
Abstract
Race-nonspecific resistance is a key to sustainable management of pathogens in bread wheat (Triticum aestivum L.) breeding. It is more durable compared to race-specific immunity, conferred by the major genes (R), which are often overcome by pathogens. The accumulation of the genes, which provide the resistance to a specific race of a pathogen, together with the introduction of race-non-specific resistance genes is the most effective strategy aimed at preventing the breakdown of genetically conditioned immunity. PCR markers improved the productivity and accuracy of classical plant breeding by means of marker-assisted selection (MAS). Multiplexing assays provide increased throughput, reduced reaction cost, and conservation of limited sample material, which are beneficial for breeding purposes. Here, we described the process of customizing multiplex PCR assay for the simultaneous identification of the major leaf rust resistance genes Lr19, Lr24, Lr26, and Lr38, as well as the slow rusting, race-nonspecific resistance genes: Lr34 and Lr68, in thirteen combinations. The adaptation of PCR markers for multiplex assays relied on: (1) selection of primers with an appropriate length; (2) selection of common annealing/extension temperature for given primers; and (3) PCR mixture modifications consisting of increased concentration of primers for the scanty band signals or decreased concentration of primers for the strong bands. These multiplex PCR protocols can be integrated into a marker-assisted selection of the leaf rust-resistant wheat genotypes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Leaf rust, caused by the causal fungus, Puccinia triticina Eriks. (syn. P. recondita Rob. ex Desmaz. f. sp. tritici), is one of the most common and damaging diseases of wheat worldwide (Huerta-Espino et al. 2011). Under optimal environmental conditions, the infection can significantly reduce the kernel weight and the number of kernels per ear, which reflects in the yield reduction, even up to 70% (Kolmer 1996; Chen et al. 2013).
More than 80 Lr (leaf rust) genes, 83 Yr (yellow rust) genes, and 63 Sr (stem rust) genes have been identified and referred in the Catalogue of Gene Symbols for Wheat (McIntosch 2019). Most of them belong to the group of major resistance genes (R genes) (Kolmer et al. 2008a, b). Several effective Lr resistance genes were successfully introduced into wheat from related species, such as: Thinopyrum elongatum (syn. Agropyron elongatum), Th. intermedium and Secale cereale (Cai et al. 2001, Zhang et al. 2010, Salina et al. 2015). The Lr19 gene was transferred into the wheat genetic background from Th. elongatum and affects the plant hypersensitivity response and increases the grain yield (Gupta et al. 2006). Th. elongatum is also a source of the Lr24 gene, which provides leaf resistance at seedling and adult plant stages (Rai et al. 2021; Schachermayr et al. 1995). Another Thinopyrum species, Th. intermedium, is a source of Lr38 gene, which ensures a stable resistance of seedlings and adult plants to many isolates of P. triticina, which appeared in Europe. Another wheat relative, rye (Secele cereale L.), is a donor of the Lr26 gene, which is present in many varieties of wheat carrying 1RS.1BL chromosome translocation (Mesterházy et al. 2000; Mebrate et al. 2008; Salina et al. 2015).
Another type of resistance genes, called race-nonspecific or horizontal resistance genes, provide the durable resistance against all races of different pathogens (Ellis et al. 2014). In case of leaf rust, this type of resistance is manifested by the slow progression of the infection (Caldwell et al. 1968). So far, eight “slow rusting” genes have been identified in the wheat genome: Lr34 (= Yr18/Sr57/Pm38), Lr46 (= Yr29/Sr58/Pm39), Lr67 (= Yr46/Sr55/Pm46), Lr68, Lr74, Lr75, Lr77, and Lr78 (Suenaga et al. 2003, Singh et al. 1998, Hiebert et al. 2010, Herrera-Foessel et al. 2012, McIntosh et al. 2015, Singla et al. 2017, Kolmer et al. 2018a, b). The Lr34 is the best known and characterized “slow rusting” loci (Dyck et al. 1987), which confers a partial resistance to leaf rust, as well as a moderate level of resistance to the stripe rust, caused by Puccinia striiformis (Yr18; McIntosh 1992, Singh 1992); powdery mildew, caused by Blumeria graminis (Pm38; Spielmeyer et al. 2005); stem rust, caused Puccinia graminis (Sr57; Dyck 1992); and barley yellow dwarf virus (Bdv1, Singh 1993). The presence of the Lr34 gene is also manifested by the appearance of leaf tip necrosis in certain environments, which can be used as a phenotypic assay for the presence of the gene (Dyck 1991; Singh 1992). The Lr34 gene encodes a pleiotropic ATP-binding cassette (ABC) transporter, of the ABCG subfamily (Krattinger et al. 2009). Resistant and susceptible haplotypes can be distinguished by three single nucleotide polymorphisms (SNP) (Lagudah et al. 2009). Other slow rusting genes, Lr46 and Lr68, were described in Pavon and Parula cultivars, respectively (Herrera-Foessel et al. 2012). Both genes showed a smaller effect than Lr34, but the combined effect of Lr34, Lr46, and Lr68 can assure near immunity (Martinez et al. 2001). The effects of these genes when appearing alone, are moderate; however, they can be used as backbone genes in combinations and interactions with other major genes, resulting in high levels of durable resistance.
The selection of individuals is an important stage in wheat breeding. Currently, the classic selection is supported by the identification of molecular markers related to valuable traits. Although molecular techniques are highly specific, sensitive, and reliable, nevertheless they are expensive, laborious, and time-consuming. Multiplex polymerase chain reaction (multiplex PCR) since its first description in 1988 (Chamberlain et al. 1988) has been successfully utilized in many areas of DNA testing. It allows the simultaneous amplification of two or more loci in one reaction (Henegariu et al. 1997a), which reduces the time and costs of investigation; hence, it has been applied in marker-assisted selection in breeding programs.
The aim of this work was to develop the multiplex PCR protocols for the simultaneous identification of the major leaf rust resistance genes Lr19, Lr24, Lr26, and Lr38 together with the “slow rusting” genes Lr34 and Lr68 in various combinations, which can be used for the selection process in wheat breeding.
Materials and methods
Plant material and DNA isolation
The experimental material consisted of 8 accessions of spring wheat (Table 1), which were reported as sources of leaf rust resistance genes, including four near-isogenic lines of ‘Thatcher’ with single genes (Lr 24, Lr26, Lr34, Lr38); one spring wheat cv. ‘Chinese Spring’ which was used as negative control for Lr19, Lr24, Lr26, and Lr38 and one spring wheat cv. ‘Artigas’ representing negative control for Lr34 and Lr68. The plant material was obtained from the National Small Grains Germplasm Facility, National Small Grains Collection in Aberdeen, Idaho, USA. Seeds were plated for germination on Petri dishes. GeneMATRIX Plant & Fungi DNA Purification Kit (EURx Ltd., Poland) was used for DNA extraction from the leaf tissue of 10-day-old seedlings. Leaf tissue samples were finely ground in liquid nitrogen and the remaining tissue structures were subsequently solubilized by lysis in the presence of a special buffer, which preserves the integrity and stimulates quantitative recovery of all traces of DNA. Further, proteinase K was used to digest contaminating proteins. “Sol-P” buffer and ethanol were added to provide selective conditions for DNA binding during brief centrifugation, while contaminants pass through the resin in the spin column. Traces of contaminants remaining on the resin were removed in two wash steps. The final DNA concentration after diluting the samples with Tris buffer (EURx Ltd., Poland) was 50 ng μl–1.
Molecular markers and PCR reactions
In order to develop freely available multiplex PCR protocols, we have used molecular markers, as well as primer sets, which are accessible and were already published. The molecular markers sequences, sizes of amplified products, and references are presented in Table 2.
The initial PCR reaction was performed using FastGene® OptimaHotStart ReadyMix (NIPPON, Germany) according to the manufacturer’s protocol. PCR mixture consisted of 1.25 µg of template DNA; PCR-grade H2O, 1 × FastGene® OptimaHotStart ReadyMix (NIPPON, Germany), and 1-µM primers (Sigma-Aldrich, USA). PCR was performed using a Labcycler thermal cycler (SensoQuest GmbH). The PCR reaction was performed with the following cycling protocol: (a) initial denaturation of 3 min at 95 °C; (b) 35 cycles of denaturation of 30 s at 95 °C, primer annealing of 30 s at 5 °C lower than the calculated melting temperature (Tm) of the given primer set; and elongation step of 1 min at 72 °C; and (c) a final elongation step of 7 min at 72 °C. The initial conditions of the PCR reaction were later developed by modifications of (1) primers’ concentrations and (2) annealing temperatures, which were crucial to design functional multiplex PCR protocols.
The PCR amplification products were electrophoresed in 2% agarose (Bioshop, Canada Inc.) gel in 1 × TBE buffer (Bioshop, Canada Inc.) stained with 4 µl of Midori Green Advanced DNA Stain (NipponGenetics Europe, Germany) per 100 ml and photographed under UV light in a Molecular Imager Gel Doc™ XR UV system with the Biorad Bio Image™ Software.
Results
The optimization of the PCR method for multiplex PCR reaction design consisted of three phases: (1) selection of effective, available molecular markers for Lr19, Lr24, Lr26, Lr34, Lr38, and Lr68, whose amplicons’ sizes allow to distinguish particular alleles in the multiplex assay; (2) adjustment of common annealing temperatures using gradient PCR; and (3) primers’ concentrations manipulation to obtain easy-to-interpret banding pattern on the gel.
Selection of markers
All markers available in the literature were analyzed. Multiplex PCR assays involve a large number of primers; hence it is required that the designed primer should be of appropriate length. Here, primers of short length, in the range of 18–22 bases were used. Primer sets, that yield amplicons with the appropriate product sizes, which can be easily distinguished using standard agarose gel electrophoresis have been selected (Table 2).
Annealing temperature
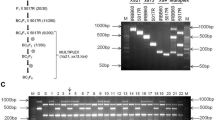
The next step was to analyze the interactions between primer sets in the course of multiplex PCR reaction. Primers with similar melting temperatures (Tm), preferably between 55 and 65 °C were used (Table 2). Hence, further approaches were conducted in order to test the suitable annealing/extension temperatures by thermal gradient (Fig. 1). A Tm variation of between 5 and 10 °C was acceptable for primers used in a pool; hence, we have used the lowest annealing temperature (55 °C) for all combinations (Table. 3). However, lower annealing temperatures yielded some unspecific products (Fig. 1), which were eliminated by the modification of primers concentrations in the following step. The common temperature for annealing allowed to multiplex thirteen marker combinations (Table 3), which could be used according to the need of the experiment or breeding program.

Gradient of annealing temperature (55–60 °C) for csGs marker linked to Lr68 gene. Arrows indicate the unspecific products, which were eliminated by the modification of primers concentrations. GeneRuler 50-bp DNA Ladder was used as DNA standard/ladder used to compare the various banding patterns
Specificity
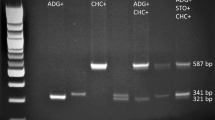
It is important to consider the specificity of primers to the target sequences, while preparing a multiplex assay, especially since competition exists when multiple target sequences are in a single reaction vessel. At first, the multiplex reactions were performed by adding primers in equimolar concentrations. Initially, equimolar primer concentrations of 1 μM each were used in the multiplex PCR. The results suggested that individual primer concentrations need further modifications. In the case of Lr24 loci, the multiplex assay adjustment process was initiated with the Xbarc71 marker that yields 103- or 85-bp products, linked with resistant or susceptible alleles, respectively (Table 2). However, the Xbarc71 primers together with primers of other markers provided false amplicons or primer-amplicon interactions (Fig. 2a) or false amplification due to primer dimers (Fig. 2b). All approaches failed, so another marker, Sr24#12, was selected for Lr24 loci identification. However, PCRs with more than two sets of primers resulted in uneven amplification, with some barely visible products or even absent (Fig. 3). To overcome these obstacles, we performed a set of PCR experiments with different proportions of primers in the reaction. The final concentration of the primers ranged between 0.4 and 1.5 μM. Generally, the PCR mixture modification consisted of increased concentration of primers for the scanty band signals or decreased concentration of primers for the strong bands (Fig. 3a, b; Table 3). The abovementioned alternations of PCR protocol allowed to perform easy-to-interpret results of multiplex PCR reactions for thirteen combinations of primers (Table 3, Figs. 4, 5, and 6).

Electropherogram of multiplex PCR reaction for Xbarc71 (Lr24) and csLv34 (Lr34) markers showing a false amplicons or primer-amplicon interactions and b false amplification due to primer dimers. GeneRuler 50 bp DNA Ladder was used as DNA standard/ladder used to compare the various banding patterns

Modification of the concentration of primers for Xbarc71 (Lr24) and csLv34 (Lr34). Initial primers concentrations of a 1 μM each and b 0.8 μM each

Electropherogram showing the presence of markers: Xwmc221 (for Lr19), Sr24#12 (Lr24), csLv34 (Lr34), and csGs (Lr68) in wheat varieties in various combinations (C1–C6)

Electropherogram showing the presence of markers: Xwmc221 (for Lr19), Sr24#12 (Lr24), P6MI2 (Lr26), and Xwmc773 (Lr38) in wheat varieties in various combinations (C7–C12)

Electropherogram showing the presence of markers: Xwmc221 (for Lr19), Sr24#12 (Lr24), P6MI2 (Lr26), csLv34 (Lr34), and csGs (Lr68) in wheat varieties in C13 combination
Discussion
In this study, we have demonstrated a set of thirteen multiplex PCR marker combinations, which can be deployed in the process of marker-assisted selection. We have developed the multiplex PCR protocols for the most effective major genes (Lr19, Lr24, Lr26, and Lr38) and slow rusting genes, including Lr34 and Lr68, which are securing the durable resistance of wheat.
Optimalization of the multiplex PCR is a challenging procedure, which is based mainly on a trial-and-error approach. In the literature, only few publications discuss the process of multiplex PCR protocol development (Chamberlain and Chamberlain 1994; Edwards and Gibbs 1994; Henegariu et al., 2018).
An initial solution to difficulties encountered in the development of multiplex PCR has been the use of hot start PCR (Chou et al. 1992). This type of enzyme eliminates nonspecific reactions (particularly production of primer dimers) caused by primer annealing at low temperatures (4 to 25 °C) before commencement of thermocycling (Kebelmann-Betzing et al., 1998). Hence, in our study, we have used the OptimaHotStart polymerase, which is activated only if the reaction mixture is heated at approximately 95 °C for 10 min (the first denaturation step).
First cycles have a substantial effect on the overall sensitivity and specificity of PCR. The success of specific amplification relies on the primers annealing to their target and the rate at which annealed primers are extended along the desired sequence. Optimal annealing depends on primer length and GC content and their concentrations, as well as annealing temperature (Chamberlain & Chamberlain 1994). Thus, the majority of modifications to improve PCR performance have been directed towards the factors affecting annealing and/or extension rates. The optimization of multiplex PCRs can raise several obstacles including poor sensitivity or specificity and/or preferential amplification of certain specific targets (Polz and Cavanaugh 1998). The presence of more than one primer pair in the multiplex PCR increases the chance of obtaining spurious amplification products, primarily because of the formation of primer dimers (Brownie et al. 1997). In the case of our experiment with Lr24 and Lr34 markers, these nonspecific products were amplified more efficiently than the desired target, consuming reaction components and producing impaired rates of annealing and extension. Thus, the optimization of multiplex PCR should aim to minimize or reduce such nonspecific interactions. This may be achieved through the utilization of primers with nearly identical optimum annealing temperatures (primer length of 18 to 30 bp or more and a GC content of 35 to 60% may prove satisfactory) and should not display significant homology either internally or to one another (Henegariu et al., 2018). Modifications including primers concentration as well as other PCR components such as PCR, dNTPs, and enzyme concentrations in multiplex PCR over those reported for most uniplex PCRs usually result in modest improvement in the specificity of the assay. Increasing the concentration of these factors may increase the likelihood of mis-priming with subsequent production of spurious nonspecific amplification products. However, optimization of these components in multiplex PCRs that are designed for simultaneous amplification of multiple targets may prove beneficial. For example, in the multiplex PCR for simultaneous detection of wheat and soybean, different ratios of primers concentrations were used for analysis of 21 different commercial food products (Shin et al. 2021).
Multiplex PCR become widely adopted within the plant breeding industry for high-throughput genotyping in a variety of applications, such as germplasm characterization and MAS (Yap et al. 2016), identification of genetically modified organisms (Bak and Emerson 2019) and pathology testing (Otti et al. 2016). It is a quick method that also allows to lower research costs and shortens the duration of the experiment. For example, Froidmont (1998) used multiplex PCR to identify 1BL/1RS translocation in wheat, together with the screening of resistance loci for yellow rust (Yr9), stem rust (Sr31), leaf rust (Lr26), and powdery mildew (Pm8). Moreover, Sumiková and Hanzalová (2010) studied rust leaf resistance genes Lr26 and Lr37 and stated that the multiplex PCR method can be a breakthrough tool in identifying varieties resistant to the disease. Tomkowiak et al. (2019) developed a multiplex PCR protocol to accelerate the identification of efficient major leaf rust resistance genes: Lr11, L13, Lr16, and Lr26 using the following molecular SSR markers: Xwmc24, Xwmc261, Xgwm630, Xwmc764, and P6M12, respectively. Multiplex PCR assays were also developed for simultaneous screening of slow rusting genes. Skowrońska et al. (2019) published a protocol for joint identification of Lr34 and Lr46, and later, they improved the protocol by adding a molecular marker linked to another slow rusting gene, Lr68 (Skowrońska et al. 2020). What is more, Lata et al. (2021) optimized a multiplex polymerase chain reaction (PCR) for simultaneous detection of two important leaf rust resistance genes: seedling resistance gene Lr24 and slow rusting gene Lr68.
In conclusion, in this study, we optimized and developed 13 combinations of multiplex PCR conditions for simultaneous identification of markers linked to both effective race-specific (Lr19, Lr24, Lr26, and Lr38) and durable, non-specific leaf resistance genes (Lr34 and Lr68). These protocols can be used to accelerate the marker-assisted resistance breeding of common wheat, which meet the recent expectations of the breeders.
References
Anikster YE, Bushnell WR, Roelfs AP, Eilam T, Manisterski J (1997) Puccinia recondita causing leaf rust on cultivated wheats, wild wheats, and rye. Can J Bot 75(12):2082–2096. https://doi.org/10.1139/b97-919
Bak A, Emerson JB (2019) Multiplex quantitative PCR for single-reaction genetically modified (GM) plant detection and identification of false-positive GM plants linked to Cauliflower mosaic virus (CaMV) infection. BMC Biotechnol 19:73. https://doi.org/10.1186/s12896-019-0571-1
Bhavani S, Singh RP, Hodson DP, Huerta-Espino J, Randhawa MS (2022) Wheat rusts: current status, prospects of genetic control and integrated approaches to enhance resistance durability. In Wheat Improvement (pp. 125–141). Springer, Cham.
Bolton MD, Kolmer JA, Garvin DF (2008) Wheat leaf rust caused by Puccinia triticina. Mol Plant Pathol 9(5):563–575. https://doi.org/10.1111/j.1364-3703.2008.00487.x
Brownie J, Shawcross S, Theaker J, Whitcombe D, Ferrie R, Newton C, Little S (1997) The elimination of primer-dimer accumulation in PCR. Nucleic Acids Res 25(16):3235–3241. https://doi.org/10.1093/NAR/25.16.3235
Büschges R, Hollricher K, Panstruga R, Simons G, Wolter M, Frijters A, Schulze-Lefert P (1997) The barley Mlo gene: a novel control element of plant pathogen resistance. Cell 88(5):695–705. https://doi.org/10.1016/S0092-8674(00)81912-1
Cai X, Jones SS, Murray TD (2001) Molecular cytogenetic characterization of Thinopyrum genomes conferring perennial growth habit in wheat-Thinopyrum amphiploids. Plant Breed 120:21–26. https://doi.org/10.1046/j.1439-0523.2001.00560.x
Caldwell RM (1968) Breeding for general and/or specific plant disease resistance. Third Int. Wheat Genetics Symposium. pp. 263–272
Chamberlain JS, Gibbs RA, Rainer JE, Nguyen PN, Thomas C (1988) Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res 16(23):11141–11156. https://doi.org/10.1093/nar/16.23.11141
Chamberlain, J. S., & Chamberlain, J. R. (1994). Optimization of multiplex PCRs. In: The polymerase chain reaction. Mullis, K.B., Ferré, F., Gibbs, R.A. (eds). Birkhäuser, Boston, MA. pp.38–46. https://doi.org/10.1007/978-1-4612-0257-8_3
Chen W, Liu T, Gao L (2013) Suppression of stripe rust and leaf rust resistances in interspecific crosses of wheat. Euphytica 192(3):339–346. https://doi.org/10.1007/s10681-012-0854-2
Chou Q, Russell M, Birch DE, Raymond J, Bloch W (1992) Prevention of pre-PCR mis-priming and primer dimerization improves low-copy-number amplifications. Nucleic Acids Res 20(7):1717–1723. https://doi.org/10.1093/NAR/20.7.1717
De Froidmont D (1998) A co-dominant marker for the 1BL/1RS wheat-rye translocation via multiplex PCR. J Cereal Sci 27(3):229–232. https://doi.org/10.1006/jcrs.1998.0194
Dodds PN, Rathjen JP (2010) Plant immunity: towards an integrated view of plant–pathogen interactions. Nat Rev Genet 11(8):539–548. https://doi.org/10.1038/nrg2812
Dyck PL (1987) The association of a gene for leaf rust resistance with the chromosome 7D suppressor of stem rust resistance in common wheat. Genome 29:467–469. https://doi.org/10.1139/g87-081
Dyck PL (1991) Genetics of adult plant leaf rust resistance and leaf tip necrosis in wheat. Crop Sci 32:874–878. https://doi.org/10.1007/BF00023766
Dyck PL (1992) Transfer of a gene for stem rust resistance from Triticum araraticum to hexaploid wheat. Genome 35(5):788–792. https://doi.org/10.1139/g92-120
Edwards MC, Gibbs RA (1994) Multiplex PCR: advantages, development, and applications. Genome Res 3(4):S65–S75. https://doi.org/10.1101/GR.3.4.S65
Ellis JG, Lagudah ES, Spielmeyer W, Dodds PN (2014) The past, present and future of breeding rust resistant wheat. Front Plant Sci 5:641. https://doi.org/10.3389/fpls.2014.00641
Erenstein O, Jaleta M, Mottaleb KA, Sonder K, Donovan J, Braun HJ. (2022). Global trends in wheat production, consumption and trade. In: Reynolds, M.P., Braun, HJ. (eds) Wheat improvement. Springer, Cham. https://doi.org/10.1007/978-3-030-90673-3_4
Ezzahiri B, Roelfs AP (1989) Inheritance and expression of adult plant resistance to leaf rust in era wheat. Plant Dis 73:549–551. https://doi.org/10.1094/PD-73-0549
Fu D, Uauy C, Distelfeld A, Blechl A, Epstein L, Chen X, Dubcovsky J (2009) A kinase-START gene confers temperature-dependent resistance to wheat stripe rust. Science 323(5919):1357–1360. https://doi.org/10.1126/science.11662
Fukuoka S, Saka N, Koga H, Ono K, Shimizu T, Ebana K, Yano M (2009) Loss of function of a proline-containing protein confers durable disease resistance in rice. Science 325(5943):998–1001. https://doi.org/10.1126/science.1175550
Gill BS, Friebe B (1998) Plant cytogenetics at the dawn of the 21st century. Curr Opin Plant Biol 1(2):109–115. https://doi.org/10.1016/S1369-5266(98)80011-3
Gupta SK, Charpe A, Prabhu KV, Haque QMR (2006) Identification and validation of molecular markers linked to the leaf rust resistance gene Lr19 in wheat. Theor Appl Genet 113(6):1027–1036. https://doi.org/10.1007/s00122-006-0362-7
Henegariu O, Heerema NA, Dlouhy SR, Vance GH, Vogt PH (1997) Multiplex PCR: critical parameters and step-by-step protocol. Biotechniques 23(3):504–511. https://doi.org/10.2144/97233rr01
Herrera-Foessel SA, Singh RP, Huerta-Espino J, Rosewarne GM, Periyannan SK, Viccars L, Lagudah ES (2012) Lr68: a new gene conferring slow rusting resistance to leaf rust in wheat. Theor Appl Genet 124(8):1475–1486. https://doi.org/10.1007/s00122-012-1802-1
Hiebert CW, Thomas JB, McCallum BD, Humphreys DG, DePauw RM, Hayden MJ, Spielmeyer W (2010) An introgression on wheat chromosome 4DL in RL6077 (Thatcher* 6/PI 250413) confers adult plant resistance to stripe rust and leaf rust (Lr67). Theor Appl Genet 121(6):1083–1091. https://doi.org/10.1007/s00122-010-1373-y
Huerta-Espino J, Singh RP, German S, McCallum BD, Park RF, Chen WQ, Bhardwaj SC, Goyeau H (2011) Global status of wheat leaf rust caused by Puccinia triticina. Euphytica 179(1):143–160. https://doi.org/10.1007/s10681-011-0361-x
Huerta-Espino J, Singh R, Crespo-Herrera LA, Villaseñor-Mir HE, Rodriguez-Garcia MF, Dreisigacker S, Barcenas-Santana D, Lagudah E (2020) Adult plant slow rusting genes confer high levels of resistance to rusts in bread wheat cultivars from Mexico. Front Plant Sci 11:824. https://doi.org/10.3389/fpls.2020.00824
Kilpatrick RA (1975) New wheat cultivars and longevity of rust resistance, 1971–75. United States Department of Agriculture Economic Research Service NE. 64:1–20
Kloppers FJ, Pretorius ZA (1997) Effects of combinations amongst genes Lr13, Lr34 and Lr37 on components of resistance in wheat to leaf rust. Plant Pathol 46:737–750. https://doi.org/10.1046/j.1365-3059.1997.d01-58.x
Kolmer JA (1996) Genetics of resistance to wheat leaf rust. Annu Rev Phytopathol 34(1):435–455. https://doi.org/10.1146/annurev.phyto.34.1.435
Kolmer JA, Singh RP, Garvin DF, Viccars L, William HM, Huerta-Espino J, Lagudah ES (2008a) Analysis of the Lr34/Yr18 rust resistance region in wheat germplasm. Crop Sci 48(5):1841–1852. https://doi.org/10.2135/cropsci2007.08.0474
Kolmer JA, Singh RP, Garvin DF, Viccars L, William HM, Huerta-Espino J, Ogbonnaya FC, Raman H, Orford S, Bariana HS, Lagudah ES (2008b) Analysis of the Lr34/Yr18 rust resistance region in wheat germplasm. Crop Sci 48:1841–1852. https://doi.org/10.2135/cropsci2007.08.0474
Kolmer JA, Su Z, Bernardo A, Bai G, Chao S (2018) Mapping and characterization of the new adult plant leaf rust resistance gene Lr77 derived from Santa Fe winter wheat. Theor Appl Genet 131:1553–1560. https://doi.org/10.1007/s00122-018-3097-3
Krattinger SG, Lagudah ES, Spielmeyer W, Singh RP, Huerta-Espino J, McFadden H, Keller B (2009) A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science 323(5919):1360–1363. https://doi.org/10.1126/science.1166453
Krattinger SG, Sucher J, Selter LL, Chauhan H, Zhou B, Tang M, Keller B (2016) The wheat durable, multipathogen resistance gene Lr34 confers partial blast resistance in rice. Plant Biotechnol J 14(5):1261–1268. https://doi.org/10.1111/pbi.12491
Lagudah ES, Krattinger SG, Herrera-Foessel S, Singh RP, Huerta-Espino J, Spielmeyer W, Keller B (2009) Gene-specific markers for the wheat gene Lr34/Yr18/Pm38 which confers resistance to multiple fungal pathogens. Theor Appl Genet 119(5):889–898. https://doi.org/10.1007/s00122-009-1097-z
Lata C, Kumar A, Gangwar OP, Prasad P, Adhikari S, Kumar S, Kulshreshtha N, Bhardwaj SC (2021) Multiplex PCR assay for the detection of Lr24 and Lr68 in salt tolerant wheat genotypes. Cereal Res Commun. https://doi.org/10.1007/s42976-021-00218-1
Ledesma-Ramírez L, Solis-Moya E, Ramírez-Pimentel JG, Dreisigacker S, Huerta-Espino J, Aguirre-Mancilla CL, Mariscal-Amaro LA (2018) Relationship between the number of partial resistance genes and the response to leaf rust in wheat genotypes. Chil J Agric Res 78(3):400–408. https://doi.org/10.4067/S0718-58392018000300400
Mago R, Bariana HS, Dundas IS, Spielmeyer W, Lawrence GJ, Pryor AJ, Ellis JG (2005a) Development of PCR markers for the selection of wheat stem rust resistance genes Sr24 and Sr26 in diverse wheat germplasm. Theor Appl Genet 111(3):496–504. https://doi.org/10.1007/s00122-005-2039-z
Mago R, Miah H, Lawrence GJ, Wellings CR, Spielmeyer W, Bariana HS, Ellis JG (2005b) High-resolution mapping and mutation analysis separate the rust resistance genes Sr31, Lr26 and Yr9 on the short arm of rye chromosome 1. Theor Appl Genet 112(1):41–50. https://doi.org/10.1007/s00122-005-0098-9
Martinez F, Niks RE, Singh RP, Rubiales D (2001) Characterization of Lr46, a gene conferring partial resistance to wheat leaf rust. Hereditas 135(2–3):111–114. https://doi.org/10.1111/j.1601-5223.2001.00111.x
McCallum BD, Hiebert CW (2022) Interactions between Lr67 or Lr34 and other leaf rust resistance genes in wheat (Triticum aestivum). Front Plant Sci 13:871970. https://doi.org/10.3389/fpls.2022.871970
McCallum BD, Hiebert CW, Cloutier S, Bakkeren G, Rosa SB, Humphreys DG, Wang X (2016) A review of wheat leaf rust research and the development of resistant cultivars in Canada. Can J Plant Pathol 38(1):1–18. https://doi.org/10.1080/07060661.2016.1145598
McCallum BD, Reimer E, McNabb W, Foster A, Rosa S, Xue A (2021) Physiologic specialization of Puccinia triticina, the causal agent of wheat leaf rust, in Canada in 2015–2019. Can J Plant Pathol 43:S333–S346. https://doi.org/10.1080/07060661.2021.1888156
McIntosh RA (1992) Pre-emptive breeding to control wheat rusts. Euphytica 63(1):3–113. https://doi.org/10.1007/978-94-017-0954-5_9
McIntosh RA, Dubcovsky J, Rogers WJ, Morris C, Appels R, Xia XC (2015) Catalogue of gene symbols for wheat: 2015–2016 supplement. https://shigen.nig.ac.jp/wheat/komugi/genes/macgene/supplement2015.pdf.
McIntosh R.A, J. Dubcovsky W.J. Rogers C. Morris i Xia X. C. (2019). Catalogue of gene symbols for wheat: 2019. https://shigen.nig.ac.jp/wheat/komugi/genes/symbolClassList.jsp.
Mebrate SA, Oerke EC, Dehne HW, Pillen K (2008) Mapping of the leaf rust resistance gene Lr38 on wheat chromosome arm 6DL using SSR markers. Euphytica 162(3):457–466. https://doi.org/10.1007/s10681-007-9615-z
Mesterházy Á, Bartoš P, Goyeau H, Niks R, Csösz M (2000) European virulence survey for leaf rust in wheat. Agronomie 20(7):793–804. https://doi.org/10.1051/agro:2000104
Moskal K, Kowalik S, Podyma W, Łapiński B, Boczkowska M (2021) The pros and cons of rye chromatin introgression into wheat genome. Agronomy 11(3):456. https://doi.org/10.3390/agronomy11030456
Otti G, Bouvaine S, Kimata B, Mkamillo G, Kumar PL, Tomlins K, Maruthi MN (2016) High-throughput multiplex real-time PCR assay for the simultaneous quantification of DNA and RNA viruses infecting cassava plants. J Appl Microbiol 120(5):1346–1356. https://doi.org/10.1111/jam.13043
Polz MF, Cavanaugh CM (1998) Bias in template-to-product ratios in multitemplate PCR. Appl Environ Microbiol 64(10):3724–3730
Pretorius ZA, Singh RP, Wagoire WW, Payne TS (2000) Detection of virulence to wheat stem rust resistance gene Sr31 in Puccinia graminis. F. sp. tritici in Uganda. Plant Dis 84(2):203–203
Rai A, Ahlawat AK, Shukla RB, Jain N, Kumar RR, Mahendru-Singh A (2021) Quality evaluation of near-isogenic line of the wheat variety HD2733 carrying the Lr24/Sr24 genomic region. 3 Biotech 11(3):1–10. https://doi.org/10.1007/s13205-021-02679-x
Salina EA, Adonina IG, Badaeva ED, Kroupin PYu, Stasyuk AI, Leonova IN, Shishkina AA, Divashuk MG, Starikova EV, Khuat TML, Syukov VV, Karlov GI (2015) A Thinopyrum intermedium chromosome in bread wheat cultivars as a source of genes conferring resistance to fungal diseases. Euphytica 204:91–101. https://doi.org/10.1007/s10681-014-1344-5
Schachermayr GM, Messmer MM, Feuillet C, Winzeler H, Winzeler M, Keller B (1995) Identification of molecular markers linked to the Agropyron elongatum-derived leaf rust resistance gene Lr24 in wheat. Theor Appl Genet 90(7):982–990. https://doi.org/10.1007/BF00222911
Schneider A, Molnár I, Molnár-Láng M (2008) Utilisation of Aegilops (goatgrass) species to widen the genetic diversity of cultivated wheat. Euphytica 163(1):1–19. https://doi.org/10.1007/s10681-007-9624-y
Shin J, Kim MJ, Kim HY (2021) Development of triplex PCR for simultaneous detection of soybean and wheat. Food Science and Biotechnology 30(6):801. https://doi.org/10.1007/S10068-021-00926-8
Singh RP (1992) Association between gene Lr34 for leaf rust resistance and leaf tip necrosis in wheat. CropSci 32(4):874–878. https://doi.org/10.2135/cropsci1992.0011183X003200040008x
Singh RP (1993) Genetic association of gene Bdv1 for tolerance to barley yellow dwarf virus with genes Lr34 and Yr18 for adult plant resistance to rusts in bread wheat. Plant Dis 77(11):1103–1106. https://doi.org/10.1094/PD-77-1103
Singh RP, Mujeeb-Kazi A, Huerta-Espino J (1998) Lr46: a gene conferring slow-rusting resistance to leaf rust in wheat. Phytopathology 88(9):890–894. https://doi.org/10.1094/PHYTO.1998.88.9.890
Singla J, Lüthi L, Wicker T, Bansal U, Krattinger SG, Keller B (2017) Characterization of Lr75: a partial, broad-spectrum leaf rust resistance gene in wheat. Theor Appl Genet 130(1):1–12. https://doi.org/10.1007/s00122-016-2784-1
Skowrońska R, Kwiatek M, Tomkowiak A, Nawracała J (2019) Development of multiplex PCR to detect slow rust resistance genes Lr34 and Lr46 in wheat. J Appl Genet 60(3):301–304. https://doi.org/10.1007/s13353-019-00520-z
Skowrońska R, Tomkowiak A, Szwarc J, Nawracała J, Kwiatek M (2020) Multiplex PCR assay for simultaneous identification of slow rust resistance genes Lr34, Lr46, and Lr68 in wheat (Triticum aestivum L.). Journal of Plant Protection Research 60(4):388–398. https://doi.org/10.24425/jppr.2020.134914
Suenaga K, Singh RP, Huerta-Espino J, William HM (2003) Microsatellite markers for genes Lr34/Yr18 and other quantitative trait loci for leaf rust and stripe rust resistance in bread wheat. Phytopathology 93(7):881–890. https://doi.org/10.1094/PHYTO.2003.93.7.881
Sumikova T, Hanzalova A (2010) Multiplex PCR assay to detect rust resistance genes Lr26 and Lr37 in wheat. Czech J Genet Plant 46(2):85–89. https://doi.org/10.17221/32/2010-CJGPB
Tomkowiak A, Skowrońska R, Buda A, Kurasiak-Popowska D, Nawracała J, Kowalczewski PŁ, Pluta M, Radzikowska D (2019) Identification of leaf rust resistance genes in selected wheat cultivars and development of multiplex PCR. Open Life Sciences 14(1):327–334. https://doi.org/10.1515/biol-2019-0036
Villareal RL, Rajaram S, Mujeeb-Kazi A, Del Toro E (1991) The effect of chromosome 1B/1R translocation on the yield potential of certain spring wheats (Triticum aestivum L.). Plant Breed 106(1):77–81. https://doi.org/10.1111/j.1439-0523.1991.tb00482.x
Wilson RA, Talbot NJ (2009) Fungal physiology–a future perspective. Microbiology 155(12):3810–3815. https://doi.org/10.1099/mic.0.035436-0
Yap R, Hsu YC, Wu YP, Lin YR, Kuo CW (2016) Multiplex PCR genotyping for five bacterial blight resistance genes applied to marker-assisted selection in rice (Oryza sativa). Plant Breed 135(3):309–317. https://doi.org/10.1111/pbr.12368
Acknowledgements
The authors would like to acknowledge and thank Harrold Bockelman at the USDA/ARS Small Grains Laboratory, Aberdeen (ID, USA) for providing the seeds samples.
Funding
This research is financed by the framework of Ministry of Agriculture and Rural Development (Poland) program: “Biological Progress in Plant Production” in years 2021–2027, task no. 5: “A molecular analysis of an adult plant slow rusting genes conferring resistance to rusts caused by Puccinia sp.” (KS.zb.802.10.2021); Project leader: M.T. Kwiatek.
Author information
Authors and Affiliations
Contributions
MK initiated the project. AN made the experiments and analyses. RB AN and MK wrote the first draft and incorporated all inputs from co-authors. RB, JS, and AT supervised the analyses. MK revised the draft and made improved the manuscript. MK is the corresponding author and revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by Izabela Pawłowicz.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Noweiska, A., Bobrowska, R., Spychała, J. et al. Multiplex PCR assay for the simultaneous identification of race specific and non-specific leaf resistance genes in wheat (Triticum aestivum L.). J Appl Genetics 64, 55–64 (2023). https://doi.org/10.1007/s13353-022-00745-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-022-00745-5