
Abstract
Genomic tools are now commonly used to assess the genetic diversity and genetic structure of species and populations, and they provide the ability to describe and address the negative effects of population declines and fragmentation. However, such studies are lacking for arboreal mammals despite their contribution to various ecosystem services, especially in uncommon and critically endangered ecosystems such as cloud forests. The aim of this work was to evaluate and compare the genetic diversity and population structure of two endemic arboreal mice from Mexican cloud forests that are associated with areas with different levels of impacts from human activities. We performed genotyping-by-sequencing in 47 Habromys schmidlyi and 17 Reithrodontomys wagneri individuals to evaluate genetic diversity and differentiation. In both species, the genetic diversity was low compared to other cricetid species, and we observed different population structure patterns, potentially linked to the different ecological associations. We detected two genetic groups in H. schmidlyi, that is a territorial species present in areas of low incline, while a single genetic group was found in R. wagneri, which forms family groups in areas with steep slopes. Overall, these results highlight how species’ genetic diversity can be differentially impacted depending on differential ecological associations within the same ecosystem. This information is essential for the development of the adequate conservation and management of these species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It is well known that the current Anthropocene period is associated with continued biodiversity loss at all ecological levels, from genetic diversity to ecosystem integrity and functionality (Ceballos et al. 2010; Otto 2018; Turvey and Crees 2019). The Convention on Biological Diversity (CBD), in their 2020 global framework, stated a need to minimize biodiversity loss in three aspects: genetic, species, and ecosystem diversity (Hoban et al. 2020). The CBD also highlighted the need to focus on the state of conservation and environmental risks in places where threatened species occur, mentioning the desirability of information on genetic diversity — important for conservation decision-making at both national and international levels (Darling et al. 2017; Breed et al. 2019; Hoban et al. 2020).
Genomic tools offer a powerful approach to assess the status of species and populations, as these tools can reveal ecological and demographic processes that shape genetic structure across the entire distribution of taxa, which is useful for conservation practitioners when dealing with threats to populations, species, and even ecosystems (Allendorf et al. 2010; Ekblom and Galindo 2011; Hoban et al. 2013; Hunter et al. 2018; Garrison et al. 2021). More importantly, these genomic tools can be used in non-model organisms with no prior genomic information or with low probability of detection or sampling — reducing the biases that may arise from small sample sizes, frequent in endangered species — since genome markers such as SNPs are not sensitive to these sampling effects (Ekblom and Galindo 2011; Hunter et al. 2018; Breed et al. 2019; Garrison et al. 2021). In this context, a set of four essential biodiversity variables have been proposed for genetic composition monitoring: (i) genetic diversity, (ii) genetic differentiation between populations, (iii) inbreeding, and (iv) effective population size (Hoban et al. 2022).
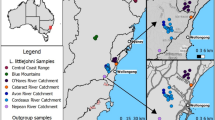
In terms of the health of ecosystems, it is also considered important to improve biodiversity protection (Hoban et al. 2020). Cloud forests are considered a relict ecosystem, which is threatened by climate change, as well as by habitat and land-use changes. They now represent less than 0.5% of Mexican territory, in isolated patches of less than 8 000 ha, and is the result of natural fragmentation of the ecosystem — due to the emergence of the Trans-Mexican Volcanic Belt, which started early- to mid-Miocene, with the last stratovolcanoes arising during the last 1.5 Myr (Ferrari et al. 2012) — but also by the replacement of half of its original distribution by human-induced landscapes (Fig. 1) (Challenger 1998; Luna-Vega et al. 1999; Ruiz-Jiménez et al. 2012). Consequently, the increasing patchiness of this ecosystem has affected the distribution of species by creating smaller and more isolated populations and reducing home-ranges due to the scarcity of resources and increased interspecific competition, particularly in species with low dispersal ability and probability of detection (Castañeda-Rico et al. 2011; Leonardi et al. 2012; Marines-Macías et al. 2018).

Sampling locations for both rodent species. Distribution of cloud forest in Mexico according to CONABIO [http://www.conabio.gob.mx/informacion/gis/] in grey, Taxco population right square and Zacualpan population left square. Geographic range based on the IUCN are shown for H. schmidlyi (dark blue) and R. wagneri (light green)
In cloud forest, arboreal rodents (together with other arboreal mammals) play a key-role in the ecosystem as seed dispersers and pollinators (Romo-Vázquez et al. 2005; Vega et al. 2017; Marines-Macías et al. 2018), and these species can also help to enhance community diversity by exploiting canopy resources, leaving ground-level resources for non-arboreal animals (Macarthur et al. 1966; August 1983). However, these arboreal species are facing strong threats due to their extreme habitat specialization, as well as low dispersal ability and detectability during sampling (Ceballos 2014). Highly specialist species are more prone to extinction due to habitat loss and fragmentation in human-modified landscapes, limiting migration and gene flow between populations and increasing inbreeding and between-population genetic divergence, which promote the reduction of effective population size and loss of genetic variability (Reed and Frankham 2003; Banks-Leite et al. 2014; Millán-Aguilar et al. 2016).
One of the largest patches of cloud forest in Mexico, and the second in priority of conservation according to national authorities, is the region of the “Upper Basin of the Amacuzac River” (CONABIO 2010; Ruiz-Jiménez et al. 2012). Within this region, three arboreal rodent species have been recorded, the Schmidlyi’s crested-tailed deer mouse (Habromys schmidlyi), the Wagner’s harvest mouse (Reithrodontomys wagneri), and the Goldman's diminutive woodrat (Nelsonia goldmani) (Pardiñas et al. 2017; Marines-Macías et al. 2018). Among these, it has been reported that H. schmidlyi and R. wagneri are sensitive to the scarcity of suitable substrates for nesting, so they depend on the local availability of arboreal substrates for the construction of their nests (Marines-Macías et al. 2018). In this way, these and other arboreal mice are useful indicator species of ecosystem change (Clavel et al. 2011; Linnell et al. 2018). Therefore, evaluating their genomic diversity could be beneficial for future conservation and management plans.
Even though the arboreal H. schmidlyi and R. wagneri are sympatric in some Mexican cloud forests, they occupy and use resources in different ways. Habromys schmidlyi shows strong association with cloud forest tree species in areas of low incline (e.g., Cleyera velutina and Carpinus caroliniana), together with Quercus sp. in areas surrounding the cloud forest patches (Marines-Macías et al. 2018). On the other hand, R. wagneri have only been found in cloud forest tree species that inhabit steep slopes, linking to the upper peaks of the mountains in the region (Marines-Macías et al. 2018). These different topographical and ecological associations likely have an impact on the distribution, and abundance of the species: H. schmidlyi is only recorded in two isolated-patches at the top of high mountains in central Mexico (Guerrero and Estado de México), where it is considered the most abundant species (Romo-Vázquez et al. 2005; Marines-Macías et al. 2018), whereas R. wagneri shows a wider range (Michoacán, Estado de México, Ciudad de México, Morelos, and Guerrero) although it is reported to be a rare species throughout its distribution (González-Cózatl and Arellano 2015; Martínez-Borrego et al. 2022). The IUCN Red List of Threatened Species classifies H. schmidlyi as Critically Endangered (Álvarez-Castañeda et al. 2018), although its conservation status at the national level has not been evaluated. On the contrary, R. wagneri (included in R. microdon by the IUCN) is classified as Least Concern by the IUCN Red List of Threatened Species (Reid and Vázquez 2016), but it is considered as Threatened [Amenazado] by Mexican law “NOM-059-SEMARNAT-2010, Anexo III 2019” [online available at: http://legismex.mty.itesm.mx/normas/ecol/semarnat059-ModAnexoIII2019_11.pdf], even though conservation status has not been formally evaluated for R. wagneri.
All these ecological and conservation characteristics make H. schmidlyi and R. wagneri interesting systems to explore how the natural fragmentation of cloud forest ecosystems translates into differences in genetic diversity and population structure. For these, we expect that H. schmidlyi will show higher levels of genetic structure because of its preference for isolated areas of low incline, whereas R. wagneri will show a weaker genetic structure due to the connectivity among steep slopes. According to this, the aim of this study was to evaluate the genomic diversity and population structure in these two ecologically distinctive arboreal rodent species living in sympatry. This study also aims to contribute to the development of conservation guidelines for the protection of cloud forest mammalian biodiversity, and our results could serve as a basis for further studies focused on the management and conservation plans for different species associated with the cloud forest, but also with other fragmented ecosystems.
Materials and methods
Sampling and DNA extraction
During 2012–2013 extensive fieldwork was conducted at the “Cerro del Huixteco” National State Park, within the Sierra de Taxco, Guerrero, Mexico (18° 36′ N 99° 36′ W), and the “Picacho de Oro y Plata” State Park, Zacualpan, Estado de México, Mexico, and surrounding areas (18° 43′ N, 99° 46′ W) (Fig. 1), where the study species are sympatric. Sampling at both locations was performed using 120 Sherman live-traps, with 60 traps placed at the ground level and the remaining 60 traps on tree trunks and branches, following the transect set on the ground. The arboreal trapping was done using climbing equipment to reach the highest possible point (20 m) [for further sampling information, see Marines-Macías et al. (2018)]. All trapped individuals were identified in situ using external characteristics, and ear clip samples were obtained using a medical punch previously disinfected with 10% chlorine and 96% ethanol. Tissues were fixed individually in 96% ethanol and then stored at − 70 °C until processing. DNA extractions were performed with the Mag-Bind® Blood & Tissue DNA OMEGA bio-tech kit (Omega Bio-tek, Inc., Norcross, US), following the manufacturer’s protocols with some modifications in incubation temperatures and time to increase DNA yield (Supplementary 1).
Genotyping-by-sequencing (GBS)
DNA was quantified with a Qubit 2.0 Fluorometer (dsDNA Broad Range Assay) for a total of 54 H. schmidlyi and 21 R. wagneri, and the samples normalized for standard concentrations required for high-throughput sequencing, using an average of 40 μl sample volumes and concentrations between 20 and 50 ng/μl. Samples were then sent to the Genomic Diversity Facility (GDF) at Cornell University for GBS library construction (Elshire et al. 2011) and sequencing services. Briefly, DNA was sheared using the PstI enzyme for genome complexity reduction, including one blank as a negative control. Individual samples were barcoded and then combined in a single pool, which was amplified through PCR reaction, purified, and quantified for sequencing on the Illumina Hi-seq 2000/2500 sequencer (Illumina Inc., USA).
Due to lack of a reference genome for both species, SNP calling was carried out with the UNEAK pipeline for each species independently (Lu et al. 2013), defining a minimum call of five reads, maximum locus and individual missing data of 20% and 25%, respectively, following the methodology of Barbosa et al. (2017). Postprocessing included further filtering in order to avoid repetitive and paralogous loci (heterozygosity above 75%) following the pipeline of White et al. (2013).
Finally, a principal coordinates analysis (PCoA) using the ade4 package (Dray and Dufour 2007) was performed individually for each rodent species to test for the genetic variation of the SNPs obtained and their distribution among populations in a model-free framework.
Genetic diversity
We used the R package diveRsity (Keenan et al. 2013) to evaluate the fit to the Hardy–Weinberg equilibrium within each sampling location using Fisher’s exact test, by calculating the rate of fixation or inbreeding (F) applying a Bonferroni correction for multiple comparisons (Rice 1989).
The SNP variation and its distribution among populations based on a model-free framework were tested using the same R package, estimating the rarefied allelic richness and private alleles, observed (Ho) and expected (He) heterozygosity, allowing 5% missing data and using 10,000 global iterations.
Genetic structure
To examine the genetic structure of each species, the software STRUCTURE (Pritchard et al. 2000) and STRUCTURE HARVESTER (Earl and vonHoldt 2012) were used to determine the most probable number of populations by calculating the ΔK value with the Evanno method (Evanno et al. 2005). STRUCTURE analyses were conducted testing from 1 to 5 populations (K), with the admixture model and correlated allele frequencies. We used an initial burn-in of 200,000, followed by 800,000 Markov Chain Monte Carlo iterations with 5 independent runs for each K value.
The geographic structure by species was also tested in Geneland (Guillot et al. 2012), with this analysis incorporating the maximum distance that an individual can move as an uncertainty value, together with the geographic position of each sample — information that was obtained according to home-range areas and movement distances in Marines-Macías et al. (2018). For this, we followed the recommendations of Guillot et al. (2012) for processing the genomic data, so we created a random subset of both datasets: four subsets for H. schmidlyi and three for R. wagneri. In each subset, the occurrence of different populations from 1 to 5 was tested, using 1 million generations with a thinning of 100 and a burn-in of 1,000. The convergence of the results over five independent runs and all subsets was determined, and the run with the highest likelihood value was chosen.
Additionally, hierarchical genetic structure was evaluated by analysis of molecular variance (AMOVA) in Arlequin 3.5.2.2 (Excoffier and Lischer 2010). To perform this analysis, both sampling sites were tested as the main groups, while the population identity of each individual was based on closeness or correspondence to overlapping home-ranges areas, according to results for each species obtained by Marines-Macías et al. (2018), which considers that these overlapping-home-ranges represent subpopulations. These analyses were performed using pairwise distance matrices (Fst) with 10,000 permutations to test the significance of our results.
To evaluate the distribution of the individual genetic distances of each species, Nei’s distances were calculated between individuals in the adegenet R package (Jombart and Ahmed 2011). Additionally, the effective population size (Ne) per population by species were estimated in the LDNe 1.31 software (Waples and Do 2008), with a random mating model, a minor allele frequency of 5% and Jackknife with parametric corrections for independence of loci.
To test for isolation-by-distance, a Mantel test was performed correlating the geographic and genetic distances. The genetic distance matrices were calculated between each sampling site (the same Fst pairwise comparisons of populations as used in the AMOVA analysis) in GenAlEx v6.503. Both Mantel tests were performed using 9,999 permutations, and as a measure of population distance, Fst / (1-Fst) was used, following the recommendations of Rousset (1997).
Finally, the effective number of migrants for both species (Nm) was calculated by the private alleles method proposed by Barton and Slatkin (1986) with a correction for population size, using Genepop 4.4 (Rousset 2008).
Results
Of the 54 individuals of H. schmidlyi, a total of 40,665 SNPs were obtained with the UNEAK pipeline, of which 4,006 were within the defined thresholds of missing data. Similarly, of the 21 individuals of R. wagneri, a total of 60,137 SNPs were obtained, of which 2,895 fulfilled the same filtering thresholds.
According to the PCoA exploratory analysis, a clear differentiation between the two populations of H. schmidlyi was observed, indicating two well-defined genetically distinct groups. These groups corresponded to the Zacualpan and the Taxco populations. In contrast, no genetic differentiation was found between the two sampling sites for R. wagneri (Fig. 2).

Principal coordinates analysis based on SNPs obtained for Habromys schmidlyi and Reithrodontomys wagneri. In both cases, light green represent SNPs present in the Taxco and dark blue those SNPs from Zacualpan population
From a total of 75 individuals (54 = H. schmidlyi and 21 = R. wagneri), seven from H. schmidlyi and four from R. wagneri were excluded due to missing data > 25%. Therefore, subsequent analyses included 47 individuals of H. schmidlyi (16 from Zacualpan and 31 from Taxco) and 17 of R. wagneri (11 from Zacualpan and 6 from Taxco).
Genetic diversity
The rate of fixation or inbreeding (F) for the 4,006 loci obtained from H. schmidlyi, were significantly different from zero in 925 loci (23%), indicating an excess of homozygotes. However, the paired value of the fixation index after Bonferroni correction (F = 0.01, P = 0.0001) indicates that both the Zacualpan and Taxco populations were in Hardy–Weinberg equilibrium, so we include all loci in further analyses (Supplementary 2). Similarly, from the 2,895 loci obtained from R. wagneri, 212 (7%) showed an excess of observed homozygotes, and the paired value of the fixation index after Bonferroni correction (F = 0.0083, P = 0.0002) showed that these populations were also in Hardy–Weinberg equilibrium (Supplementary 2).
In general, the Ho/He values were similar for both species when the populations of Taxco and Zacualpan were compared (Table 1). Furthermore, for H. schmidlyi, a rarefied mean allelic richness of 1.86 and a rarefied private allelic richness of 0.08 were obtained in the Taxco population, compared with 1.83 and 0.13 obtained in Zacualpan. On the other hand, for R. wagneri, a rarefied mean allelic richness of 1.73 and a rarefied private allelic richness of 0.17 were obtained in the Taxco population, with 1.77 and 0.08 for the Zacualpan population (Supplementary 3).
Genetic structure
The ΔK values of STRUCTURE analyses indicated that the most likely value of K was 2 for both species (Fig. 3). Considering these K values, the STRUCTURE analysis showed a clear division of two geographic clusters for H. schmidlyi (Fig. 3A; Table 2). However, for R. wagneri, there was no clear evidence of two geographic clusters (Fig. 3C; Table 2), since only two individuals from the Zacualpan population showed high values of belonging to a different genetic population; this suggests a lack of genetic subdivision for this species. Additionally, when the Geneland analysis are considered, there was evidence of genetic structure in H. schmidlyi, inferring the same two genetic clusters corresponding to the two sampled localities, Taxco and Zacualpan (Fig. 3B). Meanwhile, for R. wagneri, the Geneland analysis revealed a single genetic cluster (Fig. 3D).

Genetic structure of the 47 individuals of Habromys schmidlyi for 4,006 loci (A and B) and 17 individuals of Reithrodontomys wagneri for 2,895 loci (C and D), representing the number of detected clusters. Each individual is depicted by a radial section shaded to show its proportional membership of each genetic cluster. A and C STRUCTURE analysis; B and D Geneland analysis results. Z corresponds to Zacualpan populations and T to Taxco
The AMOVA analyses showed that the highest variation was within populations (i.e., the a priori identity of population membership according to the criterion of overlapping home-ranges [see Marines-Macías et al. (2018)]), and among groups (i.e., Zacualpan vs Taxco) for both species. In the case of R. wagneri, the variation within populations was higher (93.69%) compared with H. schmidlyi (85.31%), but the variation among groups was lower (4.03%) compared to H. schmidlyi (11.15%) (Table 3). The AMOVA revealed significant variation for all F-statistics for H. schmidlyi, but only among populations (Fct = 0.04; P = 0.007) for R. wagneri (Table 3).
Nei’s distance values show a low differentiation for both species. However, when observing the distribution of the distances in R. wagneri, a unimodal distribution is observed, which could indicate less variation between the comparisons, unlike H. schmidlyi in which there is a bimodal distribution, which suggests a greater variation between comparisons (Fig. 4). The effective population sizes were low for both species in both populations, with an estimate of 20.8 individuals in Taxco and 10.2 in Zacualpan for H. schmidlyi and 16.9 individuals in Taxco and 9.3 in Zacualpan for R. wagneri (Table 1).

Pairwise Nei’s genetic distances between individuals of Habromys schmidlyi (dark) and Reithrodontomys wagneri. (light)
Only for H. schmidlyi was geographic distance positively correlated with genetic distance (R2 = 0.679, P = 0.011), but in the case of R. wagneri the correlation was close to zero and not statistically significant (R2 = 0.288, P = 0.169). Supporting these results of isolation-by-distance, the number of migrants after size correction between Taxco and Zacualpan was lower (0.221 ± 0.15 migrants) for H. schmidlyi than for R. wagneri (0.462 ± 0.175).
Discussion
For the first time, genomic data of arboreal mammals from cloud forests were analyzed to understand how the increasing loss of connectivity between habitat patches in the cloud forest affects populations of these mammals, focusing on H. schmidlyi and R. wagneri in Central Mexico. We include all localities where H. schmidlyi has been recorded, comparing it with sympatric populations of R. wagneri, where ecological traits of each species are well described (Romo-Vázquez et al. 2005; Marines-Macías et al. 2018). This approach allowed us to evaluate the effect of the configuration of cloud forest patches on the genetic structure in these two rodent species, which are both arboreal, but which show divergent ecological traits. Even though our sample size was small compared to other studies evaluating genetic diversity in Mexican rodents (e.g., Vega et al. 2007; Garrido-Garduño and Vázquez-Domínguez 2013; Espindola et al. 2014), our conclusions were consistent across analyses and methods. This likely relates to the use of NGS tools where, for each species, we obtained thousands of genomic markers (fulfilling the minimum required reads and missing data values), thus making these results applicable for conservation approaches (Eizaguirre and Baltazar-Soares 2014; Vega et al. 2017).
Genetic diversity
The genetic diversity shown by both species seems to be lower than other endemic Mexican rodents such as Oryzomys couesi cozumelae (He = 0.690–0.864; Vega et al. 2007), Habromys simulatus (He = 0.627–0.704; Castañeda-Rico et al. 2011), and Reithrodontomys spectabilis (He = 0.659–0.765; Espindola et al. 2014). It seems also low compared with other mammal species distributed in fragmented areas and endangered habitats such as those in the Australian fragmented Eucalyptus forests, Antechinus agilis (He = 0.844–0.86; Banks et al. 2005) and Petauroides volans (He = 0.59–0.77; Taylor et al. 2007). Since not all the above He values were obtained using SNPs, future analyses using homologous loci between species are needed to corroborate if genetic diversity of H. schmidlyi and R. wagneri are truly lower.
Given the threatened status of the Mexican cloud forests, anthropogenic disturbances in the habitat could also negatively affect the connectivity between the patches, promoting the decrease in the genomic diversity of these two arboreal rodents, as has been documented in other animals inhabiting protected areas (Mohammadi et al. 2018; Fetene et al. 2019; Garrison et al. 2021). Furthermore, it has been documented that life history and natural demographic characteristics of populations could play an important role in influencing low levels of heterozygosity (Garrison et al. 2021). In this sense, the low levels of heterozygosity shown by other microendemic mammalian species that inhabit fragmented habitats have been attributed to the formation of family groups (Frère et al. 2010; Castellanos-Morales et al. 2014; Espindola et al. 2014). This specific demographic characteristic has been found in R. wagneri in both populations, where family groups were found using radiotracking information, although in the case of H. schmidlyi this characteristic was only documented in Zacualpan (Marines-Macías et al. 2018).
Genetic differentiation
Given the small number of samples for R. wagneri, the results must be taken with great caution; however, they can be interpreted as a trend. Taking into account the fact that the two sampling locations are separated by 22.7 km, which can be considered enough to expect high genetic differentiation in rodent species (Trizio et al. 2005; Bogdanov et al. 2009; Vargas et al. 2012; Castellanos-Morales et al. 2014), a genetic difference was detected between the H. schmidlyi populations, but was not detected in R. wagneri. While the clustering analyzes (Geneland and STRUCTURE) showed a “highly” structured individual identity for H. schmidlyi, the AMOVA and Nei’s distances suggest that this genetic structure is supported by low levels of genetic differentiation. This low genetic differentiation within populations of H. schmidlyi could be expected due to the recent divergence of this species from its sister taxon H. simulatus, between 1.63 and 2.33 Mya (Leon-Paniagua et al. 2007), and the fact that other Neotominae needed substantial time to reach high levels of genetic differentiation (Steppan and Schenk 2017).
Considering the habitat selection previously reported for H. schmidlyi, this species prefers areas of low incline (Marines-Macías et al. 2018), and the presence of steep slopes that divide the populations of Zacualpan and Taxco could limit the number of migrants, as shown in this study, favoring a greater genetic structure between populations. Interestingly, geographic features also shape the genetic structure of another co-generic species, H. simulatus, in which isolated populations act as genetic islands (Castañeda-Rico et al. 2011). We additionally detected an isolation-by-distance pattern in H. schmidlyi that could also be limiting the number of migrants between these two populations. So, the geography of the landscape and the distance could be promoting the subdivision of H. schmidlyi. Contrary to what was observed in H. schmidlyi, R. wagneri prefers trees located in steep slopes, habitat that could facilitate connectivity and gene flow along the Sierra de Taxco, where this species was sampled. This habitat selection could explain the lack of genetic structuring in R. wagneri, the higher number of migrants compared with H. schmidlyi, and the lack of isolation-by-distance observed. In summary, these results suggest that the steep slopes, which allow gene flow in R. wagneri, represent a local geographic barrier for H. schmidlyi, which prefers areas of low incline.
Additionally, different behaviors have been reported between the two populations of H. schmidlyi. Territorial behavior has been observed in Taxco: individuals have separate home ranges and males tend to expand that cruising area in the reproductive season, whereas in Zacualpan a gregarious social structure has been reported (Burt 1943; Coleman and Downs 2010; Marines-Macías et al. 2018). This supports the distinction of the two H. schmidlyi populations that are genetically, behaviorally, and ecologically different and also agree with the results found in R. wagneri: in this species, a gregarious social structure was reported in both Taxco and Zacualpan, in which females and males share the same areas (Burt 1943; Coleman and Downs 2010; Marines-Macías et al. 2018); showing a continuity and unity between sampled localities.
When considering estimated effective population sizes, these species show low numbers compared to other rodent species, such as Reithrodontomys raviventris (Ne = 15–131; Statham et al. 2022) or Cricetus cricetus (Ne = ~ 6000–7000; Melosik et al. 2017). However, it is important to note that rodent generations overlap due to their short life cycles, violating the sampling assumption of non-overlapping generations. As such, our Ne estimates could be lower than reality, since it is very likely that several age classes were sampled (England et al. 2006; Marandel et al. 2019; Garrison et al. 2021). Therefore, the Ne estimates reported in this study should be used with caution, and we strongly recommend further age-class cohort samplings to elucidate the actual trends over time, following the suggestions of England et al. (2006) and Marandel et al. (2019).
Implications for conservation
Habromys schmidlyi and R. wagneri are under special protection by international agencies or the Mexican National law. Both authorities agree that the main concerns are the low number of individuals and the declining trend of the populations (DOF 2010; Álvarez-Castañeda et al. 2018). We observed that H. schmidlyi was affected by local geographic features, so the continuity and connection of forest patches that this species inhabits needs to increase to avoid negative processes that could decrease genetic diversity. We believe our results highlight the importance of both H. schmidlyi populations as unique evolutionarily significant units, since they represent unique sources that should be conserved to protect the species, as well as contributing to overall conservation and management plans (Moritz 1995; Castañeda-Rico et al. 2011; Eizaguirre and Baltazar-Soares 2014). In the case of R. wagneri, the two sampling localities analyzed represent the same genetic unit for this taxon, and even though the number of migrants is low, the conservation of the natural habitats located in steep slopes is also important to maintain the genetic connectivity and avoid the negative effects of genetic drift. The conservation and management of these two species must be focused on maintaining the health and continuity of the cloud forest, which also will play an important role in the conservation and the continuity of the ecological processes in it (Mitrovski et al. 2007; Eizaguirre and Baltazar-Soares 2014; Marines-Macías et al. 2018). Furthermore, future monitoring is necessary to evaluate the conservation risk of both species in order to protect their genetic diversity (Eizaguirre and Baltazar-Soares 2014).
In this work we analyzed all known populations of H. schmidlyi and compared them with sympatric populations of R. wagneri. We showed how the complex topography in the region, which includes multiple interconnected steep slopes that isolate multiple areas of low incline, promotes different evolutionary responses in Neotominae rodents. Although both species belong to the Reithrodontomyini tribe, their contrasting social interactions and habitat selection seems to lead to a differential impact on the connectivity of their populations. This affects their genetic diversity and genetic structure and highlights that the idiosyncrasy of each species determines their evolutionary responses. Likewise, this study showcases a pervasive phenomenon across microendemic species with low population density, low detection probability, and associated with endangered ecosystems, such as Peromyscus melanurus, Reithrodontomys brevirostris, and Nelsonia goldmani (Ceballos 2014). Hence, future research should also address the entire distribution of R. wagneri and be especially concerned with the conservation of other microendemic species and the habitats that they inhabit.
Finally, the information shown here is of great importance to generate management plans and biodiversity conservation strategies not only for the species themselves, but also to be used as a proxy for evaluating the health of the ecosystem, since these arboreal rodents have key roles in the maintenance of cloud forest as seed dispersers, pollinators, and ecosystem regenerators (Schupp 1993; Williams et al. 2000; Vander-Wall et al. 2005).
Data Availability
Raw genomic data obtained from GBS is public available at: https://doi.org/10.5281/zenodo.6726673.
References
Allendorf FW, Hohenlohe PA, Luikart G (2010) Genomics and the future of conservation genetics. Nat Rev Genet 11:697–709. https://doi.org/10.1038/nrg2844
Álvarez-Castañeda ST, Castro-Arellano I, Lacher T, Vázquez E (2018) Habromys schmidlyi. In: IUCN Red List Threat. Species 2008 e.T136616A4318725. https://doi.org/10.2305/IUCN.UK.2018-2.RLTS.T136616A22376358.en. Accessed 18 Feb 2021
August PV (1983) The role of habitat complexity and heterogeneity in structuring tropical mammal communities. Ecology 64:1495–1507
Banks-Leite C, Pardini R, Tambosi LR, Pearse WD, Bueno AA, Bruscagin RT, Condez TH, Dixo M, Igari AT, Martensen AC, Metzger JP (2014) Using ecological thresholds to evaluate the costs and benefits of set-asides in a biodiversity hotspot. Science 345:1041–1045. https://doi.org/10.1126/science.1255768
Banks S, Finlayson G, Lawson S, Lindenmayer D, Paetkau D, Ward S, Taylor A (2005) The effects of habitat fragmentation due to forestry plantation establishment on the demography and genetic variation of a marsupial carnivore, Antechinus agilis. Biol Conserv 122:581–597. https://doi.org/10.1016/j.biocon.2004.09.013
Barbosa S, Paupério J, Herman JS, Ferreira CM, Pita R, Vale-Gonçalves HM, Cabral JA, Garrido-García JA, Soriguer RC, Beja P, Mira A, Alves PC, Searle JB (2017) Endemic species may have complex histories: within-refugium phylogeography of an endangered Iberian vole. Mol Ecol 26:951–967. https://doi.org/10.1111/mec.13994
Barton NH, Slatkin M (1986) A quasi-equilibrium theory of the distribution of rare alleles in a subdivided population. Heredity 56:409–415. https://doi.org/10.1038/hdy.1986.63
Bogdanov AS, Atopkin DM, Chelomina GN (2009) Analysis of genetic variation and differentiation in the pygmy wood mouse Sylvaemus uralensis (Rodentia, Muridae) aided by the RAPD-PCR method. Biol Bull 36:227–241. https://doi.org/10.1134/S1062359009030030
Breed MF, Harrison PA, Blyth C, Byrne M, Gaget V, Gellie NJC, Groom SVC, Hodgson R, Mills JG, Prowse TAA, Steane DA, Mohr JJ (2019) The potential of genomics for restoring ecosystems and biodiversity. Nat Rev Genet 20:615–628. https://doi.org/10.1038/s41576-019-0152-0
Burt WH (1943) Territoriality and home range concepts as applied to mammals. J Mammal 24:346–352
Castañeda-Rico S, León-Paniagua L, Ruedas LA, Vázquez-Domínguez E (2011) High genetic diversity and extreme differentiation in the two remaining populations of Habromys simulatus. J Mammal 92:963–973. https://doi.org/10.1644/10-MAMM-A-171.1
Castellanos-Morales G, Gasca-Pineda J, Ceballos G, Ortega J (2014) Genetic variation in a peripheral and declining population of black-tailed prairie dogs (Cynomys ludovicianus) from Mexico. J Mammal 95:467–479. https://doi.org/10.1644/12-MAMM-A-099
Ceballos G (2014) Mammals of Mexico. Johns Hopkins University Press
Ceballos G, García A, Ehrlich PR (2010) The sixth extinction crisis: loss of animal populations and species. J Cosmol 8:1821–1831
Challenger A (1998) La zona ecológica templada húmeda. In: Challenger A (ed) Utilización y Conservación de Los Ecosistemas Terrestres de México. Pasado, Presente y Futuro. CONABIO-Instituto de Biología, UNAM-Sierra Madre, México, D.F., pp 433–518
Clavel J, Julliard R, Devictor V (2011) Worldwide decline of specialist species: toward a global functional homogenization? Front Ecol Environ 9:222–228. https://doi.org/10.1890/080216
Coleman JC, Downs CT (2010) Does home range of the black-tailed tree rat (Thallomys nigricauda Thomas 1882) change with season along an aridity gradient? African Zool 45:177–188. https://doi.org/10.3377/004.045.0219
CONABIO [Comisión Nacional para el Uso y Conocimiento de la Biodiversidad] (2010) El Bosque Mesófilo de Montaña en México: Amenazas y Oportunidades para su Conservación y Manejo Sostenible. Comisión Nacional para el Conocimiento y Uso de la Biodiversidad, México D.F
Darling JA, Galil BS, Carvalho GR, Rius M, Viard F, Piraino S (2017) Recommendations for developing and applying genetic tools to assess and manage biological invasions in marine ecosystems. Mar Policy 85:54–64. https://doi.org/10.1016/j.marpol.2017.08.014
DOF [Diario Oficial de la Federación] (2010) NOM-059-SEMARNAT-2010. Protección ambiental-Especies nativas de México de flora y fauna silvestres-Categorías de riesgo y especificaciones para su inclusión, exclusión o cambio-Lista de especies en riesgo. Available at: https://dof.gob.mx/nota_detalle_popup.php?codigo=5173091. Accessed 4 Apr 2021
Dray S, Dufour A-B (2007) Journal of Statistical Software 22(4). https://doi.org/10.18637/jss.v022.i04
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Eizaguirre C, Baltazar-Soares M (2014) Evolutionary conservation-evaluating the adaptive potential of species. Evol Appl 7:963–967. https://doi.org/10.1111/eva.12227
Ekblom R, Galindo J (2011) Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 107:1–15. https://doi.org/10.1038/hdy.2010.152
Elshire RJ, Glaubitz JC, Sun Q et al (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. https://doi.org/10.1371/journal.pone.0019379
England PR, Cornuet JM, Berthier P, Tallmon DA, Luikart G (2006) Estimating effective population size from linkage disequilibrium: severe bias in small samples. Conserv Genet 7:303–308. https://doi.org/10.1007/s10592-005-9103-8
Espindola S, Cuarón AD, Gaggiotti OE, Vázquez-Domínguez E (2014) High genetic structure of the Cozumel harvest mice, a critically endangered island endemic: conservation implications. Conserv Genet 15:1393–1402. https://doi.org/10.1007/s10592-014-0625-9
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Ferrari L, Orozco-Esquivel T, Manea V, Manea M (2012) The dynamic history of the Trans-Mexican Volcanic Belt and the Mexico subduction zone. Tectonophysics 522–523:122–149. https://doi.org/10.1016/j.tecto.2011.09.018
Fetene A, Yeshitela K, Gebremariam E (2019) The effects of anthropogenic landscape change on the abundance and habitat use of terrestrial large mammals of Nech Sar National Park. Environ Syst Res 8:19. https://doi.org/10.1186/s40068-019-0147-z
Frère CH, Krützen M, Mann J, Connor RC, Bejder L, Sherwin WB, Orians GH (2010) Social and genetic interactions drive fitness variation in a free-living population. Proc Natl Acad Sci U S A 107:19949–19954. https://doi.org/10.1073/pnas
Garrido-Garduño T, Vázquez-Domínguez E (2013) Métodos de análisis genéticos, espaciales y de conectividad en genética del paisaje. Rev Mex Biodivers 84:1031–1054. https://doi.org/10.7550/rmb.32500
Garrison NL, Johnson PD, Whelan NV (2021) Conservation genomics reveals low genetic diversity and multiple parentage in the threatened freshwater mussel, Margaritifera hembeli. Conserv Genet 22:217–231. https://doi.org/10.1007/s10592-020-01329-8
González-Cózatl FX, Arellano E (2015) Notes on the ecology of Reithrodontomys microdon based on new records in the Eje Neovolcánico, México. West North Am Nat 75:377–379. https://doi.org/10.3398/064.075.0316
Guillot G, Renaud S, Ledevin R, Michaux J, Claude J (2012) A unifying model for the analysis of phenotypic, genetic, and geographic data. Syst Biol 61:897–911. https://doi.org/10.1093/sysbio/sys038
Hoban S, Archer FI, Bertola LD, Bragg JG, Breed MF, Bruford MW, Coleman MA, Ekblom R, Funk WC, Grueber CE, Hand BK, Jaffé R, Jensen E, Johnson JS, Kershaw F, Liggins L, Macdonald AJ, Mergeay J, Potter KM, Razgour O, Vernesi C (2022) Global genetic diversity status and trends: towards a suite of Essential Biodiversity Variables (EBVs) for genetic composition. Biol Rev. https://doi.org/10.1111/brv.12852
Hoban S, Arntzen JW, Bertorelle G, Bryja J, Fernandes M, Frith K, Gaggiotti O, Galbusera P, Godoy JA, Hauffe HC, Hoelzel AR, Nichols RA, Pérez-Espona S, Primmer C, Russo IRM, Segelbacher G, Siegismund HR, Sihvonen M, Sjögren-Gulve P, Vernesi C, Vilà C, Bruford MW (2013) Conservation genetic resources for effective species survival (ConGRESS): bridging the divide between conservation research and practice. J Nat Conserv 21:433–437. https://doi.org/10.1016/j.jnc.2013.07.005
Hoban S, Bruford M, Urban JD, Lopes-Fernandes M, Heuertz M, Hohenlohe PA, Paz-Vinas I, Sjögren-Gulve P, Segelbacher G, Vernesi C, Aitken S, Bertola LD, Bloomer P, Breed M, Rodríguez-Correa H, Funk WC, Grueber CE, Hunter ME, Jaffe R, Liggins L, Mergeay J, Moharrek F, Brien DO, Ogden R, Palma-Silva C, Pierson J, Ramakrishnan U, Simo-Droissart M, Tani N, Waits L, Laikre L (2020) Genetic diversity targets and indicators in the CBD post-2020 global biodiversity framework must be improved. Biol Conserv 248:108654. https://doi.org/10.1016/j.biocon.2020.108654
Hunter ME, Hoban SM, Bruford MW, Segelbacher G, Bernatchez L (2018) Next-generation conservation genetics and biodiversity monitoring. Evol Appl 11:1029–1034. https://doi.org/10.1111/eva.12661
Jombart T, Ahmed I (2011) adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071. https://doi.org/10.1093/bioinformatics/btr521
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA (2013) DiveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788. https://doi.org/10.1111/2041-210X.12067
León-Paniagua L, Navarro-Sigüenza AG, Hernández-Baños BE, Morales JC (2007) Diversification of the arboreal mice of the genus Habromys (Rodentia: Cricetidae: Neotominae) in the Mesoamerican highlands. Mol Phylogenet Evol 42:653–664. https://doi.org/10.1016/j.ympev.2006.08.019
Leonardi S, Piovani P, Scalfi M, Piotti A, Giannini R, Menozzi P (2012) Effect of habitat fragmentation on the genetic diversity and structure of peripheral populations of beech in central Italy. J Hered 103:408–417. https://doi.org/10.1093/jhered/ess004
Linnell MA, Lesmeister DB, Bailey JD, Forsman ED, Swingle JK (2018) Response of arboreal rodents to increased availability of nest substrates in young forests. J Mammal 99:1174–1182. https://doi.org/10.1093/jmammal/gyy111
Lu F, Lipka AE, Glaubitz J, Elshire R, Cherney JH, Casler MD, Buckler ES, Costich DE (2013) Switchgrass genomic diversity, ploidy, and evolution: novel insights from a network-based SNP discovery protocol. PLoS Genet 9:e1003215. https://doi.org/10.1371/journal.pgen.1003215
Luna-Vega I, Alcántara-Ayala O, Espinosa-Organista D, Morrone JJ (1999) Historical relationships of the Mexican cloud forests: a preliminary vicariance model applying parsimony analysis of endemicity to vascular plant taxa. J Biogeogr 26:1299–1305. https://doi.org/10.1046/j.1365-2699.1999.00361.x
Macarthur R, Recher H, Cody M (1966) On the relation between habitat selection and species diversity. Am Nat 100:319–332
Marandel F, Lorance P, Berthelé O, Trenkel VM, Waples RS, Lamy JB (2019) Estimating effective population size of large marine populations, is it feasible? Fish Fisheries 20:189–198. https://doi.org/10.1111/faf.12338
Marines-Macías T, Colunga-Salas P, Verde-Arregoitia LD, Naranjo EJ, León-Paniagua L (2018) Space use by two arboreal rodent species in a neotropical cloud forest. J Nat Hist 52:1417–1431. https://doi.org/10.1080/00222933.2018.1459921
Martínez-Borrego D, Arellano E, González-Cózatl FX, Castro-Arellano I, León-Paniagua L, Rogers DS (2022) Molecular systematics of the Reithrodontomys tenuirostris group (Rodentia: Cricetidae) highlighting the Reithrodontomys microdon species complex. J Mammal 103:29–44. https://doi.org/10.1093/jmammal/gyab133
Melosik I, Ziomek J, Winnicka K, Reiners TE, Banaszek A, Mammen K, Mammen U, Marciszak A (2017) The genetic characterization of an isolated remnant population of an endangered rodent (Cricetus cricetus L.) using comparative data: implications for conservation. Conserv Genet 18:759–775. https://doi.org/10.1007/s10592-017-0925-y
Millán-Aguilar O, Manzano-Sarabia M, Nettel-Hernanz A, Dodd RS, Hurtado-Oliva MA, Velázquez-Velázquez E (2016) Genetic diversity of the black mangrove Avicennia germinans (L.) Stearn in northwestern Mexico. Forests 7:1–12. https://doi.org/10.3390/f7090197
Mitrovski P, Heinze DA, Broome L, Hoffmann AA, Weeks AR (2007) High levels of variation despite genetic fragmentation in populations of the endangered mountain pygmy-possum, Burramys parvus, in alpine Australia. Mol Ecol 16:75–87. https://doi.org/10.1111/j.1365-294X.2006.03125.x
Mohammadi A, Almasieh K, Clevenger AP, Fatemizadeh F, Rezaei A, Jowkar H, Kaboli M (2018) Road expansion: a challenge to conservation of mammals, with particular emphasis on the endangered Asiatic cheetah in Iran. J Nat Conserv 43:8–18. https://doi.org/10.1016/j.jnc.2018.02.011
Moritz C (1995) Uses of molecular phylogenies for conservation. Philos Trans R Soc B Biol Sci 349:113–118. https://doi.org/10.1098/rstb.1995.0097
Otto SP (2018) Adaptation, speciation and extinction in the Anthropocene. Proc R Soc B Biol Sci 285:20182047. https://doi.org/10.1098/rspb.2018.2047
Pardiñas U, Myers P, León-Paniagua L, Ordoñez Garza N, Cook J, Kryštufek B, Haslauer R, Bradley RD, Shenbrot G, Patton J (2017) Family Cricetidae (true hamsters, voles, lemmings and new world rats and mice). In: Wilson DE, Mittermeier RA, Lacher TE (eds) Handbook of the mammals of the world, vol 7. Rodents II. Lynx Edicions, Barcelona, pp 204–279
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237
Reid F, Vázquez E (2016) Reithrodontomys microdon. In: IUCN Red List Threat. Species 2016 e.T19412A22386904. https://doi.org/10.2305/IUCN.UK.2016-2.RLTS.T19412A22386904.en. Accessed 18 Feb 2021
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Romo-Vázquez E, León-Paniagua L, Sánchez O (2005) A new species of Habromys (Rodentia: Neotominae) from México. Proc Biol Soc Wash 118:605–618. https://doi.org/10.2988/0006-324X(2005)118[605:ANSOHR]2.0.CO;2
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Rousset F (2008) GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Ruiz-Jiménez CA, Téllez-Valdés O, Luna-Vega I (2012) Clasificación de los bosques mesófilos de montaña de México: afinidades de la flora. Rev Mex Biodivers 83:1110–1144. https://doi.org/10.7550/rmb.29383
Schupp EW (1993) Quantity, quality and the effectiveness of seed dispersal by animals. In Frugivory and seed dispersal: ecological and evolutionary aspects. Vegetatio 107/108:15–29
Statham MJ, Aylward CM, Barthman-Thompson L, Kierepka EM, Sacks BN (2022) Landscape genetics of an endangered salt marsh endemic: identifying population continuity and barriers to dispersal. Conserv Genet 23:759–771. https://doi.org/10.1007/s10592-022-01446-6
Steppan SJ, Schenk JJ (2017) Muroid rodent phylogenetics: 900-species tree reveals increasing diversification rates. PLoS ONE 12(8):e0183070. https://doi.org/10.1371/journal.pone.0183070
Taylor AC, Tyndale-Biscoe H, Lindenmayer DB (2007) Unexpected persistence on habitat islands: genetic signatures reveal dispersal of a eucalypt-dependent marsupial through a hostile pine matrix. Mol Ecol 16:2655–2666. https://doi.org/10.1111/j.1365-294X.2007.03331.x
Trizio I, Crestanello B, Galbusera P, Wauters LA, Tosi G, Matthysen E, Hauffe HC (2005) Geographical distance and physical barriers shape the genetic structure of Eurasian red squirrels (Sciurus vulgaris) in the Italian Alps. Mol Ecol 14:469–481. https://doi.org/10.1111/j.1365-294X.2005.02428.x
Turvey ST, Crees JJ (2019) Extinction in the Anthropocene. Curr Biol 29:R982–R986. https://doi.org/10.1016/j.cub.2019.07.040
Vander-Wall SB, Kuhn KM, Beck MJ (2005) Seed removal, seed predation and secondary dispersal. Ecology 86:801–806. https://doi.org/10.1097/EDE.0b013e3181
Vargas V, Valenzuela-Galván D, Alcalá RE (2012) Is genetic structure of the southern pygmy mouse Baiomys musculus (Cricetidae) related to human-induced spatial landscape heterogeneity in a tropical dry forest? Genetica 140:287–295. https://doi.org/10.1007/s10709-012-9681-6
Vega R, Vázquez-Domínguez E, Mejía-Puente A, Cuarón AD (2007) Unexpected high levels of genetic variability and the population structure of an island endemic rodent (Oryzomys couesi cozumelae). Biol Conserv 137:210–222. https://doi.org/10.1016/j.biocon.2007.02.007
Vega R, Vázquez-Domínguez E, White TA, Valenzuela-Galván D, Searle JB (2017) Population genomics applications for conservation: the case of the tropical dry forest dweller Peromyscus melanophrys. Conserv Genet 18:313–326. https://doi.org/10.1007/s10592-016-0907-5
Waples RS, Do C (2008) LDNE: A program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8:753–756. https://doi.org/10.1111/j.1755-0998.2007.02061.x
White TA, Perkins SE, Heckel G, Searle JB (2013) Adaptive evolution during an ongoing range expansion: the invasive bank vole (Myodes glareolus) in Ireland. Mol Ecol 22:2971–2985. https://doi.org/10.1111/mec.12343
Williams PA, Karl BJ, Bannister P, Lee WG (2000) Small mammals as potential seed dispersers in New Zealand. Austral Ecol 25:523–532. https://doi.org/10.1111/j.1442-9993.2000.tb00057.x
Acknowledgements
We thank Miguel Ángel Salas-Téllez, Andrea Cerón-Acosta, Héctor Ortega-Salas, Martín Y. Cabrerra-Garrido, Mónica Montes, and Alfredo Gutiérrez for their assistance during field work and to José Abraham Jacobo Flores, Mayra Toledo, and Rosita Toledo for facilities during the field trip in Zacualpan. We are grateful to Ella Vázquez-Domínguez, Tania Garrido-Garduño, and Daniel Piñero for reviewing earlier versions of the manuscript.
Funding
This work was supported by grants of Consejo Nacional de Ciencia y Tecnología (CONACyT) 239482 and Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (UNAM-PAPIIT) 216713. There are no financial or commercial conflicts of interest. Pablo Colunga-Salas and Tania Marines-Macías were master’s students at the time of study, based at the Posgrado en Ciencias Biológicas, UNAM, and were supported by a fellowship from CONACyT and Apoyo a los estudios de Posgrado (UNAM-PAEP). Giovani Hernández-Canchola was supported by the Programa de Becas Posdoctorales de la Dirección General Asuntos del Personal (DGAPA, UNAM), the US National Science Foundation (DEB-1754393), and CONACyT.
Author information
Authors and Affiliations
Contributions
Conceptualization: Pablo Colunga-Salas, Tania Marines-Macías, Soraia Barbosa, Jeremy B. Searle, Livia León-Paniagua. Formal analysis: Pablo Colunga-Salas, Tania Marines-Macías, Giovani Hernández-Canchola. Funding acquisition: Giovani Hernández-Canchola, Livia León-Paniagua. Investigation: Pablo Colunga-Salas, Tania Marines-Macías, Cassandra Ramírez. Methodology: Pablo Colunga-Salas, Tania Marines-Macías, Giovani Hernández-Canchola, Soraia Barbosa, Cassandra Ramírez. Project administration: Livia León-Paniagua. Resources and supervision: Jeremy B. Searle, Livia León-Paniagua. Visualization: Pablo Colunga-Salas, Giovani Hernández-Canchola. Writing — original draft: Pablo Colunga-Salas, Tania Marines-Macías, Giovani Hernández-Canchola, Cassandra Ramírez. Writing — review and editing: Soraia Barbosa, Jeremy B. Searle, Livia León-Paniagua.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by: Jan M. Wójcik
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Colunga-Salas, P., Marines-Macías, T., Hernández-Canchola, G. et al. Population genomics reveals differences in genetic structure between two endemic arboreal rodent species in threatened cloud forest habitat. Mamm Res 68, 223–235 (2023). https://doi.org/10.1007/s13364-022-00667-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13364-022-00667-x