatment, suggesting a potential immunomodulatory role for the drug. Other cytokines stimulated by LDX are IL-4, IL-6 and IL-10, with IL-4 and mainly IL-10 showing an anti-inflammatory profile, suggesting LDX may have a role in the reduction of inflammatory response.
MPH vs LDX: Similarities and Differences
Similarities
MPH and LDH have similar targets and act in the same way: both promote the inhibition of DAT and NET [153, 156, 157]. Regarding the indirect drug-induced molecular changes, both drugs promote the activation of DRD1, DRD5, ADRA2A and ADRA2B, then modulate the dopaminergic and adrenergic signalling, therefore regulating the reuptake and release of catecholamines (primarily NE and DA) on the synaptic cleft [131, 153].
Likewise, TNF is indirectly activated by both drugs, which implies a potential immunomodulatory role for MPH and LDX. Although the role of the effect of both drugs on TNF is unknown, it has been reported that TNFα promotes astrocyte activation [161]. In an ADHD comorbid disease, major depressive disorder, activated astrocytes can reduce glutamate on the synaptic cleft via excitatory amino acid transporters (EAATs) and convert it to glutamine in the cytosol [162]. In this sense, higher glutamate levels have been observed in children with ADHD [101]. Then the activation of TNF could improve the synaptic signalling. On the other hand, in vitro studies carried out in astrocytes have shown that cytokines such as TNF, among others, support the production of neuroprotective mediators [163]. More studies are necessary to understand the role of the effect of both drugs on TNF in ADHD.
Differences
MPH and LDX have different target profiles that could underlie differences in their MoAs. MPH activates HTR1A [140, 141], which induces a partial release of DA, thereby improving the presynaptic signalling. LDX inhibits the activity of two other important proteins, not shared with MPH, involved in the monoaminergic system described in ADHD: SERT [164] and MAO [165]. The inhibition of DAT1, targeted by both drugs, leads to no reuptake of the DA to the cytosol [166]; LDX also reduces monoamine degradation by MAO inhibition [167, 168]. LDX is also a specific agonist of TAAR1 [169], a protein involved in DA pathway modulation [170]. Another target protein activated by LDX is SLC18A2 (also known as VMAT2) that contributes to regulating the reuptake and release of catecholamines (primarily NE and DA) on the synaptic cleft [153].
More differences were found among drug-induced molecular changes for both drugs. MPH has a broader range of modulation encompassing dopaminergic and adrenergic neurotransmission, inducing the activation of ADRB1 and DRD2 [143, 144] and modulating proteins involved in neuronal survival such as BCL2, BAX, CASP3 and CYC1 [147], acting as an immunoregulator owing to its effect on TNF [148] and affecting circadian rhythm through CREB1, involved in its synchronization [148]. The modulation of LDX involves dopaminergic and adrenergic neurotransmission, inducing upregulation of ADRA2A, ADRA2B, ADRA2C, DRD1 and DRD5 [155, 158, 159], and immunomodulation by modulating several interleukins, mainly IL-4 and IL-10 that present an anti-inflammatory profile [160].
When considering these differences, it is important to remark that the information available may be biased, and therefore it must be interpreted cautiously, as it only informs on data reported for one of the drugs.
Impact of Mechanisms on Clinical Observations
MPH and LDX show an impact on the aforementioned causative processes related to ADHD development (NT imbalance, neuroinflammation and defective immunoregulation, circadian system dysfunction and altered neural viability and neurodegeneration) owing to both drugs’ activity in ADHD symptomatology control.
Neurotransmitter Imbalance
MPH
MPH exerts its action promoting the downregulation of DA and NE transporter (DAT and NET) (protein targets) [139]. This leads to an increase of DA and NE levels in the prefrontal cortex (synaptic cleft) that control hyperactivity and deficits in inhibitory behaviour (improvement in attentional deficit and cognitive functioning) [139, 171]. Nevertheless, the molecular mechanisms underlying their action are poorly understood. The interaction of MPH with several proteins involved in the neurotransmission process could explain its MoA (Fig. 3).

Relationship between function and behavioural effects of monoamines. Direct targets of the stimulant drugs are indicated in each monoaminergic pathway
Regulation of Presynaptic Receptors
DA accumulation after MPH blockade of DAT induces disinhibition (upregulation) of the presynaptic receptor DRD2 (dopaminergic neuron) and activates D1 receptors on the postsynaptic neuron that uptake DA, continuing the neuronal transmission and therefore improving attention, focus and organized thoughts and actions [172, 173].
On the other hand, MPH activates HTR1A and DRD2 (and potentially other G-coupled receptors) that might have a reinforcing feedback role on MPH modulation of the NT imbalance through further regulation of DAT1 [174]. Moreover, HTR1A induces a partial DA release which in turn improves the presynaptic signalling [140, 141].
Regulation of Postsynaptic Receptors
In PFC neurons, MPH indirectly activates the postsynaptic DRD1 and DRD5 [142] that uptake DA and transmit the signal in postsynaptic neurons [175]. The stimulation of these receptors probably leads to the GRIA1 receptor phosphorylation promoted by MPH, as described by Pascoli et al. [143].
MPH also upregulates ADRA2A, ADRA2B and ADRD1B receptors located in postsynaptic neurons. The assessment of the effect of MPH on the two major signalling pathways (cAMP-dependent protein kinase [PKA] and extracellular signal-regulated kinase [ERK]) in PFC [143] showed no activation of ERK2 phosphorylation. However, the MPH effects on these signalling pathways were totally inhibited using ADRD1B blockers, showing that ADRD1B mediates the phosphorylation of PKA signalling through MPH in the PFC [143]. Moreover, it has been shown that MPH exerts excitatory actions on PFC neurons by activating ADRA2 [175, 176]. In this sense, NE neurotransmission seemed also to be modulated via ADRA1 and ADRA2 [143].
LDX
LDX controls the neuronal transmission, both at presynaptic (direct) and postsynaptic (indirect) levels. This results in an amplification of DA activity and an improvement in attentional deficit and cognitive functioning, as well as a reduction in hyperactivity (Fig. 3).
Regulation of Presynaptic Receptors
LDX inhibits DAT and NET transporters leading to an increase in DA and NE levels in the synaptic cleft [153, 156, 157]. LDX can also promote the increase of DA in the synaptic cleft by activating protein TAAR1, which produces the efflux of monoamine NTs, mainly DA, from storage sites on presynaptic neurons [155, 177]. TAAR1 activation leads to intracellular cAMP signalling that results in PKA and PKC phosphorylation and activation [178,179,180]. This PKC activation decreases DAT1 [181], NET1 [182] and SERT [183] cell surface expression, intensifying the direct blockage of monoamine transporters by LDX and improving the neurotransmission imbalance in ADHD. In addition, PKC might induce p38 MAPK-mediated inactivation of AKTs [184, 185], proteins essential for DAT cell surface redistribution due to AKT induction of actin reorganization, regulating the dopamine efflux impaired in ADHD [186].
The increase of DA promotes the dopaminergic transmission by regulating (inhibiting) other proteins: VMAT2, whose inhibition induces the DA liberation from vesicular storage and the concomitant release of cytosolic DA via DAT reverse transport [33, 187], SERT (with weaker affinity), and MAOA and MAOB [153].
Regulation of Postsynaptic Receptors
LDX amplifies dopaminergic transmission in the mesolimbic and mesocortical tracts (two major pathways in cognition and memory) [155]. The cognitive-enhancing effects of amphetamine occur through the dopaminergic transmission activation which, in turn, is mediated by both D1 dopamine receptors and α2-adrenoceptors in postsynaptic neurons in PFC [188].
The DRD1, DRD5, ADRA2A, ADRA2B and ADRA2C are activated downstream by LDX [155, 158, 159]. The effects on D1 receptors increase the locomotor activity [189], and the stimulation of ADRA2 receptors results in a profound inhibition of the spontaneous firing rate of locus coeruleus neurons [190] that could improve the efficacy of LDX on symptom control. Moreover, the indirect activation of α1-adrenoceptors and DRD1 increases the catecholaminergic neurotransmission [191].
The effect in PFC of d-amphetamine activation of DRD and ADRA2 receptors on the two major signalling pathways (PKA and ERK) has been also analysed [143]. d-Amphetamine activated the phosphorylation of both pathways through ADRD1B [143]. ADRA blockers did not inhibit the ERK phosphorylation although its effect was slightly reduced; this reflects the major role of DRD in ERK activation by d-amphetamines with a slight contribution of ADRA2 [143]. Moreover, d-amphetamine phosphorylates GRIA and, as with MPH, it could derive from the stimulation of dopamine D1/D5 receptors [143].
Probably minimal differences such as ERK2 phosphorylation and the action on locus coeruleus neurons by LDX could serve as a differentiator to determine the efficacy level of each drug.
Neuroinflammation and Defective Immunoregulation
Neuroinflammation is hypothesized to increase the risk of neurodevelopmental disorders and impaired NT activity [60].
MPH
MPH presents a pro-inflammatory profile. Several studies have exposed that the long-term use of MPH induces the DA neuron loss and microglia activation causing an increase in pro-inflammatory markers TNFα and IL-1β, among others, resulting in the activation of neuroinflammation and triggering a neurodegenerative process [192, 193]. Therefore, their long-term use must be considered the result of loss of DA transmission and, in turn, efficacy.
On the other hand, the use of MPH could create dependence or abuse [194, 195]. The chronic abuse of MPH also induces the expression of inflammatory cytokines, such as TNFα and IL-1β [148, 196, 197]. With MPH at 10 mg/kg, an increase of the inflammatory markers in the amygdale has been observed [148].
LDX
It is important to remark that the inflammatory profile of LDX has not been evaluated in an ADHD context. It has been assessed in ADHD comorbid disease, such as bipolar disorder in animal models [160], in which LDX increased the pro-inflammatory cytokines TNFα and IL-6, and also the anti-inflammatory cytokines IL-4 and IL-10 in the frontal cortex, striatum and serum [160, 198]. However, a recent study did not show LDX having an effect on IL-10 (anti-inflammatory cytokine, upregulated to control the inflammatory response duration and intensity) neither on TNFα nor on IL-1β (pro-inflammatory cytokines) [199]. On the other hand, MAOA inhibition and TAAR1 activation by LDX (both direct targets of LDX) might influence MAPK3 activation, which is crucial for the downstream modulation of a large number of proteins involved in many biological processes, such as neuroinflammation [200]. MAPK3 can induce the expression of some anti-inflammatory cytokines (e.g. IL-2, IL-10), downregulated in patients with ADHD, via NFκB (NFKB1) activation [200,201,202].
Recent evidence suggests the involvement of the immune system in several psychiatric conditions [203,204,205]; one of the most studied in this respect is depression [206,207,208]. Given the role of LDX in modulating inflammatory and immunomodulatory molecules in psychiatric condition settings, a possible role of LDX in modulating the immune system in other psychiatric conditions, such as depression or ADHD, could be expected. Thus, more studies are required to understand the inflammatory profile of LDX in ADHD.
Circadian System Dysfunction
MPH
As previously exposed, ADHD is related to a delayed sleep-phase [116]. MPH increases the activity at mid-to-late night and leads to a delay in sleep relative to the light–dark cycle [209]; but the mechanisms of MPH involved in the circadian rhythm are unknown. One study in ADHD that investigated markers of oxidative stress and inflammation showed that MPH downregulated c-FOS (gene involved in circadian rhythm) [148]. Another study that assessed the impact of MPH on CLOCK gene protein expression showed that MPH increased c-Fos expression. Although the authors did not consider its influence on the circadian functioning of the cerebral cortex, MPH causes a widespread increase in neuronal activation throughout the region [210].
Other hormones involved in circadian rhythm are serotonin and melatonin [211, 212]. Healthy children have higher 5-HT concentrations in the morning compared with patients with ADHD, with very similar concentrations in the evening [212]. In that study, MPH treatment induced a decrease of 5-HT levels in the evening (without changes in the morning); the effect of MPH on melatonin concentrations was a decrease in the morning and an increase in the evening, normalizing these values to those observed in the non-ADHD population [212].
It is interesting to note that the role of MPH as an inhibitor of DAT1 [174] and 5-HT1A [141] leads to inhibition of circadian clock regulators CRY1 and CRY2. These CLOCK components CRY1 and CRY2, implicated in the ADHD-associated dysregulation of the circadian system [213], are modelled through AMPK activity by MPH improving the impairment of this system [214]. This mechanism could explain the effect of MPH in circadian rhythm regulation.
LDX
We did not find evidence on whether the MoA of LDX induces improvements in circadian rhythm in ADHD.
Altered Neural Viability and Neurodegeneration
MPH
Neuronal Survival
Motaghinejad et al. showed that MPH downregulates CREB1 [148], a major transcription factor in brain development and neurogenesis [215], and its product BDNF, which acts downstream in the CREB pathway signalling [148]. This mechanism observed with the use of amphetamines leads to neurodegeneration and lower cell survival [216].
Other proteins related to neural viability are MAPK3 and AK1, both downregulated by MPH [148]. These proteins act upstream in the CREB pathway signalling favouring CREB/BDNF phosphorylation and the activation of this signalling pathway [148].
BDNF has a relevant role in synaptogenesis regulation and stimulates neuronal development and maintenance [122, 217]. Disruption of BDNF and its downstream signals has been found in ADHD correlating with severity of ADHD symptoms [218, 219]. MPH increases plasma concentrations of BDNF in children with ADHD, although in that study the authors found that lower baseline plasma BDNF levels have a positive effect on hyperactivity symptoms [146]. It has been postulated that higher levels of BDNF could compensate dysfunctions of DA and 5-HT systems observed in neurodevelopmental disorders [122, 220], suggesting that higher levels of BDNF in subjects with ADHD are related to more profound NT dysfunction [146]. On the other hand, more recent studies have shown that the expression of BDNF is downregulated by MPH [145, 148]. The use of neuroprotective agents reverses the effects of MPH, increasing the expression of CREB and BDNF [148, 221].
c-FOS, as does BDNF, acts downstream in the CREB pathway signalling [148]. The role of MPH on c-FOS is unclear. It has been described that c-FOS is downregulated by MPH [148]. However, it has also been described that MPH increases c-FOS level in a study that assessed the impact of MPH on circadian rhythm [210] and c-FOS expression in the medial septum (area intimately connected to the hippocampus) [222].
Apoptosis Control
Another way of acting on neural viability and neurodegeneration is through apoptosis control. BAX (pro-apoptotic) and BCL2, CASP3 and CYC1 (anti-apoptotic) proteins have been regulated by MPH, although the MoA of MPH remains elusive as MPH effects were dependent on age and brain area [147].
Oxidative Stress
Several studies have shown that MPH modulates oxidative stress in the brain [223, 224], either increasing or reducing reactive oxygen species (ROS) depending on age, drug dose and brain region, and relate that to neural viability. In this sense, Coelho-Santos et al. demonstrated that MPH promotes ROS generation via activation of RAC1-dependent NADPH oxidase (NOX) and c-Src activation [149].
Another protein related to oxidative stress is CAMK4, whose expression is activated by MPH; it has been suggested that CAMK4 increase could amplify calcium influx and be involved in the increase of oxidative stress and inflammation [148].
Taken in combination, MPH could induce apoptosis and neurodegeneration through its action on the CREB signalling pathway and oxidative stress generation through RAC1 and CAMK4. Whether these pro-neurodegenerative effects can be related to MPH misuse is not clear.
LDX
Oxidative Stress
LDX causes oxidative imbalance through increased lipid peroxidation, protein oxidation and alterations in the activity of antioxidant enzymes in some brain areas [225].
LDX increases the thiobarbituric acid-reactive substances in the cerebellum, hippocampus and cerebral cortex, although its MoA is unknown [225]. One plausible MoA related to the oxidative stress caused by LDX is the regulation of antioxidant enzymes such as catalase (CAT), glutathione peroxidase (GSH-Px) and superoxide dismutase (SOD). In this sense, two studies have shown that the levels of these enzymes were decreased by LDX in neuropsychiatric disorder models [225, 226].
Conclusions
Based on the literature published, the MoAs of both LDX and MPH (current first-line pharmacological treatments) have an effect in the network of the specific molecules involved in the described pathophysiological motives of ADHD.
Regarding NT imbalance, LDX and MPH are related to the increase of central DA and NE activity in brain regions promoting the downregulation of DAT and NET at presynaptic level. Activation of DRD and ADRA by LDX and MPH at postsynaptic level is involved in DA and NE transmission. LDX regulates more receptors (TAAR1, VMAT2, SERT, MAOA and MAOB) than MPH in neurotransmission, and minimal differences, such as ERK2 phosphorylation and the action on locus coeruleus neurons by LDX, could differentiate efficacy between both drugs. The regulation of TAAR1 is also related to MAPK3 activation, crucial in the downstream modulation of the neuroinflammation inducing the expression of some anti-inflammatory cytokines (e.g. IL-2, IL-10). In this regard, neuroinflammation and defective immunoregulation have been observed in ADHD. Long-term use of stimulant medication could cause loss of DA transmission and, therefore, efficacy. The studies published show MPH as a drug with a pro-inflammatory profile mainly due to its activation of cytokines TNFα and IL-1β. LDX promotes the activation of pro-inflammatory cytokines such as TNFα and IL-6, although it also promotes the expression of the anti-inflammatory cytokines IL-4 and IL-10 in the frontal cortex, striatum and serum. On the contrary, TNFα promotes astrocyte activation reducing glutamate on the synaptic cleft, which could improve the synaptic signalling. More studies are necessary to understand the effect of both drugs on TNF in ADHD. Also, altered neural viability and neurodegeneration have been observed in ADHD. Mechanisms of neuron survival and oxidative stress are involved in neurodegeneration. It has been shown that MPH could have an effect on neurodegeneration and oxidative stress modulation through several mechanisms. Apoptotic enzymes have been related to MPH although its MoA in that regard remains unclear. Moreover, MPH has been shown to promote ROS via RAC1and CAMK4 activation, although the evidence regarding whether MPH use indeed induces or reduces oxidative stress is not clear. The relationship between LDX and neurodegeneration has only been shown through increasing oxidative stress by downregulating the level of antioxidant enzymes CAT, GSH-Px and SOD. Lastly, circadian system dysfunction is also observed in ADHD. In this regard, the mechanisms of action of MPH involved in the circadian rhythm are unknown, although it has been shown that the use of MPH in children with ADHD normalizes serotonin and melatonin levels to those of non-ADHD population. Nevertheless, no evidence has been found on the effect of LDX on the circadian rhythm in ADHD.
Both LDX and MPH have improve the neuronal signalling that leads to an increase of DA and NE levels in the prefrontal cortex, by acting on DAT and NET and improving attentional deficit and cognitive functions. However, LDX acts on more targets than MPH. Regarding neuroinflammation and defective immunoregulation control, it seems that LDX has a greater effect related to its interaction with TAAR1 and MAOA, and its promotion of anti-inflammatory cytokines such as IL-10. MPH, as well as LDX, promotes the activation of IL-1 and TNF but the role of TNF must be studied in more depth. Regarding neural viability and neurodegeneration, it seems that both drugs have a deleterious effect resulting in increased oxidative enzyme levels. Lastly, MPH shows more evidence as a potential option for treating circadian clock impairments in patients with ADHD.
Despite the studies published on the effect of MPH and LDX on pathophysiological processes and their specific molecules involved in ADHD, there are few data published on the subject and some are based on investigations run in animal models. Further studies are necessary to improve the knowledge of the ADHD pathophysiology and to identify how the MoAs of the MPH and LDX, as current first-line pharmacological treatments, control ADHD symptomatology.
References
Franke B, Faraone SV, Asherson P, et al. The genetics of attention deficit/hyperactivity disorder in adults, a review. Mol Psychiatry. 2012;17(10):960–87.
Regier DA, Kuhl EA, Kupfer DJ. The DSM-5: classification and criteria changes. World Psychiatry. 2013;12(2):92–8.
Feldman HM, Reiff MI. Clinical practice. Attention deficit-hyperactivity disorder in children and adolescents. N Engl J Med. 2014;370(9):838–46.
Dalsgaard S, Leckman JF, Mortensen PB, Nielsen HS, Simonsen M. Effect of drugs on the risk of injuries in children with attention deficit hyperactivity disorder: a prospective cohort study. Lancet Psychiatry. 2015;2(8):702–9.
Posner J, Park C, Wang Z. Connecting the dots: a review of resting connectivity MRI studies in attention-deficit/hyperactivity disorder. Neuropsychol Rev. 2014;24(1):3–15.
Libutzki B, Ludwig S, Jacobsen RH, Reif A, Hartman CA. Direct medical costs of ADHD and its comorbid conditions on basis of a claims data analysis. Eur Psychiatry. 2019;58:38–44.
Wilens TE, Spencer TJ. Understanding attention-deficit/hyperactivity disorder from childhood to adulthood. Postgrad Med. 2010;122(5):97–109.
Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry. 2006;163(4):716–23.
Polanczyk GV, Willcutt EG, Salum GA, Kieling C, Rohde LA. ADHD prevalence estimates across three decades: an updated systematic review and meta-regression analysis. Int J Epidemiol. 2014;43(2):434–42.
Xu G, Strathearn L, Liu B, Yang B, Bao W. Twenty-year trends in diagnosed attention-deficit/hyperactivity disorder among US children and adolescents, 1997–2016. JAMA Netw Open. 2018;1(4):e181471.
Katzman MA, Bilkey TS, Chokka PR, Fallu A, Klassen LJ. Adult ADHD and comorbid disorders: clinical implications of a dimensional approach. BMC Psychiatry. 2017;17(1):302.
Centers for Disease Control and Prevention. ADHD prevalence. https://www.cdc.gov/ncbddd/adhd/. Accessed Mar 2020.
Faraone SV, Biederman J, Mick E. The age-dependent decline of attention deficit hyperactivity disorder: a meta-analysis of follow-up studies. Psychol Med. 2006;36(2):159–65.
Faraone SV, Banaschewski T, Coghill D, et al. The World Federation of ADHD International Consensus Statement: 208 evidence-based conclusions about the disorder. Neurosci Biobehav Rev. 2021;128:789–818.
Vahia VN. Diagnostic and statistical manual of mental disorders 5: a quick glance. Indian J Psychiatry. 2013;55(3):220–3.
Klein MO, Battagello DS, Cardoso AR, Hauser DN, Bittencourt JC, Correa RG. Dopamine: functions, signaling, and association with neurological diseases. Cell Mol Neurobiol. 2019;39(1):31–59.
Cortese S. Pharmacologic treatment of attention deficit-hyperactivity disorder. N Engl J Med. 2020;383(11):1050–6.
tínez-Núñez B, Quintero J. Update the multimodal treatment of ADHD (MTA): twenty years of lessons. Actas Esp Psiquiatr. 2019;47(1):16–22.
Sikirica V, Flood E, Dietrich CN, et al. Unmet needs associated with attention-deficit/hyperactivity disorder in eight European countries as reported by caregivers and adolescents: results from qualitative research. Patient. 2015;8(3):269–81.
Guideline N. Attention deficit hyperactivity disorder: the NICE guideline on diagnosis and management of ADHD in children, young people and adults. London: The British Psychological Society and the Royal College of Psychiatrists; 2009.
Pliszka S, AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894–921.
Subcommittee on Attention-Deficit/Hyperactivity Disorder, Steering Committee on Quality Improvement and Management, Wolraich M, et al. ADHD: clinical practice guideline for the diagnosis, evaluation, and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Pediatrics. 2011;128(5):1007–22.
Shalev L, Tsal Y, Mevorach C. Computerized progressive attentional training (CPAT) program: effective direct intervention for children with ADHD. Child Neuropsychol. 2007;13(4):382–8.
Sonuga-Barke EJ, Coghill D. The foundations of next generation attention-deficit/hyperactivity disorder neuropsychology: building on progress during the last 30 years. J Child Psychol Psychiatry. 2014;55(12):e1-5.
Vinogradov S, Fisher M, de Villers-Sidani E. Cognitive training for impaired neural systems in neuropsychiatric illness. Neuropsychopharmacology. 2012;37(1):43–76.
Arns M, Conners CK, Kraemer HC. A decade of EEG theta/beta ratio research in ADHD: a meta-analysis. J Atten Disord. 2013;17(5):374–83.
Moriyama TS, Polanczyk G, Caye A, Banaschewski T, Brandeis D, Rohde LA. Evidence-based information on the clinical use of neurofeedback for ADHD. Neurotherapeutics. 2012;9(3):588–98.
Sitaram R, Ros T, Stoeckel L, et al. Closed-loop brain training: the science of neurofeedback. Nat Rev Neurosci. 2017;18(2):86–100.
Qian Y, Chen M, Shuai L, Cao QJ, Yang L, Wang YF. Effect of an ecological executive skill training program for school-aged children with attention deficit hyperactivity disorder: a randomized controlled clinical trial. Chin Med J. 2017;130(13):1513–20.
Prevatt F, Yelland S. An empirical evaluation of ADHD coaching in college students. J Atten Disord. 2015;19(8):666–77.
Kubik JA. Efficacy of ADHD coaching for adults with ADHD. J Atten Disord. 2010;13(5):442–53.
Nelwan M, Vissers C, Kroesbergen EH. Coaching positively influences the effects of working memory training on visual working memory as well as mathematical ability. Neuropsychologia. 2018;113:140–9.
Faraone SV. The pharmacology of amphetamine and methylphenidate: relevance to the neurobiology of attention-deficit/hyperactivity disorder and other psychiatric comorbidities. Neurosci Biobehav Rev. 2018;87:255–70.
Cortese S, Adamo N, Del Giovane C, et al. Comparative efficacy and tolerability of medications for attention-deficit hyperactivity disorder in children, adolescents, and adults: a systematic review and network meta-analysis. Lancet Psychiatry. 2018;5(9):727–38.
Caye A, Swanson JM, Coghill D, Rohde LA. Treatment strategies for ADHD: an evidence-based guide to select optimal treatment. Mol Psychiatry. 2019;24(3):390–408.
Leucht S, Hierl S, Kissling W, Dold M, Davis JM. Putting the efficacy of psychiatric and general medicine medication into perspective: review of meta-analyses. Br J Psychiatry. 2012;200(2):97–106.
Faraone SV, Newcorn JH, Cipriani A, et al. Placebo and nocebo responses in randomised, controlled trials of medications for ADHD: a systematic review and meta-analysis. Mol Psychiatry. 2022;27(1):212–9.
Posner J, Greenhill LL. Attention-deficit/hyperactivity disorder. 2nd ed. Arlington: American Psychiatric Association; 2013.
Ficha Tecnica Elvanse 30 mg capsulas duras. https://cima.aemps.es/cima/dochtml/ft/77642/FT_77642.html.
VYVANSE® (lisdexamfetamine dimesylate) capsules, for oral use, CII. VYVANSE ® (lisdexamfetamine dimesylate) chewable tablets, for oral use, CII. Initial U.S. Approval: 20. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208510lbl.pdf.
Kam H, Jeong H. Pharmacogenomic biomarkers and their applications in psychiatry. Genes. 2020;11(12):1445.
FDA. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling. Accessed Aug 2021.
da Silva BS, Leffa DT, Beys-da-Silva WO, et al. Integrative proteomics and pharmacogenomics analysis of methylphenidate treatment response. Transl Psychiatry. 2019;9(1):308.
Angyal N, Horvath EZ, Tarnok Z, et al. Association analysis of norepinephrine transporter polymorphisms and methylphenidate response in ADHD patients. Prog Neuropsychopharmacol Biol Psychiatry. 2018;84(Pt A):122–8.
Gomez-Sanchez CI, Carballo JJ, Riveiro-Alvarez R, et al. Pharmacogenetics of methylphenidate in childhood attention-deficit/hyperactivity disorder: long-term effects. Sci Rep. 2017;7(1):10391.
Joensen B, Meyer M, Aagaard L. Specific genes associated with adverse events of methylphenidate use in the pediatric population: a systematic literature review. J Res Pharm Pract. 2017;6(2):65–72.
Myer NM, Boland JR, Faraone SV. Pharmacogenetics predictors of methylphenidate efficacy in childhood ADHD. Mol Psychiatry. 2018;23(9):1929–36.
Yuan D, Zhang M, Huang Y, Wang X, Jiao J, Huang Y. Noradrenergic genes polymorphisms and response to methylphenidate in children with ADHD: a systematic review and meta-analysis. Medicine. 2021;100(46):e27858.
Patel JN, Mueller MK, Guffey WJ, Stegman J. Drug prescribing and outcomes after pharmacogenomic testing in a developmental and behavioral health pediatric clinic. J Dev Behav Pediatr. 2020;41(1):65–70.
Elsayed NA, Yamamoto KM, Froehlich TE. Genetic influence on efficacy of pharmacotherapy for pediatric attention-deficit/hyperactivity disorder: overview and current status of research. CNS Drugs. 2020;34(4):389–414.
Mohan P, Gupta YK, Prakash J. Application of pharmacogenomics in psychiatric practice: the road ahead. Ind Psychiatry J. 2021;30(1):4–5.
Tang Girdwood SC, Rossow KM, Van Driest SL, Ramsey LB. Perspectives from the Society for Pediatric Research: pharmacogenetics for pediatricians. Pediatr Res. 2022;91(3):529–38.
Wehry AM, Ramsey L, Dulemba SE, Mossman SA, Strawn JR. Pharmacogenomic testing in child and adolescent psychiatry: an evidence-based review. Curr Probl Pediatr Adolesc Health Care. 2018;48(2):40–9.
Curatolo P, D’Agati E, Moavero R. The neurobiological basis of ADHD. Ital J Pediatr. 2010;36(1):79.
Cabral MDI, Liu S, Soares N. Attention-deficit/hyperactivity disorder: diagnostic criteria, epidemiology, risk factors and evaluation in youth. Transl Pediatr. 2020;9(Suppl 1):S104–13.
Faraone SV, Larsson H. Genetics of attention deficit hyperactivity disorder. Mol Psychiatry. 2019;24(4):562–75.
Lee SH, Ripke S, Neale BM, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45(9):984–94.
Thapar A, Cooper M, Eyre O, Langley K. What have we learnt about the causes of ADHD? J Child Psychol Psychiatry. 2013;54(1):3–16.
Grimm O, Kranz TM, Reif A. Genetics of ADHD: what should the clinician know? Curr Psychiatry Rep. 2020;22(4):18.
Verlaet AA, Noriega DB, Hermans N, Savelkoul HF. Nutrition, immunological mechanisms and dietary immunomodulation in ADHD. Eur Child Adolesc Psychiatry. 2014;23(7):519–29.
Ribasés M, Ramos-Quiroga JA, Sánchez-Mora C, et al. Contribution of LPHN3 to the genetic susceptibility to ADHD in adulthood: a replication study. Genes Brain Behav. 2011;10(2):149–57.
Lange M, Norton W, Coolen M, et al. The ADHD-susceptibility gene lphn3.1 modulates dopaminergic neuron formation and locomotor activity during zebrafish development. Mol Psychiatry. 2012;17(9):946–54.
Bijlenga D, Vollebregt MA, Kooij JJS, Arns M. The role of the circadian system in the etiology and pathophysiology of ADHD: time to redefine ADHD? Atten Defic Hyperact Disord. 2019;11(1):5–19.
Wallis D, Russell HF, Muenke M. Review: genetics of attention deficit/hyperactivity disorder. J Pediatr Psychol. 2008;33(10):1085–99.
D’Onofrio BM, Van Hulle CA, Waldman ID, Rodgers JL, Rathouz PJ, Lahey BB. Causal inferences regarding prenatal alcohol exposure and childhood externalizing problems. Arch Gen Psychiatry. 2007;64(11):1296–304.
Alwan NA, Hamamy H. Maternal iron status in pregnancy and long-term health outcomes in the offspring. J Pediatr Genet. 2015;4(2):111–23.
San Maurotín I, Blumenfeld Olivares JA, Garicano Vilar E, et al. Nutritional and environmental factors in attention-deficit hyperactivity disorder (ADHD): a cross-sectional study. Nutr Neurosci. 2018;21(9):641–7.
Tong L, Xiong X, Tan H. Attention-deficit/hyperactivity disorder and lifestyle-related behaviors in children. PLoS ONE. 2016;11(9): e0163434.
Tseng PT, Cheng YS, Yen CF, et al. Peripheral iron levels in children with attention-deficit hyperactivity disorder: a systematic review and meta-analysis. Sci Rep. 2018;8(1):788.
Biederman J, Faraone SV. Attention-deficit hyperactivity disorder. Lancet. 2005;366(9481):237–48.
Rothenberger LG. Molecular genetics research in ADHD: ethical considerations concerning patients’ benefit and resource allocation. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(8):885–95.
Wu J, Xiao H, Sun H, Zou L, Zhu LQ. Role of dopamine receptors in ADHD: a systematic meta-analysis. Mol Neurobiol. 2012;45(3):605–20.
Aureli A, Del Beato T, Sebastiani P, et al. Attention-deficit hyperactivity disorder and intellectual disability: a study of association with brain-derived neurotrophic factor gene polymorphisms. Int J Immunopathol Pharmacol. 2010;23(3):873–80.
Ivanchak N, Fletcher K, Jicha GA. Attention-deficit/hyperactivity disorder in older adults: prevalence and possible connections to mild cognitive impairment. Curr Psychiatry Rep. 2012;14(5):552–60.
Sharma A, Couture J. A review of the pathophysiology, etiology, and treatment of attention-deficit hyperactivity disorder (ADHD). Ann Pharmacother. 2014;48(2):209–25.
Arnsten AF, Pliszka SR. Catecholamine influences on prefrontal cortical function: relevance to treatment of attention deficit/hyperactivity disorder and related disorders. Pharmacol Biochem Behav. 2011;99(2):211–6.
Valera EM, Faraone SV, Murray KE, Seidman LJ. Meta-analysis of structural imaging findings in attention-deficit/hyperactivity disorder. Biol Psychiat. 2007;61(12):1361–9.
Shaw P, Eckstrand K, Sharp W, et al. Attention-deficit/hyperactivity disorder is characterized by a delay in cortical maturation. Proc Natl Acad Sci USA. 2007;104(49):19649–54.
Del Campo N, Chamberlain SR, Sahakian BJ, Robbins TW. The roles of dopamine and noradrenaline in the pathophysiology and treatment of attention-deficit/hyperactivity disorder. Biol Psychiat. 2011;69(12):e145–57.
Tripp G, Wickens JR. Neurobiology of ADHD. Neuropharmacology. 2009;57(7–8):579–89.
Volkow ND, Wang GJ, Newcorn JH, et al. Motivation deficit in ADHD is associated with dysfunction of the dopamine reward pathway. Mol Psychiatry. 2011;16(11):1147–54.
Staller JA, Faraone SV. Targeting the dopamine system in the treatment of attention-deficit/hyperactivity disorder. Expert Rev Neurother. 2007;7(4):351–62.
Engert V, Pruessner JC. Dopaminergic and noradrenergic contributions to functionality in ADHD: the role of methylphenidate. Curr Neuropharmacol. 2008;6(4):322–8.
Gizer IR, Ficks C, Waldman ID. Candidate gene studies of ADHD: a meta-analytic review. Hum Genet. 2009;126(1):51–90.
Yamamoto K, Shinba T, Yoshii M. Psychiatric symptoms of noradrenergic dysfunction: a pathophysiological view. Psychiatry Clin Neurosci. 2014;68(1):1–20.
Dahl MJ, Mather M, Sander MC, Werkle-Bergner M. Noradrenergic responsiveness supports selective attention across the adult lifespan. J Neurosci. 2020;40(22):4372–90.
De tino B, Strange BA, Dolan RJ. Noradrenergic neuromodulation of human attention for emotional and neutral stimuli. Psychopharmacology. 2008;197(1):127–36.
Ulke C, Rullmann M, Huang J, et al. Adult attention-deficit/hyperactivity disorder is associated with reduced norepinephrine transporter availability in right attention networks: a (S,S)-O-[11C]methylreboxetine positron emission tomography study. Transl Psychiatry. 2019;9(1):301.
Kent L, Doerry U, Hardy E, et al. Evidence that variation at the serotonin transporter gene influences susceptibility to attention deficit hyperactivity disorder (ADHD): analysis and pooled analysis. Mol Psychiatry. 2002;7(8):908–12.
Hawi Z, Dring M, Kirley A, et al. Serotonergic system and attention deficit hyperactivity disorder (ADHD): a potential susceptibility locus at the 5-HT(1B) receptor gene in 273 nuclear families from a multi-centre sample. Mol Psychiatry. 2002;7(7):718–25.
Wang CH, Liu C, Cong EZ, et al. Association of tryptophan hydroxylase-2 polymorphisms with oppositional defiant disorder in a Chinese Han population. Behav Brain Funct. 2016;12(1):30.
Dark C, Homman-Ludiye J, Bryson-Richardson RJ. The role of ADHD associated genes in neurodevelopment. Dev Biol. 2018;438(2):69–83.
Bruce A, Johnson A, Ian L, Tin R, Keith R, Peter W. Internal organization of the cell; channels and the electrical properties. In: Molecular biology of the cell. 4th ed. New York: Garland Science; 2002.
Medin T, Jensen V, Skare O, Storm-Mathisen J, Hvalby O, Bergersen LH. Altered alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor function and expression in hippocampus in a rat model of attention-deficit/hyperactivity disorder (ADHD). Behav Brain Res. 2019;15(360):209–15.
Puts NA, Ryan M, Oeltzschner G, Horska A, Edden RAE, Mahone EM. Reduced striatal GABA in unmedicated children with ADHD at 7T. Psychiatry Res Neuroimaging. 2020;30(301): 111082.
Edden RA, Crocetti D, Zhu H, Gilbert DL, Mostofsky SH. Reduced GABA concentration in attention-deficit/hyperactivity disorder. Arch Gen Psychiatry. 2012;69(7):750–3.
Hammerness P, Biederman J, Petty C, Henin A, Moore CM. Brain biochemical effects of methylphenidate treatment using proton magnetic spectroscopy in youth with attention-deficit hyperactivity disorder: a controlled pilot study. CNS Neurosci Ther. 2012;18(1):34–40.
Hu Y, Chen X, Gu H, Yang Y. Resting-state glutamate and GABA concentrations predict task-induced deactivation in the default mode network. J Neurosci. 2013;33(47):18566–73.
Arnsten AF. The emerging neurobiology of attention deficit hyperactivity disorder: the key role of the prefrontal association cortex. J Pediatr. 2009;154(5):I-S43.
Gilbert DL, Ridel KR, Sallee FR, Zhang J, Lipps TD, Wassermann EM. Comparison of the inhibitory and excitatory effects of ADHD medications methylphenidate and atomoxetine on motor cortex. Neuropsychopharmacology. 2006;31(2):442–9.
Naaijen J, Bralten J, Poelmans G, et al. Glutamatergic and GABAergic gene sets in attention-deficit/hyperactivity disorder: association to overlapping traits in ADHD and autism. Transl Psychiatry. 2017;7(1): e999.
Ma L, Chen YH, Chen H, Liu YY, Wang YX. The function of hypothalamus-pituitary-adrenal axis in children with ADHD. Brain Res. 2011;12(1368):159–62.
Buske-Kirschbaum A, Trikojat K, Tesch F, et al. Altered hypothalamus-pituitary-adrenal axis function: a relevant factor in the comorbidity of atopic eczema and attention deficit/hyperactivity disorder? Psychoneuroendocrinology. 2019;105:178–86.
Kaneko M, Hoshino Y, Hashimoto S, Okano T, Kumashiro H. Hypothalamic-pituitary-adrenal axis function in children with attention-deficit hyperactivity disorder. J Autism Dev Disord. 1993;23(1):59–65.
Isaksson J, Nilsson KW, Lindblad F. Early psychosocial adversity and cortisol levels in children with attention-deficit/hyperactivity disorder. Eur Child Adolesc Psychiatry. 2013;22(7):425–32.
Stephens MA, Wand G. Stress and the HPA axis: role of glucocorticoids in alcohol dependence. Alcohol Res. 2012;34(4):468–83.
Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin Neurosci. 2006;8(4):383–95.
Bao AM, Swaab DF. The human hypothalamus in mood disorders: the HPA axis in the center. IBRO Rep. 2019;6:45–53.
Buitelaar JK. The role of the HPA-axis in understanding psychopathology: cause, consequence, mediator, or moderator? Eur Child Adolesc Psychiatry. 2013;22(7):387–9.
Miyazaki C, Koyama M, Ota E, et al. Allergic diseases in children with attention deficit hyperactivity disorder: a systematic review and meta-analysis. BMC Psychiatry. 2017;17(1):120.
Pelsser LM, Buitelaar JK, Savelkoul HF. ADHD as a (non) allergic hypersensitivity disorder: a hypothesis. Pediatr Allergy Immunol. 2009;20(2):107–12.
Scahill L, de Graft-Johnson A. Food allergies, asthma, and attention deficit hyperactivity disorder. J Child Adolesc Psychiatr Nurs. 1997;10(2):36–40 (quiz 1–2).
Lee YA, Goto Y. Habenula and ADHD: convergence on time. Neurosci Biobehav Rev. 2013;37(8):1801–9.
Xie Y, Tang Q, Chen G, et al. New insights into the circadian rhythm and its related diseases. Front Physiol. 2019;10:682.
Mogavero F, Jager A, Glennon JC. Clock genes, ADHD and aggression. Neurosci Biobehav Rev. 2018;91:51–68.
Corkum P, Tannock R, Moldofsky H. Sleep disturbances in children with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 1998;37(6):637–46.
Spruyt K, Gozal D. Sleep disturbances in children with attention-deficit/hyperactivity disorder. Expert Rev Neurother. 2011;11(4):565–77.
Masi G, Fantozzi P, Villafranca A, et al. Effects of melatonin in children with attention-deficit/hyperactivity disorder with sleep disorders after methylphenidate treatment. Neuropsychiatr Dis Treat. 2019;15:663–7.
Cortese S, Konofal E, Lecendreux M. Alertness and feeding behaviors in ADHD: does the hypocretin/orexin system play a role? Med Hypotheses. 2008;71(5):770–5.
Lewis TL Jr, Polleux F. Neuronal morphogenesis: Golgi outposts, acentrosomal microtubule nucleation, and dendritic branching. Neuron. 2012;76(5):862–4.
Donev R, Thome J. Inflammation: good or bad for ADHD? Atten Defic Hyperact Disord. 2010;2(4):257–66.
Shim SH, Hwangbo Y, Kwon YJ, et al. Increased levels of plasma brain-derived neurotrophic factor (BDNF) in children with attention deficit-hyperactivity disorder (ADHD). Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1824–8.
Yan YH, Li SH, Gao Z, et al. Neurotrophin-3 promotes proliferation and cholinergic neuronal differentiation of bone marrow-derived neural stem cells via notch signaling pathway. Life Sci. 2016;1(166):131–8.
Elia J, Glessner JT, Wang K, et al. Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat Genet. 2011;44(1):78–84.
Joseph N, Zhang-James Y, Perl A, Faraone SV. Oxidative stress and ADHD: a meta-analysis. J Atten Disord. 2015;19(11):915–24.
Sezen H, Kandemir H, Savik E, et al. Increased oxidative stress in children with attention deficit hyperactivity disorder. Redox Rep. 2016;21(6):248–53.
Homman-Ludiye J, Kwan WC, de Souza MJ, Rodger J, Bourne JA. Ephrin-A2 regulates excitatory neuron differentiation and interneuron migration in the developing neocortex. Sci Rep. 2017;7(1):11813.
Henderson NT, Le chand SJ, Hruska M, Hippenmeyer S, Luo L, Dalva MB. Ephrin-B3 controls excitatory synapse density through cell-cell competition for EphBs. Elife. 2019;21:8.
Calo L, Cinque C, Patane M, et al. Interaction between ephrins/Eph receptors and excitatory amino acid receptors: possible relevance in the regulation of synaptic plasticity and in the pathophysiology of neuronal degeneration. J Neurochem. 2006;98(1):1–10.
Sheleg M, Yochum CL, Wagner GC, Zhou R, Richardson JR. Ephrin-A5 deficiency alters sensorimotor and monoaminergic development. Behav Brain Res. 2013;236(1):139–47.
Volkow ND, Fowler JS, Wang G, Ding Y, Gatley SJ. Mechanism of action of methylphenidate: insights from PET imaging studies. J Atten Disord. 2002;6(Suppl 1):S31-43.
National Institute for Health and Care Excellence. Attention deficit hyperactivity disorder: diagnosis and management. https://www.nice.org.uk/guidance/NG87. NICE guideline NG87. 2018.
Kooij JJS, Bijlenga D, Salerno L, et al. Updated European consensus statement on diagnosis and treatment of adult ADHD. Eur Psychiatry. 2019;56:14–34.
Pitzianti MB, Spiridigliozzi S, Bartolucci E, Esposito S, Pasini A. New insights on the effects of methylphenidate in attention deficit hyperactivity disorder. Front Psychiatry. 2020;11: 531092.
Semrud-Clikeman M, Filipek PA, Biederman J, et al. Attention-deficit hyperactivity disorder: magnetic resonance imaging morphometric analysis of the corpus callosum. J Am Acad Child Adolesc Psychiatry. 1994;33(6):875–81.
Connor DF, Steingard RJ. New formulations of stimulants for attention-deficit hyperactivity disorder: therapeutic potential. CNS Drugs. 2004;18(14):1011–30.
Maldonado R. Comparison of the pharmacokinetics and clinical efficacy of new extended-release formulations of methylphenidate. Expert Opin Drug Metab Toxicol. 2013;9(8):1001–14.
CIMA. Rubicrono Summary of Product Characteristics. https://cima.aemps.es/cima/dochtml/ft/81347/fichatecnica_81347.html. AEMPS. 2021.
Rajala AZ, Populin LC, Jenison RL. Methylphenidate affects task-switching and neural signaling in non-human primates. Psychopharmacology. 2020;237(5):1533–43.
kowitz JS, DeVane CL, Pestreich LK, Patrick KS, Muniz R. A comprehensive in vitro screening of d-, l-, and dl-threo-methylphenidate: an exploratory study. J Child Adolesc Psychopharmacol. 2006;16(6):687–98.
kowitz JS, DeVane CL, Ramamoorthy S, Zhu HJ. The psychostimulant d-threo-(R,R)-methylphenidate binds as an agonist to the 5HT(1A) receptor. Pharmazie. 2009;64(2):123–5.
Zhang CL, Feng ZJ, Liu Y, et al. Methylphenidate enhances NMDA-receptor response in medial prefrontal cortex via sigma-1 receptor: a novel mechanism for methylphenidate action. PLoS ONE. 2012;7(12): e51910.
Pascoli V, Valjent E, Corbille AG, et al. cAMP and extracellular signal-regulated kinase signaling in response to d-amphetamine and methylphenidate in the prefrontal cortex in vivo: role of beta 1-adrenoceptors. Mol Pharmacol. 2005;68(2):421–9.
Motaghinejad M, Motevalian M, Fatima S, Beiranvand T, Mozaffari S. Topiramate via NMDA, AMPA/kainate, GABAA and Alpha2 receptors and by modulation of CREB/BDNF and Akt/GSK3 signaling pathway exerts neuroprotective effects against methylphenidate-induced neurotoxicity in rats. J Neural Transm. 2017;124(11):1369–87.
Wetzell BB, Muller MM, Cobuzzi JL, Hurwitz ZE, DeCicco-Skinner K, Riley AL. Effect of age on methylphenidate-induced conditioned taste avoidance and related BDNF/TrkB signaling in the insular cortex of the rat. Psychopharmacology. 2014;231(8):1493–501.
Amiri A, Torabi Parizi G, Kousha M, et al. Changes in plasma brain-derived neurotrophic factor (BDNF) levels induced by methylphenidate in children with attention deficit-hyperactivity disorder (ADHD). Prog Neuropsychopharmacol Biol Psychiatry. 2013;2(47):20–4.
Reus GZ, Scaini G, Jeremias GC, et al. Brain apoptosis signaling pathways are regulated by methylphenidate treatment in young and adult rats. Brain Res. 2014;2(1583):269–76.
Motaghinejad M, Motevalian M, Falak R, Heidari M, Sharzad M, Kalantari E. Neuroprotective effects of various doses of topiramate against methylphenidate-induced oxidative stress and inflammation in isolated rat amygdala: the possible role of CREB/BDNF signaling pathway. J Neural Transm. 2016;123(12):1463–77.
Coelho-Santos V, Socodato R, Portugal C, et al. Methylphenidate-triggered ROS generation promotes caveolae-mediated transcytosis via Rac1 signaling and c-Src-dependent caveolin-1 phosphorylation in human brain endothelial cells. Cell Mol Life Sci. 2016;73(24):4701–16.
Frampton JE. Lisdexamfetamine dimesylate: a review in paediatric ADHD. Drugs. 2018;78(10):1025–36.
Frampton JE. Lisdexamfetamine: a review in ADHD in adults. CNS Drugs. 2016;30(4):343–54.
Ford N. Tackling female genital cutting in Somalia. Lancet. 2001;358(9288):1179.
Hutson PH, Pennick M, Secker R. Preclinical pharmacokinetics, pharmacology and toxicology of lisdexamfetamine: a novel d-amphetamine pro-drug. Neuropharmacology. 2014;87:41–50.
Dew RE, Kollins SH. Lisdexamfetamine dimesylate: a new option in stimulant treatment for ADHD. Expert Opin Pharmacother. 2010;11(17):2907–13.
Ekstrand E, Murphy HM, Wideman CH. The effects of the prodrug Vyvanse on spatial working memory and adiposity in rats. Pharmacol Biochem Behav. 2019;186: 172765.
Heal DJ, Cheetham SC, Smith SL. The neuropharmacology of ADHD drugs in vivo: insights on efficacy and safety. Neuropharmacology. 2009;57(7–8):608–18.
Strajhar P, Vizeli P, Patt M, et al. Effects of lisdexamfetamine on plasma steroid concentrations compared with d-amphetamine in healthy subjects: a randomized, double-blind, placebo-controlled study. J Steroid Biochem Mol Biol. 2019;186:212–25.
Heal DJ, Hallam M, Prow M, et al. Dopamine and mu-opioid receptor dysregulation in the brains of binge-eating female rats - possible relevance in the psychopathology and treatment of binge-eating disorder. J Psychopharmacol. 2017;31(6):770–83.
Solanto MV. Neuropsychopharmacological mechanisms of stimulant drug action in attention-deficit hyperactivity disorder: a review and integration. Behav Brain Res. 1998;94(1):127–52.
Valvassori SS, Resende WR, Varela RB, et al. The effects of histone deacetylase inhibition on the levels of cerebral cytokines in an animal model of mania induced by dextroamphetamine. Mol Neurobiol. 2018;55(2):1430–9.
Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28(3):138–45.
Pitsillou E, Bresnehan SM, Kagarakis EA, et al. The cellular and molecular basis of major depressive disorder: towards a unified model for understanding clinical depression. Mol Biol Rep. 2020;47(1):753–70.
Liberto CM, Albrecht PJ, Herx LM, Yong VW, Levison SW. Pro-regenerative properties of cytokine-activated astrocytes. J Neurochem. 2004;89(5):1092–100.
Guerdjikova AI, Mori N, Casuto LS, McElroy SL. Novel pharmacologic treatment in acute binge eating disorder - role of lisdexamfetamine. Neuropsychiatr Dis Treat. 2016;12:833–41.
Popovic B, Bhattacharya P, Sivaswamy L. Lisdexamfetamine: a prodrug for the treatment of attention-deficit/hyperactivity disorder. Am J Health Syst Pharm. 2009;66(22):2005–12.
Vaughan RA, Foster JD. Mechanisms of dopamine transporter regulation in normal and disease states. Trends Pharmacol Sci. 2013;34(9):489–96.
Gaweska H, Fitzpatrick PF. Structures and mechanism of the monoamine oxidase family. Biomol Concepts. 2011;2(5):365–77.
KEGG icon KEGG PATHWAY Database. Dopaminergic synapse—Homo sapiens (human). https://www.genome.jp/kegg-bin/show_pathway?hsa04728. Accessed May 2021.
Moore CF, Sabino V, Cottone P. Trace amine associated receptor 1 (TAAR1) modulation of food reward. Front Pharmacol. 2018;9:129.
Sotnikova TD, Caron MG, Gainetdinov RR. Trace amine-associated receptors as emerging therapeutic targets. Mol Pharmacol. 2009;76(2):229–35.
Volkow ND, Fowler JS, Wang GJ, Ding YS, Gatley SJ. Role of dopamine in the therapeutic and reinforcing effects of methylphenidate in humans: results from imaging studies. Eur Neuropsychopharmacol. 2002;12(6):557–66.
Seeman P, Madras BK. Anti-hyperactivity medication: methylphenidate and amphetamine. Mol Psychiatry. 1998;3(5):386–96.
Wilens TE. Effects of methylphenidate on the catecholaminergic system in attention-deficit/hyperactivity disorder. J Clin Psychopharmacol. 2008;28(3 Suppl 2):S46-53.
Contini V, Victor MM, Marques FZC, et al. Response to methylphenidate is not influenced by DAT1 polymorphisms in a sample of Brazilian adult patients with ADHD. J Neural Transm. 2010;117(2):269–76.
Gronier B. In vivo electrophysiological effects of methylphenidate in the prefrontal cortex: involvement of dopamine D1 and alpha 2 adrenergic receptors. Eur Neuropsychopharmacol. 2011;21(2):192–204.
Andrews GD, Lavin A. Methylphenidate increases cortical excitability via activation of alpha-2 noradrenergic receptors. Neuropsychopharmacology. 2006;31(3):594–601.
Miller GM. The emerging role of trace amine-associated receptor 1 in the functional regulation of monoamine transporters and dopaminergic activity. J Neurochem. 2011;116(2):164–76.
Babusyte A, Kotthoff M, Fiedler J, Krautwurst D. Biogenic amines activate blood leukocytes via trace amine-associated receptors TAAR1 and TAAR2. J Leukoc Biol. 2013;93(3):387–94.
Underhill SM, Hullihen PD, Chen J, et al. Amphetamines signal through intracellular TAAR1 receptors coupled to Gα13 and GαS in discrete subcellular domains. Mol Psychiatry. 2021;26(4):1208–23.
Rutigliano G, Accorroni A, Zucchi R. The case for TAAR1 as a modulator of central nervous system function. Front Pharmacol. 2017;8:987.
Peterson SM, Pack TF, Wilkins AD, et al. Elucidation of G-protein and beta-arrestin functional selectivity at the dopamine D2 receptor. Proc Natl Acad Sci USA. 2015;112(22):7097–102.
Bermingham DP, Blakely RD. Kinase-dependent regulation of monoamine neurotransmitter transporters. Pharmacol Rev. 2016;68(4):888–953.
Jayanthi LD, Samuvel DJ, Blakely RD, Ramamoorthy S. Evidence for biphasic effects of protein kinase C on serotonin transporter function, endocytosis, and phosphorylation. Mol Pharmacol. 2005;67(6):2077–87.
Shimizu T, Kato T Jr, Tachibana A, Sasaki MS. Coordinated regulation of radioadaptive response by protein kinase C and p38 mitogen-activated protein kinase. Exp Cell Res. 1999;251(2):424–32.
Yokota T, Wang Y. p38 MAP kinases in the heart. Gene. 2016;575(2 Pt 2):369–76.
Kitagishi Y, Minami A, Nakanishi A, Ogura Y, Matsuda S. Neuron membrane trafficking and protein kinases involved in autism and ADHD. Int J Mol Sci. 2015;16(2):3095–115.
Riddle EL, Hanson GR, Fleckenstein AE. Therapeutic doses of amphetamine and methylphenidate selectively redistribute the vesicular monoamine transporter-2. Eur J Pharmacol. 2007;571(1):25–8.
Spencer RC, Devilbiss DM, Berridge CW. The cognition-enhancing effects of psychostimulants involve direct action in the prefrontal cortex. Biol Psychiatry. 2015;77(11):940–50.
Ferger B, Kropf W, Kuschinsky K. Studies on electroencephalogram (EEG) in rats suggest that moderate doses of cocaine or d-amphetamine activate D1 rather than D2 receptors. Psychopharmacology. 1994;114(2):297–308.
Graham AW, Aghajanian GK. Effects of amphetamine on single cell activity in a catecholamine nucleus, the locus coeruleus. Nature. 1971;234(5324):100–2.
Vickers SP, Hackett D, Murray F, Hutson PH, Heal DJ. Effects of lisdexamfetamine in a rat model of binge-eating. J Psychopharmacol. 2015;29(12):1290–307.
Sadasivan S, Pond BB, Pani AK, Qu C, Jiao Y, Smeyne RJ. Methylphenidate exposure induces dopamine neuron loss and activation of microglia in the basal ganglia of mice. PLoS ONE. 2012;7(3): e33693.
Yamamoto BK, Raudensky J. The role of oxidative stress, metabolic compromise, and inflammation in neuronal injury produced by amphetamine-related drugs of abuse. J Neuroimmune Pharmacol. 2008;3(4):203–17.
Motaghinejad M, Karimian SM, Motaghinejad O, Shabab B, Asadighaleni M, Fatima S. The effect of various morphine weaning regimens on the sequelae of opioid tolerance involving physical dependency, anxiety and hippocampus cell neurodegeneration in rats. Fundam Clin Pharmacol. 2015;29(3):299–309.
Tagaya H. Methylphenidate: pharmacology, indication and potential of abuse. Nihon Rinsho. 2010;68(8):1550–5.
Jones Z, Dafny N. Acute and chronic dose-response effect of methylphenidate on ventral tegmental area neurons correlated with animal behavior. J Neural Transm. 2014;121(3):327–45.
Motaghinejad M, Motevalian M, Shabab B. Effects of chronic treatment with methylphenidate on oxidative stress and inflammation in hippocampus of adult rats. Neurosci Lett. 2016;21(619):106–13.
Pinsonneault JK, Han DD, Burdick KE, et al. Dopamine transporter gene variant affecting expression in human brain is associated with bipolar disorder. Neuropsychopharmacology. 2011;36(8):1644–55.
Bristot G, Ascoli BM, Scotton E, Géa LP, Pfaffenseller B, Kauer-Sant'Anna M. Effects of lithium on inflammatory and neurotrophic factors after an immune challenge in a lisdexamfetamine animal model of mania. Braz J Psychiatry. 2019;41(5):419–27.
Ramalingam P, Poulos MG, Lazzari E, et al. Chronic activation of endothelial MAPK disrupts hematopoiesis via NFKB dependent inflammatory stress reversible by SCGF. Nat Commun. 2020;11(1):666.
Los M, Schenk H, Hexel K, Baeuerle PA, Droge W, Schulze-Osthoff K. IL-2 gene expression and NF-kappa B activation through CD28 requires reactive oxygen production by 5-lipoxygenase. EMBO J. 1995;14(15):3731–40.
Cao S, Zhang X, Edwards JP, Mosser DM. NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J Biol Chem. 2006;281(36):26041–50.
Muller N. Inflammation in schizophrenia: pathogenetic aspects and therapeutic considerations. Schizophr Bull. 2018;44(5):973–82.
Perry BI, Upthegrove R, Kappelmann N, Jones PB, Burgess S, Khandaker GM. Associations of immunological proteins/traits with schizophrenia, major depression and bipolar disorder: a bi-directional two-sample mendelian randomization study. Brain Behav Immun. 2021;97:176–185.
Sakrajda K, Szczepankiewicz A. Inflammation-related changes in mood disorders and the immunomodulatory role of lithium. Int J Mol Sci. 2021;22(4):1532.
Alvarez-Mon MA, Gomez-Lahoz AM, Orozco A, et al. Blunted expansion of regulatory T lymphocytes is associated with increased bacterial translocation in patients with major depressive disorder. Front Psychiatry. 2020;11: 591962.
Sakamoto S, Zhu X, Hasegawa Y, et al. Inflamed brain: targeting immune changes and inflammation for treatment of depression. Psychiatry Clin Neurosci. 2021;75(10):304–11.
Troubat R, Barone P, Leman S, et al. Neuroinflammation and depression: a review. Eur J Neurosci. 2021;53(1):151–71.
Antle MC, van Diepen HC, Deboer T, Pedram P, Pereira RR, Meijer JH. Methylphenidate modifies the motion of the circadian clock. Neuropsychopharmacology. 2012;37(11):2446–55.
Baird AL, Coogan AN, Kaufling J, Barrot M, Thome J. Daily methylphenidate and atomoxetine treatment impacts on clock gene protein expression in the mouse brain. Brain Res. 2013;4(1513):61–71.
Hardeland R, Madrid JA, Tan DX, Reiter RJ. Melatonin, the circadian multioscillator system and health: the need for detailed analyses of peripheral melatonin signaling. J Pineal Res. 2012;52(2):139–66.
Molina-Carballo A, Naranjo-Gómez A, Uberos J, et al. Methylphenidate effects on blood serotonin and melatonin levels may help to synchronise biological rhythms in children with ADHD. J Psychiatr Res. 2013;47(3):377–83.
Lamia KA, Sachdeva UM, DiTacchio L, et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326(5951):437–40.
Onat OE, Kars ME, Gul S, et al. Human CRY1 variants associate with attention deficit/hyperactivity disorder. J Clin Investig. 2020;130(7):3885–900.
Blendy JA. The role of CREB in depression and antidepressant treatment. Biol Psychiat. 2006;59(12):1144–50.
Guo W, Crossey EL, Zhang L, et al. Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS ONE. 2011;6(5):e19351.
Xu L, Hu Y, Huang L, et al. The association between attention deficit hyperactivity disorder and general anaesthesia - a narrative review. Anaesthesia. 2019;74(1):57–63.
Bramham CR, Panja D. BDNF regulation of synaptic structure, function, and plasticity. Neuropharmacology. 2014;76(Pt C):601–2.
Liu DY, Shen XM, Yuan FF, et al. The physiology of BDNF and its relationship with ADHD. Mol Neurobiol. 2015;52(3):1467–76.
Corominas-Roso M, Ramos-Quiroga JA, Ribases M, et al. Decreased serum levels of brain-derived neurotrophic factor in adults with attention-deficit hyperactivity disorder. Int J Neuropsychopharmacol. 2013;16(6):1267–75.
Ebrahimzadeh A, Moghadam SY, Rahimi H, et al. Crocin acts as a neuroprotective mediator against methylphenidate-induced neurobehavioral and neurochemical sequelae: possible role of the CREB-BDNF signaling pathway. Acta Neurobiol Exp. 2019;79(4):352–66.
Garcia-Aviles A, Albert-Gasco H, Arnal-Vicente I, et al. Acute oral administration of low doses of methylphenidate targets calretinin neurons in the rat septal area. Front Neuroanat. 2015;9:33.
Gomes KM, Inacio CG, Valvassori SS, et al. Superoxide production after acute and chronic treatment with methylphenidate in young and adult rats. Neurosci Lett. 2009;465(1):95–8.
tins MR, Reinke A, Petronilho FC, Gomes KM, Dal-Pizzol F, Quevedo J. Methylphenidate treatment induces oxidative stress in young rat brain. Brain Res. 2006;1078(1):189–97.
Eger GA, Ferreira VV, Batista CR, et al. Antioxidant effect of simvastatin throught oxidative imbalance caused by lisdexamfetamine dimesylate. An Acad Bras Cienc. 2016;88(1):335–48.
Macêdo DS, de Lucena DF, Queiroz AI, et al. Effects of lithium on oxidative stress and behavioral alterations induced by lisdexamfetamine dimesylate: relevance as an animal model of mania. Prog Neuropsychopharmacol Biol Psychiatry. 2013;3(43):230–7.
Acknowledgements
Funding
Dr. Gutiérrez-Casares received honoraria from Takeda Farmacéutica España S.A. as a scientific advisor. Dr. Javier Quintero and Dr. Cecilio Álamo did not receive honoraria for their contributions to this study. The journal’s Rapid Service Fee was funded by Takeda Farmacéutica España S.A.
Medical Writing, Editorial, and Other Assistance
The authors want to thank Helena Bartra (Anaxomics Biotech, Barcelona) for her assistance in graphic support and support in the structured search, Cristina Segú-Vergés (Anaxomics Biotech, Barcelona) for project coordination and proofreading, and Sergio Alonso-Orgaz (Lidesec, Madrid) for formatting, and medical writing support, editorial assistance and graphic support. The graphic support, literature search support, medical writing support, and editorial assistance were funded by Takeda Farmacéutica España S.A.
Author Contributions
José R. Gutiérrez-Casares conceptualised the review, performed the literature search, wrote the first draft and revised the final manuscript. Javier Quintero performed the literature search and reviewed, managed the project, criticised, completed and revised the final manuscript. Cecilio Álamo performed the literature search and reviewed, criticised, completed and revised the final manuscript.
Disclosures
Javier Quintero: Speaker and/or scientific advisory boards for Takeda, Janssen, and Rubió Research funding: Instituto de Salud Carlos III. Collaborations: none. José R. Gutiérrez-Casares: Scientific advisor within the scope of the present study: Takeda Farmacéutica España S.A. Speaker or advisory board: Janssen (Johnson&Johnson), Takeda; Research funding: Lumbeck, Takeda. Collaborations: Laboratoires Servier. Cecilio Álamo: Speaker or advisory board: Janssen (Johnson&Johnson), Takeda, Rubió, Rovi, Lunbeck, Laboratoires Servier; Research funding: none. Collaborations: none.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Quintero, J., Gutiérrez-Casares, J.R. & Álamo, C. Molecular Characterisation of the Mechanism of Action of Stimulant Drugs Lisdexamfetamine and Methylphenidate on ADHD Neurobiology: A Review. Neurol Ther 11, 1489–1517 (2022). https://doi.org/10.1007/s40120-022-00392-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-022-00392-2