
Abstract
This paper presents the analytical and numerical comparison of two methods of estimation of additive × additive × additive (aaa) interaction of QTL effects. The first method takes into account only the plant phenotype, while in the second we also included genotypic information from molecular marker observation. Analysis was made on 150 doubled haploid (DH) lines of barley derived from cross Steptoe × Morex and 145 DH lines from Harrington × TR306 cross. In total, 153 sets of observation was analyzed. In most cases, aaa interactions were found with an exert effect on QTL. Results also show that with molecular marker observations, obtained estimators had smaller absolute values than phenotypic estimators.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The analysis of inheritance of quantitative traits, due to their polygenic nature, requires the use of appropriate statistical and genetic methods. Among these methods, the most interesting are those that enable the determination of the mode of action of genes in the studied population.
The concept of genetic interactions is known for more than a hundred years (Bateson and Mendel 1902). Considering that a complex phenotype may be the effect of a combination of multiple loci, various statistical methods have been developed for identifying genetic epistasis effects (Chen et al. 2011). Most studies are focused on single locus analysis, which directly tests the association between individual genes and phenotypic variants. Pairwise interactions are often used in modern genetics (Brem et al. 2005; Jarvis and Cheverud 2011; Gaertner et al. 2012), but higher-order interactions are often neglected. This kind of more complex interaction requires complete, precise data to be successfully included, but this type of data was rarely available since more recent times (Carlborg et al. 2006; Cordell 2009). There is no denying that we do not fully understand all of the mechanics of heritability and the higher-order interactions may be the missing element of explaining the relationship between genotype and phenotype (Hartman et al. 2001; Manolio et al. 2009).
Quantitative traits are not only one of the most important in the viewpoint of breeding programs but also can be influenced by a multiplicity of polymorphic genes, environmental conditions, and genetic interactions, making them extremely difficult to fully understand (Members of the Complex Trait Consortium 2003; Mackay 2014).
The purpose of the research reported in this article is to compare two methods of estimation of the parameter connected with the additive × additive × additive (aaa) interaction gene effect: the phenotypic method and the genotypic method. The comparison was made by analytical methods and with analyses of data sets of barley doubled haploid lines. To our knowledge, this is the first report about aaa interaction.
Material and methods
If in the experiment we observed n homozygous (doubled haploid, DH; recombinant inbred, RI) plant lines, we get an n-vector of phenotypic mean observations y = [y1 y2 ... yn]’ and q n-vectors of marker genotype observations ml, l = 1, 2, …, q. The i-th element (i = 1, 2, …, n) of vector ml is equal − 1 or 1, depending on the parent’s genotype exhibited by the i-th line.
Estimation based on the phenotype
Estimation of the additive × additive × additive interaction of homozygous loci (three-way epistasis) effect aaa on the basis of phenotypic observations y requires identification of groups of extreme lines, i.e., lines with the minimal and maximal expression of the observed trait (Choo and Reinbergs 1982). The group of minimal lines consists of the lines which contain, theoretically, only alleles decreasing the value of the trait. Analogously, the group of maximal lines contains the lines which have only alleles increasing the trait value. In this paper, we identify the groups of extreme lines as minimal and maximal, respectively, lines of the empirical distribution of means. The total three-way epistasis interaction effect aaa can be estimated by the following formula:
where \({\overline{L} }_{\mathrm{min}}\) and \({\overline{L} }_{\mathrm{max}}\) denote the means for the groups of minimal and maximal lines, respectively, \(\overline{L }\) denotes the mean for all lines. The number of genes (number of effective factors) obtained on the basis of phenotypic observations only was calculated using the formula presented by Kaczmarek et al. (1988).
Estimation based on the genotypic observations
Estimation of aaa is based on the assumption that the genes responsible for the trait are closely linked to the observed molecular marker. By choosing from all observed markers p, we can explain the variability of the trait, and model observations for the lines as follows:
where 1 denotes the n-dimensional vector of ones, μ denotes the general mean, X denotes (n × p)-dimensional matrix of the form \({\varvec{X}}=\left[\begin{array}{cc}\begin{array}{cc}{{\varvec{m}}}_{{l}_{1}}& {{\varvec{m}}}_{{l}_{2}}\end{array}& \begin{array}{cc}\cdots & {{\varvec{m}}}_{{l}_{p}}\end{array}\end{array}\right]\), l1, l2, ..., lp \(\in\) {1, 2, ..., q}, β denotes the p-dimensional vector of unknown parameters of the form \({\varvec{\beta}}\boldsymbol{^{\prime}}=\left[\begin{array}{cc}\begin{array}{cc}{a}_{{l}_{1}}& {a}_{{l}_{2}}\end{array}& \begin{array}{cc}\cdots & {a}_{{l}_{p}}\end{array}\end{array}\right]\), Z denotes matrix which columns are products of some columns of matrix X, γ denotes the vector of unknown parameters of the form \({\varvec{\gamma}}\boldsymbol{^{\prime}}=\left[\begin{array}{cc}\begin{array}{cc}{aa}_{{l}_{1}{l}_{2}}& {aa}_{{{l}_{1}l}_{3}}\end{array}& \begin{array}{cc}\cdots & {aa}_{{{l}_{p-1}l}_{p}}\end{array}\end{array}\right]\), W denotes matrix which columns are three-way products of some columns of matrix X, δ denotes the vector of unknown parameters of the form \({\varvec{\delta}}\boldsymbol{^{\prime}}=\left[\begin{array}{cc}\begin{array}{cc}{aaa}_{{l}_{1}{l}_{2}{l}_{3}}& {aaa}_{{{l}_{1}{l}_{2}l}_{4}}\end{array}& \begin{array}{cc}\cdots & {aaa}_{{{{l}_{p-2}l}_{p-1}l}_{p}}\end{array}\end{array}\right]\), and e denotes the n-dimensional vector of random variables such that E(ei) = 0, Cov(ei, ej) = 0 for i ≠ j, i, j = 1, 2, …, n. The parameters \({a}_{{l}_{1}}\), \({a}_{{l}_{2}}\), ..., \({a}_{{l}_{p}}\) are the additive effects of the genes controlling the trait, parameters \({aa}_{{l}_{1}{l}_{2}}\), \({aa}_{{l}_{1}{l}_{3}}\), ..., \({aa}_{{l}_{p-1}{l}_{p}}\) are the additive × additive interaction effects and parameters \({aaa}_{{l}_{1}{l}_{2}{l}_{3}}\), \({aaa}_{{l}_{1}{l}_{2}{l}_{4}}\), ..., \({aaa}_{{{l}_{p-2}l}_{p-1}{l}_{p}}\) are the additive × additive × additive interaction effects. We assume that the epistatic and three-way epistatic interaction effects show only loci with significant additive gene action effects. This assumption significantly decreases the number of potential significant effects and causes the regression model to be more useful.
Denoting by \(\boldsymbol\alpha\boldsymbol'=\lbrack\mu\;\boldsymbol\beta\boldsymbol'\boldsymbol\;\boldsymbol\gamma\boldsymbol'\;\boldsymbol\delta\boldsymbol'\rbrack\) and \({\varvec{G}}=[\begin{array}{ccc}{\varvec1}& {\varvec{X}}& \begin{array}{cc}{\varvec{Z}}& {\varvec{W}}\end{array}\end{array}]\) we obtain the model
If G is of full rank, the estimate of \({\varvec{\upalpha}}\) is given by (Searle 1982)
The total three-way epistasis aaa effect of genes influencing the trait can be found as follows:
For the marker selection of model (2), we used a stepwise feature selection by Akaike information criteria (Akaike 1998). The procedure consisted of two steps: first, we divided markers into groups based on chromosomes they were located on and performed stepwise feature selection by AIC; after that, we combined the remaining markers into one group and we repeated selection as above. All of the remaining markers were combined into the final group and the last feature selection was performed on a model with additive × additive × additive interaction effect included. To counteract the multiple comparisons problem, we used the Bonferroni correction.
Examples
To compare the estimates of aaa obtained by different methods, the following data sets were used.
Example 1
The first set of data we used in our experiment comes from North American Barley Genome Mapping Project (NABGMP) and consists of 150 doubled haploid (DH) lines of barley tested in sixteen environments [Crookston, MN, 1992; Ithaca, NY, 1992; Guelph, Ontario, 1992; Pullman, WA, 1992; Brandon, Manitoba, 1992; Outlook, Saskatchewan, 1992; Goodale, Saskatchewan, 1992; Saskatoon, Saskatchewan, 1992; Tetonia, ID, 1992; Bozeman, MT (irrigated), 1992; Bozeman, MT (dryland), 1992; Aberdeen, ID, 1991; Klamath Falls, OR, 1991; Pullman, WA, 1991; Bozeman, MT (irrigated), 1991; and Bozeman, MT (dryland),1991]. Steptoe × Morex cross was developed by the Oregon State University Barley Breeding Program by crossing “Steptoe” and “Morex” barley varieties (Kleinhofs et al. 1993; Romagosa et al. 1996; http://wheat.pw.usda.gov/ggpages/SxM). The linkage map used consisted of 223 molecular markers, mostly RFLP, with mean distance between markers equal to 5.66 cM. Lines were analyzed for eight phenotypic traits (alpha amylase, AA; diastatic power, DP; grain protein, GP; grain yield, GY; height, H; heading date, HD; lodging, L; malt extract, ME; Hayes et al. 1993). Missing marker values were estimated with non-missing data of flanking markers (Martinez and Curnow 1994) and GP, L, and ME traits data were transformed by \(arcsin\sqrt{x/100}\).
Example 2
The second data set also comes from the NABGM project and consist of 145 doubled haploid (DH) lines of barley (cross of two-rowed varieties Harrington × TR306) analyzed for seven phenotypic traits (weight of grain harvested per unit area, WG; number of days from planting until emergence of 50% of heads on main tillers, NH; number of days from planting until physiological maturity, NM; plant height, H; lodging transformed by \(arcsin\sqrt{x/100}\), L; 1000 kernel weight, KW; test weight, TW) and tested in five environments (in four environments, observations were made over two years: Brandon, Manitoba, 1992 and 1993; Ailsa Craig, Ontario, 1992 and 1993; Elora, Ontario, 1992 and 1993; Outlook, Saskatchewan, 1992 and 1993; Ste-Anne-de-BeUevue, Quebec, 1993) (Tinker et al. 1996, http://wheat.pw.usda.gov/ggpages/HxT). We used the map composed of 127 molecular markers (mostly RFLP) with the mean distance between markers equal to 10.62 cm.
Considering that each trait and environment was classified as an independent variable in both cases, in total of 153 sets of observations were deemed. Trait data was transformed to achieve normal distribution of the observed features. In all cases, transformation was successful and normal distribution was obtained.
Results
Analytical comparison
The estimators, (1) and (5), of the three-way epistasis effect aaa can be analyzed and compared under simplified assumptions: (i) that the markers are unlinked and (ii) that the segregation of each marker is compatible with the genetic model appropriate for the analyzed population, which in our case means that the probability of observing “1” is the same as observing “ − 1”. This is true if we consider that model (2) treats the marker observations as fixed. In fact, the vectors ml, l = 1, 2, ..., q, constitute observations of some random variables. If the marker data satisfied exactly assumptions (i) and (ii) we would have
where \(\overline y^{(l_kl_{k'}l_{k''},-)}\) and \(\overline y^{(l_kl_{k'}l_{k''},+)}\) denote the means for lines with observations of k-th, k’-th, and k’’-th markers equal − 1 and 1, respectively.
In practice, the marker data do not accurately meet the following conditions for model (6). Taking into consideration that markers chosen for model (2) are far apart from each other on the linkage map, assumption (i) is true. To test the assumption (ii) \({\chi }^{2}\), the test is used before any analysis is performed.
Numerical comparison
Obtained results for estimates of total additive × additive × additive interaction effect was presented in Tables 1, 2, 3, and 4. Tables 1 and 2 contain phenotypic and genotypic analysis, respectively, for the 150 doubled haploid lines of barley from the Steptoe × Morex cross; Tables 3 and 4 for the 145 doubled haploid lines of barley from the Harrington × TR306 cross. Figures 1 and 2 show the relative comparison of phenotypic and genotypic estimates of the total additive × additive × additive interaction effect in the form of a box-and-whisker diagram of the values \(\left(\widehat{{aaa}_{g}}/\widehat{{aaa}_{p}}\right)\bullet 100\), classified by the observed phenotypic traits.

Relative comparison of phenotypic and genotypic estimates of the total additive × additive × additive interaction effect for the 150 doubled haploid lines of barley obtained from the Steptoe × Morex cross: box-and-whisker diagram of the values \(\left(\widehat{{aaa}_{g}}/\widehat{{aaa}_{p}}\right)\bullet 100\), classified by the observed phenotypic traits (AA, alpha amylase; DP, diastatic power; GP, grain protein; GY, grain yield; H, height; HD, heading date; L, lodging; ME, malt extract)

Relative comparison of phenotypic and genotypic estimates of the total additive × additive × additive interaction effect for the 145 doubled haploid lines of barley obtained from the cross Harrington × TR306: box-and-whisker diagram of the values \(\left(\widehat{{aaa}_{g}}/\widehat{{aaa}_{p}}\right)\bullet 100\), classified by the observed phenotypic traits (H, plant height; KW, 1000 kernel weight; L, lodging; NH, number of days from planting until emergence of 50% of heads on main tillers; NM, number of days from planting until physiological maturity; TW, test weight; WG, weight of grain harvested per unit area)
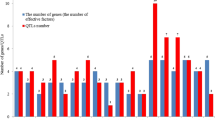
Results show that in 90 cases (70%) we found statistically significant additive × additive × additive interaction effects (Table 1). The same amount of interactions was found for marker observation, but only in 72 cases, where we confirmed results statistically (Table 2). Comparisons of genotypic and phenotypic estimates of the total additive × additive × additive interaction effect show that in the majority of cases (79%), the effect was smaller than the total aaa interaction effect from phenotypic observations alone (Fig. 1). However, the scope of calculated estimates is quite large ranging from − 1590.91% for HD to 1800.00% for H in the same environment (WA92). In a total of five cases, we observed estimate values higher than |1000|%. The smallest range of estimates was observed for the trait DP. Number of genes (effective factors) ranged from 3–10 with average of 3.4 (Table 1). Minimal number of included markers equals 12, where maximum number was 32, with an average of 19.5 markers per model. The number of three-way interactions ranged from 0–35 with an average of 8.3 (Table 2).
For the Harrington × TR306, cross results show that in 63 cases (100%), we found statistically significant additive × additive × additive interaction effects (Table 3). The same amount of interactions was found for marker observation, but only in 35 cases, where we confirmed results statistically (Table 4). Comparisons of genotypic and phenotypic estimates of the total additive × additive × additive interaction effect show that in majority of cases (79%), the effect was smaller than the total aaa interaction effect from phenotypic observations alone (Fig. 2). Same as above, the scope of calculated estimates is quite large ranging from − 2194.31% for WG in environment QC93 to 2866.67% for KW in ON93a. In a total of four cases, we observed estimate values higher than |1000|%. The smallest range of estimates was observed for the trait NM. The number of genes (effective factors) ranged from 0–13 with an average of 5.6 (Table 3). A minimal number of included markers equals 7, where the maximum number was 21, with an average of 13.9 markers per model. The number of three-way interactions ranged from 0–36 with an average of 4.8 (Table 2).
In total, we analyzed 153 sets of observations, independently for each trait and each environment. Both examples were considered separately.
Discussion
Breeding programs aim to enhance the most desirable traits. Actions based solely on phenotypic observations and gene effects are likely to miss the potentially huge impact of interaction and higher-order interaction effects (Taylor and Ehrenreich 2015). Analytical and numerical comparisons of methods of estimation of the total additive × additive × additive interaction effects are presented in this paper. The numerical comparison was conducted on 153 sets of observations from two examples of barley doubled haploid lines.
The analytic comparison shows that, under the assumption of correct segregation and no linkage between markers, the formulae for the phenotypic and genotypic estimators are comparable and that the additive × additive × additive interaction effect of each QTLs triad is smaller than the phenotypic effect.
The numerical comparison of estimates of additive × additive × additive interaction effect shows that in most cases (79% for both examples), genotypic estimate of aaa interaction is smaller than the phenotypic. This sentence is true due to the reason that phenotypic estimate consists of total additive × additive × additive interaction effects of all genes, unlike the genotypic estimate which includes only selected genes. For the rest of the cases that show lower values of phenotypic than genotypic estimates, it may be the result of a high genetic diversity with a lesser phenotypic diversity of the DH lines. High ranges of differences for the calculated estimates are most likely the result of a lot of different experimental variants such as different traits, environments, and experimental situations (Bocianowski and Krajewski 2009). The number of genes (effective factors) in phenotypic estimation does not directly influence the number of markers, as well as the number of aaa interaction included in genotypic models. Both the number of effective factors and number of markers are pretty consistent with few outliers, which makes sense considering that our method tries to include the maximum amount of best-fitted factors. On the contrary, the number of aaa interactions ranged quite widely which may be the result of omitting markers that by themselves do not improve the model but can create the best threes.
In this paper, stepwise feature selection by Akaike information criteria was used. We received comparable results to the previous paper using the same datasets (Bocianowski 2012) with backward stepwise regression as well as to the method of inclusive interval mapping (ICIM) (described by Li et al. 2008). The presented results show that the inclusion of higher-order (aaa) interactions in multiple regression models can have an exert influence on QTL effect.
An important assumption to make is that aaa interaction effects show only loci connected to markers with significant effects. Including additional markers may reveal additional interaction but with significant increase of data quantity requirement (Manolio et al. 2009). Further studies are necessary with respect to additive × additive × additive interaction effects conducted by machine learning methods and by simulation analysis that would make possible consideration of different experimental situations. Current data was not sufficient enough to use machine learning for feature selection. For data containing more markers, we suggest the use of LASSO and SHAP values methods.
Conclusions
Higher-order interactions are usually neglected due to extensive data requirements, although this does not mean they are irrelevant, on the contrary —— higher-order interactions occur often and can have a huge impact on phenotype.
The presented methods were useful statistical tools for QTL characteristics and allow estimating aaa interactions.
On the basis of available literature, this is the first report concerning the presence of analytical and numerical comparisons of two methods of estimation of additive × additive × additive interaction of QTL effects.
Further studies of higher-order interactions and methods of their estimation are necessary.
Availability of data and material
The data presented in this study are available on request from the corresponding authors.
References
Akaike H (1998) Information theory and an extension of the maximum likelihood principle. In: Parzen E, Tanabe K, Kitagawa G (Eds.) Selected Papers of Hirotugu Akaike. Springer Series in Statistics (Perspectives in Statistics). Springer: New York, NY, pp. 199–213. https://doi.org/10.1007/978-1-4612-1694-0_15
Bateson W, Mendel G (1902) Mendel’s Principles of Heredity: a defence, with a translation of Mendel’s original papers on hybridisation. Cambridge University Press, Cambridge. https://doi.org/10.1017/CBO9780511694462
Bocianowski J (2012) Analytical and numerical comparisons of two methods of estimation of additive × additive interaction of QTL effects. Sci Agric 69:240–246. https://doi.org/10.1590/S0103-90162012000400002
Bocianowski J, Krajewski P (2009) Comparison of the genetic additive effect estimators based on phenotypic observations and on molecular marker data. Euphytica 165:113–122. https://doi.org/10.1007/s10681-008-9770-x
Brem RB, Storey JD, Whittle J, Kruglyak L (2005) Genetic interactions between polymorphisms that affect gene expression in yeast. Nature 436:701–703. https://doi.org/10.1038/nature03865
Carlborg Ö, Jacobsson L, Åhgren P, Siegel P, Andersson L (2006) Epistasis and the release of genetic variation during long-term selection. Nat Genet 38(4):418–420. https://doi.org/10.1038/ng1761
Chen CCM, Schwender H, Keith J, Nunkesser R, Mengersen K, Macrossan P (2011) Methods for identifying SNP interactions: a review on variations of Logic Regression, Random Forest and Bayesian logistic regression. IEEE/ACM Trans Comput Biol Bioinform 8(6):1580–1591. https://doi.org/10.1109/TCBB.2011.46
Choo TM, Reinbergs E (1982) Analyses of skewness and kurtosis for detecting gene interaction in a doubled haploid population. Crop Sci 22(2):231–235. https://doi.org/10.2135/cropsci1982.0011183X002200020008x
Cordell HJ (2009) Detecting gene–gene interactions that underlie human diseases. Nat Rev Genet 10(6):392–404. https://doi.org/10.1038/nrg2579
Gaertner BE, Parmenter MD, Rockman MV, Kruglyak L, Phillips PC (2012) More than the sum of its parts: a complex epistatic network underlies natural variation in thermal preference behavior in Caenorhabditis elegans. Genetics 192(4):1533–1542. https://doi.org/10.1534/genetics.112.142877
Hartman JL, Garvik B, Hartwell L (2001) Principles for the buffering of genetic variation. Science 291:1001–1004. https://doi.org/10.1126/science.1056072
Hayes PM, Liu BH, Knapp SJ, Chen F, Jones B, Blake T, Franckowiak J, Rasmusson D, Sorrells M, Ullrich SE, Wesenberg D, Kleinhofs A (1993) Quantitative trait locus effects and environmental interaction in a sample of North American barley germ plasm. Theor Appl Genet 87(3):392–401. https://doi.org/10.1007/BF01184929
Jarvis JP, Cheverud JM (2011) Mapping the epistatic network underlying murine reproductive fatpad variation. Genetics 187(2):597–610. https://doi.org/10.1534/genetics.110.123505
Kaczmarek Z, Surma M, Adamski T (1988) Epistatic effects in estimation of the number of genes on the basis of doubled haploid lines. Genetica Polonica 29:353–359
Kleinhofs A, Kilian A, Saghai Maroof MA, Biyashev RM, Hayes P, Chen FQ, Lapitan N, Fenwick A, Blake TK, Kanazin V, Ananiev E, Dahleen L, Kudrna D, Bollinger J, Knapp SJ, Liu B, Sorrells M, Heun M, Franckowiak JD, Hoffman D, Skadsen R, Steffenson BJ (1993) A molecular, isozyme and morphological map of the barley (Hordeum vulgare) genome. Theor Appl Genet 86(6):705–712. https://doi.org/10.1007/BF00222660
Li H, Ribaut JM, Li Z, Wang J (2008) Inclusive composite interval mapping (ICIM) for digenic epistasis of quantitative traits in biparental populations. Theor Appl Genet 116(2):243–260. https://doi.org/10.1007/s00122-007-0663-5
Mackay TFC (2014) Epistasis and quantitative traits: using model organisms to study gene–gene interactions. Nat Rev Genet 15(1):22–33. https://doi.org/10.1038/nrg3627
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TFC, McCarroll SA, Visscher PM (2009) Finding the missing heritability of complex diseases. Nature 461(7265):747–753. https://doi.org/10.1038/nature08494
Martinez O, Curnow RN (1994) Missing markers when estimating quantitative trait loci using regression mapping. Heredity 73:198–206. https://doi.org/10.1038/hdy.1994.120
Members of the Complex Trait Consortium (2003) The nature and identification of quantitative trait loci: a community’s view. Nat Rev Genet 4(11):911–916. https://doi.org/10.1038/nrg1206
Romagosa I, Ullrich SE, Han F, Hayes PM (1996) Use of the additive main effects and multiplicative interaction model in QTL mapping for adaptation in barley. Theor Appl Genet 93(1):30–37. https://doi.org/10.1007/BF00225723
Searle SR (1982) Matrix algebra useful for statistics. Wiley, New York
Taylor MB, Ehrenreich IM (2015) Higher-order genetic interactions and their contribution to complex traits. Trends Genet 31(1):34–40. https://doi.org/10.1016/j.tig.2014.09.001
Tinker NA, Mather DE, Rossnagel BG, Kasha KJ, Kleinhofs A, Hayes PM, Falk DE, Ferguson T, Shugar LP, Legge WG, Irvine RB, Choo TM, Briggs KG, Ullrich SE, Franckowiak JD, Blake TK, Graf RJ, Dofing SM, Saghai Maroof MA, Scoles GJ, Hoffman D, Dahleen LS, Kilian A, Chen F, Biyashev RM, Kudrna DA, Steffenson BJ (1996) Regions of the genome that affect agronomic performance in two-row barley. Crop Sci 36(4):1053–1062. https://doi.org/10.2135/cropsci1996.0011183X003600040040x
Author information
Authors and Affiliations
Contributions
Conceptualization, JB; methodology, AC and JB; software, AC; validation, AC and JB; formal analysis, AC; investigation, AC and JB; resources, AC and JB; data curation, AC and JB; writing—original draft preparation, AC; writing—review and editing, AC and JB; visualization, AC; supervision, JB; all authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the author.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflicts of interest
Authors declare no competing interests.
Additional information
Communicated by Izabela Pawłowicz
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cyplik, A., Bocianowski, J. Analytical and numerical comparisons of two methods of estimation of additive × additive × additive interaction of QTL effects. J Appl Genetics 63, 213–221 (2022). https://doi.org/10.1007/s13353-021-00676-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-021-00676-7