
Abstract
The blood–spinal cord barrier (BSCB) plays important roles in the recovery of spinal cord injury (SCI), and caveolin-1 is essential for the integrity and permeability of barriers. Basic fibroblast growth factor (bFGF) is an important neuroprotective protein and contributes to the survival of neuronal cells. This study was designed to investigate whether bFGF is beneficial for the maintenance of junction proteins and the integrity of the BSCB to identify the relations with caveolin-1 regulation. We examined the integrity of the BSCB with Evans blue dye and fluorescein isothiocyanate–dextran extravasation, measured the junction proteins and matrix metalloproteinases, and evaluated the locomotor function recovery. Our data indicated that bFGF treatment improved the recovery of BSCB and functional locomotion in contusive SCI model rats, reduced the expression and activation of matrix metalloproteinase-9, and increased the expressions of caveolin-1 and junction proteins, including occludin, claudin-5, p120-catenin, and β-catenin. In the brain, in microvascular endothelial cells, bFGF treatment increased the levels of junction proteins, caveolin-1 small interfering RNA abolished the protective effect of bFGF under oxygen–glucose deprivation conditions, and the expression of fibroblast growth factor receptor 1 and co-localization with caveolin-1 decreased significantly, which could not be reversed by bFGF treatment. These findings provide a novel mechanism underlying the beneficial effects of bFGF on the BSCB and recovery of SCI, especially the regulation of caveolin-1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Traumatic spinal cord injury (SCI) is a devastating disease that results in permanent disability. Previous research focuses largely on improving neurological manifestations through the improvement of sensory function and locomotor function [1–4], but study of the blood–spinal cord barrier (BSCB) still lacks sufficient investigation. Similarly to the blood–brain barrier (BBB), BSCB plays a protective and regulatory role in spinal cord parenchyma. Endothelial cells, basement membranes, pericytes, and astrocytic end-feet processes constitute a specialized system for this functional barrier [5]. Early microvascular reactions and BSCB disruption are instrumental in the pathophysiology of SCI and repair [6, 7].
Caveolin-1 (Cav-1), a major structural protein of caveolae that is known to be involved in endocytosis, vesicular trafficking, and signal transduction [8, 9], regulates the permeability of the BBB. In several models of adult brain injuries, Cav-1 is increased in the endothelium for several days following cold cortical injury and brain ischemia [10, 11]. The knockdown of Cav-1 reduces matrix metalloproteinase (MMPs) activity in endothelial cells and their angiogenic response to vascular endothelial growth factor (VEGF) [12]. On the contrary, the loss of Cav-1 is crucial in the activation of MMPs and BBB breakdown in stroke animal models [13]. Cav-1 regulates expression of junction-associated proteins in brain microvascular endothelial cells, and attenuated Cav-1 levels are correlated with heightened permeability of endothelia [14]. In addition, Cav-1 knockout mice present higher vascular permeability, which has been found to aggravate disorders in rodent models [15, 16]. Although the exact role of Cav-1 in SCI remains unclear, it is plausible that Cav-1 is a critical mediator of BSCB integrity.
Basic fibroblast growth factor (bFGF) is a neurotrophic factor that functions as a neuroprotective agent and rescues neurons from various insults in vitro and in vivo. Administration of exogenous growth factors following SCI promotes functional recovery [17, 18]; intrathecal administration of bFGF also significantly enhances functional recovery after moderate and severe contusions in SCI rats [19, 20], but the precise mechanism underlying the observed therapeutic effects has not been elucidated. In our latest studies, bFGF was demonstrated to inhibit excessive autophagy and endoplasmic reticulum stress in SCI, which contributed to functional recovery [21, 22]. In addition, it has been reported that bFGF preserves BBB integrity after intracerebral hemorrhage in mice [23]. However, the effect of bFGF on BSCB has not been reported.
In this study, a contusive SCI rat model was constructed using an Infinite Horizon Impactor Device, to evaluate the effects of bFGF on BSCB integrity in vivo. Furthermore, an oxygen-glucose deprivation (OGD) cell model was established to clarify the role of Cav-1 in bFGF-mediated endothelial barrier function in vitro. Our data indicate, for the first time, that Cav-1 is essential for the function of bFGF in the recovery of the BSCB and correlates with fibroblast growth factor receptor 1 (FGFR1) regulation that contributes substantially to the repair of SCI.
Materials and Methods
Experimental Animals and Surgical Procedures
Eighty adult female Sprague–Dawley rats (weighing 220–250 g) were obtained from the Animal Center of Chinese Academy of Science. All experimental procedures were approved by the ethics committee of Wenzhou Medical University and performed in accordance with the Guide for the Care and Use of Laboratory Animals. Rats were anesthetized with 10% chloral hydrate (3.5 ml/kg) before a laminectomy. The vertebral column was stabilized by clamping the T8 and T10 vertebral bodies with forceps fixed to the base of an Infinite Horizon Impact Device. Rats were situated on the platform, and the 2.5-mm stainless steel impactor tip was positioned over the midpoint of T9 and impacted with 150 kdyn force with no dwell time. The force/displacement graph was used to monitor impact consistency and any animals that exhibited an abnormal impact graph or > 10% deviation from 150 kdyn were immediately excluded from the study. The incision sites were then closed in layers and a topical antibiotic (cefazolin sodium salt, 50 mg/kg, i.p.) was applied to the incision site. For the sham-operated controls, the animals underwent a T9 laminectomy without contusion injury. Postoperative care involved manually emptying the urinary bladder twice a day (until the return of bladder function). The SCI rats were randomly divided into 2 groups, a bFGF-treated group (bFGF) and a vehicle group (SCI). Based on our previous study [22], recombinant human bFGF purchased from Sigma-Aldrich (St. Louis, MO, USA) was injected subcutaneously near the back wound at a dose of 80 μg/kg at 30 min post injury; after this, the recombinant human bFGF was administered every 2 days until the animals were euthanized. The death rate of our model was zero.
Tissue Preparation
At specific time points after SCI, animals were anesthetized with 10% chloral hydrate (3.5 ml/kg) and perfused via cardiac puncture initially with 0.9% saline solution. For immunohistochemistry, animals were perfused with 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS) after saline solution. A 0.5-cm section of the spinal cord, centered at the lesion site, was dissected out, postfixed by immersion in 4% paraformaldehyde for 24 h. and then placed in 30% sucrose in 0.1 M PBS for 2 days. The segments were embedded in optimal cutting temperature for frozen sections, and longitudinal or transverse sections were then cut at 10 or 20 μm on a cryostat (Leica Microsystems Wetzlar GmbH, Hesse-Darmstadt, Germany). For Western blot, a spinal cord segment (0.5 cm length) at the contusion epicenter was dissected and immediately stored at –80°C.
Measurement of BSCB Disruption
The integrity of BSCB was investigated with Evans Blue dye and fluorescein isothiocyanate (FITC)–dextran (MW 70 kDa; Sigma-Aldrich) extravasation, according to previous reports [24, 25]. At 1 day after SCI, animals were injected with 4 ml/kg 2% Evans Blue (Sigma-Aldrich) into the tail vein. Two hours later, animals were anesthetized and killed by intracardiac perfusion with saline. For qualitative examination of Evans Blue extravasation, the animals were perfused with PBS and subsequently with 4% formaldehyde, as described above. The spinal cords were separated into 20-μm thick sections with a cryostat. The fluorescence of Evans Blue in spinal tissues was observed with a fluorescence microscope and the relative fluorescence intensity was determined by Image Pro-Plus (Media Cybernetics, Rockville, MD, USA). In order to quantify leakage of larger molecular weight molecules, FITC–dextran (4 mg/kg) was injected into the tail vein and allowed to circulate for 2 h. The animals were killed and perfused with saline. The T8–T10 segment was removed, homogenized in PBS, and centrifuged. The supernatant fluorescence (excitation at 493 nm and emission at 517 nm) was then measured.
Locomotion Recovery Assessment
In order to examine the locomotor function after injury, the Basso, Beattie, and Bresnahan scale were scored by trained investigators who were blind to the experimental conditions [26]. It is performed in an open-field scale everyday postoperation. Briefly, the Basso, Beattie, and Bresnahan locomotion rating scale ranges from 0 points (complete paralysis) to 21 points (normal locomotion). The scale is based upon the natural progression of locomotion recovery in rats with thoracic SCIs.
Cell Culture and Treatment
Human brain microvascular endothelial cells (HBMEC) and endothelial cell medium were purchased from ScienCell Research Laboratories (ScienCell Research Laboratories, San Diego, CA, USA). HBMEC were grown as a monolayer in endothelial cell medium. All cells were incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air. The cells were subcultured into 60-mm or 35-mm dishes coated with fibronectin and confluent cells were exposed to a hypoxia chamber (1029; Thermo Fisher Scientific, Waltham, MA, USA) for 12 h after overnight starvation with 0.5% fetal calf serum. Before OGD, the medium was then changed to sugar-free basic medium. The oxygen concentration was < 0.2%, as monitored by an oxygen analyzer. bFGF (50 ng/ml) was treated for 30 min before OGD. The bFGF powder was dissolved in PBS to make the stock solution (50 μg/μl). For the control group, PBS without drug was used. Primary astrocytes and the SHSY-5Y cell line were purchased from the Cell Storage Center of Wuhan University (Wuhan, Hubei, China). Both cell types were cultured with 10% F12/DMEM medium (Gibco, Carlsbad, CA, USA) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. The OGD conditions and procedure of these cells were same with HBMEC.
Primary Hippocampal Neuron Culture
Primary hippocampal neurons were established from the brains of neonatal Sprague–Dawley rats (<4 h of age). Hippocampi were dissected from the brains and rinsed in ice-cold dissection buffer. Blood vessels and white matter were removed and tissues were treated with 0.125% trypsin in Hank’s balanced salt solution for 20 min at 37°C. The whole solution was filtered through stainless steel (200 mesh; BD Biosciences, San Jose, CA, USA). Cell suspension was centrifuged twice at 198 g for 5 min and the cell pellets resuspended in DMEM/F-12 with 10% fetal bovine serum, 100 U/l penicillin, 100 mg/l streptomycin and 0.5 mM glutamine. Cells were seeded at a density of 1–5 × 105/ml in 6-well plates kept at 37°C in a 5% CO2 incubator. After 24 h, the culture medium was changed to Neurobasal Medium (Gibco) with 2% B27 and changed every 2–3 days. Arabinosylcytosin (10 mg/l; Sigma-Aldrich) was added at 72 h to prevent the growth of non-neuronal cells. All experiments were performed at 8–11 days after seeding. The procedure of OGD is same with other cells.
Small Interfering RNA Transfection
HBMEC at 70–80% confluence were transfected with 100 pmol Cav-1 small interfering RNA (siRNA) or negative-control siRNA (Bioneer, Daejeon, Korea) using lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA), according to the manufacturer’s instructions. Twenty-four hours after transfection, cells were subjected to OGD treatment. Specific silencing was confirmed by Western blot.
HBMEC Monolayer Permeability Assay
The effect of OGD on endothelial monolayer permeability to FITC–dextran was assessed using polyethylene terephthalate membrane 24-well cell culture inserts with a 0.4-μm pore size (Corning Life Sciences, Corning, NY, USA). Cells were placed on the upper side of the insert and allowed to grow to confluence. FITC–dextran at a concentration of 1 mg/ml was added to the endothelial monolayer after exposure to OGD [27]. Two hours later, relative fluorescence passed through the chamber was determined using a fluorescence plate reader at an excitation wavelength at 493 nm and an emission wavelength at 517 nm (SpectraMax M2e; Molecular Devices, Sunnyvale, CA, USA). Endothelial monolayer permeability was assessed by the intensity of FITC–dextran in the lower chamber. To test whether bFGF could protect the integrity of endothelial monolayer permeability, cells were pretreated with bFGF (40 ng/ml) for 1 h. In addition, cells were pretreated with Cav-1 siRNA for 24 h, to clarify the role of Cav-1 in endothelial cell.
Gelatin Zymography
In vivo, the activity of MMP-2 and MMP-9 at 1 day after injury was examined by gelatin zymography. Briefly, the protein concentration of the homogenates was determined by the bicinchoninic acid method (BCA protein assay kit; Thermo Fisher Scientific, Rockford, IL, USA). After determination of protein concentration of the homogenates, equal amounts of protein (30 μg) were loaded on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, co-polymerized with 1 mg/ml gelatin (Sigma-Aldrich). Gels were washed in 2.5% Triton X-100 (Sigma-Aldrich) for 1 h and then incubated for 24 h in a developing buffer, including Tris 50 mM/l (pH 7.6), CaCl 25 mM, NaCl 0.2 mM, and 0.02% (w/v) Brij-35 (Sigma-Aldrich) at 37°C, followed by staining with Coomassie blue (Sigma-Aldrich). Gels were destained to visualize gelatinolytic bands (MMP-2/MMP-9) on a dark blue background. In vitro, after OGD treatment, MMP-2/MMP-9 in conditioned media was analyzed by gelatin zymography as described above.
Western Blot
For in vivo protein analysis, a spinal cord segment (0.5 cm length) at the contusion epicenter was dissected at 6 h, and 1, 3 and 7 days and immediately stored at –80°C for Western blotting. For protein extraction, the tissue was homogenized in modified buffer [50 mM Tris-HCl, 1% NP-40, 20 mM dithiothreitol, 150 mM NaCl (pH 7.4)] containing protease inhibitor cocktail (10 μl/ml; GE Healthcare Biosciences, Pittsburgh, PA, USA). The complex was then centrifuged at 13,362 g and the supernatant obtained for protein assay. In vitro, HBMEC were lysed in RIPA buffer (25 mM Tris-HCl, 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) with protease and phosphatase inhibitors. The extracts above were quantified with BCA reagents. We separated proteins on a 10% or 12% gel and transferred them onto a polyvinylidene fluoride membrane (Bio-Rad, Hercules, CA, USA). The membrane was blocked with 5% milk (Bio-Rad) in TBS with 0.05% Tween 20 (TBST) for 1.5 h and incubated with the antibodies p120-catenin (1:1000), β-catenin (1:1000), occludin (1:300), claudin-5 (1:300), Cav-1 (1:300) in TBST for 2 h at room temperature or overnight at 4°C. The membranes were washed with TBST 3 times and treated with horseradish peroxidase-conjugated secondary antibodies (1:3000) for 1 h at room temperature. Signals were visualized by ChemiDocXRS + Imaging System (Bio-Rad). β-Actin (1:300) was used as an internal control. Experiments were repeated 3 times and the densitometric values of the bands on Western blots obtained by Image J software (National Institutes of Health, Bethesda, MD, USA) were subjected to statistical analysis. Anti-p120-catenin and β-catenin were from Abcam (Cambridge, UK); all other antibodies were from Santa Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Real-Time Polymerase Chain Reaction
Total RNA was isolated using Trizol reagents according to manufacturer's protocol. RNA (0.5 μg) was used as a template for first-strand cDNA synthesis using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Life Technologies, Carlsbad, CA, USA). Reverse transcription products were amplified with the 7900HT Fast Real-Time PCR System in a 10-μl final reaction volume using SYBR Green PCR Master Mix (Bio-Rad, Hercules, CA, USA) under the following conditions: 2 min at 50°C and 10 min at 95°C, followed by a total of 40 cycles of 2 temperature cycles (15 s at 95°C and 1 min at 60°C). Primers for MMP-2, MMP-9, and actin were designed against known rat sequences and human sequences as follows: rat, MMP-2 forward: 5’- GGACAGTGACACCACGTGACA,-3’, reverse: 5’-ACTCATTCCCTG CGAAGAACA-3’; MMP-9 forward: 5’-AACCCTGGTCACCGGACTTC-3’, reverse: 5’-CACCCGGTTGTGGAAACTCAC-3’; β-actin forward: 5’-AAGATCCTGACCGAGCGTGGC-3’, reverse: 5’-CAGCACTGTGTTGGCATAGAGG-3’; Cav-1 forward: 5’-GCCCTCACAGGGACATCTCTACA-3’, reverse: 5’-CCGCAATCACATCTTCAAAGTCA-3’; human, MMP-2 forward: 5’-CCCAGACAGGTGATCTTGACC-3’, reverse: 5’-CTTGCGAGGGAAGAAGTTGTAG-3’; MMP-9 forward: 5’-ATCCGGCACCTCTATGGTC-3’, reverse: 5’-CTGAGGGGTGGACAGTGG-3’; β-actin forward: 5’-CCTGGCACCCAGCACAAT-3’, reverse: 5’-GCCGATCCACACGGAGTACT - 3’. The fluorescence threshold value (Ct value) was calculated using the SDS Enterprise Database software. The relative value of mRNA expression was calculated by the comparative ΔΔCt method. In brief, mean Ct values were normalized to the internal control actin and the difference was defined as ΔCt. The difference between the mean ΔCt values of SCI group and bFGF treatment group was calculated and defined as ΔΔCt. The comparative mRNA expression level was expressed as 2^ (–ΔΔCt). All agents mentioned were from Life Technologies.
Immunohistochemistry
Frozen sections were processed for double labeling with antibodies against claudin-5 (1:100; Santa Cruz), p120-catenin (1:200; Santa Cruz), NeuN (1:500; Abcam), glial fibrillary acidic protein (1:100; Santa Cruz), and CD31 (1:100, Santa Cruz). Alexa Fluor 488 (1:1000; Abcam) or Texas Red-conjugated secondary antibodies (1:200; Santa Cruz) were used. Nuclei were labeled with Hoechst (Beyotime Institute of Biotechnology, Shanghai, China). The control study was performed by using PBS instead of primary antibody. For cell immunostaining, the HBMEC grown to confluence on fibronectin-coated coverslips were subjected to the indicated treatments. Cells were washed 3 times with PBS, fixed in 4% paraformaldehyde for 30 min, and blocked for 30 min at 37°C with 5% bovine serum albumin. The cells were then incubated with anti-p120-catennin (1:200), anti-β-catenin (1:200), anticlaudin-5 (1:50), antioccludin (1:50), anti-Cav-1 (1:100), or anti-FGFR1 (1:100) overnight at 4°C. Alexa Fluor 488 (1:1000) or TR-conjugated secondary antibodies were used. Anti-CD31, anti-FGFR1, and TR-conjugated secondary antibodies were from Santa Cruz, Alexa Fluor 488 was from Abcam, and others antibodies are same as used in the Western blot experiments.
Statistical Analysis
All quantitative data are expressed as means ± SEM if normally distributed. Student’s t test was used between 2 groups if data were normally distributed. Otherwise, we used the Wilcoxon rank sum test instead. For analyzing 3 or more groups, differences were assessed by 1-way ANOVA if data were normally distributed and variance homogeneous; otherwise, the Kruskal–Wallis test was performed, followed by post-hoc adjustment using Bonferroni’s multiple comparisons test. Normality was checked using the Kolmogorov–Smirnov test and the homogeneity of variance was checked by the Levene test. A probability value p < 0.05 was considered to show statistical significance.
Results
bFGF Attenuates BSCB Disruption After SCI
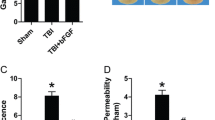
To determine the effect of bFGF on BSCB integrity, we assessed the degree of BSCB disruption after SCI by Evans Blue and FITC–dextran assay. As is shown in Fig. 1A, we assessed the intensity of Evans Blue dye extravasation at 6 h, 1 d, 3 d, 7 d, and 14 d postinjury with or without bFGF. We found that the extravasation of Evans Blue significantly increased after injury, and showed maximal BSCB disruption at 1 d after injury. bFGF showed a remarkable reduction of the intensity of Evans Blue at 6 h, 1 d, 3 d, and 7 d postinjury. As 1 d is the peak of BSCB permeability and the effect of bFGF at 1 d is most obvious, we suggest that there is a relatively large time window in which to exploit the disruption of the BSCB. From Fig. 1(B, C), it is easy to see that the fluorescence intensity of Evans Blue in spinal cord sections is much weaker in the bFGF treatment group than the SCI group, which suggests that bFGF increased the integrity of BSCB after SCI. To quantify the leakage of larger molecular weight molecules, FITC–dextran was injected into the tail vein. The intensity of FITC–dextran extravasation was obviously enhanced and bFGF reduced the fluorescence intensity compared with the SCI group (Fig. 1D). To further support the role of bFGF in increasing BSCB integrity, we analyzed immune cell infiltration. CD68, the marker of macrophages, was detected at 1 d postinjury with or without bFGF. As is shown in Fig. 1(E, F), bFGF reduced the expression of CD68.

Basic fibroblast growth factor (bFGF) reduces blood–spinal cord barrier (BSCB) permeability after contusive spinal cord injury (SCI). After SCI, rats were treated with bFGF and barrier permeability was measured at 1 d after injury, using Evans Blue dye and fluorescein isothiocyanate (FITC)–dextran. (A) Quantification of Evans Blue dye extracted from spinal cord at 6 h–14 d postinjury with or without bFGF. Data were analyzed by Student’s t test (6 h: t = 4.444, p = 0.011; 1 d: t = 10.398, p < 0.001; 5 d: t = 5.013, p = 0.007; 7 d: t = 2.895, p = 0.044). (B) Representative whole spinal cords showing Evans Blue dye permeabilized into the spinal cord at 1 d. (C) Representative confocal images of sham, SCI, and bFGF-treatment groups. Scale bar = 1 mm. (D) Quantification of the FITC–dextran extravasation (excitation at 490 nm and emission at 520 nm). Data were analyzed by ANOVA (F[2, 9] = 79.682, p < 0.001). Post-hoc analyses were done using Bonferroni’s multiple comparison test (***p < 0.001). (E) Western blot and densitometric analysis of CD68 at 1 d with or without bFGF. Data were analyzed by Student’s t test (t = 6.444, p = 0.023). (F) Immunochemical staining of CD68. Scale bar = 50 μm. *p < 0.05, **p < 0.01, ***p < 0.001
Furthermore, to verify the protective effect of bFGF on functional recovery post-SCI, the functional recovery and pathologic morphology after bFGF treatment were evaluated (see Supplementary Fig. 1). The Basso, Beattie, and Bresnahan scores of SCI were assessed at 1, 3, 5, 7, and 14 d postinjury (Supplementary Fig. 1A). There was no obvious difference in the Basso, Beattie, and Bresnahan scores between the SCI model and the bFGF groups at 1 d after injury. However, the Basso, Beattie, and Bresnahan scores of the bFGF group were increased from d 3 to d 14 after injury, indicating that bFGF stimulated the recovery of locomotor activity. As shown in Supplementary Fig. 1(B), footprint analyses for bFGF-treated rats at 14 d post-SCI disclosed fairly consistent hindlimb coordination and some toe dragging, while the SCI group showed inconsistent coordination and extensive drags, as revealed by ink streaks extending from both hindlimbs. From Supplementary Fig. 1(C, D), the results of hematoxylin and eosin stain, and NeuN stain, also demonstrated that bFGF ameliorated the pathological morphology of tissues and increased the number of neurons at 3 d postinjury (Supplementary Fig. 1E). All these data indicate that bFGF attenuates BSCB disruption in the early stages after SCI, which is beneficial for the functional recovery of SCI.
bFGF Inhibits the Expression and Activation of MMP-9 After SCI
Although many factors are known to contribute to BBB/BSCB disruption, MMPs play a critical role [28]. Therefore, we measured the expression and activation of MMP-2 and MMP-9 after bFGF treatment. First, we examined the expression of MMP-2/MMP-9 mRNA at 6 h–7 d after injury by real-time polymerase chain reaction (n = 5/group). As shown in Fig. 2(A, B), the mRNA levels of MMP-9 markedly increased at 1 d after injury, whereas the level of MMP-2 exhibited no significant changes (Fig. 2B). In addition, bFGF treatment significantly inhibited MMP-9 mRNA expression at 1 d after injury compared with the vehicle controls (Fig. 2C). Next, the activities of MMP-2/MMP-9 were analyzed by gelatin zymography. Figure 2(E) shows that MMP-9 activation was markedly enhanced at 1 d after injury, consistent with previous reports [29], with bands corresponding to the active form of MMP-9 and the inactive zymogen (pro-MMP-9). Although pro-MMP-2 appeared in the SCI group, there was no active MMP-2, which is consistent with the report of Lee et al. reports [25]. bFGF significantly decreased the level of active MMP-9 and pro-MMP-2 compared with the vehicle controls at 1 d after injury. In addition, we also performed double-labeling immunofluorescence between MMP-9 and other cell markers. As shown in Supplementary Fig. 2, the results of the Western blot suggest that the expression of MMP-9 was upregulated after SCI, and bFGF significantly reduced its expression. Further staining indicated that MMP-9 was mainly expressed in endothelial cells and neurons (Supplementary Fig. 2B, C); however, in astrocytes, there is no co-localization with GFAP, but some positive points were found in the nuclear of astrocytes (Supplementary Fig. 2D). These data suggest that bFGF might prevent BSCB disruption by inhibiting the expression and activation of MMP-9 after SCI.

Basic fibroblast growth factor (bFGF) inhibits the expression and activation of matrix metalloproteinase (MMP)-9 after spinal cord injury. Spinal cord tissues were isolated at 6 h–7 d after injury. Real-time polymerase chain reaction and gelatin zymography were processed as described in the “Materials and Methods” (n = 5/group). (A, B) mRNA level of MMP-2/MMP-9 at 6 h–7 d after injury. Data were analyzed by Kruskal–Wallis test (MMP-9: p = 0.002; MMP-2: p = 0.002). Post-hoc analyses were done using Bonferroni’s multiple comparison test (*p < 0.05). (C, D) mRNA level of MMP-2/MMP-9 at 1 d after injury with administration of bFGF. MMP-9 data were analyzed by Kruskal–Wallis test (MMP-9: p = 0.005). MMP-2 data were analyzed by ANOVA (F[2, 9] = 3.828, p = 0.063). Post-hoc analyses were done using Bonferroni’s multiple comparison test (*p < 0.05). (E) Gelatin zymography at 1 d after injury. (F) Densitometric analysis of zymography. Data were analyzed by Student’s t test (active MMP-9: t = 6.819, p = 0.002; pro-MMP-2: t = 4.394, p = 0.039)
bFGF Inhibits the Disruption of Tight and Adherens Junction After SCI
It was previously discovered the tight junction (TJ) proteins are critical structural proteins in the BSCB [21]. To determine whether SCI-induced hyperpermeability is caused by TJ alterations, the expression of claudin-5 and occludin, the major TJ proteins present in the BBB [30], were examined by Western blot in spinal cord lysates. After injury, the expression of occludin and claudin-5 decreased, and the decrease was especially prominent at 1 day after injury (Fig. 3A, C). However, the level of occludin returned nearly to the level of the sham group after 7 days. Furthermore, we examined the levels of adherens junctions (AJ) proteins. Catenins, a family of proteins found in complexes with adhesion molecules of animal cells, were degraded and were present in significant numbers at 1 d postinjury (Fig. 3A, B). However, bFGF-treated groups showed significantly higher levels of occludin, claudin-5, p120-catenin, and β-catenin at 1 d after injury (Fig. 3D, E, F). In addition, a double-labeling immunofluorescence was performed in blood vessels. The fluorescence intensity of claudin-5 and p120-catenin immunoreactivities were decreased in endothelial cells (labeled by CD31) after injury compared with sham controls, and bFGF treatment attenuated the decrease in its intensity (Fig. 3G, H). These data indicate that bFGF prevents BSCB disruption by inhibiting the degradation of TJ and AJ molecules after SCI.

Basic fibroblast growth factor (bFGF) prevents the loss of tight junction (TJ) and adherens junction (AJ) proteins after spinal cord injury (SCI). (A) The protein levels of p120-catenin, β-catenin,occludin, and claudin-5 at 6 h–7 d after injury. (B) Densitometric analyses of p120-catenin and β-catenin. Data were analyzed by ANOVA (β-catenin: F[4, 10] = 158.458, p < 0.001; p120-catenin: F[4, 10] = 39.625, p < 0.001). Post-hoc analyses were done using Bonferroni’s multiple comparison test (**p < 0.01, ***p < 0.001). (C) Densitometric analyses of occludin and claudin-5. Data were analyzed by ANOVA (claudin-5: F[4, 10] = 360.991, p < 0.001; occludin: F[4, 10] = 70.827, p < 0.001). Post-hoc analyses were done using Bonferroni’s multiple comparison test (**p < 0.01, ***p < 0.001). (D) Protein levels of TJ and AJ proteins 1 d after bFGF treatment. (E) Densitometric analyses of p120-catenin and β-catenin. Data were analyzed by ANOVA (β-catenin: F[2, 6] = 68.213, p < 0.001; p120-catenin: F[2, 6] = 15.736, p = 0.004). Post-hoc analyses were done using Bonferroni’s multiple comparison test (**p < 0.01, ***p < 0.001 vs Sham, # p < 0.05, ## p < 0.01 vs SCI). (F) Densitometric analyses of occludin and claudin-5. Data were analyzed by ANOVA (claudin-5: F[2, 6] = 36.956, p < 0.001; occludin: F[2, 6] = 32.484, p = 0.001). Post-hoc analyses were done using Bonferroni’s multiple comparison test (**p < 0.01 vs sham, ## p < 0.01 vs SCI). (G) Double immunofluorescence shows that claudin-5 (green) co-localizes in endothelial cells (CD31, red). The background is staining of the blood cells. (H) Double immunofluorescence of p120-catenin (green) and CD31 (red). Scale bar = 25 μm
bFGF Prevents Loss of Cav-1 in Endothelial Cells after SCI
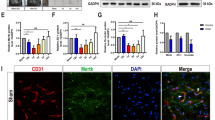
It is reported that reduced Cav-1 accompanied the diminished expression of TJ-associated proteins following stimulation of HBMECs with the chemokine CCL2, which contributes to BBB disruption [14]. However, the relationship of Cav-1 and BSCB disruption is still unclear. In our study, we found that in addition to the loss of TJ and AJ proteins following SCI, Cav-1 expression significantly decreased at 1 d after injury, shown by both mRNA and protein levels (Fig. 4A–C). In addition, bFGF increased the expression of Cav-1 (Fig. 4D, E). Interestingly, as shown in Fig. 4(F), Cav-1 was co-localized with CD31 (marker of endothelial cells) but not with NeuN (marker of neurons) or GFAP (marker of astrocytes), suggesting that Cav-1 was only located in endothelial cells in normal spinal cord. Therefore, we presume that bFGF exerts its beneficial role in BSCB integrity in relation to its regulation of Cav-1.

Basic fibroblast growth factor (bFGF) increases the level of caveolin-1 (Cav-1) in endothelial cells after spinal cord injury. (A) mRNA levels of Cav-1 at 6 h–7 d after injury. Data were analyzed by ANOVA (F[4, 19] = 4.816, p = 0.007). Post-hoc analyses were done using Bonferroni’s multiple comparison test (*p < 0.05). (B) Protein level of Cav-1 at 6 h–7 d after injury. (C) Densitometric analyses of Western blots. Data were analyzed by ANOVA (F[4, 10] = 71.280, p < 0.001). Post-hoc analyses were done using Bonferroni’s multiple comparison test (*p < 0.05). (D) Protein level of Cav-1 at 1 d after bFGF treatment. (E) Densitometric analyses of Western blots. Data were analyzed by ANOVA (F[2, 6] = 74.499, p < 0.001). Post-hoc analyses were done using Bonferroni’s multiple comparison test (***p < 0.001 vs Sham, # p < 0.05 vs SCI). (F) Double immunofluorescence of Cav-1 (green) with CD31 (red, marker of endothelial cell), NeuN (red, marker of neuron), and glial fibrillary acidic protein (red, marker of astrocyte). Scale bar = 50 μm
bFGF Inhibits the Levels of TJ and the Activation of MMP-9 in HBMEC After OGD
Endothelial monolayer is the main component of the BBB or BSCB. Its partitioning, underlying tissue from blood components in the vessel wall, maintains the tissue fluid balance and host defense through dynamically opening intercellular junctions. To investigate the effect of bFGF on barrier integrity, we exposed HBMEC to OGD conditions. First, we examined whether bFGF affects the alteration of TJ and AJ proteins. The expression levels of p120-catenin, β-catenin, occludin, and claudin-5 decreased at 12 h after exposure to OGD conditions, and this effect was significantly reversed by bFGF (Fig. 5A, C, D). Moreover, expression and activation of MMP-9 were detected in HBMEC. We found that bFGF inhibited MMP-9 expression and activation, which was stimulated under OGD conditions, but MMP-2 presented no significant changes (Fig. 5F). As is known, astrocytes and neurons are important sources of MMP-2/MMP-9. Therefore, it would be interesting to determine whether bFGF can affect MMP-2/MMP-9 activation in these 2 cell types. We found that the rapid activation of MMP-2/MMP-9 was inhibited by bFGF in astrocytes under OGD conditions (Fig. 5; upper panel). However, there was no apparent MMP-9 activation in SH-SY5Y (middle panel; Fig. 5H). As our in vivo results have shown that MMP-9 was also expressed in neurons (Supplementary Fig. 2C), to clarify the inconsistent results, we performed the same experiments in cultured primary hippocampal neurons. As is shown in Fig. 5H (bottom panel), bFGF inhibited the activation of MMP-9 induced by OGD. These data imply that bFGF inhibits the loss of junction proteins, and reduces the expression and activation of MMP-9, but not MMP-2, in endothelial cells under OGD conditions.

Basic fibroblast growth factor (bFGF) inhibits the loss of caveolin-1 (Cav-1) and junction proteins, and attenuates the elevation of extracellular matrix metalloproteinase (MMP)-9 levels in endothelial cells after oxygen–glucose deprivation (OGD). (A) Protein levels of Cav-1, p120-catenin, β-catenin, occludin, and claudin-5 after 12 h of OGD. Densitometric analyses of (B) Cav-1, (C) adherens junction proteins, (D) and tight junction proteins. Data were analyzed by ANOVA (Cav-1: F[2, 6] = 26.702, p = 0.001; p120-catenin: F[2, 6] = 91.763, p < 0.001; β-catenin: F[2, 6] = 472.576, p < 0.001; occludin: F[2, 6] = 53.484, p < 0.001; claudin-5: F[2, 6] = 27.251, p = 0.001). Post-hoc analyses were done using Bonferroni’s multiple comparison test [**p < 0.01, ***p < 0.001 vs control (CON), # p < 0.05, ## p < 0.01 vs OGD]. (E) Immunocytochemistry of Cav-1 from fixed human brain microvascular endothelial cells after OGD for 12 h. Scale bar = 50 μm. (F) The mRNA levels of MMP-2/MMP-9 after 12 h of OGD in each group. Data were analyzed by ANOVA (MMP-9: F[2, 6] = 23.723, p = 0.001). Post-hoc analyses were done using Bonferroni’s multiple comparison test (**p < 0.01 vs CON, ## p < 0.01 vs OGD). (G) Gelatin zymography and densitometric analyses for cell medium in endothelial cell after OGD. Data were analyzed by Student’s t test (MMP-9: t = 7.623, p = 0.002, ## p < 0.01 vs OGD). (H) Gelatin zymography for cell medium in astrocytes (upper panel), neuronal cells line SH-SY5Y (middle panel), and primary hippocampal neuron (bottom panel). * P < 0.05, ** P < 0.01 vs CON, # P < 0.05 vs OGD
The Repair Effect of bFGF on Endothelial Barrier is Partly Mediated by Cav-1
To support our hypothesis that the effect of bFGF is related to Cav-1 in vitro, Cav-1 siRNA was transfected to endothelial cells. bFGF attenuated the reduction of Cav-1 expression by OGD in both Western blot and immunofluorescence analyses (Fig. 5A, B, E), which was consistent with our in vivo results . To assess the full level of knockdown, we checked the effects of Cav-1 siRNA on Cav-1 protein under control conditions. The results showed the rate of knockdown is about 65% and a scrambled siRNA control gives no significant difference compared with normal control. Figure 6(B–E) shows that the effect of bFGF on junction proteins was significantly reversed by Cav-1 siRNA. The results of immunofluorescence further confirmed that bFGF mediating junction protein was partly through Cav-1 (Fig. 7A, B). To study the roles of bFGF and Cav-1 on endothelial barriers, cell monolayer permeability was examined by FITC–dextran permeating to the lower chamber. As illustrated in Fig. 6(F), OGD remarkably elevated the quantity of FITC–dextran across the endothelial barrier, and the knockdown of Cav-1 enhanced this disruption, demonstrating the essential role of Cav-1 in the maintenance of barrier integrity. bFGF treatment alone significantly reduced the permeability of the OGD-treated cell monolayer to FITC–dextran, suggesting the protective role of bFGF in endothelial barrier integrity, which further confirmed our results in vivo. Moreover, Cav-1 siRNA strongly reversed the effect of bFGF. These data demonstrate that the repair function of bFGF on the endothelial barrier is partly mediated by Cav-1.

Knockdown of caveolin-1 (Cav-1) reversed the protective effect of basic fibroblast growth factor (bFGF). (A) The effect of Cav-1 small interfering RNA (siRNA) under normal conditions. Data were analyzed by ANOVA (F[2, 6] = 122.751, p < 0.001). Post-hoc analyses were done using Bonferroni’s multiple comparison test (***p < 0.01). (B) The protein levels of Cav-1, p120-catenin, β-catenin, occludin and claudin-5. Densitometric analyses of (C) Cav-1, (D) adherens junction proteins and (E) tight junction proteins. Data were analyzed by ANOVA (Cav-1: F[4, 10] = 313.787, p < 0.001; p120-catenin: F[4, 10] = 65.428, p < 0.001; β-catenin: F[4, 10] = 70.302, p < 0.001; occludin: F[4, 10] = 76.124, p < 0.001; claudin-5: F[4, 10] = 320.234, p < 0.001 ). Post-hoc analyses were done using Bonferroni’s multiple comparison test [*p < 0.05, **p < 0.01, ***p < 0.001 vs control (CON), # p < 0.05, ## p < 0.01, ### p < 0.001 vs OGD, &&p < 0.01, &&&p < 0.001 vs OGD + bFGF]. (F) The permeability of FITC–dextran across endothelial cells monolayers in the endothelial cells transfected with Cav-1 siRNA under oxygen–glucose deprivation conditions. Data were analyzed by ANOVA (F[4, 10] = 100.817, p < 0.001). Post-hoc analyses were done using Bonferroni’s multiple comparison test (***p < 0.001 vs CON, # p < 0.05, ### p < 0.001 vs OGD, &&&p < 0.001 vs OGD + bFGF)

Immunofluorescence of adherens junction and tight junction proteins. (A) Immunofluorescence of p120-catenin and β-catenin. (B) Immunofluorescence of occludin and claudin-5. Scale bar = 50 μm. All results represent 3 independent experiments
Interaction of Cav-1 and FGFR1 is Essential for the Function of bFGF in Endothelial Cells
To further investigate how Cav-1 exerts its role, we examined the expression of FGFR1, a major receptor of bFGF. As shown in Fig. 8(A, B), the expression of Cav-1 decreased under OGD conditions, while FGFR1 showed no obvious change. The transfection of Cav-1 siRNA reduced the level of Cav-1 significantly, and inhibited FGFR1 expression, which could not be reversed by bFGF treatment, suggesting that Cav-1 was closely related to the regulation of FGFR1. To elucidate the interaction of Cav-1 and FGFR1, immunofluorescence detection was applied. We found the co-localization of Cav-1 and FGFR1 on the membranes of endothelial cells in normal conditions, and the level of Cav-1 and FGFR1 decreased significantly in the siRNA transfected cells. bFGF treatment could not preserve FGFR1 compared with untransfected cells (Fig. 8C). Collectively, these data suggest that Cav-1 might be essential for the protective effect of bFGF, largely owing to its co-localization with FGFR1, which might relate to the activation of downstream FGF signal pathways.

Expression and co-localization of caveolin-1 (Cav-1) and fibroblast growth factor receptor 1 (FGFR1_ in the endothelial cells. (A) Protein levels of Cav-1 and FGFR1 in the endothelial cells transfected with Cav-1 small interfering RNA under oxygen–glucose deprivation (OGD) conditions for 12 h, treated by basic fibroblast growth factor (bFGF). (B) Densitometric analyses of Cav-1 and FGFR1. Data were analyzed by ANOVA (FGFR1: F[4, 10] = 37.415, p < 0.001; Cav-1: F[4, 10] = 313.787, p < 0.001). Post-hoc analyses were done using Bonferroni’s multiple comparison test (*p < 0.05 vs CON, # p < 0.05, ## p < 0.01 vs OGD, &&p < 0.01, &&&p < 0.001 vs OGD + bFGF). (C) Co-localization detection by immunofluorescence staining of Cav-1 (green) and FGFR1 (red), and the nucleus is labeled by Hoechst (blue). Scale bar = 20 μm
Discussion
BSCB disruption occurs under various pathological conditions, such as SCI and amyotrophic lateral sclerosis. Traumatic SCI results in dramatic alteration of the spinal cord blood flow and causes systemic hypotension as a result of the interruption of descending sympathetic circuits [31]. SCI pathology resulted in rapid, permanent changes to the structure and function of the microvessels at the cellular level [32, 33]. In our study, the BSCB was significantly disrupted at 1 d after injury, via the evaluation of the extravasation of Evans Blue dye and FITC–dextran (Fig. 1), which is in agreement with previous reports [29, 34–36]. bFGF is demonstrated to preserve blood-brain barrier (BBB) integrity through RhoA inhibition after intracerebral hemorrhage in mice [23]. Moreover, in the in vitro BBB model, initial contact of glioblastoma cells with normal brain endothelial cells strengthens the barrier function via bFGF secretion, and a neutralization antibody for bFGF inhibits the recovery of BBB function [37]. Our previous reports show that bFGF treatment improves functional recovery after SCI, in part by inhibiting the apoptosis of neurons by inhibiting excessive autophagy and endoplasmic reticulum stress [21, 22]. However, whether bFGF has a beneficial role in BSCB recovery remains unclear. In this study, we found that bFGF treatment effectively prevented BSCB disruption both in vivo and in vitro. Changes in the expression and distribution of TJ and AJ proteins are closely related to the permeability of BSCB during SCI [38]. The levels of occludin, or zonula occludens 1 (ZO-1), are decreased at 1 or 3 d after SCI [25, 39]. It has been reported that occludin, claudin-5, and ZO-1 are lost or degraded at 48 h postinjury, which aggravates the disruption of BSCB integrity [40]. Our data showed that the expression of occludin and claudin-5 apparently decreased at 1 d after injury, while bFGF significantly inhibited the reduction of these molecules in vivo and in vitro (Figs. 3 and 5). Moreover, bFGF treatment enhanced the expression of AJ protein in endothelial cells. Consistent with our previous study, we concluded that bFGF had a therapeutic effect in the early stage of SCI via improving the BBB scores and pathological morphology at 3 d postinjury.
Upregulation of MMP-9 mediates BSCB disruption by degrading the basal components of TJ junctions and AJ junctions, thereby facilitating the infiltration of immune cells and initiating SCI-induced secondary damage [41]. Valproic acid inhibits ischemia-induced BBB disruption and brain edema by inhibiting MMP-9 induction and TJ breakdown. In our study, the expression and enzyme activity of MMP-9 were upregulated at 1 d after SCI, and inhibited by bFGF significantly (Fig. 2 and Supplementary Fig. 2). Moreover, the upregulation of MMP-2 also contributes to the initial opening of the BBB by degrading the basal lamina leading to neuronal injury [42]. Several studies have reported that active MMP-2 appeared at 5 d after SCI [25]. Interestingly, our results showed no obvious activation of MMP-2 at 1 d after injury (Fig. 2). In addition, the expression and activity of MMP-9 was markedly inhibited by bFGF treatment in OGD-induced endothelial cells. The mechanism of bFGF inhibiting MMP-9 activity in SCI has no related investigation, which needs to be further discussed in our next projects.
After ischemia-reperfusion in the brain, Cav-1 is remarkably downregulated in brain microvessels [43]. Cav-1 deficiency in mice led to higher MMPs activities and BBB permeability than in wild-type mice [44]. Cav-1 regulates the expression of junction-associated proteins in the brain microvascular endothelial cells; attenuated Cav-1 level is correlated with heightened permeability of endothelia [14]. In addition, Cav-1 knockout mice present higher microvascular permeability in the tumor barrier, and the loss of Cav-1 increases tumor permeability and growth, which may relate to enhanced VEGF signaling or decreased vascular endothelial cadherin [45]. The opening of AJs observed in Cav-1 knockout endothelium suggests that Cav-1 is necessary for AJ assembly or maintenance, which contributes to endothelial barrier function [15]. Our results showed that Cav-1 expression was reduced after SCI, as shown by both mRNA and protein levels (Fig. 4), similar to the progress of TJ and AJ protein expression in SCI. The expression of Cav-1 was highly decreased at 1 d after SCI, which was increased with bFGF administration (Fig. 4), suggesting that degradation of TJ and AJ proteins may be associated with the expression of Cav-1. Our data showed that the upregulation of TJ and AJ proteins by bFGF were reversed by Cav-1 siRNA. Furthermore, Cav-1 siRNA strongly increased the permeability, but with bFGF (Fig. 6). These results indicated that the repair effect of bFGF on the endothelial barrier was partially mediated by Cav-1. However, views of the role of Cav-1 in different pathologies are contradictory. In several models of adult brain injuries, Cav-1 is increased in the endothelium for several days following cold cortical injury and brain ischemia [10, 11]. In the cortical cold injury model in rats, an increased expression of Cav-1 precedes the decreased expression of occludin and claudin-5 at 2 d postinjury [11]. The Cav-1 protein in the membrane fraction of microvessels begins to upregulate at 5 min and reached a peak 10 min after treatment, which was associated with diminished expression of several TJ-associated proteins (ZO-1, occludin, and claudin-5), which led to increased BBB permeability [10]. This evidence suggests that the functions of Cav-1 are differential in different models or different organizations.
A recent study has illustrated that Cav-1 mediated growth factor-induced disassembly of adherens junctions to support tumor cell dissociation [46]. In the blood–tumor barrier, Cav-1 also participates in VEGF-mediated permeability [47]. Many studies have shown that Cav-1 is closely involved in the regulation of FGFR [48, 49]. Herein, we found the co-localization of Cav-1 and FGFR1 on the membranes of endothelial cells in normal conditions. However, the levels of Cav-1 decreased under OGD conditions, but FGFR1 did not change. Cav-1 siRNA dramatically reduced the expression of FGFR1, which could not be reversed by bFGF treatment (Fig. 8). A recent study has claimed that Cav-1 orchestrated bFGF downstream signaling control of angiogenesis in placental artery endothelial cells [50], but the interaction mechanism of Cav-1 and FGFR1 needs to be further investigated.
However, although abundant evidence suggests that bFGF has a therapeutic effect on SCI in rats [19, 22, 51], there has been no clinical trial investigating this. In acute stroke, a 24-h intravenous infusion of 5 mg bFGF has been confirmed as safe for patients and results in an improved outcome [52]. As the therapeutic potential of bFGF has been well recognized for decades, and the neuroprotection of FGF in SCI has already been translated to clinical trials [53, 54], it is reasonable to believe that bFGF will be applied to the clinic. In addition, the combination of a delivery system or special biomaterials to increase its stability and prolong the half-life might contribute to the utility of bFGF.
In conclusion, this study has demonstrated that bFGF improves the recovery of BSCB in an SCI model by increasing junction proteins and the key element Cav-1, inhibiting the expression and activation of MMP-9. In endothelial cells, bFGF treatment also increases the levels of junction proteins; Cav-1 siRNA abolished the effect of bFGF under OGD conditions, which might be related to the co-localization of Cav-1 and FGFR1. Taken together, our results suggest that bFGF may provide an effective therapeutic intervention by preventing BSCB disruption via the critical determinant Cav-1 after SCI, which involves interactions with FGFR1.
References
Kaneko S, Iwanami A, Nakamura M, et al. A selective Sema3A inhibitor enhances regenerative responses and functional recovery of the injured spinal cord. Nat Med 2006;12:1380-1389.
Mikami Y, Toda M, Watanabe M, Nakamura M, Toyama Y, Kawakami Y. A simple and reliable behavioral analysis of locomotor function after spinal cord injury in mice. Technical note. J Neurosurg 2002;97:142-147.
Jeong SR, Kwon MJ, Lee HG, et al. Hepatocyte growth factor reduces astrocytic scar formation and promotes axonal growth beyond glial scars after spinal cord injury. Exp Neurol 2012;233:312-322.
Giszter SF. Spinal cord injury: Present and future therapeutic devices and prostheses. Neurotherapeutics 2008;5:147-162.
Bartanusz V, Jezova D, Alajajian B, Digicaylioglu M. The blood–spinal cord barrier: morphology and clinical implications. Ann Neurol 2011;70:194-206.
Sharma HS. Early microvascular reactions and blood–spinal cord barrier disruption are instrumental in pathophysiology of spinal cord injury and repair: novel therapeutic strategies including nanowired drug delivery to enhance neuroprotection. J Neural Transm (Vienna) 2011;118:155-176.
Fassbender JM, Whittemore SR, Hagg T. Targeting Microvasculature for neuroprotection after SCI. Neurotherapeutics 2011;8:240-251.
Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing "preassembled signaling complexes" at the plasma membrane. J Biol Chem 1998;273:5419-5422.
Lisanti MP, Scherer PE, Tang Z, Sargiacomo M. Caveolae, caveolin and caveolin-rich membrane domains: a signalling hypothesis. Trends Cell Biol 1994;4:231-235.
Wang P, Liu Y, Shang X, Xue Y. CRM197-induced blood-brain barrier permeability increase is mediated by upregulation of caveolin-1 protein. J Mol Neurosci 2011;43:485-492.
Nag S, Venugopalan R, Stewart DJ. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol 2007;114:459-469.
Madaro L, Antonangeli F, Favia A, et al. Knock down of caveolin-1 affects morphological and functional hallmarks of human endothelial cells. J Cell Biochem 2013;114:1843-1851.
Shen J, Ma S, Chan P, et al. Nitric oxide down-regulates caveolin-1 expression in rat brains during focal cerebral ischemia and reperfusion injury. J Neurochem 2006;96:1078-1089.
Song L, Ge S, Pachter JS. Caveolin-1 regulates expression of junction-associated proteins in brain microvascular endothelial cells. Blood 2007;109:1515-1523.
Siddiqui MR, Komarova YA, Vogel SM, et al. Caveolin-1-eNOS signaling promotes p190RhoGAP-A nitration and endothelial permeability. J Cell Biol 2011;193:841-850.
Gu Y, Dee CM, Shen J. Interaction of free radicals, matrix metalloproteinases and caveolin-1 impacts blood–brain barrier permeability. Front Biosci 2011;3:1216-1231.
Schnell L, Schneider R, Kolbeck R, Barde YA, Schwab ME. Neurotrophin-3 enhances sprouting of corticospinal tract during development and after adult spinal cord lesion. Nature 1994;367:170-173.
Cheng H, Cao Y, Olson L. Spinal cord repair in adult paraplegic rats: partial restoration of hind limb function. Science 1996;273:510-513.
Rabchevsky AG, Fugaccia I, Fletcher-Turner A, Blades DA, Mattson MP, Scheff SW. Basic fibroblast growth factor (bFGF) enhances tissue sparing and functional recovery following moderate spinal cord injury. J Neurotrauma 1999;16:817-830.
Rabchevsky AG, Fugaccia I, Turner AF, Blades DA, Mattson MP, Scheff SW. Basic fibroblast growth factor (bFGF) enhances functional recovery following severe spinal cord injury to the rat. Exp Neurol 2000;164:280-291.
Zhang HY, Wang ZG, Wu FZ, et al. Regulation of autophagy and ubiquitinated protein accumulation by bFGF promotes functional recovery and neural protection in a rat model of spinal cord injury. Mol Neurobiol 2013;48:452-464.
Zhang HY, Zhang X, Wang ZG, et al. Exogenous basic fibroblast growth factor inhibits ER stress-induced apoptosis and improves recovery from spinal cord injury. CNS Neurosci Ther 2013;19:20-29.
Huang B, Krafft PR, Ma Q, et al. Fibroblast growth factors preserve blood-brain barrier integrity through RhoA inhibition after intracerebral hemorrhage in mice. Neurobiol Dis 2012;46:204-214.
Figley SA, Khosravi R, Legasto JM, Tseng YF, Fehlings MG. Characterization of vascular disruption and blood-spinal cord barrier permeability following traumatic spinal cord injury. J Neurotrauma 2014;31:541-552.
Lee JY, Kim HS, Choi HY, Oh TH, Yune TY. Fluoxetine inhibits matrix metalloprotease activation and prevents disruption of blood–spinal cord barrier after spinal cord injury. Brain 2012;135:2375-2389.
Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 1995;12:1-21.
Ma X, Zhang H, Pan Q, et al. Hypoxia/Aglycemia-induced endothelial barrier dysfunction and tight junction protein downregulation can be ameliorated by citicoline. PLOS ONE 2013;8:e82604.
Zhang HQ, Chang M, Hansen CN, Basso DM, Noble-Haeusslein LJ. Role of matrix metalloproteinases and therapeutic benefits of their inhibition in spinal cord injury. Neurotherapeutics 2011;8:206-220.
Lee JY, Choi HY, Na WH, Ju BG, Yune TY. Ghrelin inhibits BSCB disruption/hemorrhage by attenuating MMP-9 and SUR1/TrpM4 expression and activation after spinal cord injury. Biochim Biophys Acta 2014;184:2403-2412.
Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 2010;468:562-566.
Furlan JC, Fehlings MG. Cardiovascular complications after acute spinal cord injury: pathophysiology, diagnosis, and management. Neurosurg Focus 2008;25:E13.
Benton RL, Maddie MA, Minnillo DR, Hagg T, Whittemore SR. Griffonia simplicifolia isolectin B4 identifies a specific subpopulation of angiogenic blood vessels following contusive spinal cord injury in the adult mouse. J Comp Neurol 2008;507:1031-1052.
Utepbergenov DI, Mertsch K, Sporbert A, et al. Nitric oxide protects blood-brain barrier in vitro from hypoxia/reoxygenation-mediated injury. FEBS Lett 1998;424:197-201.
Popovich PG, Horner PJ, Mullin BB, Stokes BT. A quantitative spatial analysis of the blood-spinal cord barrier. I. Permeability changes after experimental spinal contusion injury. Exp Neurol 1996;142:258-275.
Noble LJ, Wrathall JR. Distribution and time course of protein extravasation in the rat spinal cord after contusive injury. Brain Res 1989;482:57-66.
Lee JY, Choi HY, Ahn HJ, Ju BG, Yune TY. Matrix metalloproteinase-3 promotes early blood–spinal cord barrier disruption and hemorrhage and impairs long-term neurological recovery after spinal cord injury. Am J Pathol 2014;184:2985-3000.
Toyoda K, Tanaka K, Nakagawa S, et al. Initial contact of glioblastoma cells with existing normal brain endothelial cells strengthen the barrier function via fibroblast growth factor 2 secretion: a new in vitro blood-brain barrier model. Cell Mol Neurobiol 2013;33:489-501.
Liebner S, Czupalla CJ, Wolburg H. Current concepts of blood-brain barrier development. Int J Develop Biol 2011;55:467-476.
Lee JY, Kim HS, Choi HY, Oh TH, Ju BG, Yune TY. Valproic acid attenuates blood-spinal cord barrier disruption by inhibiting matrix metalloprotease-9 activity and improves functional recovery after spinal cord injury. J Neurochem 2012;121:818-829.
Wu Q, Jing Y, Yuan X, et al. Melatonin treatment protects against acute spinal cord injury-induced disruption of blood spinal cord barrier in mice. J Mol Neurosci 2014;54:714-722.
Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci 2002;22:7526-7535.
Dang AB, Tay BK, Kim HT, Nauth A, Alfonso-Jaume MA, Lovett DH. Inhibition of MMP2/MMP9 after spinal cord trauma reduces apoptosis. Spine 2008;33:E576-E579.
Fu S, Gu Y, Jiang JQ, et al. Calycosin-7-O-beta-D-glucoside regulates nitric oxide /caveolin-1/matrix metalloproteinases pathway and protects blood–brain barrier integrity in experimental cerebral ischemia–reperfusion injury. J Ethnopharmacol 2014;155:692-701.
Gu Y, Zheng G, Xu M, et al. Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J Neurochem 2012;120:147-156.
Lin MI, Yu J, Murata T, Sessa WC. Caveolin-1-deficient mice have increased tumor microvascular permeability, angiogenesis, and growth. Cancer Res 2007;67:2849-2856.
Orlichenko L, Weller SG, Cao H, et al. Caveolae mediate growth factor-induced disassembly of adherens junctions to support tumor cell dissociation. Mol Biol Cell 2009;20:4140-4152.
Zhao LN, Yang ZH, Liu YH, Ying HQ, Zhang H, Xue YX. Vascular endothelial growth factor increases permeability of the blood-tumor barrier via caveolae-mediated transcellular pathway. J Mol Neurosci 2011;44:122-129.
Citores L, Wesche J, Kolpakova E, Olsnes S. Uptake and intracellular transport of acidic fibroblast growth factor: evidence for free and cytoskeleton-anchored fibroblast growth factor receptors. Mol Biol Cell 1999;10:3835-3848.
Citores L, Khnykin D, Sorensen V, et al. Modulation of intracellular transport of acidic fibroblast growth factor by mutations in the cytoplasmic receptor domain. J Cell Sci 2001;114:1677-1689.
Feng L, Liao WX, Luo Q, et al. Caveolin-1 orchestrates fibroblast growth factor 2 signaling control of angiogenesis in placental artery endothelial cell caveolae. J Cell Physiol 2012;227:2480-2491.
Liu WG, Wang ZY, Huang ZS. Bone marrow-derived mesenchymal stem cells expressing the bFGF transgene promote axon regeneration and functional recovery after spinal cord injury in rats. Neurol Res 2011;33:686-693.
Bogousslavsky J, Victor SJ, Salinas EO, et al. Fiblast (trafermin) in acute stroke: results of the European-Australian phase II/III safety and efficacy trial. Cerebrovasc Dis 2002;14:239-251.
Wu JC, Huang WC, Chen YC, et al. Acidic fibroblast growth factor for repair of human spinal cord injury: a clinical trial. J Neurosurg Spine 2011;15:216-227.
Wu JC, Huang WC, Tsai YA, Chen YC, Cheng H. Nerve repair using acidic fibroblast growth factor in human cervical spinal cord injury: a preliminary Phase I clinical study. J Neurosurg Spine 2008;8:208-214.
Acknowledgments
This study was supported, in part, by research grants from the Zhejiang Provincial Natural Science Funding (LY14H150010, LY14H170002, LY14H090013), National Natural Science Funding of China (81472165, 81200958, 81572237, 81372112, 81501953), Ninbo City Natural Science Funding (2015A610208), Zhejiang Pharmaceutical Association Project (2014ZYY36), Zhejiang Provincial Program for the Cultivation of High-level Innovative Health talents (to J.X.), Zhejiang Provincial Program of Medical and Health Science (2014KYA131), Wenzhou Program of Science and Technology (Y20140003), and State Key Basic Research Development Program (2012CB518105).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Li-Bing Ye and Xi-Chong Yu contributed equally to this work.
An erratum to this article is available at http://dx.doi.org/10.1007/s13311-016-0495-6.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Basic fibroblast growth factor (bFGF) improves functional recovery after spinal cord injury (SCI). (A) Basso, Beattie, and Bresnahan scores of each group. Data were analyzed by MANOVA (Ftime = 92.338, p < 0.001; Ftime.group = 34.662, p < 0.001; Fgroup = 5.783, p = 0.017). Intergroup comparison analyses were done using Student’s t test (**p < 0.01, ***p < 0.001, SCI vs SCI + bFGF). (B) Representative footprints obtained from each group at 14 d postinjury. (C) Hematoxylin and eosin staining of each group at 3 d after injury; 40×: scale bar = 1 mm, 200×: scale bar = 100 μm. (D) NeuN staining of each group at 3 d after injury. 40×: scale bar = 1 mm, 200×: scale bar = 100 μm. The 200× is the magnified images of anterior horn in 40×. (E) Quantitative analysis of neurons in NeuN staining. Data were analyzed by ANOVA (F[2, 6] = 41.059, p < 0.001). Post-hoc analyses were done using Bonferroni’s multiple comparison test (*p < 0.05, ***p < 0.001) (GIF 339 kb)
Supplementary Fig. 2
Basic fibroblast growth factor inhibits the expression of matrix metalloproteinase (MMP)-9 after spinal cord injury. (A) Western blot and densitometric analysis of MMP-9 at 1 d after injury. Data were analyzed by ANOVA (F[2, 6] = 69.174, P < 0.001). Post-hoc analyses were done using Bonferroni’s multiple comparison test (***p < 0.001 vs sham, ## p < 0.01 vs SCI). (B) Double immunofluorescence of MMP-9 (red) with CD31 (green) at each group. (C) Double immunofluorescence of MMP-9 (red) with NeuN (green) at each group. (D) Double immunofluorescence of MMP-9 (red) with glial fibrillary acidic protein (green) at each group. Scale bar = 50 μm (GIF 328 kb)
ESM 3
(PDF 1225 kb)
ESM 4
(PDF 1225 kb)
Rights and permissions
About this article

Cite this article
Ye, LB., Yu, XC., Xia, QH. et al. Regulation of Caveolin-1 and Junction Proteins by bFGF Contributes to the Integrity of Blood–Spinal Cord Barrier and Functional Recovery. Neurotherapeutics 13, 844–858 (2016). https://doi.org/10.1007/s13311-016-0437-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-016-0437-3