
Abstract
The aims of this work is to scrutinize the structural, physical and electrical properties of nitroamine (NH2NO2) adsorption on the outer and inner surface of pristine and C-replacing boron nitride nanosheet (BN nanosheet), using density functional theory methods at cam-B3LYP/6-31G (d) level of theory. Inspections of determined results represent that the adsorption of nitramide on the outer surface of pristine and C-replaced BN nanosheet is exothermic and on the inner surface it is endothermic. The deformation energy of system displays that the geometry and structure of BN nanosheet and nitramide in the BN nanosheet/NH2NO2 complex change significantly from the original state, whereas the quantum parameters and gap energy of the BN nanosheet/NH2NO2 system alter slightly from the original state. The nuclear magnetic resonance and molecular electrostatic potential consequences exhibit that in the BN nanosheet/NH2NO2 complex, the highest density of electrons is concentrated surrounding the NH2NO2 molecule.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
After the discovery of the carbon nanotube, extensive researches have been done to find other nanomaterials and nanoconfigurations. One of the most interesting configurations of carbon is graphene. It is a crystalline allotrope of carbon with two-dimensional properties. Due to its fascinating structural and electronic properties, it is suitable for providing electronic nanosheets or nanosensors [1,2,3,4,5,6,7]. The band gap of graphene sheet is almost zero and this property is useful to making electronic devices. In recent years, functionalizing and doping methods have been extensively used to increase the efficiency of graphene sheet in electronic and nanodetector applications [8,9,10,11]. In recent years, many researches have focused on finding other nanosheet compounds such as silicon carbide, boron phosphide, aluminum nitride, zinc oxide, gallium nitride, aluminum phosphide, beryllium oxide and boron nitride [12,13,14,15,16,17,18]. Boron nitride nanosheet was discovered in 2005 and, due to its structural and fundamental properties, it is useful in making nanochips, nanosensors, optoelectronic devices, nanoscale device technology and nanoadsorbent [19,20,21,22,23,24,25,26,27,28,29,30]. On the other hand, BN nanosheet with a band gap in the range of 4.2–6 eV is notably used to make a sensitive sensor of toxic and hazardous compounds in industries [12, 27, 28, 31,32,33,34,35].
Nitroamine (NH2NO2) is the simplest nitramine compound that is greatly used in military explosive, propellant and fuel applications. In recent years, extensive theoretical and experimental investigations have been done on the interaction of NH2NO2 compound with nanomaterials and nanotubes [36, 37]. In the current work, the structural, physical, and electrical properties of BN nanosheet in the presence of C-replaced and NH2NO2 molecule were studied using the DFT theory. To find the appropriate and suitable adsorption sites, many different configurations and orientations of NH2NO2 molecule on the B and N sites of nanosheet were examined, and all configuration models were optimized using B3LYP/3-21G level of theory. From all optimized models, the ten (10) stable and suitable models were selected for this work and then the selected models were optimized again using cam-B3LYP/6-31G (d) level of theory. The geometrical, chemical reactivity, quantum parameters, adsorption, deformation energy, and molecular electrostatic potential for all selected models are determined and analyzed. The obtained results may be useful for performing an adsorbent or nanosensor for detecting nitramine molecule.
Computational section
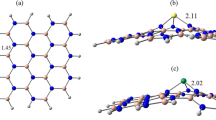
For denoting the adsorption of NH2NO2 molecule on the surface of pristine and C-replaced BN nanosheet, we define the A and B models, respectively. The a, b, c, d and e indexes are used for adsorption of NH2NO2 from H, N(NH2), O, N(NO2) head and parallel orientation on the boron position of the BN nanosheet, respectively (see Fig. 1). The f, g, h, i and j indexes are used for adsorption of NH2NO2 molecule from H, N(NH2), O, N(NO2) head and parallel orientation on the nitrogen position of the BN nanosheet, respectively.

2D views of the pristine and C-doped BN nanosheets
The A-a to B-j models are optimized by using the DFT method at cam-B3LYP level of theory using the 6-31G (d) base set [38] when performing the GAMESS suite of programs [39]. The pristine and C-replaced BN nanosheet and the A-a to B-j models before and after the optimizing process are given in Figs. 1 and 2. The ends of the nanosheet at all systems are saturated by hydrogen atoms for preventing the dangling bonds at the edges of the nanosheet. The adsorption energy (E ads) [40,41,42] of NH2NO2 molecule on the surface of pristine, C-replaced BN nanosheet is calculated by:
\(E_{{{\text{BN - nano}}\,{\text{sheet}}/{\text{NH}}_{2} {\text{NO}}_{2} }} ,\) \(E_{{{\text{BN - nano}}\,{\text{sheet}}}}\) and \(E_{{{\text{NH}}_{2} {\text{NO}}_{2} }}\) are the total potential energy of the BN nanosheet/NH2NO2 system, BN nanosheet and NH2NO2, respectively, and the BSSE is the base set superposition error. The deformation energy (E def) of the BN nanosheet, NH2NO2 molecule, BN nanosheet/NH2NO2 complex and binding energy (E bin) for the A-a to B-j systems are calculated by Eqs. (2–5) [43].
where E BN-nanosheet in system and \(E_{{{\text{NH}}_{2} {\text{NO}}_{2} }}\) are the energy of the BN nanosheet and NH2NO2 molecule in the BN nanosheet/NH2NO2 system, when NH2NO2 and BN nanosheet are absent, respectively:
The E totaldef, E defBN-nanosheet and \(E_{{{\text{def \,NH}}_{2} {\text{NO}}_{2} }}\) are the deformation energy of the BN nanosheet/NH2NO2 system, BN nanosheet and NH2NO2 molecule in their optimized structure.


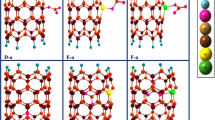
2D views of NH2NO2 adsorption on the surface of pristine and C-doped BN nanosheets (A-a to B-j models); in all models the left configuration is before optimization
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energies are used for determining the gap energy (E g), density of state (DOS) plots, Fermi level energy (E FL) and work functions (∆φ) (see Eqs. 6–8):
The chemical potential (μ) and electronegativity of nanosheet (χ), global hardness (η) and softness (S) of nanosheet, index of electrophilicity (ω) and charge transfer parameters (ΔN) of the A-a to B-j adsorption systems are calculated using Eqs. (9–14):
where A = −E LUMO and I = −E HOMO are the electron affinity and ionization potential, respectively [40,41,42].
Results and discussion
Structural and geometrical properties
Herein, the A model of the BN nanosheet contains 24B, 24N and 18H atoms (B24N24H18, see Fig. 1) and in the B model 24C atoms are superseded in the layers 1, 3, 5, 7 and 9 in spite of B and N sites of nanosheet (B12N12C24H18, see Fig. 1). From the optimized results, the B–N bond length of the A model is 1.44 Å and the bond lengths of B–N, B–C, N–C and C–C in the B model are 1.45, 1.39, 1.54 and 1.38 Å, respectively (see Fig S1 in supplementary data).
As seen in Fig S2 in supplementary data, the B–N, B–C and N–C bond lengths and the <B–C–N bond angle of the A-a to B-j models after adsorbing NH2NO2 molecule change slightly from the unadorned models.
The optimized results display that in the B-e model, the NH2NO2 molecule dissociates to NH2 and NO2 parts, and in this form the NH2 part is adsorbed on the nanosurface and the NO2 part goes away from the nanosheet. In the A-j model, the NH2NO2 molecule dissociates into 2O atoms and NNH2 part, and also in this model the 2O atoms are chemically adsorbed on the surface of the nanosheet and the NNH2 part goes away from the nanosheet. Also in the B-j model, the NH2NO2 molecule dissociates into NH2, NO and O parts; the O atom is chemically connected on the surface of the nanosheet and other parts go away from the nanosheet. On the other hand, in the other adsorption models after optimizing the NH2NO2/BN nanosheet complex, the NH2NO2 molecule goes away from the nanosheet and so the type of adsorption process of these systems is physorption.
According to calculated results in Table 1, the average distance between the NH2NO2 molecule and the BN nanosheet in the B-e, A-j and B-j models is 1.51, 1.77 and 1.63 Å, respectively. By replacing C atoms, the geometrical structure of the BN nanosheet is dramatically deformed and so the average distance of the NH2NO2/nanosheet decreases.
Comparison results show that the adsorption energy of the B-e, A-j and B-j models is positive due to dissociation of the NH2NO2 molecule on the surface of the nanosheet, whereas the E ads values of the other models are negative and these adsorbed models are exothermic. Also, the B-g model with E ads = −10.48 kcal/mol is the most stable adsorption model (see Table 1). The obtained results demonstrate that the E ads of the NH2NO2/nanosheet system depends on C-replaced positions and NH2NO2 molecule orientation. The C-replacing atoms increase the adsorption of the NH2NO2 molecule on the surface of the nanosheet. These results confirm that C-replaced BN nanosheet is a good compound for adsorbing NH2NO2 molecule. In all studied models (the A-a to B-j models), the basis set superposition error (BSSE) is in the range 0.0005–0.001 kcal/mol. In the A-a to B-j models, the binding energy (E bin) values are negative and the NH2NO2 molecule is adsorbed on the surface of the BN nanosheet. The maximum binding energy is seen in the B-e, A-j and B-j models with −173.22, −214.22 and −257.87 kcal/mol, respectively.
To understand the deforming structures of the BN nanosheet and NH2NO2 molecule in the BN nanosheet/NH2NO2 complex, the deformation energy of the BN nanosheet, NH2NO2 molecule and BN nanosheet/NH2NO2 complex is calculated using Eqs. (2–4) and the results are listed in Table 1.
Comparisons of structural results indicate that significant curvature in the geometry of nanosheet and NH2NO2 molecule occurs in the B-e, A-j and B-j adsorption models. On the other hand, the deformation energy of the BN nanosheet (E def BN nanosheet) for the B-a, B-b, B-c, B-d, B-f, B-g, B-h and B-i models is negative and for the A-a to A-j models positive. The negative value of deformation energy shows that the structures of NH2NO2 molecule and BN nanosheet are deformed spontaneously from the original state and all deformed structures are stable from the thermodynamic point of view. However, the deformation energies of the B-e, A-j and B-j models for NH2NO2 molecule, BN nanosheet and NH2NO2/BN nanosheet complex are positive and so the deformation of these models is endothermic and needs more energy. The order of deformation energy in the B-e, A-j and B-j models is: E def (A-j) > E def (B-j) > E def (B-e). Furthermore, the deformation energy of the NH2NO2 molecule and total deformation energy of the A-a, A-b, A-c, A-d, A-e, A-f, B-a, B-b, B-c, B-d and B-f models are negative. The total deformation energy of the B model is lower than that of the A models, thereby replacing of carbon atoms alter the structure of the BN nanosheet/NH2NO2 complex.
Dipole moment is one another properties of system that it used to detect the nature of reactivity and impurity atoms effect of system. The dipole moment of the C-replaced models (B-a to B-j models) are more than pristine models (A-a to A-j models). The replacing carbon atoms increase dipole moment and reactivity of nanosheet (see Table 1), and also the orientations of NH2NO2 molecule on the surface of BN nanosheet change the dipole moment values. The dipole moment of A-g model (6.33 debye) and B-e model (13.84 debye) are significantly more than other those models. It is notable that with increasing the dipole moment at the A-a to B-j models the adsorption and interaction energy of system increase.
Analysis HOMO and LUMO orbitals
For understanding the structural and electrical properties of BN nanosheet/NH2NO2 complex, the HOMO and the LUMO orbitals of the A-a to B-j models are determined and results are given in Fig. 3.




Plots of HOMO and LUMO orbital structures for NH2NO2 adsorption on the surface of pristine and C-doped BN nanosheets (A-a to B-j models, see Fig. 2)
The HOMO orbital density of the A-a, A-b, A-c, A-d, A-e, A-f, A-g, A-h and A-i models are uniformly localized surrounding NH2NO2 molecule, whereas at the A-j model the HOMO orbital density is localized around NH2NO2 molecule and near adsorption position of nanosheet. While with replacing C atoms in the B-a, B-b, B-c, B-d, B-e, B-f, B-g, B-h and B-i models the HOMO orbital density are distributed on the end layers surface of nanosheet, and also in the B-j model the HOMO orbital density is localized surrounding NH2NO2 and end layers of nanosheet.
It is remarkable that the LUMO orbital densities at all adsorption models except A-j and B-j models are distributed uniformly around nanosheet. These results confirm that the surface of nanosheet is a good surface for electrophilic reaction. The negative values of ∆ρ(\(\Delta \rho = \rho_{2} - \rho_{1}\)) of NBO and Mulliken charge for NH2NO2 molecule confirm that the NH2NO2 molecule is an electron acceptor and decrease the charge densities around nanosheet from original state (see Table 2).
The calculated gap energy (E g) for the A-a to B-j models are tabulated in Table 3. The E g energy of the A-a to A-j models is in range 4.06–5.23 eV; these results are in agreement with other research [32]. The E g energy of the B-a to B-j models is in range 0.94–1.33 eV and is lower than the A-a to A-j models. Due to replacing carbon atoms, the conductivity and reactivity of the B-a to B-j models are more than the A-a to A-j models, and also the B-a to B-j models are a good semiconductor in electronic applications.
The density of state (DOS) plots for the A-a to B-j adsorption models display that at the A-a, A-b, A-c, A-d, A-e, A-f, A-g and A-h models in the HOMO and LUMO regions there are 12 and 14 peaks, respectively (see Fig S3). In the B-a, B-b, B-c, B-d, B-f, B-g, B-h and B-i in the HOMO and LUMO region, there are 16 and 17 peaks, respectively. In the A-j, B-e and B-j models due to dissociation of NH2NO2 molecule in the HOMO and LUMO regions, we observe 18 and 19 peaks, respectively. Comparison results exhibit that in the LUMO region, more maxima peaks are observed than in the HOMO region.
By replacing C atoms, two small peaks are seen in the gap region and then due to these unfolded peaks the E g values of the BN nanosheet decrease significantly compared to pristine BN nanosheet models. So, the conductivity of the C-replaced BN nanosheet is more than that of the pristine model of BN nanosheet.
According to the calculated quantum parameters of the BN nanosheet/NH2NO2 in Table 3, the global hardness (η) values of the A-a to A-j models and the B-a to B-j models are in the range 2.03–2.61 and 0.47–0.66 eV, respectively. The global hardness of C-replaced BN nanosheet is lower than that of pristine BN nanosheet and so the reactivity of C-replaced BN nanosheet is more than that of pristine BN nanosheet. This fact confirms that the C-replaced BN nanosheet is more active than the pristine models for adsorbing and detecting NH2NO2 molecules.
The electrochemical potential (μ) results reveal that on replacing C atoms and adsorbing NH2NO2 molecules, the electrochemical potential of the system changes slightly from the original state.
The NBO and Milliken charge around the NH2NO2 molecule (Δρ NBO and Δρ mulk) and the maximum amount of electronic charge (ΔN) for the A-a to B-j models are negative (see Tables 2, 3). The negative values of Δρ NBO, Δρ mulk and ΔN at all adsorption models demonstrate that in the BN nanosheet/NH2NO2 system, the BN nanosheet has a donor electron effect and NH2NO2 molecule has an acceptor electron effect. Due to this effect, the charge density is localized around the NH2NO2 molecule.
The work function (ΔΦ) is the minimum energy to remove an electron from a solid surface to outside of the compound and it is used to describe the emitted electron current densities.
The work function values of the A-a to A-j models and the B-a to B-j models are in the range of −2.03 to −2.61 eV and −0.47 to −0.66 eV, respectively (see Table 3). A comparison of results shows that the work function of C-replaced BN nanosheet/NH2NO2 complex is lower than that of pristine models and so the emitted electron current densities of the B-a to B-j models are lower than those of the A-a to A-j models.
Nuclear magnetic resonance parameters
In this section, the evaluated nuclear magnetic resonance (NMR) parameters are used to understand the charge density of the material. The nuclear magnetic resonance parameters are generated by the response of electrons to an external uniform magnetic field and the spin–spin interaction. By using the gauge included atomic orbital (GIAO) approach [40,41,42], the chemical shielding isotropic (CSI) and chemical shielding anisotropic (CSA) parameters for 11B, 15N and 13C sites are calculated using Eqs. (15, 16):
The calculated CSI and CSA parameters of the A-a to B-j models are given in Tables S3–S6 in supplementary data. The NMR spectrum and the CSI plots for the A-a to B-j models are shown in Fig. 4 and Fig S4 in supplementary data. It is evident that the CSI values of N atoms are more than those of C and B atoms due to more electronegativity of the N atom with respect to B and C atoms. In the A-a to B-j models, the CSI values of N42, N51, N61, B32, B52, C12, C14, C53, C72, C74, C91, C94, N61, N101 and B43 sites are more than those of other sites of the nanosheet, whereas the CSI values of N32, N43, N52, N62, N83, N52, B73, C13, C35, C54, C75, C95, N42 and N63 sites are lower than those of other sites of the nanosheet. Therefore, the electrostatic behavior of the BN nanosheet surface varies significantly from the unabsorbed system. Comparison NMR spectrum of the A-a to B-j models (see Fig S4) shows that the shielding parameters of the A-j, B-e and B-j models are different from those of other adsorption models due to dissociation of the NH2NO2 molecule.

The CSI plots for NH2NO2 adsorption on the surface of pristine and C-doped BN nanosheet (A-a to B-j models, see Fig. 2)
The natural bond orbital (NBO)
The natural bonding orbital (NBO) is one of the important parameters used to understand the effects of NH2NO2 adsorption on the structural and orbital properties of the nanosheet. Using the natural bonding orbital results, the interaction energy or second-order perturbation interaction energy E (2) [44] between the bond orbitals with antibond orbitals or Rydberg orbitals can be calculated with delocalization \(i \to j\):
where F ij and q i are off-diagonal and donor orbital occupancy, respectively, and \(\varepsilon_{i} \,\,\) and \(\varepsilon_{j} \,\,\) are orbital energies. According to the NBO calculated results of Tables S7–S10 in supplementary data, the E (2) values of the A-a to A-j models are greater than those of B-a to B-j models. This result confirms that the stabilization energy between the donor and acceptor orbitals in the C-replaced BN nanosheet is significantly lower than that of the pristine models. Thereby by replacing C atoms, the activities and polarizability of the BN nanosheet increases significantly from the original state.
A comparison of the NBO results shows that the highest stabilization energy (E (2)) in the A-a, A-b, A-c, A-d, A-e models is shown at transition between the donor orbital to acceptor orbital \(\sigma B62 - N52 \to \sigma^{*} B52 - N52\) and for the A-f, A-g, A-h, A-i and A-j models at the transition between the donor orbital and acceptor orbital \(\sigma B62 - N52 \to \sigma^{*} B52 - N52\). On the other hand, the highest stabilization energy (E (2)) in the B-a to B-j models is shown at the transition between the donor orbital and acceptor orbital \(\sigma \,C53 - B42 \to \sigma^{*} C54 - \, C53\).
Molecular electrostatic potential
The molecular electrostatic potential (MEP) calculated results are used to determine the size and shape, and the positive, negative and neutral regions of the molecule [45, 46]. These results are useful for determining the charge distribution around the compound surface and nucleophilic or electrophilic region of the compound. This means that for all A-a to B-j models, the MEP and contour potential plots are calculated around the adsorption position and all calculated results are shown in Fig S5 in supplementary data. Herein, the green and red colors represent the negative and positive charges or the electrophilic and nucleophilic regions, respectively. The calculated results point out in all A-a to B-j models a negative potential, green color, is shown around the NH2NO2 molecule, whereas a positive potential, red color, is localized around the BN nanosheet. These results are completely in agreement with the NBO, quantum and NMR results. Therefore in the A-a to B-e models, due to donor electron effect of the BN nanosheet, a low charge transfer occurs between the nanosheet and the NH2NO2 molecule. Comparison of MEP and contour potential plots of the A and B models demonstrate that with C-replacing atoms the charge transfer between the BN nanosheet and NH2NO2 decreases slightly compared with the pristine models.
Conclusions
In this work, we investigated the structural, physical and electrical properties of adsorption NH2NO2 molecule on the surface of the pristine and C-replaced BN nanosheet. In the A-a to B-j models, the adsorption energy of the NH2NO2/BN nanosheet system strongly depends on the adsorption orientation of the NH2NO2 molecule and C-replaced atoms. The adsorption energy of NH2NO2 molecule on the outer surface of the pristine and C-replaced BN nanosheet is negative, exothermic and favorable.
The E g, NBO and global hardness results of C-replaced BN nanosheet are lower than those of the pristine models, and so the conductivity, polarizability and activity of C-replaced BN nanosheets are greater than those of pristine BN nanosheets. These results demonstrate that the C-replaced BN nanosheet models are good compounds to act as sensitive sensors and absorbers of the NH2NO2 molecule.
References
Novoselov, K.S., Geim, A.K., Morozov, S.V., Jiang, D., Zhang, Y., Dubonos, S.V., Grigorieva, I.V., Firsov, A.A.: Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004)
Geim, A.K., Novoselov, K.S.: The rise of graphene. Nat. Mater. 6, 183–191 (2007)
Zhang, Y.B., Tan, Y.W., Stormer, H.L., Kim, P.: Experimental observation of the quantum Hall effect and Berry’s phase in graphene. Nature 438, 201–204 (2005)
Morozov, S.V., Novoselov, K.S., Katsnelson, M.I., Schedin, F., Elias, D.C., Jaszczak, J.A., Geim, A.K.: Giant intrinsic carrier mobilities in graphene and its bilayer. Phys. Rev. Lett. 100, 016602–016604 (2008)
Novoselov, K.S., Jiang, Z., Zhang, Y., Morozov, S.V., Stormer, H., Zeitler, U., Maan, J.C., Boebinger, G.S., Kim, P., Geim, A.K.: Room-temperature quantum Hall effect in graphene. Science 315, 1379 (2007)
Zhou, J., Wang, Q., Sun, Q., Jena, P.: Stability and electronic structure of bilayer graphone. Appl. Phys. Lett. 98, 063108–5 (2011)
Pujari, B.S., Kanhere, D.G.: Density functional investigations of defect-induced mid-gap states in graphane. J. Phys. Chem. C 113, 21063–21067 (2009)
Lu, G., Ocula, E.L., Chen, J.: Gas detection using low-temperature reduced graphene oxide sheets. Appl. Phys. Lett. 94, 083111–083113 (2009)
Pashangpour, M., Bagheri, Z., Ghaffari, V.: A comparison of electronic transport properties of graphene with hexagonal boron nitride substrate and graphene, a first principle study. Eur. Phys. J. B 86(269), 1–6 (2013)
Robinson, J.T., Perkins, F.K., Snow, E.S., Wei, Z.Q., Sheehan, P.E.: NO2 and humidity sensing characteristics of few-layer graphene. Nano Lett. 8, 3137–3140 (2008)
Yoon, H.J., Jun, D.H., Yang, J.H., Zhou, Z., Yang, S.S., Cheng, M.M.: Carbon dioxide gas sensor using a graphene sheet. Sens. Actuators B. Chem. 157, 310–331 (2011)
Sakhavand, N., Shahsavari, R.: Synergistic behavior of tubes, junctions, and sheets imparts mechano-mutable functionality in 3D porous boron nitride nanostructures. J. Phys. Chem. C 118(39), 22730–22738 (2014)
Zhou, J., Wang, Q., Sun, Q., Jena, P.: Stability and electronic structure of bilayer graphone. Appl. Phys. Lett. 98, 063108/1–3 (2011)
Ahin, H., Ataca, C., Ciraci, S.: Magnetization of graphane by dehydrogenation. Appl. Phys. Lett. 95, 222510/1–4 (2009)
Boukhvalov, D.W., Katsnelson, M.I., Lichtenstein, A.I.: Hydrogen on graphene: Electronic structure, total energy, structural distortions and magnetism from first-principles calculations. Phys. Rev. B 77, 035427/1–3 (2008)
Nair, R.R., Ren, W., Jalil, R., Riaz, I., Kravets, V.G., Britnell, L., Blake, P., Schedin, F., Mayorov, A.S., Yuan, S., Katsnelson, M.I., Cheng, H.M., Strupinski, W., Bulusheva, L.G., Okotrub, A.V., Grigorieva, I.V., Grigorenko, A.N., Novoselov, K.S., Geim, A.K.: Fluorographene: a two-dimensional counterpart of teflon. Small 6, 2877–2884 (2010)
Ahin, H., Topsakal, M., Ciraci, S.: Structures of fluorinated graphene and their signatures. Phys. Rev. B. 83, 115432–115438 (2011)
Withers, F., Dubois, M., Savchenko, A.K.: Electron properties of fluorinated single-layer graphene transistors. Phys. Rev. B. 82, 073403/1–6 (2010)
Li, J., Jin, P., Dai, W., Wang, C., Li, R., Wu, T., Tang, C.: Excellent performance for water purification achieved by activated porous boron nitride nanosheets. Math. Chem. Phys. 196, 186–193 (2017)
Li, J., Jin, P., Tang, C.: Cr(III) adsorption by fluorinated activated boron nitride: a combined experimental and theoretical investigation. RSC. Adv. 4, 14815–14821 (2014)
Park, C.H., Louie, S.G.: Energy gaps and Stark effect in boron nitride nanoribbons. Nano Lett. 8, 2200–2203 (2008)
Zhang, Z., Guo, W.: Energy-gap modulation of BN ribbons by transverse electric fields: first-principles calculations. Phys. Rev. B. 77, 075403–075405 (2008)
Sun, L., Li, Y., Li, Z., Li, Q., Zhou, Z., Chen, Z., Yang, Z., Hou, J.G.: Electronic structures of SiC nanoribbons. J. Chem. Phys. 129, 174114/1–15 (2008)
Botello-Méndez, A.R., Lpez-Ur, F., Terrones, M., Terrones, H.: Magnetic behavior in zinc oxide zigzag nanoribbons. Nano Lett. 8, 1562–1565 (2008)
Li, H., Dai, J., Li, J., Zhang, S., Zhou, J., Zhang, L., Chu, W., Chen, D., Zhao, H., Yang, J., Wu, Z.: Electronic structures and magnetic properties of GaN sheets and nanoribbons. J. Phys. Chem. C 114, 11390–11395 (2010)
Zhang, X., Liu, Z., Hark, S.: Synthesis and optical characterization of single-crystalline AlN nanosheets. Solid. Stat. Commun. 143, 317–320 (2007)
Lopez, A., Bezanilla, H.J., Terrones, H., Sumpter, B.H.: Electronic structure calculations on edge functionalised armchair boron nitride nanoribbons. J. Phys. Chem. C 116(29), 15675–15681 (2012)
Anota, E.C., Juarez, A.R., Castro, M., Cocoletzi, H.H.: A density functional theory analysis for adsorption of the amine group on graphene and BN nanosheets. J. Mol. Model. 19, 321–328 (2013)
Barsan, N., Koziej, D., Weimar, U.: Metal oxide-based gas sensor research: how to? Sci. Direct. 121(1), 18–35 (2007)
Lin, Y.M., Valdes, G.A., Han, S.J., Farmer, D.B., Meric, I., Sun, Y., Wu, Y., Dimitrakopoulos, C., Grill, A., Avouris, P., Jenkins, K.A.: 100-GHz transistors from Wafer-scale epitaxial graphene. Science 332, 1294–1297 (2010)
Zhao, J.Y., Zhao, F.Q., Ju, X.H., Gao, H.X., Zhou, S.Q.: Density functional theory studies on the adsorption of NH2NO2 on Al13 cluster. J. Clust. Sci. 23, 395–410 (2012)
Bhattacharya, A., Bhattacharya, S., Das, G.P.: Band gap functionalization of BN sheet. Phys. Rev. B. 85, 035415 (2012)
Zhang, Z., Guo, W., Dai, Y.: Stability and electronic properties of small boron nitride nanotubes. J. Appl. Phys. 105, 084312/1–8 (2009)
Zheng, F., Zhou, G., Liu, Z., Wu, J., Duan, W., Gu, B.L., Zhang, S.B.: Half metallicity along the edges of zigzag boron nitride nanoribbons. Phys. Rev. B. 78, 205415/1–4 (2008)
Neek-Amal, M., Beheshtian, J., Sadeghi, A., Michel, K.H., Peeters, F.M.: Boron nitride monolayer: a strain tunable nanosensor. J. Phys. Chem. C 117(25), 13261–13267 (2013)
Li, Y., Zhou, Z., Zhao, J.: Functionalization of BN nanotubes with dichlorocarbenes. Nanotechnology 19, 015202 (2008)
Wang, L., Yi, C., Zou, H., Xu, J., Xu, W.: On the isomerization and dissociation of nitramide encapsulated inside an armchair (5,5) single-walled carbon nanotube. Mater. Chem. Phys. 127, 232–238 (2011)
Zhang, M.L., Ning, T., Zhang, S.Y., Li, Z.M., Yuan, Z.H., Cao, Q.X.: Response time and mechanism of Pd modified TiO2 gas sensor. Mater. Sci. Semi. Proc. 17, 149–154 (2014)
Schmidt, M.W., Baldridge, K.K., Boatz, J.A., Elbert, S.T., Gordon, M.S., Jensen, J.H., Koseki, S., Matsunaga, N., Nguyen, K.A., Su, S.J., Windus, T.L., Dupuis, M., Montgomery, J.A.: General atomic and molecular electronic structure system. J. Comp. Chem. 14, 1347–1363 (1993)
Rezaei-Sameti, M., Yaghoobi, S.: Theoretical study of adsorption of CO gas on pristine and AsGa-doped (4,4) armchair models of BPNTs. Comput. Cond. Mater. 3, 21–29 (2015)
Rezaei Sameti, M.: The effect of doping three Al and N atoms on the chemical shielding tensor parameters of the boron phosphide nanotubes: a DFT study. Phys. B 407, 22–26 (2012)
Rezaei-Sameti, M., Samadi Jamil, E.: The adsorption of CO molecule on pristine, As, B, BAs doped (4,4) armchair AlNNTs: a computational study. J. Nanostr. Chem. 3, 1–9 (2016)
James, C., Amalraj, A., Reghunathan, R., Hubert Joe, I., Jaya Kumar, V.S.: Structural conformation and vibrational spectroscopic studies of 2, 6-bis (p-N,N-dimethyl benzylidene) cyclohexanone using density functional theory. J. Raman Spect. 37, 1381–1392 (2006)
Glendening, E., Reed, A., Carpenter, J., Weinhold, F.: NBO Version 31. GaussianInc., Pittsburg (2003)
Bulat, F.A., Toro-Labbé, A., Brinck, T., Murray, J.S., Politzer, P.: Quantitative analysis of molecular surfaces: areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 16(11), 1679–1691 (2010)
Bulat, F.A., Burgess, J.S., Matis, B.R., Baldwin, J.W., Macaveiu, L., Murray, J.S., Politzer, P.: Hydrogenation and fluorination of graphene models: analysis via the average local ionization energy. J. Phys. Chem. A 116(33), 8644–8652 (2012)
Acknowledgements
The authors thank the Computational Information Center of Malayer University for providing the necessary facilities to carry out the research.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rezaei-Sameti, M., Jukar, N.J. A computational study of nitramide adsorption on the electrical properties of pristine and C-replaced boron nitride nanosheet. J Nanostruct Chem 7, 293–307 (2017). https://doi.org/10.1007/s40097-017-0237-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40097-017-0237-2