
Abstract
Attention-Deficit/Hyperactivity Disorder (ADHD) is a prevalent psychiatric condition characterized by developmentally inappropriate symptoms of inattention and/or hyperactivity/impulsivity, which leads to impairments in the social, academic, and professional contexts. ADHD diagnosis relies solely on clinical assessment based on symptom evaluation and is sometimes challenging due to the substantial heterogeneity of the disorder in terms of clinical and pathophysiological aspects. Despite the difficulties imposed by the high complexity of ADHD etiology, the growing body of research and technological advances provide good perspectives for understanding the neurobiology of the disorder. Such knowledge is essential to refining diagnosis and identifying new therapeutic options to optimize treatment outcomes and associated impairments, leading to improvements in all domains of patient care. This review is intended to be an updated outline that addresses the etiological and neurobiological aspects of ADHD and its treatment, considering the impact of the “omics” era on disentangling the multifactorial architecture of ADHD.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Attention-Deficit/Hyperactivity Disorder (ADHD) is characterized by developmentally inappropriate symptoms of inattention and/or hyperactivity/impulsivity, which leads to impairments in the social, school/academic, and professional contexts. The worldwide prevalence of ADHD is among the highest for neurodevelopmental disorders, estimated at ≈5–7% for children/adolescents [1, 2] and ≈2.5% for adults [3, 4].
ADHD diagnosis requires a careful clinical assessment based on symptom evaluation and functional impairments. Diagnostic classification practices such as the Diagnostic and Statistical Manual of Mental Disorders—5th edition (DSM-5; APA 2013) offer critical standard criteria to guide clinicians for an accurate diagnosis. According to DSM-5, to fulfill the diagnosis criteria, patients should present: (a) at least six (for children and adolescents) or five (for adults) out of the nine symptoms of inattention and/or hyperactivity/impulsivity for a minimum of 6 months; (b) symptoms onset before 12 years of age; (c) symptoms that cause impairments in living; (d) symptoms that affect different life contexts (e.g., home and school). Notably, the manifestation of symptoms should not be better explained by other conditions (e.g., schizophrenia, bipolar disorder, etc.). DSM-5 describes three clinical presentations of ADHD: predominantly inattentive, predominantly hyperactive, or combined. In general, the inattentive and combined presentations are the most frequently observed among patients, except for young children (3–5 years old), for which hyperactivity is more pronounced [5, 6].
ADHD diagnosis is sometimes challenging due to the substantial heterogeneity of the disorder in terms of clinical and pathophysiological aspects, a fact that impacts research on the etiological and neurobiological specificities of ADHD [7]. The high rates of psychiatric comorbidities observed in patients with ADHD and the considerable proportion of overlap in symptoms and causes with other mental disorders are key factors to be considered in this sense. More than 60% of individuals with ADHD present at least one comorbid psychiatric disorder, frequently including depressive, anxiety, and disruptive behavioral disorders [8,9,10]. Autism spectrum disorder (ASD) and ADHD are also frequently comorbid [11, 12]. According to the current DSM-5 diagnostic criteria, ADHD diagnosis should no longer be excluded in the presence of ASD, as in previous versions. Patients with ADHD also present higher rates of obesity, sleep disturbances, asthma, autoimmune and inflammatory diseases, and other somatic and metabolic-related problems [13,14,15].
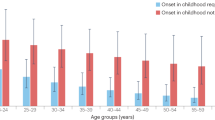
Another source of heterogeneity is the diverse clinical profile of children and adults with ADHD. For example, the comorbidity profile mentioned above also varies across development. In children, the most common comorbidities include externalizing (conduct and oppositional defiant) and learning disorders. In contrast, mood, anxiety, and especially personality and substance use disorders become more prevalent in adult life [13, 14]. In addition, while in children and adolescents ADHD is more frequent in boys than in girls, ranging from 3:1 to 10:1 ratios, in adulthood, a more balanced distribution of prevalence across genders is observed [16, 17]. Beyond the age-dependent decline of symptoms, especially in the hyperactive/impulsive domain [18], clinical follow-ups have shown that ADHD can fluctuate or remit throughout the lifespan, including during adulthood [19,20,21,22]. Population cohort studies have also demonstrated that the adult ADHD population may not be composed only of individuals with ADHD initiating in childhood and persisting into adulthood but also comprised by at least 20% of late-onset cases occurring in late adolescence and young adulthood [23,24,25,26], challenging the classical neurodevelopmental conceptualization of the disorder [27].
Considering the consequences of ADHD symptomatology in different life contexts and its high social and economic impact, the clinical, epidemiological, and neurobiological aspects of this disorder have been widely studied. Despite the challenges related to clinical heterogeneity, as mentioned above, promising perspectives to understanding the neurobiological basis of ADHD have emerged in the last few years. In this literature review, we provide an updated appraisal of the etiological and neurobiological aspects of ADHD and its treatment in light of the “omics” era for disentangling the multifactorial architecture of ADHD. The search was based on published research collected from the PubMed database using general search keywords for each topic followed by a critical evaluation of existing research to include the most relevant evidence (i.e., supported by meta-analysis, highlighted by systematic reviews and/or large recent studies presenting robust and unbiased methodologies).
2 Environmental correlates of ADHD
Among the environmental variables proposed as risk factors predisposing to ADHD, pre-, peri- and postnatal factors are the most frequently associated, including low birth weight and prematurity [28], maternal exposure to stressful events, or other health-related maternal conditions [29, 30]. Significant effects of maternal (during pregnancy) and/or early childhood exposures to cigarettes [31], alcohol [32], medications [33,34,35], and other substances such as lead and manganese [36] on the risk of ADHD have been reported. See Additional file 1: Tables S1 and S2 for a comprehensive overview of meta-analyses reporting the effects of prenatal, newborn, and lifespan-related factors and exposures. Nutritional aspects and diet across the lifespan have also been considered as factors that potentially influence ADHD susceptibility [37,38,39]. See Additional file 1: Table S3 for more detailed information from meta-analyses results investigating biomarkers and nutritional factors. Other risk factors for ADHD include psychosocial conditions such as low income, hostile parental care, and life adversities. However, it is important to consider that these factors might have more prominent effects as course modifiers of prognosis rather than in the predisposition of the disorder [40].
Although investigations on environmental risk factors for ADHD have yielded some reproducible findings, causality inferences remain elusive since unmeasured factors may confound the results [41]. The potential effects of gene-environment interactions and correlations should be considered. In this regard, a possible effect of the genetic variation on the predisposition of exposure to specific risk or protective environmental contexts may indirectly affect ADHD susceptibility, imposing difficulties in interpreting associations [42]. Maternal-related risk factors, for example, may be confounded by familial or genetic factors. On the other hand, some environmental exposures can also result in biological modifications, such as DNA methylation, which are reversible changes in genomic function independent of the DNA sequence that has also been associated with ADHD [43, 44]. Thus, environment and genetics act together in the etiology of ADHD, making it challenging to disentangle several sources of bias/confounding, possibly affecting the observed associations of environmental risk factors.
3 Genetics of ADHD
ADHD has a multifactorial etiology, in which genetic and environmental factors are involved in its development. The genetic contribution to ADHD is among the highest for psychiatric disorders. A high family aggregation is reported for ADHD and data from a large population sample demonstrated that the hazard ratio for ADHD relatives, compared to the frequency of ADHD in relatives of unaffected people, is 70 for monozygotic twins, around 8 for dizygotic twins and full siblings, between 2 and 3 for half-siblings, 2.2 for full cousins, and 1.5 for half cousins [45]. The total heritability of ADHD has been estimated at 70–80% [46, 47]. These estimates seem stable throughout childhood and adolescence [48] and similar for both males and females [49]. For adults, some studies, especially those based on self-report measures, describe a drop in heritability estimates; however, studies that used cross-informant data and/or clinical diagnosis report that heritability estimates in adults could be as high as those observed in children, suggesting that phenotype measurement biases could play a larger role than true age-dependent decline [50]. Although additive effects are the major component of heritability, available evidence suggests that nonadditive effects (genetic interactions) also contribute to a large proportion of phenotypic variance and that the effects of a shared family environment are minimal [49, 51].
The understanding of the genetic architecture of ADHD has significantly evolved with the technological advances in the molecular genetics field, especially with the availability of large-scale genome-wide association studies (GWAS). Based on these studies, the proportion of the phenotypic variation attributed to common genetic variants (known as h2SNP) has been estimated for ADHD. The ADHD GWAS that reported the first genome-wide significant findings (2019-ADHD) estimated the ADHD h2SNP at ≈22% [52]. In an updated GWAS meta-analysis of ADHD (2022-ADHD), the h2SNP was lower than in the previous GWAS, valued at 14% [53]. The authors attributed this difference to varying ascertainment strategies and designs or lower effective sample sizes on specific individual cohorts that comprised the study. Nevertheless, the results indicate that around 7000 common variants can explain 90% of the h2SNP, confirming the extensive polygenic architecture of ADHD and that many hits, in addition to those already discovered, will become significant as the sample size increases. Although the h2SNP estimated for ADHD is far from reaching the heritability estimated in twin studies, it should be noticed that the h2SNP of psychiatric phenotypes is usually much lower than the total heritability because of unmeasured factors in GWAS, such as rare and structural variants, gene–gene and gene-environmental interactions, and epigenomics.
The 2019-ADHD GWAS comprised a sample of 20,183 cases and 35,191 controls (the first wave of data from the Danish iPSYCH15 cohort (iPSYCH1) plus 11 ADHD cohorts collected by the Psychiatric Genomics Consortium (PGC)) and revealed 12 significant hits. The 2022-ADHD GWAS, including the extended Danish iPSYCH cohort (iPSYCH1 plus new iPSYCH2 data), the Icelandic deCODE cohort, and the PGC, totalizing a sample of 38,691 individuals with ADHD and 186,843 controls, showed 27 genome-wide loci associated with ADHD, of which 21 were new [53]. The associated loci include variants located around genes involved in a variety of processes, such as neurodevelopmental mechanisms and neurogenesis (e.g., PPP1R16A, B4GALT2, PTPRF, SORCS3, DUSP6, FOXP2, MEF2C, and METTL15 genes).
Initially, it was expected the first significant hits to be revealed in these GWAS would be dopaminergic and noradrenergic genes, which were previously implicated in ADHD by meta-analysis of candidate gene studies, such as those encoding the dopamine receptors (e.g. DRD2, DRD4), the dopamine transporter (DAT1) and the enzymes monoamine oxidase (MAOA), catechol-O-methyltransferase (COMT) and dopamine beta-hydroxylase (DBH) [54, 55]. Interestingly, these GWAS pointed out none of such candidate genes previously implicated in ADHD. However, as mentioned above, the h2SNP is explained by a much higher number of SNPs than those significantly associated. Therefore, genetic variants pointed out by candidate genes can still become significant as the sample size increases, as observed for other disorders, such as schizophrenia and depression, for which the significance in variants of candidate genes only emerged when GWAS reached huge sample sizes (> 150 thousand individuals) [56,57,58].
Genomic studies revealed an extensive genetic overlap of ADHD with a wide range of phenotypes, including psychiatric, cognitive, behavioral, and metabolic domains, suggesting that the clinically observed association of ADHD with these phenotypes can be explained, at least in part, by shared genetic factors [52, 53, 59]. The cross-trait genetic correlation is usually estimated using the linkage disequilibrium score regression (LDSC) or the polygenic scores (PGS) approaches. These analyses have revealed negative relationships between ADHD and different neurocognitive domains, including attention (beta = − 0.07, P = 3,25E-7) and working memory (beta = − 0.05, P = 2,45E-3) [53]. The same direction of results is reported for educational attainment (rg = − 0.55, P = 1,42E-151), which provides further support to the relationship between ADHD and impairments in cognitive and executive functioning [53, 60]. On the other hand, ADHD presents positive genetic correlations with other psychiatric disorders (e.g., major depressive disorder (rg = 0.31 to 0.52, P = 2,96E-06 to 2,18E-20), autism spectrum disorder (rg = 0.42, P = 1,19E-18), bipolar disorder (rg = 0.26, P = 7E-6), and schizophrenia (rg = 0.17 to 0.22, P = 1,72E-09 to 1,22E-06) and with other psychiatric-related conditions, such as neuroticism (rg = 0.23 to 0.33, P = 0.0008 to 2,61E-13), symptoms of depression (rg = 0.39 to 0.53, P = 1,25E-6 to 1,63E-32), insomnia (rg = 0.46, p = 3,73E-20), smoking initiation (rg = 0.48, P = 1,78E-11) and cannabis use (rg = 0.29 to 0.61, P = 1,63E-5 to 1,27E-42) [53, 59, 61]. Interestingly, ADHD also presents positive genetic correlations with metabolic and immune-related diseases such as overweight/obesity (rg = 0.21 to 0.27, P = 7,5E-7 to 5,46E-20), type 2 diabetes (rg = 0.18, P = 1,14E-5), asthma (rg = 0.12, P = 0,011), and psoriasis (rg = 0.23, P = 1,0E − 3) [53, 62], suggesting the possibility of a shared genetic etiology between these phenotypes. Notably, there are no genetic correlations between ADHD and most neurological diseases, such as Alzheimer’s [53, 59, 63, 64]. Even though these phenotypes are not genetically correlated, it is important to notice that there are traits, such as intelligence and cognitive performance, presenting the same direction of association to both ADHD and Alzheimer’s disease, suggesting the possibility of shared traits that contribute to both phenotypes.
The genomic studies of ADHD also reinforce the high polygenic nature of ADHD and the dose-dependent effect of the PGS for ADHD, where the increase in PGS deciles was associated with a corresponding increase in the odds ratio for ADHD in the target samples [52]. Besides the association with ADHD diagnosis, PGS for ADHD is associated with quantitative symptom measures of inattention and hyperactivity/impulsivity (even in the general population), reinforcing the view that ADHD is one extreme of a symptom distribution curve [65,66,67].
Relevant findings from genome-wide studies have also helped to disentangle the clinical heterogeneity of ADHD observed in different stages of life and provided insights into the debate about the neurodevelopmental conceptualization of the disorder. A GWAS comparing the genetic background between children and adults showed a shared genetic contribution to ADHD in these groups, supporting the neurodevelopmental nature of persistent ADHD in adults [68]. A subsequent study found a similar level of genetic correlation between childhood ADHD and persistent ADHD (rg = 0.82), but lower with late-diagnosed ADHD (rg = 0.65), suggesting this group might present some differences in its genetic architecture when compared to childhood ADHD [69]. Although similar patterns of cross-trait genetic correlations for children and adults were reported by Rovira et al., 2020 [68], Rajagopal et al., 2022 [69] found different patterns across ADHD subgroups for some traits, including a higher genetic correlation for childhood ADHD with autism than late-diagnosed ADHD, and for depression and alcohol use disorders for late-diagnosed ADHD compared to childhood ADHD. This suggests possible differential genetic susceptibility for common comorbidities across ADHD subgroups.
3.1 Mendelian randomization
Despite the great advances in understanding the biological aspects related to ADHD, as summarized above, the causal effects underlying the reported associations are still largely unknown. In this regard, Mendelian Randomization (MR) has been used as a promising approach to investigate potential causal relationships between an exposure (e.g., brain measures or environmental exposures) and an outcome (e.g., ADHD susceptibility) by using genetic variants as instrumental variables. Since genetic variants are randomly determined at conception, using them as instruments has the potential to eliminate confounding effects behind associations while evaluating causal effects between a risk factor and a clinical phenotype [70, 71].
Several associations with ADHD have been explored through MR. Concerning the links with other psychiatric phenotypes, it has been suggested a causal effect of ADHD genetic liability with an increased risk for posttraumatic stress disorder [72], autism spectrum disorder (potentially a bidirectional effect [73, 74]), major depression [75, 76], substance use disorder [77, 78], including smoking [76, 78, 79], cannabis use [61, 78], and a weaker effect on alcohol dependence [78]. On the other hand, genetics of neurocognitive, such as general intelligence [80,81,82], and brain features (e.g. total brain volume, functional connectivity between the default mode and salience networks [80], and white matter microstructure of anterior limb of internal capsule [83] seem to represent a relevant causal role in ADHD pathogenesis, distinguishing important brain aspects playing etiological roles from those that are simply correlated with or consequences of ADHD. A bidirectional causality has also been observed between ADHD and obesity-related traits [84,85,86]. These findings help to disentangle the previous associations observed in cross-sectional studies, providing insights into the biological mechanisms underlying ADHD pathophysiology that can be useful for the research on potential biomarkers and preventive strategies for ADHD.
Still, the limitations of this approach should be considered while interpreting the results. MR relies on major assumptions that when violated can introduce bias to the analyses, especially regarding the absence of horizontal pleiotropy and potential confounders affecting the exposure-outcome relationship [87]. This is especially challenging for psychiatric disorders, considering their highly complex and polygenic architecture, and the involvement of pleiotropic genetic variation in their etiology. Nevertheless, alternative methodologies have been proposed to overcome these limitations, improving the interpretability and reliability of the findings, such as Mendelian Randomization Pleiotropy RESidual Sum and Outlier (MR-PRESSO) [88] and Causal Analysis Using Summary Effect estimates (CAUSE) [89].
4 Neurotransmission alterations in ADHD
Despite evidence demonstrating high polygenicity in ADHD, it is possible that many variants at different loci may converge on common mechanisms. A putative pathophysiological mechanism that has been long implicated in ADHD involves the dysregulation of monoaminergic neurotransmission systems, mainly dopaminergic and noradrenergic. These neurotransmitters exert their functions by a mechanism with a curve pattern in an ‘inverted U’ shape, that is, both very intense activity (for example, during stressful situations) and very low activity (for example, states of somnolence) impair the functioning of these systems [90].
The physiological changes induced by the binding of dopamine and norepinephrine to their respective receptors involve the modulation of several cognitive and executive processes usually impaired in ADHD (see Table 1), corroborating the monoaminergic hypothesis for ADHD pathophysiology [91]. For example, dopamine receptors of subtypes D1 and D2 are abundant in brain regions mainly involved in signaling reward circuits, learning and memory, and locomotor activity [90]. Also, patients with ADHD have a higher density of the dopamine transporter (DAT), responsible for the reuptake of DA into presynaptic neurons [92], which could lead to alterations in dopamine levels in the synaptic cleft. The binding of norepinephrine with adrenergic receptors has been shown to modulate working memory processes, for which moderate or high levels of this neurotransmitter present differential binding affinities to each type of receptor and consequently different physiological effects on working memory [93]. Furthermore, the effects of methylphenidate treatment on working memory appear to be dependent on α2 noradrenergic receptors, whereas the improvement in sustained attention involves the α1 subtype [94].
Although not as extensively as the dopaminergic and noradrenergic systems, the involvement of serotonergic [95], glutamatergic [96], and GABAergic [97] systems have also been implicated in ADHD by studies associating altered levels of these neurotransmitters with ADHD (Table 1). Experimental models also demonstrated that methylphenidate is able to restore impaired glutamatergic transmission at the prefrontal cortex and improve behavioral symptoms of ADHD, a process that appears to be mediated by AMPA glutamatergic receptors [98].
Besides the above-mentioned evidence, the involvement of neurotransmitter systems in ADHD is reinforced by the mechanism of action of most drugs used for its treatment that involves targets related to these biological pathways (e.g., methylphenidate, which targets DAT and NET). Additional evidence for the involvement of these neurotransmission systems in ADHD is summarized in Table 1. Nevertheless, ADHD involves a complex neurobiology that results from the interaction between various dysfunctional neurophysiological systems, as evidenced by genomic studies.
5 Brain structural and functional alterations in ADHD
Pathophysiological insights can also be inferred from neuroimaging studies, which has reported differences in structural and functional brain architecture between patients with ADHD and neurotypical individuals, especially in children. Large meta-analyses from the ADHD working group of the ENIGMA (Enhancing NeuroImaging Genetics through Meta-Analysis) consortium reported that children with ADHD present smaller volumes in different subcortical brain regions (i.e., nucleus accumbens, amygdala, caudate, hippocampus, and putamen) and total intracranial volume [118], as well as decreased cortical surface area (mainly in frontal, cingulate, and temporal regions) and thickness (in the fusiform gyrus and temporal pole) [119]. Such alterations are not observed when restricting the analyses to adults and no effects of psychostimulants were found [118, 119]. Through adopting an independent component analysis of whole-brain morphometry images, associations of ADHD with reduced volumes in frontal lobes, striatum, and their interconnecting white matter microstructure were reported for both children and adults, supporting a role of the frontostriatal circuit regardless of age [120]. A large cross-sectional brain imaging study, the Adolescent Brain and Cognitive Development (ABCD), also suggests reduced structural brain measures in children with ADHD, although with modest effect sizes [121], indicating additional investigation is needed to support specific cortical and subcortical alterations as potential biomarkers for ADHD.
Structural connectivity differences in ADHD have been investigated using Diffusion Tensor Imaging (DTI), a technique based on the observation of the directionality and coherence of water diffusion that allows inferences on the microstructure of brain white matter (WM). Meta-analyses based on fractional anisotropy (FA) measures, a common DTI parameter that is considered a marker of white matter microstructural integrity, support that the most robust findings related to ADHD involve the frontostriatal circuitry and the interhemispheric connection through the corpus callosum [122, 123]. Microstructural alterations in the cingulum and corticospinal tracts are also commonly associated with ADHD. However, contrasting DTI findings have been reported, with either reduced or increased FA measures being observed for the same brain circuitry. These disparities are observed for different methodological approaches, for example, voxelwise or tractography-based, and might reflect potential confounders as sources of heterogeneity among studies, such as non-controlled head motion group differences [122]. A recent large multi-cohort mega-analysis that used original DTI data attempting to overcome these issues showed that lower FA of inferior longitudinal (ILF) and left uncinate (UNC) fasciculi were associated with both ADHD diagnosis and symptoms (attention problem scores) [124]. These findings are consistent with a pathophysiological mechanism for ADHD that involves brain networks related to cognitive functions (ILF) and emotion/reward processing (UNC). Although the general landscape for the role of the implicated regions is congruent with functions usually impaired in ADHD, such as cognition, behavioral plasticity, and emotional and motor control, larger and more homogeneous studies are necessary to validate them as key factors in the neurobiology of ADHD.
Functional Magnetic Resonance Imaging (fMRI) studies associate ADHD with alterations in connectivity in multiple brain networks involved mainly in executive functions, attention, emotions, and sensory and motor activities. A broadly studied model for ADHD suggests an impaired synchronization between the DMN (Default Mode Network), which is normally more active when tasks are not performed, and the TPN (Task-Positive Network), which is activated while performing tasks. In individuals with ADHD, inadequate DMN hyperactivity and TPN hypoactivity while performing tasks have been suggested as an explanation for ADHD symptoms [125]. In resting-state fMRI (rsfMRI) studies, the frontoparietal network, involved in executive functions, has demonstrated hyperconnectivity with regions of the DNM and affective network, while hypoactivity with regions of the ventral attention and somatosensory networks in individuals with ADHD [126]. ADHD has also been associated with disrupted connectivity between DNM and systems involved in cognitive control and affective/motivational and salience networks [127]. A large mega-analysis of rsfMRI supports the findings suggesting that ADHD is associated with stronger coactivation/weaker anticorrelation between DNM and TPN networks, including the dorsal and salience/ventral attention networks, which is thought to result in attention lapses/mind-wandering and worse performance on attention and executive function tasks [128]. Impaired functional connectivity in these networks has been supported by task-based studies across several cognitive domains [129]. Congruently, methylphenidate treatment is associated with the suppression of DNM activity during cognitive tasks and the improvement of resting state functional connectivity within DMN in patients with ADHD [130, 131]. However, a recent meta-analysis of rsfMRI reported no significant spatial convergence of hyperconnectivity or hypoconnectivity related to ADHD across studies [132]. These results reflect once again the high heterogeneity of neuroimaging studies, including differences in experimental methods, characteristics of participants, and analytic design, as well as the heterogeneous presentation of ADHD and its complex pathophysiology.
Although methodological issues challenge the identification of brain aspects related to ADHD, such associations are supported by genomic studies showing a high genetic correlation between ADHD and several neuroimaging measures, including intracranial and total brain volumes and cortical surface area [133,134,135,136]. Additionally, it was suggested the neuroimaging associations observed in children may also be detected in adults depending on the genotype profile [137]. Therefore, the genetic architecture of ADHD may be shared to some extent with brain structure variation in both children and adults.
6 Epigenomics
Some studies evaluating differential methylation in candidate genes were conducted in ADHD [43, 138]. However, as occurred for GWAS, the number of epigenome-wide association studies (EWAS) is growing. EWAS aims to identify epigenetic modifications involved in the susceptibility and course of phenotypes in a large-scale analysis, which evaluates methylation sites spread throughout the entire genome. In the case of ADHD, at least 11 EWASs have already been published, two of which are meta-analyses [139,140,141,142,143,144,145,146,147,148,149]. Unfortunately, several single-site EWAS did not reveal positions (differentially methylated positions—DMPs) in the epigenome-wide significance level. However, some promising results can already be summarized, mainly from analyzes involving differentially methylated regions (DMRs). For example, three studies identified VIPR2 (Vasoactive Intestinal Peptide Receptor 2) as a differentially methylated gene in ADHD [139, 140, 147]. In addition, one study demonstrated differential methylation in the ST3GAL3 gene (ST3 Beta-Galactoside Alpha-2,3-Sialyltransferase 3) in the epigenome-wide significance level [143], which was also associated with ADHD by GWASs [52, 53].
The two meta-analyses involved different study designs [145, 148]. One meta-EWAS included only children and adolescents and looked at ADHD trajectories. Significant genes in the case–control analysis involving cord blood (n = 2477) were not implicated in ADHD symptomatology during childhood and adolescence (n = 2374). At birth, nine CpGs predicted later ADHD symptoms in genes that are not GWAS hits for psychiatric disorders yet (ERC2—ELKS/RAB6-interacting/CAST family member; CREB5—cAMP responsive element binding protein 5; ZBTB38—zinc finger and BTB domain containing 38; PPIL1—peptidylprolyl isomerase like 1; and TRERF1—transcriptional regulating factor 1) [148]. The second one [145] was a meta-EWAS of ADHD symptoms in adults that included three individual cohorts (the Netherlands Twin Register, Dunedin Multidisciplinary Health and Development Study, and Environmental Risk Longitudinal Twin Study). The combined sample size was 4,689 individuals. One epigenome-wide significant DMP was detected in the Dunedin study (chromosome 8 intergenic), but this finding was not observed in the meta-EWAS of the three cohorts. In region-based analyses, six DMRs were identified in the Netherlands Twin Register, 19 in the Dunedin study, and none in the Environmental Risk Longitudinal Twin Study. No significant DMR was associated with more than one cohort. The top single-cohort gene was TNXB, a genome-wide hit in the schizophrenia and neuroticism GWASs [150,151,152].
Although the results summarized here are promising, this set of studies is still small, especially involving samples of adult individuals and looking at clinical aspects of ADHD (only three EWASs). Further studies with even larger samples are needed to elucidate the role of epigenetic mechanisms in ADHD throughout life. Additionally, the relationship between epigenomics and genomics should be better studied since epigenetic markers also present significant heritability estimates [153]. A recent study evaluating global DNA methylation (GMe) showed that the GMe levels observed in ADHD cases were lower than controls. Furthermore, the ADHD PGS was negatively correlated with GMe in ADHD cases [154]. On a locus-specific level, the ADHD PGS was associated with differential methylation in an epigenome-wide significant level with two genes that were not implicated in ADHD or other psychiatric disorders by GWASs yet (GART—phosphoribosylglycinamide formyltransferase, phosphoribosylglycinamide synthetase, phosphoribosylaminoimidazole synthetase, and SON—DNA and RNA binding protein) [147]. Therefore, these findings demonstrated a degree of genetic control over methylation driven by the underlying genomics of ADHD, which should be further explored as a potential mechanism for gene-environment interactions.
7 Metagenomics
The bidirectional communication between the gut microbiota and the nervous system is known as the gut-brain axis (GBA). The main mechanisms through which the gut microbiota could affect the nervous system involve the production and metabolism of neurotransmitters, the vagus nerve, the hypothalamic–pituitary–adrenal axis, and inflammation pathways [155, 156]. The unbalance of microbiota composition—dysbiosis—can lead to systemic inflammation, dysregulation of neurotransmitter levels, and increased oxidative stress, affecting brain functions via the gut-brain axis.
The role of microbiota in the pathogenesis of ADHD has been suggested by studies showing differences in microbiota composition and/or abundance between patients with ADHD and controls [157]. An increased abundance of Enterococcus, Bifidobacterium, and Odoribacter has been reported in ADHD patients. These genera are involved in pathways related to the production and release of the neurotransmitters, such as GABA and dopamine, suggesting that such gut dysbiosis could be related to the pathogenesis of ADHD through neurotransmitter dysregulation [158,159,160]. On the other hand, the abundance of Faecalibacterium is decreased in ADHD and is inversely related to ADHD severity [159, 161, 162]. Faecalibacterium has been related to anti-inflammatory properties [163]. Unbalanced levels of this genus could influence the production of pro-inflammatory cytokines and induce systemic inflammation, a mechanism that has been proposed to contribute to ADHD pathogenesis [164, 165].
The specific differences in gut microbiota mentioned above describe the most common findings that have been highlighted by recent reviews; however, a meta-analysis that assessed the relationship between gut microbiota and ADHD found no significant differences at the phylum and family levels, reporting as the only significant association the higher within-group diversity of Blautia in patients with ADHD compared to healthy controls [166]. Blautia is known to interfere with the regulation and function of inflammation and immunity pathways, reinforcing the hypothesis that the link between gut microbiota and ADHD pathogenesis might involve inflammation mechanisms.
Regarding the general composition of the microbiota, while some individual studies have found differences in patients with ADHD in diversity indices (alpha and beta) [167, 168], the overall evidence and meta-analyses point to high heterogeneity across studies and do not support a consistent association of diversity indices with ADHD [160, 166]. In general, the studies evaluating the microbiota effects on ADHD are limited, and contradictory/inconsistent findings have been reported, likely due to methodological differences and small sample sizes [157].
8 ADHD treatment
Early and adequate intervention has the potential to reduce the risk of negative ADHD-related outcomes in mental and physical health [169]. The treatment options can be non-pharmacological, pharmacological, or a combination of both. The first comprises psychosocial, cognitive, and behavioral training that stimulates specific neuropsychological domains associated with ADHD, such as cognitive and executive functions. According to meta-analyses, patients may experience some benefits of such psychotherapies compared to placebo, and they are especially recommended as first-line or adjunct treatment for young children (aged 6 years or less) and less severe cases [170,171,172]. However, for most of these psychotherapeutic interventions, meta-analyses point to low certainty and inferior efficacy in reducing the core symptomatology of ADHD compared to the pharmacological treatment (e.g., with stimulants) [170].
Therapeutic guidelines, including those from the British Association of Psychopharmacology [173] and the National Institute for Health and Care Excellence [171], generally recommend stimulant medications, namely lisdexamfetamine and methylphenidate, as the first-choice pharmacological treatment for moderate to severe ADHD cases and patients aged 6 years or older. For patients who neither tolerate nor respond to treatment with these drugs, non-stimulants (e.g., atomoxetine) are considered the second choice, followed by adrenergic agents (e.g., clonidine and guanfacine) or alternative non-stimulant drugs, including antidepressants such as tricyclics and bupropion [171, 173,174,175]. As detailed below, the intervention options have varying properties in terms of effect size, mechanism of action, as well as different profiles of side effects. The treatment strategy depends on these factors and should be carefully evaluated on an individual basis according to the clinical profile of the patients, especially considering the presence of comorbidities, which are frequently observed in patients with ADHD.
8.1 ADHD medications
The safety and efficacy of commonly used ADHD medications are supported by metanalyses (Cortese et al., 2018), which provide the basis for the guidelines’ recommendations mentioned above. Besides being efficacious in reducing the core symptomatology of ADHD, there is congruent evidence indicating their potential to improve related outcomes, such as quality of life, academic performance, social aspects, comorbid neuropsychiatric conditions, as well as reduce the risk of other functional impairments, including criminality, incidents of self-harm, suicidality, and risk-taking behaviors [169, 176,177,178,179].
8.1.1 Stimulants
In general, stimulants present superior efficacy compared to other medications for all age ranges. Among the stimulants, amphetamines have shown greater effect sizes in reducing ADHD symptoms than methylphenidate [180]. Although the benefit of methylphenidate on ADHD symptoms is also observed in the long-term when compared to discontinuation or switching to a placebo, the long-term effect sizes are usually smaller than those reported in short-term studies of methylphenidate treatment [174].
Methylphenidate, the most used medication worldwide, is derived from piperidine and structurally similar to amphetamine. Both methylphenidate and amphetamines produce similar effects in increasing the availability of dopamine and norepinephrine in the synaptic cleft, leading to enhanced neurotransmission, but their mechanisms of action differ in some respects. The pharmacological activity of methylphenidate results mainly from blocking the dopamine and norepinephrine transporters (DAT and NET, respectively), thus inhibiting the reuptake of these neurotransmitters into presynaptic neurons [104]. On the other hand, besides the inhibition of DAT and NET through the direct binding to them as a false substrate, amphetamines also promote increased presynaptic efflux of dopamine into the extracellular space. The regulation of this process involves the modulation of functions such as attention, pleasure, and motor activity [105]. Stimulants also improve executive functions frequently impaired in individuals with ADHD, such as inhibitory control, working memory, and sustained attention in an age-independent manner [181,182,183].
Amphetamines and methylphenidate present with a similar profile of adverse effects, which are usually mild and transient. The most common are decreased appetite, dry mouth, irritability, sleep disturbances, tachycardia, and headache; however, amphetamines are associated with a higher rate of side effects, such as weight loss and insomnia [170]. Concerns about stimulant use and adverse cardiovascular outcomes, including increases in heart rate or blood pressure, have also been raised [180]. However, the relationship between these medications and severe cardiovascular events has been controversially discussed since large registry studies do not support a clear association between serious cardiovascular outcomes and stimulant use [184, 185]. Additional caution should be taken when using stimulants to treat patients with ADHD in comorbidity with substance use disorders because of the addictive potential of these medications, especially the short-acting formulations [186]. In these cases, alternative non-stimulant treatment options or long-acting stimulant formulations are most suitable [187]. Treatment of ADHD with comorbid bipolar disorder also requires special care because of the risk of stimulants inducing mixed/manic episodes in these patients [188, 189].
8.1.2 Non-stimulants
For patients who do not respond, do not tolerate, or present specific conditions for which stimulants are contraindicated, the non-stimulant medications atomoxetine, clonidine, and guanfacine are alternative FDA-approved treatment options. Meta-analyses confirm that all these medications are safe and superior to placebo in reducing ADHD symptoms, and the specificities of each drug are described below.
In clinical assessments, atomoxetine presents efficacy of modest effect size [180] but is still superior to placebo in reducing the severity of ADHD symptoms. Compared to stimulants, atomoxetine is less effective and usually has a slower initial response, with symptom improvement being noticed only after several weeks of treatment. Besides, a considerable percentage (~ 40%) of patients persist with impairing symptoms while being treated with atomoxetine, demanding additional management [109]. Nevertheless, atomoxetine might be particularly indicated in cases of comorbid ADHD and anxiety or for patients with a history of substance abuse, for which it may be considered the first treatment option [190, 191]. Regarding tolerability, the most observed adverse events include nausea, loss of appetite, fatigue, dizziness, irritability, abdominal pain, and sleep disturbances [109]. Considering the potential risk of suicidal behavior reported by clinical trials in children receiving atomoxetine, although not supported by meta-analyses [192], it is recommended to monitor closely any notable behavior shift mainly at the beginning of treatment or dose adjustment in children.
Atomoxetine is a potent and selective inhibitor of norepinephrine reuptake that acts through the blockade of the NET. Such presynaptic NET inhibition leads to increased norepinephrine extracellular levels and occurs mainly in the prefrontal cortex but not in the mesolimbic and mesocortical pathways up to the nucleus accumbens. Therefore, atomoxetine presents minimal abuse or dependence potential [191]. Although atomoxetine presents a low affinity for DAT, it also promotes increased dopamine concentrations in the prefrontal cortex, which may result from the nonspecific modulation of dopamine reuptake via NET, which is highly expressed in this brain region. Atomoxetine has little or no affinity for noradrenergic receptors or any other type of receptor [193].
Extended-release formulations of the α2-adrenergic agonists clonidine and guanfacine are also second-choice alternatives for ADHD treatment, both as monotherapy and as an adjunct to stimulants. Although monotherapy with these agents has shown superior efficacy to placebo, when compared to stimulant medications, they have smaller effect sizes and their treatment effects take longer to be observed [110, 180, 194, 195]. However, the administration of these drugs as an add-on therapy to stimulant medications has been recommended since the combined mechanism of action of both drug classes may result in clinical improvement in ADHD symptomatology or a decrease in adverse effects [110, 196].
Guanfacine and clonidine have similar pharmacological properties and, differently from atomoxetine, their actions to potentiate the noradrenergic transmission occur through the direct agonism of the postsynaptic α2-adrenergic receptors rather than via NET blockade [196]. However, they differ in terms of potency and affinity. Guanfacine is a much less potent agonist than clonidine and seems to have a higher affinity for α2A receptors, while clonidine binds less selectively to all three subtypes of α2 receptors. In addition, differently from guanfacine, clonidine has the potential to act on imidazoline receptors as well, which would explain the more pronounced sedative and hypotensive effects of clonidine [110, 197]. Common adverse effects of both clonidine and guanfacine include somnolence/sedation, fatigue, irritability, insomnia, and nightmares, and they are usually more frequent and more pronounced than those observed for atomoxetine [195].
8.1.3 Other therapeutic alternatives
Although not included in clinical guidelines, therapeutic alternatives, such as the antidepressant bupropion, have also been used in clinical practice to treat ADHD symptoms. In this sense, there is evidence of its effectiveness, especially at high doses, although the consistency across studies is not too strong [198, 199]. The prescription of this medication might be specially considered as an option when comorbid conditions such as mood disorders or nicotine dependence are present, considering its primary antidepressant effect and its contribution to smoking cessation [200, 201]. Bupropion, unlike other typical antidepressants, such as tricyclics, promotes the inhibition of dopamine and norepinephrine reuptake only, and not serotonin. This drug also acts as a non-competitive antagonist of nicotinic acetylcholine receptors. Its mechanism of action is similar to that of stimulants, but the inhibition of neurotransmitter reuptake induced by bupropion is weaker than that produced by stimulants [199].
8.2 Pharmacogenomics
Although the approved medications are generally safe, efficacious, and lead to an important relief of ADHD core symptoms, they are not curative, and a considerable proportion of patients do not reach an optimal response. Besides, their effectiveness is further reduced due to high rates of poor drug adherence and persistence [202]. Such heterogeneity in treatment response, which may reflect the clinical heterogeneity of ADHD itself, affects treatment decision-making and challenges the definition of concise recommendations to guide clinicians. Thus, addressing the risks and benefits of the available medications for ADHD, the possibility of drug repurposing of approved compounds and the development of new therapeutic alternatives are the focus of current research to improve the treatment outcomes of patients with ADHD.
As briefly reviewed above, the known pharmacological actions of common medications used to treat ADHD involve only a few biological pathways, which seems inconsistent with the high complexity of the pathophysiological mechanisms proposed for ADHD. In this sense, GWAS arises as an important tool to advance knowledge for a more comprehensive understanding of the molecular underpinnings of ADHD neurobiology and the medications used for its treatment, helping to guide the development of better strategies for patient care [203].
As GWAS of ADHD susceptibility increases in power, they can reveal additional potential pathways involved with the disorder’s pathophysiology and yield new candidates for prospective drug development. Nonetheless, no significant genome-wide results have been found in GWAS for the response to treatment in patients with ADHD [204, 205]. The lack of success can be explained by the small sample sizes with genomic and treatment data available worldwide. However, promising insights can be inferred by applying strategies such as polygenic risk scores to test the genetic overlap between ADHD treatment response and the genetic liability for ADHD or other psychiatric traits. For example, higher polygenic risk scores for ADHD were associated with symptomatic improvement following ADHD medication in a small study using a Chinese sample [206], suggesting shared molecular mechanisms between ADHD and treatment response.
On the other hand, in a study investigating the druggable genome, the genes targeted by the FDA-approved medications for ADHD were not associated with ADHD at the genome-wide level, indicating that these drugs act through alternative pathways different from those underlying ADHD neurobiology [207]. Another interesting finding from the Lundbeck Foundation Initiative for Integrative Psychiatric Research (iPSYCH2012) case-cohort [208] suggests that ADHD cases with higher polygenic liability for mood and/or psychotic disorders might be at increased risk of discontinuing or switching stimulant treatment. Other studies applying alternative strategies taking advantage of integrative genome-wide, expression, and proteomic data from experimental studies have also reported some insights into the genetic background possibly involved with treatment response. They have revealed pathways involved in nervous system development and function, neurotransmitter release, GABA transmission, as well as other categories related to neurological diseases, psychological disorders, and behavior [205, 209]. Interestingly, they did not point specifically to the pathways largely implicated in ADHD, namely the dopaminergic and/or noradrenergic transmission, suggesting that additional mechanisms might underlie ADHD neurobiology and drug actions.
Although the evidence from candidate gene studies suggests a genetic contribution related to the dopaminergic and noradrenergic pathways in the treatment response of ADHD, especially in children, few robust and replicated results have been observed across these studies. A meta-analysis conducted by Myer et al. (2017) suggests polymorphisms in the SLC6A2/NET, COMT, ADRA2A, SLC6A3/DAT1 and DRD4 genes as potential predictors of the efficacy of methylphenidate in children [210]. For adults, a meta-analysis carried out by Bonvicini et al. (2016) concluded that for most polymorphisms, there is not enough data to apply this methodology. The only polymorphism for which it was possible to perform the meta-analysis was the 40-base pair VNTR in the DAT1 gene, for which no significant association with the response to methylphenidate was found [54].
The current limited knowledge provided by genetic studies of ADHD treatment so far reinforces the need to improve the understanding of the molecular basis of ADHD treatments. As further insights on the molecular biology of ADHD and medication used to treat it emerge, advancements in the pharmacotherapeutic options for ADHD improve, potentially benefiting the care of patients with ADHD. The above-mentioned exploration of the druggable genome in ADHD based on GWAS yielded processes of signal transduction and cell adhesion as novel possibilities for intervention in ADHD treatment, presenting possibilities of drug repurposing and novel targets for drug development [207]. A potentially prosperous example of drug repurposing based on molecular knowledge is the use of fasoracetam monohydrate, originally tested in clinical trials for vascular dementia, to treat ADHD. Fasoracetam is a metabotropic glutamate receptor activator and a clinical trial demonstrated it is efficacious in reducing symptoms of ADHD in cases of impaired glutamatergic signaling, a common pathway implicated in ADHD by genetic associations studies [211].
9 Concluding remarks
ADHD is an important impairing condition of public health due to its prevalence and persistence across the lifespan, and because it leads to a higher risk of adverse outcomes, such as academic underachievement, substance use and abuse, other psychiatric disorders, somatic diseases, risky behaviors, and premature death. Although the existing medications are considered safe and efficacious in relieving symptoms and preventing many negative consequences, there are groups of individuals with inappropriate responses, and these drugs’ long-term therapeutic effects are not robust. Research aiming to provide a more comprehensive understanding of the disorder’s pathophysiology is warranted to improve the diagnosis and management of the disorder.
Hundreds of studies have attempted to identify the underlying neurobiology or biomarkers for ADHD. These studies have faced several challenges, especially those related to the high heterogeneity of the disorder. Regardless, there is no doubt about the role of biology in the etiology and course of ADHD, and some facts are already a consensus in the area [7] (Fig. 1). For example, ADHD is substantially influenced by genetics with a polygenic architecture, there is extensive biological and phenotypic overlap between ADHD and other psychiatric traits, and the correlations observed in clinical and epidemiological studies have been confirmed by studies of genetic correlations. However, it is still necessary to increase sample sizes through large international collaborations to reveal additional genetic loci involved in ADHD susceptibility, increase the accuracy of polygenic scores, and ultimately understand the biological meaning of the findings and their utility in clinical practice.

Multifactorial complexity of Attention-Deficit/Hyperactivity Disorder (ADHD). ADHD presents substantial heritability in children, adolescents, and adults (the polled average is nearly 80%). Genetic and environmental factors modulate brain features (e.g., regional volumes, cortical thickness, connectivity, etc.) that are potentially in the causal pathway of the disorder
The study of brain biology has been somehow boosted by the development of neuroimaging techniques. The combined effects of risk factors may affect brain regions and networks that control cognitive, executive, and emotional processes usually impaired in ADHD, which can be evaluated through neuroimaging. These studies have helped to understand the functional and structural aspects of ADHD and added to our understanding of which brain areas are modulated and/or modified during the pharmacological treatment of ADHD. Despite the growing progress, the findings should be interpreted with caution due to the small sample sizes evaluated in these studies added to the high heterogeneity of both neuroimaging study designs and ADHD presentation.
The role of the environment in ADHD has also been the focus of several studies. Still, the reported associations should be viewed as correlated rather than causal risk factors. Since both environment and genetics interact in a complex manner in the contribution to the etiology of ADHD and the high probability of unmeasured factors in the studies, it is a challenge to identify environmental risk factors robustly associated with ADHD without the interference of confounders. Epigenomics and metagenomics will certainly become important tools in studying the role of the environment in the etiology and course of ADHD. While these approaches are promising, the available sample sizes are still very small. From this perspective, international consortia will be essential to meet this need for larger sample sizes. An important example is Eat2BeNice, a multicentric and international project whose main goal is to investigate the effects of nutrition and lifestyle on ADHD using genomics, epigenomics, and metagenomics as research tools (https://newbrainnutrition.com).
It is also a consensus that pharmacogenomics, at this first moment, will be useful for understanding the biological mechanisms involved in the therapeutic response rather than providing cost-effective tests. The application of pharmacogenetic tests is still far from benefiting clinical practice. While advancing the knowledge in ADHD neurobiology and treatment response, the development or repurposing of drugs with different targets from those of the current medications is pursued to improve patient care.
10 Future directions
Considering ADHD dimensionality and multifactorial etiology, several measures have been related to ADHD and its treatment, including clinical symptomatology, cognitive functioning, environmental factors, brain aspects, and biological signatures (Fig. 1). However, no unique measure is proven to guide diagnosis or treatment strategies. Promising biomarkers will probably combine numerous domains of measurement.
Although the knowledge provided by epidemiological and biological studies has significantly improved in the last few years, it is still a long way to comprehensively understand the causes and disentangle all the heterogeneity of this disorder. It is necessary to increase sample sizes to improve the power of genomic studies, which will also allow investigating the genetic influence on the trajectories of the disorder, helping to elucidate, for example, the occurrence of late-onset ADHD that has been demonstrated in population studies, and other sources of heterogeneity. In addition, most studies have focused on European ancestry samples, and extrapolating the findings to other ethnicities is not guaranteed. Future research should investigate more diverse samples and consider diverse cultural contexts. In this sense, international collaborations such as the Latin American Genomics Consortium (LAGC) are important initiatives to advance psychiatric genetics research in populations other than European.
The growing body of research and technological advances provide good perspectives for understanding the neurobiology of ADHD, refining diagnosis, and identifying new therapeutic options to optimize treatment outcomes and associated impairments, leading to improvements in all domains of patient care.
Data availability
Not applicable.
References
Polanczyk G, de Lima MS, Horta BL, et al. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry. 2007;164:942–8. https://doi.org/10.1176/ajp.2007.164.6.942.
Thomas R, Sanders S, Doust J, et al. Prevalence of attention-deficit/hyperactivity disorder: a systematic review and meta-analysis. Pediatrics. 2015;135:e994-1001. https://doi.org/10.1542/peds.2014-3482.
Fayyad J, Sampson NA, Hwang I, et al. The descriptive epidemiology of DSM-IV Adult ADHD in the world health organization world mental health surveys. Atten Defic Hyperact Disord. 2017;9:47–65. https://doi.org/10.1007/s12402-016-0208-3.
Song P, Zha M, Yang Q, et al. The prevalence of adult attention-deficit hyperactivity disorder: a global systematic review and meta-analysis. J Glob Health. 2021;11:04009. https://doi.org/10.7189/jogh.11.04009.
Vitola ES, Bau CHD, Salum GA, et al. Exploring DSM-5 ADHD criteria beyond young adulthood: phenomenology, psychometric properties and prevalence in a large three-decade birth cohort. Psychol Med. 2017;47:744–54. https://doi.org/10.1017/S0033291716002853.
Willcutt EG. The prevalence of DSM-IV attention-deficit/hyperactivity disorder: a meta-analytic review. Neurotherapeutics. 2012;9:490–9. https://doi.org/10.1007/s13311-012-0135-8.
Faraone SV, Banaschewski T, Coghill D, et al. The world federation of adhd international consensus statement: 208 evidence-based conclusions about the disorder. Neurosci Biobehav Rev. 2021;128:789–818. https://doi.org/10.1016/j.neubiorev.2021.01.022.
Seo J-C, Jon D-I, Shim S-H, et al. Prevalence and comorbidities of attention deficit hyperactivity disorder among adults and children/adolescents in Korea. Clin Psychopharmacol Neurosci. 2022;20:126–34. https://doi.org/10.9758/cpn.2022.20.1.126.
Gnanavel S, Sharma P, Kaushal P, Hussain S. Attention deficit hyperactivity disorder and comorbidity: a review of literature. World J Clin Cases. 2019. https://doi.org/10.12998/wjcc.v7.i17.2420.
Spencer TJ, Biederman J, Mick E. Attention-deficit/hyperactivity disorder: diagnosis, lifespan, comorbidities, and neurobiology. J Pediatr Psychol. 2007;32:631–42. https://doi.org/10.1093/jpepsy/jsm005.
Jensen CM, Steinhausen H-C. Comorbid mental disorders in children and adolescents with attention-deficit/hyperactivity disorder in a large nationwide study. Atten Defic Hyperact Disord. 2015;7:27–38. https://doi.org/10.1007/s12402-014-0142-1.
Lai M-C, Kassee C, Besney R, et al. Prevalence of co-occurring mental health diagnoses in the autism population: a systematic review and meta-analysis. Lancet Psychiatry. 2019;6:819–29. https://doi.org/10.1016/S2215-0366(19)30289-5.
Akmatov MK, Ermakova T, Bätzing J. Psychiatric and nonpsychiatric comorbidities among children with ADHD: an exploratory analysis of nationwide claims data in Germany. J Atten Disord. 2021;25:874–84. https://doi.org/10.1177/1087054719865779.
Chen Q, Hartman CA, Haavik J, et al. Common psychiatric and metabolic comorbidity of adult attention-deficit/hyperactivity disorder: a population-based cross-sectional study. PLoS ONE. 2018. https://doi.org/10.1371/journal.pone.0204516.
Instanes JT, Klungsøyr K, Halmøy A, et al. Adult ADHD and comorbid somatic disease: a systematic literature review. J Atten Disord. 2018;22:203–28. https://doi.org/10.1177/1087054716669589.
Simon V, Czobor P, Bálint S, et al. Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry. 2009;194:204–11. https://doi.org/10.1192/bjp.bp.107.048827.
Faraone SV, Asherson P, Banaschewski T, et al. Attention-deficit/hyperactivity disorder. Nat Rev Dis Primers. 2015;1:15020. https://doi.org/10.1038/nrdp.2015.20.
Biederman J, Mick E, Faraone SV. Age-dependent decline of symptoms of attention deficit hyperactivity disorder: impact of remission definition and symptom type. Am J Psychiatry. 2000;157:816–8. https://doi.org/10.1176/appi.ajp.157.5.816.
Edvinsson D, Ekselius L. Six-year outcome in subjects diagnosed with attention-deficit/hyperactivity disorder as adults. Eur Arch Psychiatry Clin Neurosci. 2018;268:337–47. https://doi.org/10.1007/s00406-017-0850-6.
Karam RG, Breda V, Picon FA, et al. Persistence and remission of ADHD during adulthood: a 7-year clinical follow-up study. Psychol Med. 2015;45:2045–56. https://doi.org/10.1017/S0033291714003183.
Sibley MH, Rohde LA, Swanson JM, et al. Late-onset ADHD reconsidered with comprehensive repeated assessments between ages 10 and 25. Am J Psychiatry. 2018;175:140–9. https://doi.org/10.1176/appi.ajp.2017.17030298.
Grevet EH, Bandeira CE, Vitola ES, et al. The course of attention-deficit/hyperactivity disorder through midlife. Eur Arch Psychiatry Clin Neurosci. 2022. https://doi.org/10.1007/s00406-022-01531-4.
Agnew-Blais JC, Polanczyk GV, Danese A, et al. Evaluation of the persistence, remission, and emergence of attention-deficit/hyperactivity disorder in young adulthood. JAMA Psychiat. 2016;73:713–20. https://doi.org/10.1001/jamapsychiatry.2016.0465.
Breda V, Rohde LA, Menezes AMB, et al. The neurodevelopmental nature of attention-deficit hyperactivity disorder in adults. Br J Psychiatry. 2021;218:43–50. https://doi.org/10.1192/bjp.2020.200.
Caye A, Swanson J, Thapar A, et al. Life span studies of ADHD-conceptual challenges and predictors of persistence and outcome. Curr Psychiatry Rep. 2016;18:111. https://doi.org/10.1007/s11920-016-0750-x.
Moffitt TE, Houts R, Asherson P, et al. Is adult ADHD a childhood-onset neurodevelopmental disorder? evidence from a four-decade longitudinal cohort study. Am J Psychiatry. 2015;172:967–77. https://doi.org/10.1176/appi.ajp.2015.14101266.
Thapar A, Cooper M, Rutter M. Neurodevelopmental disorders. Lancet. Psychiatry. 2017;4:339–46. https://doi.org/10.1016/S2215-0366(16)30376-5.
Anderson PJ, de Miranda DM, Albuquerque MR, et al. Psychiatric disorders in individuals born very preterm / very low-birth weight: an individual participant data (IPD) meta-analysis. EClinicalMedicine. 2021. https://doi.org/10.1016/j.eclinm.2021.101216.
Zeng Y, Tang Y, Yue Y, et al. Cumulative evidence for association of parental diabetes mellitus and attention-deficit/hyperactivity disorder. Neurosci Biobehav Rev. 2020;117:129–39. https://doi.org/10.1016/j.neubiorev.2019.11.003.
Sanchez CE, Barry C, Sabhlok A, et al. Maternal pre-pregnancy obesity and child neurodevelopmental outcomes: a meta-analysis. Obes Rev. 2018;19:464–84. https://doi.org/10.1111/obr.12643.
Huang L, Wang Y, Zhang L, et al. Maternal smoking and attention-deficit/hyperactivity disorder in offspring: a meta-analysis. Pediatrics. 2018. https://doi.org/10.1542/peds.2017-2465.
Wetherill L, Foroud T, Goodlett C. Meta-analyses of externalizing disorders: genetics or prenatal alcohol exposure? Alcohol Clin Exp Res. 2018;42:162–72. https://doi.org/10.1111/acer.13535.
Masarwa R, Platt RW, Filion KB. Acetaminophen use during pregnancy and the risk of attention deficit hyperactivity disorder: a causal association or bias? Paediatr Perinat Epidemiol. 2020;34:309–17. https://doi.org/10.1111/ppe.12615.
Leshem R, Bar-Oz B, Diav-Citrin O, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin norepinephrine reuptake inhibitors (SNRIs) during pregnancy and the risk for autism spectrum disorder (ASD) and attention deficit hyperactivity disorder (ADHD) in the offspring: a true effect or a bias? a systematic review & meta-analysis. Curr Neuropharmacol. 2021;19:896–906. https://doi.org/10.2174/1570159X19666210303121059.
Ai Y, Zhao J, Shi J, Zhu TT. Antibiotic exposure and childhood attention-deficit/hyperactivity disorder: systematic review and meta-analysis. Psychopharmacology. 2021;238:3055–62. https://doi.org/10.1007/s00213-021-05989-3.
Nilsen FM, Tulve NS. A systematic review and meta-analysis examining the interrelationships between chemical and non-chemical stressors and inherent characteristics in children with ADHD. Environ Res. 2020. https://doi.org/10.1016/j.envres.2019.108884.
Shareghfarid E, Sangsefidi ZS, Salehi-Abargouei A, Hosseinzadeh M. Empirically derived dietary patterns and food groups intake in relation with attention deficit/hyperactivity disorder (ADHD): a systematic review and meta-analysis. Clin Nutr ESPEN. 2020;36:28–35. https://doi.org/10.1016/j.clnesp.2019.10.013.
Prades N, Varela E, Flamarique I, et al. Water-soluble vitamin insufficiency, deficiency and supplementation in children and adolescents with a psychiatric disorder: a systematic review and meta-analysis. Nutr Neurosci. 2022. https://doi.org/10.1080/1028415X.2021.2020402.
Kotsi E, Kotsi E, Perrea DN. Vitamin D levels in children and adolescents with attention-deficit hyperactivity disorder (ADHD): a meta-analysis. Atten Defic Hyperact Disord. 2019;11:221–32. https://doi.org/10.1007/s12402-018-0276-7.
Thapar A, Cooper M, Eyre O, Langley K. What have we learnt about the causes of ADHD? J Child Psychol Psychiatry. 2013;54:3–16. https://doi.org/10.1111/j.1469-7610.2012.02611.x.
Kim JH, Kim JY, Lee J, et al. Environmental risk factors, protective factors, and peripheral biomarkers for ADHD: an umbrella review. Lancet Psychiatry. 2020;7:955–70. https://doi.org/10.1016/S2215-0366(20)30312-6.
Stergiakouli E, Hamshere M, Holmans P, et al. Investigating the contribution of common genetic variants to the risk and pathogenesis of ADHD. Am J Psychiatry. 2012;169:186–94. https://doi.org/10.1176/appi.ajp.2011.11040551.
Dall’Aglio L, Muka T, Cecil CAM, et al. The role of epigenetic modifications in neurodevelopmental disorders: a systematic review. Neurosci Biobehav Rev. 2018;94:17–30. https://doi.org/10.1016/j.neubiorev.2018.07.011.
Müller D, Grevet EH, da Silva BS, et al. The neuroendocrine modulation of global DNA methylation in neuropsychiatric disorders. Mol Psychiatry. 2021;26:66–9. https://doi.org/10.1038/s41380-020-00924-y.
Chen Q, Brikell I, Lichtenstein P, et al. Familial aggregation of attention-deficit/hyperactivity disorder. J Child Psychol Psychiatry. 2017;58:231–9. https://doi.org/10.1111/jcpp.12616.
Faraone SV, Perlis RH, Doyle AE, et al. Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57:1313–23. https://doi.org/10.1016/j.biopsych.2004.11.024.
Larsson H, Chang Z, D’Onofrio BM, Lichtenstein P. The heritability of clinically diagnosed attention deficit hyperactivity disorder across the lifespan. Psychol Med. 2014;44:2223–9. https://doi.org/10.1017/S0033291713002493.
Bergen SE, Gardner CO, Kendler KS. Age-related changes in heritability of behavioral phenotypes over adolescence and young adulthood: a meta-analysis. Twin Res Hum Genet. 2007;10:423–33. https://doi.org/10.1375/twin.10.3.423.
Polderman TJC, Benyamin B, de Leeuw CA, et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. 2015;47:702–9. https://doi.org/10.1038/ng.3285.
Brikell I, Kuja-Halkola R, Larsson H. Heritability of attention-deficit hyperactivity disorder in adults. Am J Med Genet B Neuropsychiatr Genet. 2015;168:406–13. https://doi.org/10.1002/ajmg.b.32335.
Burt SA. Rethinking environmental contributions to child and adolescent psychopathology: a meta-analysis of shared environmental influences. Psychol Bull. 2009;135:608–37. https://doi.org/10.1037/a0015702.
Demontis D, Walters RK, Martin J, et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet. 2019;51:63–75. https://doi.org/10.1038/s41588-018-0269-7.
Demontis D, Walters GB, Athanasiadis G, et al. Genome-wide analyses of ADHD identify 27 risk loci, refine the genetic architecture and implicate several cognitive domains. Psychiatry Clin Psychol. 2022. https://doi.org/10.1016/j.euroneuro.2022.07.018.
Bonvicini C, Faraone SV, Scassellati C. Attention-deficit hyperactivity disorder in adults: A systematic review and meta-analysis of genetic, pharmacogenetic and biochemical studies. Mol Psychiatry. 2016;21:872–84. https://doi.org/10.1038/mp.2016.74.
Gizer IR, Ficks C, Waldman ID. Candidate gene studies of ADHD: a meta-analytic review. Hum Genet. 2009;126:51–90. https://doi.org/10.1007/s00439-009-0694-x.
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7. https://doi.org/10.1038/nature13595.
Howard DM, Adams MJ, Clarke T-K, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22:343–52. https://doi.org/10.1038/s41593-018-0326-7.
Wray NR, Ripke S, Mattheisen M, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50:668–81. https://doi.org/10.1038/s41588-018-0090-3.
Brainstorm Consortium, Anttila V, Bulik-Sullivan B, et al. Analysis of shared heritability in common disorders of the brain. Science. 2018. https://doi.org/10.1126/science.aap8757.
Green A, Baroud E, DiSalvo M, et al. Examining the impact of ADHD polygenic risk scores on ADHD and associated outcomes: a systematic review and meta-analysis. J Psychiatr Res. 2022;155:49–67. https://doi.org/10.1016/j.jpsychires.2022.07.032.
Soler Artigas M, Sánchez-Mora C, Rovira P, et al. Attention-deficit/hyperactivity disorder and lifetime cannabis use: genetic overlap and causality. Mol Psychiatry. 2020;25:2493–503. https://doi.org/10.1038/s41380-018-0339-3.
Tylee DS, Sun J, Hess JL, et al. Genetic correlations among psychiatric and immune-related phenotypes based on genome-wide association data. Am J Med Genet B Neuropsychiatr Genet. 2018;177:641–57. https://doi.org/10.1002/ajmg.b.32652.
Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51:404–13. https://doi.org/10.1038/s41588-018-0311-9.
Wightman DP, Jansen IE, Savage JE, et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet. 2021;53:1276–82. https://doi.org/10.1038/s41588-021-00921-z.
Li JJ, He Q. Polygenic scores for ADHD: a meta-analysis. Res Child Adolesc Psychopathol. 2021;49:297–310. https://doi.org/10.1007/s10802-021-00774-4.
Stojanovski S, Felsky D, Viviano JD, et al. Polygenic risk and neural substrates of attention-deficit/hyperactivity disorder symptoms in youths with a history of mild traumatic brain injury. Biol Psychiatry. 2019;85:408–16. https://doi.org/10.1016/j.biopsych.2018.06.024.
Taylor MJ, Martin J, Lu Y, et al. Association of genetic risk factors for psychiatric disorders and traits of these disorders in a swedish population twin sample. JAMA Psychiat. 2019;76:280–9. https://doi.org/10.1001/jamapsychiatry.2018.3652.
Rovira P, Demontis D, Sánchez-Mora C, et al. Shared genetic background between children and adults with attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2020;45:1617–26. https://doi.org/10.1038/s41386-020-0664-5.
Rajagopal VM, Duan J, Vilar-Ribó L, et al. Differences in the genetic architecture of common and rare variants in childhood, persistent and late-diagnosed attention-deficit hyperactivity disorder. Nat Genet. 2022;54:1117–24. https://doi.org/10.1038/s41588-022-01143-7.
Davies NM, Holmes MV, Davey Smith G. Reading mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018. https://doi.org/10.1136/bmj.k601.
Sanderson E, Glymour MM, Holmes MV, et al. Mendelian randomization. Nat Rev Methods Primers. 2022;2:1–21. https://doi.org/10.1038/s43586-021-00092-5.
Wendt FR, Garcia-Argibay M, Cabrera-Mendoza B, et al. The relationship of attention-deficit/hyperactivity disorder with posttraumatic stress disorder: a two-sample mendelian randomization and population-based sibling comparison study. Biol Psychiatry. 2022;3223(22):01521–9. https://doi.org/10.1016/j.biopsych.2022.08.012.
Peyre H, Schoeler T, Liu C, et al. Combining multivariate genomic approaches to elucidate the comorbidity between autism spectrum disorder and attention deficit hyperactivity disorder. J Child Psychol Psychiatry. 2021;62:1285–96. https://doi.org/10.1111/jcpp.13479.
Baranova A, Wang J, Cao H, et al. Shared genetics between autism spectrum disorder and attention-deficit/hyperactivity disorder and their association with extraversion. Psychiatry Res. 2022. https://doi.org/10.1016/j.psychres.2022.114679.
Riglin L, Leppert B, Dardani C, et al. ADHD and depression: investigating a causal explanation. Psychol Med. 2021;51:1890–7. https://doi.org/10.1017/S0033291720000665.
Soler Artigas M, Sánchez-Mora C, Rovira P, et al. Mendelian randomization analysis for attention deficit/hyperactivity disorder: studying a broad range of exposures and outcomes. Int J Epidemiol dyac. 2022. https://doi.org/10.1093/ije/dyac128.
Vilar-Ribó L, Sánchez-Mora C, Rovira P, et al. Genetic overlap and causality between substance use disorder and attention-deficit and hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2021;186:140–50. https://doi.org/10.1002/ajmg.b.32827.
Treur JL, Demontis D, Smith GD, et al. Investigating causality between liability to ADHD and substance use, and liability to substance use and ADHD risk, using mendelian randomization. Addict Biol. 2021. https://doi.org/10.1111/adb.12849.
Fluharty ME, Sallis H, Munafò MR. investigating possible causal effects of externalizing behaviors on tobacco initiation: a mendelian randomization analysis. Drug Alcohol Depend. 2018;191:338–42. https://doi.org/10.1016/j.drugalcdep.2018.07.015.
Ahn K, Norman LJ, Justice CM, Shaw P. ADHD and its neurocognitive substrates: a two sample mendelian randomization study. Transl Psychiatry. 2022;12:378. https://doi.org/10.1038/s41398-022-02139-x.
Rao S, Baranova A, Yao Y, et al. Genetic relationships between attention-deficit/hyperactivity disorder, autism spectrum disorder, and intelligence. Neuropsychobiology. 2022. https://doi.org/10.1159/000525411.
Savage JE, Jansen PR, Stringer S, et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet. 2018;50:912–9. https://doi.org/10.1038/s41588-018-0152-6.
de Tavares ME, A, Carpena MX, Vitola ES, et al. White Matter microstructure effect in ADHD: a two-sample mendelian randomization study. medRxiv. 2022;2022(210):731.
Leppert B, Riglin L, Wootton RE, et al. The effect of attention deficit/hyperactivity disorder on physical health outcomes: a 2-sample mendelian randomization study. Am J Epidemiol. 2021;190:1047–55. https://doi.org/10.1093/aje/kwaa273.
Karhunen V, Bond TA, Zuber V, et al. The link between attention deficit hyperactivity disorder (ADHD) symptoms and obesity-related traits: genetic and prenatal explanations. Transl Psychiatry. 2021;11:455. https://doi.org/10.1038/s41398-021-01584-4.
Liu C-Y, Schoeler T, Davies NM, et al. Are there causal relationships between attention-deficit/hyperactivity disorder and body mass index? Evidence from multiple genetically informed designs. Int J Epidemiol. 2021;50:496–509. https://doi.org/10.1093/ije/dyaa214.
Burgess S, Davey Smith G, Davies NM, et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res. 2020. https://doi.org/10.12688/wellcomeopenres.15555.2.
Verbanck M, Chen C-Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–8. https://doi.org/10.1038/s41588-018-0099-7.
Morrison J, Knoblauch N, Marcus JH, et al. Mendelian randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics. Nat Genet. 2020;52:740–7. https://doi.org/10.1038/s41588-020-0631-4.
Beaulieu J-M, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. https://doi.org/10.1124/pr.110.002642.
Del Campo N, Chamberlain SR, Sahakian BJ, Robbins TW. The roles of dopamine and noradrenaline in the pathophysiology and treatment of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2011;69:e145-157. https://doi.org/10.1016/j.biopsych.2011.02.036.
Fusar-Poli P, Rubia K, Rossi G, et al. Striatal dopamine transporter alterations in ADHD: pathophysiology or adaptation to psychostimulants? A meta-analysis. Am J Psychiatry. 2012;169:264–72. https://doi.org/10.1176/appi.ajp.2011.11060940.
Berridge CW, Spencer RC. Differential cognitive actions of norepinephrine a2 and a1 receptor signaling in the prefrontal cortex. Brain Res. 2016;1641:189–96. https://doi.org/10.1016/j.brainres.2015.11.024.
Spencer RC, Devilbiss DM, Berridge CW. The cognition-enhancing effects of psychostimulants involve direct action in the prefrontal cortex. Biol Psychiatry. 2015;77:940–50. https://doi.org/10.1016/j.biopsych.2014.09.013.
Wang L-J, Yu Y-H, Fu M-L, et al. Attention deficit-hyperactivity disorder is associated with allergic symptoms and low levels of hemoglobin and serotonin. Sci Rep. 2018;8:10229. https://doi.org/10.1038/s41598-018-28702-5.
Bauer J, Werner A, Kohl W, et al. Hyperactivity and impulsivity in adult attention-deficit/hyperactivity disorder is related to glutamatergic dysfunction in the anterior cingulate cortex. World J Biol Psychiatry. 2018;19:538–46. https://doi.org/10.1080/15622975.2016.1262060.
Bollmann S, Ghisleni C, Poil SS, et al. Developmental changes in gamma-aminobutyric acid levels in attention-deficit/hyperactivity disorder. Transl Psychiatry. 2015. https://doi.org/10.1038/tp.2015.79.
Cheng J, Liu A, Shi MY, Yan Z. Disrupted glutamatergic transmission in prefrontal cortex contributes to behavioral abnormality in an animal model of ADHD. Neuropsychopharmacology. 2017;42:2096–104. https://doi.org/10.1038/npp.2017.30.
Speranza L, di Porzio U, Viggiano D, et al. Dopamine: the neuromodulator of long-term synaptic plasticity. Reward and Movement Control Cells. 2021;10:735. https://doi.org/10.3390/cells10040735.
Ilgin N, Senol S, Gucuyener K, et al. Is increased D2 receptor availability associated with response to stimulant medication in ADHD. Dev Med Child Neurol. 2001;43:755–60. https://doi.org/10.1017/s0012162201001384.
Volkow ND, Wang G-J, Newcorn J, et al. Brain dopamine transporter levels in treatment and drug naïve adults with ADHD. Neuroimage. 2007;34:1182–90. https://doi.org/10.1016/j.neuroimage.2006.10.014.
Volkow ND, Wang G-J, Newcorn J, et al. Depressed dopamine activity in caudate and preliminary evidence of limbic involvement in adults with attention-deficit/hyperactivity disorder. Arch Gen Psychiatry. 2007;64:932–40. https://doi.org/10.1001/archpsyc.64.8.932.
Rosa-Neto P, Lou HC, Cumming P, et al. Methylphenidate-evoked changes in striatal dopamine correlate with inattention and impulsivity in adolescents with attention deficit hyperactivity disorder. Neuroimage. 2005;25:868–76. https://doi.org/10.1016/j.neuroimage.2004.11.031.
Zimmer L. Contribution of clinical neuroimaging to the understanding of the pharmacology of methylphenidate. Trends Pharmacol Sci. 2017;38:608–20. https://doi.org/10.1016/j.tips.2017.04.001.
Faraone SV. The pharmacology of amphetamine and methylphenidate: Relevance to the neurobiology of attention-deficit/hyperactivity disorder and other psychiatric comorbidities. Neurosci Biobehav Rev. 2018;87:255–70. https://doi.org/10.1016/j.neubiorev.2018.02.001.
Stahl SM, Pradko JF, Haight BR, et al. A review of the neuropharmacology of bupropion, a dual norepinephrine and dopamine reuptake inhibitor. Prim Care Companion J Clin Psychiatry. 2004;6:159–66.
Arnsten AFT, Pliszka SR. Catecholamine influences on prefrontal cortical function: relevance to treatment of attention deficit hyperactivity disorder and related disorders. Pharmacol Biochem Behav. 2011;99:211–6. https://doi.org/10.1016/j.pbb.2011.01.020.
Llorente AM, Voigt RG, Jensen CL, et al. Performance on a visual sustained attention and discrimination task is associated with urinary excretion of norepineprhine metabolite in children with attention-deficit/hyperactivity disorder (AD/HD). Clin Neuropsychol. 2006;20:133–44. https://doi.org/10.1080/13854040490888495.
Childress AC. A critical appraisal of atomoxetine in the management of ADHD. Ther Clin Risk Manag. 2016;12:27–39. https://doi.org/10.2147/TCRM.S59270.
Hirota T, Schwartz S, Correll CU. Alpha-2 agonists for attention-deficit/hyperactivity disorder in youth: a systematic review and meta-analysis of monotherapy and add-on trials to stimulant therapy. J Am Acad Child Adolesc Psychiatry. 2014;53:153–73. https://doi.org/10.1016/j.jaac.2013.11.009.
Olivier B. Serotonin: a never-ending story. Eur J Pharmacol. 2015;753:2–18. https://doi.org/10.1016/j.ejphar.2014.10.031.
Castellanos FX, Elia J, Kruesi MJ, et al. Cerebrospinal fluid monoamine metabolites in boys with attention-deficit hyperactivity disorder. Psychiatry Res. 1994;52:305–16. https://doi.org/10.1016/0165-1781(94)90076-0.
Banerjee E, Nandagopal K. Does serotonin deficit mediate susceptibility to ADHD? Neurochem Int. 2015;82:52–68. https://doi.org/10.1016/j.neuint.2015.02.001.
Huang X, Wang M, Zhang Q, et al. The role of glutamate receptors in attention-deficit/hyperactivity disorder: from physiology to disease. Am J Med Genet B Neuropsychiatr Genet. 2019;180:272–86. https://doi.org/10.1002/ajmg.b.32726.
Silveri MM, Sneider JT, Crowley DJ, et al. Frontal lobe GABA levels during adolescence: associations with impulsivity and response inhibition. Biol Psychiatry. 2013;74:296–304. https://doi.org/10.1016/j.biopsych.2013.01.033.
Edden RAE, Crocetti D, Zhu H, et al. Reduced GABA concentration in attention-deficit/hyperactivity disorder. Arch Gen Psychiatry. 2012;69:750–3. https://doi.org/10.1001/archgenpsychiatry.2011.2280.
Ende G, Cackowski S, Van Eijk J, et al. Impulsivity and aggression in female BPD and ADHD patients: association with ACC glutamate and GABA concentrations. Neuropsychopharmacology. 2016;41:410–8. https://doi.org/10.1038/npp.2015.153.
Hoogman M, Bralten J, Hibar DP, et al. Subcortical brain volume differences of participants with ADHD across the lifespan: an ENIGMA collaboration. Lancet Psychiatry. 2017;4:310–9. https://doi.org/10.1016/S2215-0366(17)30049-4.
Hoogman M, Muetzel R, Guimaraes JP, et al. Brain imaging of the cortex in ADHD: a coordinated analysis of large-scale clinical and population-based samples. Am J Psychiatry. 2019;176:531–42. https://doi.org/10.1176/appi.ajp.2019.18091033.
Cupertino RB, Soheili-Nezhad S, Grevet EH, et al. Reduced fronto-striatal volume in attention-deficit/hyperactivity disorder in two cohorts across the lifespan. Neuroimage Clin. 2020. https://doi.org/10.1016/j.nicl.2020.102403.
Bernanke J, Luna A, Chang L, et al. Structural brain measures among children with and without ADHD in the adolescent brain and cognitive development study cohort: a cross-sectional US population-based study. The Lancet Psychiatry. 2022;9:222–31. https://doi.org/10.1016/S2215-0366(21)00505-8.
Aoki Y, Cortese S, Castellanos FX. Research review: diffusion tensor imaging studies of attention-deficit/hyperactivity disorder: meta-analyses and reflections on head motion. J Child Psychol Psychiatry. 2018;59:193–202. https://doi.org/10.1111/jcpp.12778.
Bu X, Cao M, Huang X, He Y. The structural connectome in ADHD. Psychoradiology. 2021;1:257–71. https://doi.org/10.1093/psyrad/kkab021.
Sudre G, Norman L, Bouyssi-Kobar M, et al. A mega-analytic study of white matter microstructural differences across five cohorts of youth with attention deficit hyperactivity disorder. Biol Psychiat. 2022. https://doi.org/10.1016/j.biopsych.2022.09.021.
Cortese S, Kelly C, Chabernaud C, et al. Towards systems neuroscience of ADHD: A meta-analysis of 55 fMRI studies. Am J Psychiatry. 2012. https://doi.org/10.1176/appi.ajp.2012.11101521.
Gao Y, Shuai D, Bu X, et al. Impairments of large-scale functional networks in attention-deficit/hyperactivity disorder: a meta-analysis of resting-state functional connectivity. Psychol Med. 2019;49:2475–85. https://doi.org/10.1017/S003329171900237X.
Sutcubasi B, Metin B, Kurban MK, et al. Resting-state network dysconnectivity in ADHD: A system-neuroscience-based meta-analysis. World J Biol Psychiatry. 2020;21:662–72. https://doi.org/10.1080/15622975.2020.1775889.
Norman LJ, Sudre G, Price J, et al. Evidence from “big data” for the default-mode hypothesis of ADHD: a mega-analysis of multiple large samples. Neuropsychopharmacology. 2022. https://doi.org/10.1038/s41386-022-01408-z.
Kowalczyk OS, Mehta MA, O’Daly OG, Criaud M. Task-based functional connectivity in attention-deficit/hyperactivity disorder: a systematic review. Biol Psychiatry Global Open Sci. 2021. https://doi.org/10.1016/j.bpsgos.2021.10.006.
Peterson BS, Potenza MN, Wang Z, et al. An FMRI study of the effects of psychostimulants on default-mode processing during Stroop task performance in youths with ADHD. Am J Psychiatry. 2009;166:1286–94. https://doi.org/10.1176/appi.ajp.2009.08050724.
Picon FA, Sato JR, Anés M, et al. Methylphenidate alters functional connectivity of default mode network in drug-naive male adults with ADHD. J Atten Disord. 2020;24:447–55. https://doi.org/10.1177/1087054718816822.
Cortese S, Aoki YY, Itahashi T, et al. Systematic review and meta-analysis: resting-state functional magnetic resonance imaging studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2021;60:61–75. https://doi.org/10.1016/j.jaac.2020.08.014.
Grasby KL, Jahanshad N, Painter JN, et al. The genetic architecture of the human cerebral cortex. Science. 2020. https://doi.org/10.1126/science.aay6690.
Klein M, Walters RK, Demontis D, et al. Genetic markers of ADHD-related variations in intracranial volume. AJP. 2019;176:228–38. https://doi.org/10.1176/appi.ajp.2018.18020149.
Liu S, Smit DJA, Abdellaoui A, et al. Brain structure and function show distinct relations with genetic predispositions to mental health and cognition. Biol Psychiatry Cogn Neurosci Neuroimaging. 2022;S2451–9022(22):00186. https://doi.org/10.1016/j.bpsc.2022.08.003.
Zhao B, Li T, Smith SM, et al. Common variants contribute to intrinsic human brain functional networks. Nat Genet. 2022;54:508–17. https://doi.org/10.1038/s41588-022-01039-6.
Bandeira CE, Grevet EH, Cupertino RB, et al. The role of glucocorticoid receptor gene in the association between attention deficit-hyperactivity disorder and smaller brain structures. J Neural Transm. 2021;128:1907–16. https://doi.org/10.1007/s00702-021-02425-w.
Wiers CE, Lohoff FW, Lee J, et al. Methylation of the dopamine transporter gene in blood is associated with striatal dopamine transporter availability in ADHD: a preliminary study. Eur J Neurosci. 2018;48:1884–95. https://doi.org/10.1111/ejn.14067.
Peter CJ, Fischer LK, Kundakovic M, et al. DNA methylation signatures of early childhood malnutrition associated with impairments in attention and cognition. Biol Psychiatry. 2016;80:765–74. https://doi.org/10.1016/j.biopsych.2016.03.2100.
Wilmot B, Fry R, Smeester L, et al. Methylomic analysis of salivary DNA in childhood ADHD identifies altered DNA methylation in VIPR2. J Child Psychol Psychiatry. 2016;57:152–60. https://doi.org/10.1111/jcpp.12457.
Gervin K, Nordeng H, Ystrom E, et al. Long-term prenatal exposure to paracetamol is associated with DNA methylation differences in children diagnosed with ADHD. Clin Epigenetics. 2017;9:77. https://doi.org/10.1186/s13148-017-0376-9.
Heinrich H, Grunitz J, Stonawski V, et al. Attention, cognitive control and motivation in ADHD: Linking event-related brain potentials and DNA methylation patterns in boys at early school age. Sci Rep. 2017;7:3823. https://doi.org/10.1038/s41598-017-03326-3.
Walton E, Pingault J-B, Cecil C, a. M, et al. Epigenetic profiling of ADHD symptoms trajectories: a prospective, methylome-wide study. Mol Psychiatry. 2017;22:250–6. https://doi.org/10.1038/mp.2016.85.
Chen Y-C, Sudre G, Sharp W, et al. Neuroanatomic, epigenetic and genetic differences in monozygotic twins discordant for attention deficit hyperactivity disorder. Mol Psychiatry. 2018;23:683–90. https://doi.org/10.1038/mp.2017.45.
van Dongen J, Zilhão NR, Sugden K, et al. Epigenome-wide association study of attention-deficit/hyperactivity disorder symptoms in adults. Biol Psychiatry. 2019;86:599–607. https://doi.org/10.1016/j.biopsych.2019.02.016.
Meijer M, Klein M, Hannon E, et al. Genome-wide DNA methylation patterns in persistent attention-deficit/hyperactivity disorder and in association with impulsive and callous traits. Front Genet. 2020;11:16. https://doi.org/10.3389/fgene.2020.00016.
Mooney MA, Ryabinin P, Wilmot B, et al. Large epigenome-wide association study of childhood ADHD identifies peripheral DNA methylation associated with disease and polygenic risk burden. Transl Psychiatry. 2020;10:8. https://doi.org/10.1038/s41398-020-0710-4.
Neumann A, Walton E, Alemany S, et al. Association between DNA methylation and ADHD symptoms from birth to school age: a prospective meta-analysis. Transl Psychiatry. 2020;10:398. https://doi.org/10.1038/s41398-020-01058-z.
Rovira P, Sánchez-Mora C, Pagerols M, et al. Epigenome-wide association study of attention-deficit/hyperactivity disorder in adults. Transl Psychiatry. 2020;10:199. https://doi.org/10.1038/s41398-020-0860-4.
Pardiñas AF, Holmans P, Pocklington AJ, et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet. 2018;50:381–9. https://doi.org/10.1038/s41588-018-0059-2.
Nagel M, Jansen PR, Stringer S, et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat Genet. 2018;50:920–7. https://doi.org/10.1038/s41588-018-0151-7.
Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium. Electronic address: douglas.ruderfer@vanderbilt.edu, Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium. Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell. 2018;173:1705-1715.e16. https://doi.org/10.1016/j.cell.2018.05.046.
Min JL, Hemani G, Hannon E, et al. Genomic and phenotypic insights from an atlas of genetic effects on DNA methylation. Nat Genet. 2021;53:1311–21. https://doi.org/10.1038/s41588-021-00923-x.
Müller D, Grevet EH, Figueira da Silva NA, et al. Global DNA methylation changes in adults with attention deficit-hyperactivity disorder and its comorbidity with bipolar disorder: links with polygenic scores. Mol Psychiatry. 2022;27:2485–91. https://doi.org/10.1038/s41380-022-01493-y.
Boonchooduang N, Louthrenoo O, Chattipakorn N, Chattipakorn SC. Possible links between gut-microbiota and attention-deficit/hyperactivity disorders in children and adolescents. Eur J Nutr. 2020;59:3391–403. https://doi.org/10.1007/s00394-020-02383-1.
Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015;28:203–9.
Sukmajaya AC, Lusida MI, Soetjipto null, Setiawati Y,. Systematic review of gut microbiota and attention-deficit hyperactivity disorder (ADHD). Ann Gen Psychiatry. 2021;20:12. https://doi.org/10.1186/s12991-021-00330-w.
Aarts E, Ederveen THA, Naaijen J, et al. Gut microbiome in ADHD and its relation to neural reward anticipation. PLoS ONE. 2017. https://doi.org/10.1371/journal.pone.0183509.
Gkougka D, Mitropoulos K, Tzanakaki G, et al. Gut microbiome and attention deficit/hyperactivity disorder: a systematic review. Pediatr Res. 2022. https://doi.org/10.1038/s41390-022-02027-6.
Shirvani-Rad S, Ejtahed H-S, Ettehad Marvasti F, et al. The role of gut microbiota-brain axis in pathophysiology of ADHD: a systematic review. J Atten Disord. 2022;26:1698–710. https://doi.org/10.1177/10870547211073474.
Jiang H-Y, Zhou Y-Y, Zhou G-L, et al. Gut microbiota profiles in treatment-naïve children with attention deficit hyperactivity disorder. Behav Brain Res. 2018;347:408–13. https://doi.org/10.1016/j.bbr.2018.03.036.
Wan L, Ge W-R, Zhang S, et al. Case-control study of the effects of gut microbiota composition on neurotransmitter metabolic pathways in children with attention deficit hyperactivity disorder. Front Neurosci. 2020;14:127. https://doi.org/10.3389/fnins.2020.00127.
Qiu X, Zhang M, Yang X, et al. Faecalibacterium prausnitzii upregulates regulatory T cells and anti-inflammatory cytokines in treating TNBS-induced colitis. J Crohns Colitis. 2013;7:e558-568. https://doi.org/10.1016/j.crohns.2013.04.002.
Almeida PGC, Nani JV, Oses JP, et al. Neuroinflammation and glial cell activation in mental disorders. Brain Behav Immu. 2020. https://doi.org/10.1016/j.bbih.2019.100034.
Anand D, Colpo GD, Zeni G, et al. Attention-deficit/hyperactivity disorder and inflammation: what does current knowledge tell us? Sys Rev Front Psychiatry. 2017;8:228. https://doi.org/10.3389/fpsyt.2017.00228.
Wang N, Gao X, Zhang Z, Yang L. Composition of the gut microbiota in attention deficit hyperactivity disorder: a systematic review and meta-analysis. Front Endocrinol. 2022. https://doi.org/10.3389/fendo.2022.838941.
Lee M-J, Lai H-C, Kuo Y-L, Chen VC-H. Association between gut microbiota and emotional-behavioral symptoms in children with attention-deficit/hyperactivity disorder. J Personalized Med. 2022;12:1634. https://doi.org/10.3390/jpm12101634.
Prehn-Kristensen A, Zimmermann A, Tittmann L, et al. Reduced microbiome alpha diversity in young patients with ADHD. PLoS ONE. 2018. https://doi.org/10.1371/journal.pone.0200728.
Boland H, DiSalvo M, Fried R, et al. A literature review and meta-analysis on the effects of ADHD medications on functional outcomes. J Psychiatr Res. 2020;123:21–30. https://doi.org/10.1016/j.jpsychires.2020.01.006.
Catalá-López F, Hutton B, Núñez-Beltrán A, et al. The pharmacological and non-pharmacological treatment of attention deficit hyperactivity disorder in children and adolescents: a systematic review with network meta-analyses of randomised trials. PLoS ONE. 2017. https://doi.org/10.1371/journal.pone.0180355.
NICE guideline (2018) Attention deficit hyperactivity disorder: diagnosis and management
Nimmo-Smith V, Merwood A, Hank D, et al. Non-pharmacological interventions for adult ADHD: a systematic review. Psychol Med. 2020;50:529–41. https://doi.org/10.1017/S0033291720000069.
Bolea-Alamañac B, Nutt DJ, Adamou M, et al. Evidence-based guidelines for the pharmacological management of attention deficit hyperactivity disorder: update on recommendations from the british association for psychopharmacology. J Psychopharmacol. 2014;28:179–203. https://doi.org/10.1177/0269881113519509.
Cortese S. Pharmacologic treatment of attention deficit-hyperactivity disorder. N Engl J Med. 2020;383:1050–6. https://doi.org/10.1056/NEJMra1917069.
Fawns T. Attention deficit and hyperactivity disorder. Prim Care. 2021;48:475–91. https://doi.org/10.1016/j.pop.2021.05.004.
Adamo N, Seth S, Coghill D. Pharmacological treatment of attention-deficit/hyperactivity disorder: assessing outcomes. Expert Rev Clin Pharmacol. 2015;8:383–97. https://doi.org/10.1586/17512433.2015.1050379.
Chang Z, Ghirardi L, Quinn PD, et al. Risks and benefits of attention-deficit/hyperactivity disorder medication on behavioral and neuropsychiatric outcomes: a qualitative review of pharmacoepidemiology studies using linked prescription databases. Biol Psychiatry. 2019;86:335–43. https://doi.org/10.1016/j.biopsych.2019.04.009.
Coghill DR, Banaschewski T, Soutullo C, et al. Systematic review of quality of life and functional outcomes in randomized placebo-controlled studies of medications for attention-deficit/hyperactivity disorder. Eur Child Adolesc Psychiatry. 2017;26:1283–307. https://doi.org/10.1007/s00787-017-0986-y.
Krinzinger H, Hall CL, Groom MJ, et al. Neurological and psychiatric adverse effects of long-term methylphenidate treatment in ADHD: a map of the current evidence. Neurosci Biobehav Rev. 2019;107:945–68. https://doi.org/10.1016/j.neubiorev.2019.09.023.
Cortese S, Adamo N, Del Giovane C, et al. Comparative efficacy and tolerability of medications for attention-deficit hyperactivity disorder in children, adolescents, and adults: a systematic review and network meta-analysis. Lancet Psychiatry. 2018;5:727–38. https://doi.org/10.1016/S2215-0366(18)30269-4.
Adler LA, Dirks B, Deas PF, et al. Lisdexamfetamine dimesylate in adults with attention-deficit/ hyperactivity disorder who report clinically significant impairment in executive function: results from a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74:694–702. https://doi.org/10.4088/JCP.12m08144.
Brown TE, Chen J, Robertson B. Relationships between executive function improvement and ADHD symptom improvement with lisdexamfetamine dimesylate in adults with ADHD and executive function deficits: a post hoc analysis. Prim Care Companion CNS Disord. 2020. https://doi.org/10.4088/PCC.19m02559.
Tamminga HGH, Reneman L, Huizenga HM, Geurts HM. Effects of methylphenidate on executive functioning in attention-deficit/hyperactivity disorder across the lifespan: a meta-regression analysis. Psychol Med. 2016;46:1791–807. https://doi.org/10.1017/S0033291716000350.
Cooper WO, Habel LA, Sox CM, et al. ADHD drugs and serious cardiovascular events in children and young adults. N Engl J Med. 2011;365:1896–904. https://doi.org/10.1056/NEJMoa1110212.
Habel LA, Cooper WO, Sox CM, et al. ADHD medications and risk of serious cardiovascular events in young and middle-aged adults. JAMA. 2011;306:2673–83. https://doi.org/10.1001/jama.2011.1830.
Chamakalayil S, Strasser J, Vogel M, et al. Methylphenidate for attention-deficit and hyperactivity disorder in adult patients with substance use disorders: good clinical practice. Front Psychiatry. 2020. https://doi.org/10.3389/fpsyt.2020.540837.
McIntosh D, Kutcher S, Binder C, et al. Adult ADHD and comorbid depression: a consensus-derived diagnostic algorithm for ADHD. Neuropsychiatr Dis Treat. 2009;5:137–50.
Perugi G, Vannucchi G, Bedani F, Favaretto E. Use of stimulants in bipolar disorder. Curr Psychiatry Rep. 2017;19:7. https://doi.org/10.1007/s11920-017-0758-x.
Viktorin A, Rydén E, Thase ME, et al. The risk of treatment-emergent mania with methylphenidate in bipolar disorder. Am J Psychiatry. 2017;174:341–8. https://doi.org/10.1176/appi.ajp.2016.16040467.
Cortese S, Holtmann M, Banaschewski T, et al. Practitioner review: current best practice in the management of adverse events during treatment with ADHD medications in children and adolescents. J Child Psychol Psychiatry. 2013;54:227–46. https://doi.org/10.1111/jcpp.12036.
Upadhyaya HP, Desaiah D, Schuh KJ, et al. A review of the abuse potential assessment of atomoxetine: a nonstimulant medication for attention-deficit/hyperactivity disorder. Psychopharmacology. 2013;226:189–200. https://doi.org/10.1007/s00213-013-2986-z.
Bangs ME, Wietecha LA, Wang S, et al. Meta-analysis of suicide-related behavior or ideation in child, adolescent, and adult patients treated with atomoxetine. J Child Adolesc Psychopharmacol. 2014;24:426–34. https://doi.org/10.1089/cap.2014.0005.
Clemow DB, Bushe CJ. Atomoxetine in patients with ADHD: a clinical and pharmacological review of the onset, trajectory, duration of response and implications for patients. J Psychopharmacol. 2015;29:1221–30. https://doi.org/10.1177/0269881115602489.
Brown KA, Samuel S, Patel DR. Pharmacologic management of attention deficit hyperactivity disorder in children and adolescents: a review for practitioners. Transl Pediatr. 2018;7:36–47. https://doi.org/10.21037/tp.2017.08.02.
Mechler K, Banaschewski T, Hohmann S, Häge A. Evidence-based pharmacological treatment options for ADHD in children and adolescents. Pharmacol Ther. 2022. https://doi.org/10.1016/j.pharmthera.2021.107940.
Huss M, Chen W, Ludolph AG. Guanfacine extended release: a new pharmacological treatment option in Europe. Clin Drug Investig. 2016;36:1–25. https://doi.org/10.1007/s40261-015-0336-0.
Giovannitti JA, Thoms SM, Crawford JJ. Alpha-2 adrenergic receptor agonists: a review of current clinical applications. Anesth Prog. 2015;62:31–8. https://doi.org/10.2344/0003-3006-62.1.31.
Ng QX. A systematic review of the use of bupropion for attention-deficit/hyperactivity disorder in children and adolescents. J Child Adolesc Psychopharmacol. 2017;27:112–6. https://doi.org/10.1089/cap.2016.0124.
Verbeeck W, Bekkering GE, Van den Noortgate W, Kramers C. Bupropion for attention deficit hyperactivity disorder (ADHD) in adults. Cochrane Database Syst Rev. 2017. https://doi.org/10.1002/14651858.CD009504.pub2.
Bond DJ, Hadjipavlou G, Lam RW, et al. The Canadian network for mood and anxiety treatments (CANMAT) task force recommendations for the management of patients with mood disorders and comorbid attention-deficit/hyperactivity disorder. Ann Clin Psychiatry. 2012;24:23–37.
Hamedi M, Mohammdi M, Ghaleiha A, et al. Bupropion in adults with attention-deficit/hyperactivity disorder: a randomized, double-blind study. Acta Med Iran. 2014;52:675–80.
Gajria K, Lu M, Sikirica V, et al. Adherence, persistence, and medication discontinuation in patients with attention-deficit/hyperactivity disorder—a systematic literature review. Neuropsychiatr Dis Treat. 2014;10:1543–69. https://doi.org/10.2147/NDT.S65721.
Zayats T, Neale BM. Recent advances in understanding of attention deficit hyperactivity disorder (ADHD): how genetics are shaping our conceptualization of this disorder. F1000Res. 2020. https://doi.org/10.12688/f1000research.18959.2.
Mick E, Neale B, Middleton FA, et al. Genome-wide association study of response to methylphenidate in 187 children with attention-deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1412–8. https://doi.org/10.1002/ajmg.b.30865.
Pagerols M, Richarte V, Sánchez-Mora C, et al. Integrative genomic analysis of methylphenidate response in attention-deficit/hyperactivity disorder. Sci Rep. 2018;8:1881. https://doi.org/10.1038/s41598-018-20194-7.
Zhong Y, Yang B, Su Y, et al. The association with quantitative response to attention-deficit/hyperactivity disorder medication of the previously identified neurodevelopmental network genes. J Child Adolesc Psychopharmacol. 2020;30:348–54. https://doi.org/10.1089/cap.2018.0164.
Hegvik T-A, Waløen K, Pandey SK, et al. Druggable genome in attention deficit/hyperactivity disorder and its co-morbid conditions new avenues treatment. Mol Psychiatry. 2021;26:4004–15. https://doi.org/10.1038/s41380-019-0540-z.
Brikell I, Wimberley T, Albiñana C, et al. Genetic, clinical, and sociodemographic factors associated with stimulant treatment outcomes in ADHD. Am J Psychiatry. 2021;178:854–64. https://doi.org/10.1176/appi.ajp.2020.20121686.
da Silva BS, Leffa DT, Beys-da-Silva WO, et al. Integrative proteomics and pharmacogenomics analysis of methylphenidate treatment response. Transl Psychiatry. 2019;9:308. https://doi.org/10.1038/s41398-019-0649-5.
Myer NM, Boland JR, Faraone SV. Pharmacogenetics predictors of methylphenidate efficacy in childhood ADHD. Mol Psychiatry. 2018;23:1929–36. https://doi.org/10.1038/mp.2017.234.
Elia J, Ungal G, Kao C, et al. Fasoracetam in adolescents with ADHD and glutamatergic gene network variants disrupting mGluR neurotransmitter signaling. Nat Commun. 2018;9:4. https://doi.org/10.1038/s41467-017-02244-2.
Funding
The author(s) received financial support from the Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq (CHDB, grant number 424041/2016-2), (EHG, grant number 426905/2016-4), from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES—Finance Code 001 and FIPE-HCPA 160600), from the Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (EHG, grant number PqG-19/2551-0001731-6), (CHDB, grant number PqG-19/2551-001668-9), from the São Paulo Research Foundation—FAPESP (grant number 05652-0/2020).
Author information
Authors and Affiliations
Contributions
BSS wrote the first draft of the manuscript and performed the literature search. DLR performed the literature search and made substantial contributions to the conception of the manuscript. JKNR and LF contributed to the literature review and writing processes. EHG and CHDB critically revised the manuscript for important intellectual content. All authors reviewed the final version of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following potential conflict of interest: Dr. Grevet was on the speaker’s bureau for Novartis and Shire for three years. He also received travel awards (air tickets and hotel accommodations) for participating in two psychiatric meetings from Shire and Novartis. All other authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Additional file 1: Table S1.
Meta-analyses summarizing effects from exposures to several substances (prenatal and postnatal). Table S2. Meta-analyses summarizing associations with biomarkers and nutritional factors. Table S3. Meta-analyses summarizing associations with parental, newborn, and lifespan-related factors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
da Silva, B.S., Grevet, E.H., Silva, L. et al. An overview on neurobiology and therapeutics of attention-deficit/hyperactivity disorder. Discov Ment Health 3, 2 (2023). https://doi.org/10.1007/s44192-022-00030-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44192-022-00030-1