
Abstract
In this article, we propose a novel mechanism for the atomic-level processes that lead to oxide formation and eventually Pt dissolution at an oxidized Pt(111) surface. The mechanism involves a Pt extraction step followed by the substitution of chemisorbed oxygen to the subsurface. The energy diagrams of these processes have been generated using density functional theory and were analyzed to determine the critical coverages of chemisorbed oxygen for the Pt extraction and Oads substitution steps. The Pt extraction process depends on two essential conditions: (1) the local coordination of a Pt surface atom by three chemisorbed oxygen atoms at nearest-neighboring fcc adsorption sites; (2) the interaction of the buckled Pt atom with surface water molecules. Results are discussed in terms of surface charging effects caused by oxygen coverage, surface strain effects, as well the contribution from electronic interaction effects. The utility of the proposed mechanism for the understanding of Pt stability at bimetallic surfaces will be demonstrated by evaluating the energy diagram of a CuML/Pt(111) near-surface alloy.

Atomistic mechanism of Pt extraction at oxidized surfaces
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Understanding the mechanisms of noble metal dissolution under electrochemical conditions is a subject of unfading interest in corrosion science as well as energy and environmental science [1–3]. A crucial step in the comprehension of metal dissolution is to understand surface oxidation, a topic that has captivated interest for decades [4]. Experimental studies have found the formation of subsurface oxygen on oxidized transition metal surfaces [5–8]; yet, so far no coherent theoretical picture of the atomistic mechanism underlying these processes has emerged [4].
Platinum is the electrocatalytic material of choice in porous electrodes of electrochemical energy technologies such as low-temperature polymer electrolyte fuel cells (PEFC) and electrolysis cells [9]. However, it is known that typical operational conditions such as high temperature, high potential, low pH and, in particular, extensive potential cycling lead to unacceptable rates of Pt dissolution [10–12]. The kinetics of Pt dissolution in turn is correlated with loss of catalyst material, transformation of porous composite structure, decrease of active surface area, change in wettability of pore walls, decline of electrochemical performance, and ultimately the lifetime reduction of the device [13].
In spite of tremendous efforts in both experiment and theory focusing on understanding degradation issues and improving catalyst stability [14–16], the understanding of materials properties that control Pt stability and mechanisms involved in catalyst dissolution remains a controversial topic [17, 18]. Especially, the comprehension of the elementary surface processes involved is still elusive.
Pt dissolution may proceed through both electrochemical and chemical pathways [19, 20]. In a recent detailed experimental study, Furuya et al. [12] examined the influence of electrolyte composition and pH on Pt dissolution. Analysis of potential versus pH diagrams (Pourbaix diagrams) suggests that the anion nature has negligible impact on Pt dissolution; the pH on the other hand exerts a significant impact on the dissolution kinetics [12].
The works of Topalov et al. [10], Xing et al. [21], as well as Rinaldo et al. [22] revealed a staggering enhancement in Pt dissolution rate by a factor ∼2000 during voltage cycling in accelerated stress tests when the upper potential limit of the voltage cycle was increased above 1.2 VRHE. It was found consistently in these independent studies that the reduction of Pt oxide during the reduction part of the voltage cycle is responsible for the hugely increased rate of Pt dissolution. In addition, based on the in-situ electrochemical scanning tunneling microscopy (STM) observation, Wakisaka et al. [23] have found that the irreversible morphological changes at Pt(111) occur predominantly during the reduction phase of oxide. The underlying process is sensitive to the formation of the so-called as α-oxide phase [24] that forms as a surface layer upon ramping up the potential to values above ∼1.1 VRHE [11].
On the other hand, there is also a much smaller amount of Pt dissolution that is found in potential hold experiments at potentials below 1.1 VRHE. The rate of this process does not depend directly on the electrode potential, but it is affected by the amount of surface oxide, which in turn determines the surface excess energy or surface tension of Pt nanoparticles as well as the rate constant of dissolution [19, 20].
Analysis of the cyclic voltammetry (CV) data of polycrystalline Pt in 0.5 M aqueous H2SO4, correlated with electrochemical quartz-crystal nanobalance data revealed that the onset potential of surface oxidation lies at E ≈ 0.85 VRHE [5]. The adsorbed surface species at Pt that is formed by water oxidation is usually identified as chemisorbed oxygen (Oads at 0.85 < E < 1.10 VRHE), which forms up to half of a monolayer (ML) coverage [5]. As the potential increases, further discharge of water molecules forms additional Oads that is susceptible to undergoing interfacial exchange with Pt surface atoms; this so-called place-exchange is a basic process involved in irreversible oxide growth [17, 25–27]. In situ X-ray scattering [28], as well as STM studies under ultrahigh vacuum conditions [29] confirmed the existence of interfacial place-exchange at Pt(111). The formation of subsurface oxygen has also been observed in experiments on other oxidized transition metal surfaces [6–8].
Modeling studies have been performed to rationalize the kinetics of the various stages of Pt oxide formation, growth and reduction, as well, its correlation to Pt dissolution at extended surfaces and in nanoparticle systems [3, 18–22, 30, 31]. A recent kinetic model of Rinaldo et al. provides a comprehensive picture of surface electrochemical processes during oxide formation and reduction at Pt(111) in the voltage range of 0.65–1.15 VRHE [31]. Density functional theory (DFT), molecular dynamics (MD), and ab initio MD have also been employed to study involved reaction mechanisms at atomistic scales [32–37].
Fantauzzi et al. [32] have performed reactive force field (ReaxFF) MD calculations to determine the surface energies for structures with up to one monolayer of oxygen on Pt(111). Their results reveal four stable surface phases corresponding to pure adsorbate, low-coverage and high-coverage buckled, and subsurface-oxygen structures, respectively. They have found that stable phases of Pt(111) with oxygen coverage beyond 0.56 ML contain subsurface oxygen [32]. The work of Gu and Balbuena [36] suggests that the tetra-I site is the energetically preferred site for adsorption of atomic oxygen in the subsurface of Pt(111). However, the large activation barrier of ∼2.5 eV for the transfer of surface oxygen to this site seemed to render this place-exchange a highly unlikely event [36].
In an earlier work, Hawkins et al. [34] had used DFT to study the buckling of Pt atoms at oxygenated Pt(111) surfaces in vacuum conditions. They obtained minimum energy pathways (MEPs) for the processes of Oads atom diffusing from the surface fcc site to the Pt buckled hcp site, followed by diffusion to the subsurface tetra-I site at the coverages of 0.25, 0.5, and 0.5625 ML. In their work, it was found that Pt buckling by ∼1.7 Å is energetically favorable at oxygen coverage of 0.5625 ML, but not at 0.50 or 0.25 ML O. However, as mentioned in their article, the MEP for higher Oads coverages was not found due to the convergence failure of nudged elastic band calculations at 0.75 and 1.0 ML [34]. In addition, the possible role of surface water molecules in this process was not explored.
Considering that the formation of subsurface oxygen at high coverages has also been seen experimentally on other oxidized transition metal surfaces [6–8], we strive to find a universal mechanism of metal atom extraction at an oxidized Pt(111) surface and subsequent transfer of an adsorbed surface oxygen atom to the subsurface. We will discuss that the Pt extraction process depends on two essential conditions: (1) local coordination of Pt by three chemisorbed oxygen atoms in neighboring fcc adsorption sites; (2) the interaction of the buckled Pt surface atom with surface water molecules. From a geometrical point of view, the first condition is fulfilled above 0.50 ML O at Pt(111). Substitution of Oads into the vacancy created by the extracted Pt atom becomes energetically favorable above 0.75 ML O, as next nearest neighbor hollow fcc sites around the Pt atom start to be occupied. We will demonstrate that the extracted state is energetically more stable than the buckled state proposed in [34].
Computational Methods
All electronic structure calculations were carried out with the periodic DFT package VASP [38]. The ionic cores were represented by projected augmented waves [39]. The Kohn-Sham one-electron wave functions were expanded in a plane wave basis set up to an energy cutoff of 400 eV. Exchange-correlation effects were incorporated within the generalized gradient approximation (GGA), using the exchange-correlation functional by Perdew, Burke, and Ernzerhof (PBE) [40]. This functional consistently describes the properties of water at metal surfaces [41, 42]. Geometry optimization studies were terminated when all forces on ions were less than 0.05 eV Å−1. Minimum energy structures were determined using Monkhorst-Pack k‐point sampling grids with 3 × 3 × 1 k points [43].
The surface was modeled by a super-cell containing a four-layer slab of Pt(111) and a 2√3 × 2√3-R30° water layer with hexagonal structure on one side of the slab. Our model included 48 Pt atoms per slab and 8 water molecules in the unit cell. Repeated slabs were separated by a vacuum region of 15 Å. In all geometry optimizations, the two bottom layers of the slab were fixed, while the two top layers together with adsorbates and the water layer were allowed to relax. A standard dipole correction scheme [44], implemented in VASP, compensated the unphysical interaction between opposite surfaces that correspond to neighboring images across the vacuum region of the periodically repeated slab.
To account for the orientation of water molecules at the interface, we evaluated two mixed-phase water structures with (1) three H-up and one H-down as well as four flat-lying water molecules and (2) three H-down and one H-up as well as four flat-lying water molecules (structures were shown in SI). We note that the preferred orientation of water molecules depends on electrode potential, oxygen adsorption state, and total surface charge density; the positively (or negatively) charged surface stabilizes the H-up (or H-down) structures [45, 46]. In this article, we compare minimum energy pathways of the extraction mechanism for the two interfacial water structures with fixed orientation.
Results and Discussion
Mechanism of Pt atom extraction at oxidized (111) surfaces
We were interested in generating the energy diagram of the following sequence of elementary surface processes: (1) extraction of a Pt surface atom along the surface normal (Z-coordinate) and (2) transfer of an adsorbed oxygen atom into the vacant site left by the Pt atom. For this purpose, we utilized the constrained optimization method [47–50]. In this technique, the Z variable of the transferring atomic nucleus is constrained in defined steps, between values corresponding to the fully optimized initial and final states. At each of the Z values, a full geometry optimization is performed for all other degrees of the freedom. This method allows the molecules to rotate and translate subject to the above constraint. Using this procedure, we chose equal increments of Z for the Pt atom in the Pt extraction step, i.e., between the fully optimized structures with Pt atom in the initial lattice position and in the extracted state by two water molecules. Thereafter, we translated the Oads atom in equal steps to also create the trajectory along Z for the Oads substitution step. This method is efficient and reliable under the premise that the Z-coordinate represents a suitable reaction coordinate.
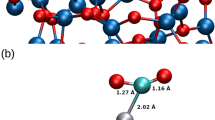
Figure 1 illustrates the proposed mechanism of Pt atom extraction and Oads substitution into the sublayer. In the buckling step, the threefold Oads-coordinated Pt (labeled as Pt45) is initially lifted by ∼1.7 Å from the original lattice position to assume an energetically more favorable position above the surface. Formation of this intermediate state, referred to as buckled hcp in [34], is accompanied by a lateral move of one of the neighboring oxygen atoms (labeled as O1) from its initial fcc position to the hcp site. Thereafter, two surface water molecules facilitate the complete extraction of Pt atom, with an overall displacement of ∼2.6 Å from the initial lattice position, to reach an energetically more stable state. Following Pt extraction by ∼2.6 Å, one of the neighboring oxygen atoms, which was not bonded to the detached Pt atom (shown as O6 in Fig. 1), substitutes into the subsurface filling the tetra-I site underneath the hcp site. The energetics of these processes is sensitive to the Oads coverage as will be discussed.

Illustration of the mechanism of Pt extraction and Oads substitution at oxygen-covered Pt(111); gray, red, blue, white, and green colors show atoms of Pt, chemisorbed oxygen, oxygen in water layer, hydrogen, and the local Pt site involved in this mechanism, respectively. Active atoms are labeled with numbers: Pt45 is the extraction site; O1, O6, and O14 are the chemisorbed oxygen atoms at fcc sites around Pt45; O1 is the diffused chemisorbed oxygen atom in the buckling step; O6 is the transferred oxygen atom in the Oads substitution step; O7 and O15 are the oxygen atoms in water molecules involved in the extraction step (color figure online)
Figure 2a, b shows the energy diagrams of the processes illustrated in Fig. 1 as a function of Oads coverage and for the two opposite orientations of water layer. Comparing the energy diagrams, it is clear that the impact of Oads coverage on the MEP is dominant, while the water orientation plays a minor role. By increasing the coverage above 0.5 ML O, geometrically threefold Oads coordinated local Pt sites start to form, which as will be shown, is the decisive local criterion for the Pt extraction; above this critical coverage, concerted sequences of Pt extraction and Oads substitution at the threefold coordinated Pt site leads to an energetically more favorable place-exchanged state. It should be noted that our unit cell size only allows us to change the coverage with an increment of 8.33 %; thus, simulations with larger unit cell sizes would unequivocally show this effect for any coverage above 0.5 ML O. The activation barrier energies for the Pt buckling step (with ΔZ ∼1.7 Å) were found to be ∼0.37 eV for 0.58 ML O, and ∼0.50 eV for 0.75 and 1.0 ML O. For this step, the value of Pt displacement is in very good agreement with STM images under ultrahigh vacuum conditions showing ‘protrusions’ interpreted as buckled Pt atoms with an apparent height of 1.7 Å [29].

Minimum energy pathways for Pt extraction and Oads substitution at coverages above 0.50 ML O, and for two opposite orientations of water layer: a three H-up and one H-down, b three H-down and one H-up
Following the buckling step (with ΔZ ∼1.7 Å), the Pt atom is completely extracted from the surface and lifted by ∼2.6 Å relative to its initial position at the surface in order to relax into even more stable state which corresponds to the second minimum in Fig. 2. For the extraction step, two flat-lying surface water molecules are necessary to be involved in the process of forming a chemical complex of [PtO2(H2O)2] at the interface. Notably, we did not find a stable state with only one water molecule involved in the extraction process. The 3D charge density difference isosurfaces for the [PtO2(H2O)2] are shown in Fig. 3, illustrating the electropositive (blue) and electronegative (yellow) zones that occurs between the oxygen atoms of two water molecules (shown as O7 and O15) and the extracted Pt, as well as the extracted Pt and two neighboring chemisorbed oxygen atoms (shown as O1 and O14).

The 3D charge density difference isosurfaces of Pt45, extracted by two water molecules at 1.0 ML O coverage; blue shows negative isosurfaces (charge depletion zones); yellow shows positive isosurfaces (charge accumulation zones); the isosurfaces value is taken as 0.002 e per Å−3 (color figure online)
In the final step, the neighboring oxygen atom that was not bonded to the extracted Pt (shown as O6 in Figs. 1 and 3) transfers to the subsurface to become adsorbed on the tetra-I site underneath the hcp site. This final step is also sensitive to the oxygen coverage and is only energetically favorable at coverages above 0.75 ML O, as the second nearest neighbor hollow fcc sites around the Pt atom start to be occupied with Oads (also see Fig. 5 for clarification). This finding is in agreement with the results presented in [32]. The reaction energies corresponding to Fig. 2 and Pt displacement from surface for all the studied systems are provided in Table 1.
We further explored the effect of Pt coordination with one and two chemisorbed oxygen at lower coverages on the buckling and extraction processes. Figure 4a shows the energy diagrams of Pt displacement at θO = 0.42 ML O (where Pt surface atom is coordinated with one Oads) and at θO = 0.50 ML O (as Pt surface atom is coordinated with two Oads, also see Fig. 5), in comparison with that for triply coordinated Pt (at θO = 0.58 ML O). As indicated, the formation of threefold Oads-coordinated Pt atom is a critical condition for the Pt atom extraction at Pt(111), while Pt detachment from surface is energetically unfavorable for lower coordination with Oads.

a Comparison between the minimum energy pathways for Pt extraction at coverages below and above 0.50 ML O. b Comparison between the minimum energy pathways for Pt extraction and Oads substitution with and without water layer at 0.58 ML O

a–e Top views of the most stable O surface atom configurations for coverage of a 0.42 ML, b 0.50 ML, c 0.58 ML, d 0.75 ML and e 1.0 ML; gray and red colors are used to represent Pt and Oads, respectively. Green shows the local Pt atom, labeled as Pt45, which participates in the mechanism of extraction; f–j the corresponding top views of the charge density difference isosurfaces. Blue shows negative isosurfaces (charge depletion zones); yellow shows positive isosurfaces (charge accumulation zones); the isosurfaces value is taken as 0.002 e per Å−3 (color figure online)
Interestingly, the role of surface water molecules is indispensable to stabilize the extracted Pt atom and provide a minimum energy pathway for Oads substitution. In Fig. 4b, the mechanism is evaluated in the absence of a water layer. In this case, although the Pt buckling step remains energetically viable at θO > 0.50 ML, the extraction step is prohibited without water molecules, i.e., there is no stable state for the Pt extraction; moreover, without surface water molecules Oads substitution involves a large activation barrier of ∼1 eV, which renders the substitution step unlikely.
Surface Charging Effects at Oxygen-Covered Pt(111)
We discuss results in the context of surface charging behavior at oxidized Pt(111) slightly below and above the critical coverage of 0.50 ML. Figure 5a–e illustrates the most stable oxygen surface atom configurations for Oads coverages, ranging from 0.42 to 1.0 ML. As θO increases in this range, the calculated average Bader charge [51] per atom of the surface Pt layer grows from 0.27 e0 per atom to 0.67 e0 per atom (see Table S1 in SI), where e0 is the elementary charge.
As discussed, for θO > 0.50 ML, local Pt sites appear (shown as Pt45 in Fig. 5) that are coordinated by chemisorbed oxygen atoms in each adjacent three-fold hollow fcc site. This state of high local Oads coordination induces electronic charge depletion at triply coordinated Pt surface atoms that exceeds ∼0.7 e0. The magnitude of the electronic charge depletion is larger for triply coordinated Pt as compared to doubly (0.49 e0/Pt) or singly (0.26 e0/Pt) coordinated Pt (see Table S1 in SI).
Plots in Fig. 5f–j show isosurfaces of the charge density difference, defined as
where ρtotal is the charge density of the total system; ρslab and ρads are separately calculated charge density for the bare slab and adsorbed oxygen atoms fixed in their positions in the overall system, respectively. The isosurfaces in Fig. 5f–j illustrate the charge transfer that has occurred between Pt surface and chemisorbed oxygen atoms. Electronegative zones (yellow) are created by electron transfer from Pt atoms to chemisorbed oxygen atoms, which lead to the electropositive zones at surface Pt atoms (blue). As shown, as the Pt site is coordinated with three oxygen atoms for coverages above 0.50 ML, it becomes more electropositive inducing a larger local polarization at the site.
To rationalize the local driving force for the Pt extraction process, we estimated the z-component of the local dipole moment at Pt45. The simulated slab is asymmetric with a chemisorbed oxygen adlayer formed only at one side. This asymmetry causes a redistribution of charge between the two slab surfaces. The net amount of the total surface charge can be determined after application of the dipole correction to eliminate the electric field between periodic images of the slab. We determined the total surface charge, Q, induced by this correction for the Oads-free backside of the simulated slab. For electroneutrality, an equal but opposite total amount of charge, −Q, must be deposited at the Oads-side of the slab. For simplicity, we assume that this total charge is distributed in equal amounts over the surface Pt atoms at the oxygen-side of the slab, corresponding to a value of –Q/n per surface atom, where n = 12 is the number of surface Pt atoms per unit cell. We are interested in the local dipole moment induced by chemisorbed oxygen at the Pt45 site. We determine the Bader charge [51] for this atom relative to the corresponding uncharged atom, qPt45, and obtain the induced Oads-dipole charge as δq = qPt45 + Q/n. As for the dipole length we use the distance dz along z from the center of Pt45 to the plane of the adsorbed oxygen atoms coordinated to this site. The dipole moment is calculated as μ45 = δq × dz. Using the same method, we calculated the local dipole moment between the extracted Pt site and two involved surface water molecules after completion of the Pt extraction and Oads substitution steps, in order to estimate the net dipole moment for these states (see Table S1 in SI). Values of μ45 are shown in Table 2 for the different oxygen coverages considered.
The calculated dipole moments are consistent with values reported in [17]. Upon coordination of Pt45 with three Oads, a strong local dipole moment (>3.5 D) directed toward the surface occurs, which destabilizes Pt45 and leads to its extraction from the surface. As a result of Pt extraction and O substitution, the local dipole moment, μ45, drops to much smaller values (∼0.1 D) and eventually changes orientation at high oxygen coverage of 1.0 ML, thereby stabilizing the surface at the site at which Pt extraction and Oads substitution have occurred.
Furthermore, we generated the planar average of the induced charge density along the surface normal for various oxygen coverages, shown in Fig. 6a. The plots were obtained by calculating the difference between the total plane-averaged charge density of the interfacial system and separately obtained charge densities of the Pt slab and the adsorbates (defined as Δ\( \boldsymbol{\lambda} \)) as a function of z. The adsorption of oxygen induces a large polarization at the interface with the dipole moment directed towards the surface. However, as shown in Fig. 6b, Pt extraction and Oads substitution steps modify the surface dipole moment, which, as discussed, results in obtaining an energetically more stable state.

Comparison between the planar average of the induced charge density along the surface normal for a Pt(111) with oxygen coverages above 0.50 ML; and b Pt(111) with surface coverage of 1.0 ML O and that for the Oads substituted system. c Calculated work function as a function of surface oxygen coverage for a 2 × 2 Pt(111) system
The surface potential energy is a function of the charge density distribution, which varies with θO. We calculated the work function of the surface, Φ, which is the energy required to bring an electron from inside of the slab to the vacuum. Φ is calculated as the difference between the potential energy of an electron in the middle of the vacuum and in the middle of the Pt slab. Figure 6c shows Φ as a function of oxygen coverage for the 2 × 2 Pt(111) reference systems; the work function increases with increasing θO due to the positive charge carried on the surface layer of Pt atoms.
Coverage by oxygen also affects the surface strain as well the electronic interactions. We studied the effect of coverage on the lateral strain using a 2 × 2 Pt(111) system. For this aim, structural optimizations of the slab for varying unit cell parameter were carried out in order to find the energy profiles as a function of the lateral strain. As shown in Fig. 7, results suggest that for coverage >0.50 ML surface contraction is favored, with a maximum of ∼3 % at 1.0 ML, but below <0.50 ML O lateral expansion is favored. This result is also in agreement with the recent DFT study by Li et al. [52].

Percent surface strain caused by oxygen coverage at Pt(111). The energy profile is relative to the slab optimized in its bulk lattice constant. Coverage above 0.5 ML O results in surface contraction and coverage below 0.5 ML O leads to surface expansion
To understand the effect of oxygen coverage on the electronic interactions, we utilized the d-band model proposed by Hammer and Nørskov [53]. In this model, an upshift in the average energy of the d-states relative to the Fermi level, so-called as the d-band center, results in a stronger interaction between surface atoms and adsorbates. We first calculated the binding energy of oxygen as a function of coverage for a 2 × 2 Pt(111) slab system using the following equation:
where n is the number of oxygen atoms in the unit cell, and \( {\mathrm{E}}_{O_2} \) was considered as 5.87 eV [54]. As shown in Fig. 8a, increasing the oxygen coverage lowers the strength of the interaction of oxygen with the surface and results in decreasing the binding energy. Figure 8b shows the d-projected density of states (DOS) of surface Pt atoms as a function of coverage for a 2 × 2 Pt(111) system. The d-band is narrower and higher in energy for clean surface and as the coverage increases the d-band center shifts down to more negative values. Consistently, as shown in Fig. 8c, downshift in the d-band center with coverage lowers the strength of interaction of oxygen with the surface.

a Calculated binding energy of oxygen for varying coverages at Pt(111). b d-band projected density of states of surface Pt atoms for varying oxygen coverages at Pt(111). c Variations in the binding energy of oxygen as a function of the d-band center of Pt surface atoms
Furthermore, we examined the proposed mechanism of Pt extraction for a CuML/Pt(111) near-surface alloy system. In this model system, a monolayer of Cu atoms was considered in the subsurface of the Pt(111) slab. Figure 9 compares the energy diagrams for the Pt extraction and Oads substitution processes on CuML/Pt(111) with that of Pt(111) at θO = 0.58 ML. For this coverage, the extraction energy for CuML/Pt(111) was found to be −0.17 eV while that for Pt(111) was obtained as −0.82 eV. In addition, the activation barrier for the buckling step at CuML/Pt(111) was obtained as 0.63 eV which is larger as compared to 0.36 eV for Pt(111). Results suggest that the extraction is both kinetically and energetically less favored for CuML/Pt(111) system. Indeed, a recent study has reported the noticeable enhancement in the Pt stability of CuML/Pt(111) [55]. In the future, we will continue this study to obtain a relation between the extraction energy and segregation energy for near-surface alloys with a range of different metals in the sublayer, in order to unravel systematic trend in the stability of these systems.

Comparison between the energy diagrams of Pt extraction and Oads substitution mechanism for CuML/Pt(111) and Pt(111) systems at 0.58 ML O
Conclusions
We proposed a new mechanism of Pt extraction and Oads substitution that depends on stringent local selection criteria: (i) the destabilization of surface Pt atoms by the formation of chemisorbed Oads in three adjacent fcc adsorption sites and (ii) the presence of surface water molecules to stabilize the extracted Pt. Results suggest that the extraction mechanism is favored above 0.5 ML O, where the first criteria is fulfilled. Oads substitution to the vacancy left by the extracted Pt, on the other hand, is energetically favorable above 0.75 ML O, as the next fcc nearest neighbors of the Pt site start to be occupied by oxygen. We discussed that the strong induced local dipole moment is the main driving force for Pt extraction; in addition, the role of surface water molecules is essential to stabilize the extracted Pt atom and provide a minimum energy path for the Oads substitution. Moreover, we studied the effect of oxygen coverage on surface strain and electronic interactions. Increase in the coverage above 0.5 ML O results in surface contraction, while for coverages below 0.5 ML O surface expansion is favored. Increase in the coverage also shifts the d-band center down and leads to lowering the binding energy of oxygen. Furthermore, we generated the energy diagram of Pt extraction and Oads substitution mechanism for the CuML/Pt(111) system. As compared to Pt(111), it was found that the Pt extraction from surface is both kinetically and energetically less favored for this system suggesting the stronger interaction between Pt surface atom and Cu subsurface atoms.
The approach of anionic species from the solution could fulfill a similar role as interfacial water dipoles in enabling the Pt extraction process. We expect that the proposed re-interpretation of interfacial place-exchange as a local process will make it possible to extend the proposed mechanism to different Pt surface structures, ordered or disordered, toward understanding the mechanisms of Pt dissolution and predicting the stability of various surface structures.
References
D.A.J. Rand, R. Woods, J. Electroanal. Chem. Interfacial Electrochem. 35, 209 (1972)
H. Angerstein-Kozlowska, B.E. Conway, W.B.A. Sharp, J. Electroanal. Chem. Interfacial Electrochem. 43, 9 (1973)
B.E. Conway, Prog. Surf. Sci. 49, 331 (1995)
S. Cherevko, N. Kulyk, K.J. Mayrhofer, Nano Energy (2016). doi:10.1016/j.nanoen.2016.03.005
G. Jerkiewicz, G. Vatankhah, J. Lessard, M.P. Soriaga, Y.S. Park, Electrochim. Acta 49, 1451 (2004)
A.R. Kortan, R.L. Park, Phys. Rev. B 23, 6340 (1981)
F.P. Leisenberger, G. Koller, M. Sock, S. Surnev, M.G. Ramsey, F.P. Netzer, B. Klötzer, K. Hayek, Surf. Sci. 445, 380 (2000)
Y.B. He, M. Knapp, E. Lundgren, H. Over, J. Phys. Chem. B 109, 21825 (2005)
M.K. Debe, Nature 486, 43 (2012)
A.A. Topalov, I. Katsounaros, M. Auinger, S. Cherevko, J.C. Meier, S.O. Klemm, K.J.J. Mayrhofer, Angew. Chem. Int. Ed. 51, 12613 (2012)
A.A. Topalov, S. Cherevko, A.R. Zeradjanin, J.C. Meier, I. Katsounaros, K.J.J. Mayrhofer, Chem. Sci. 5, 631 (2014)
Y. Furuya, T. Mashio, A. Ohma, M. Tian, F. Kaveh, D. Beauchemin, G. Jerkiewicz, ACS Catal. 5, 2605 (2015)
M. Eikerling, A. Kulikovsky, Polymer Electrolyte Fuel Cells: Physical Principles of Materials and Operation (CRC Press, 2014)
M.A. Hoque, F.M. Hassan, D. Higgins, J.-Y. Choi, M. Pritzker, S. Knights, S. Ye, Z. Chen, Adv. Mater. 27, 1229 (2015)
Y. Kang, J. Snyder, M. Chi, D. Li, K.L. More, N.M. Marković, V.R. Stamenković, Nano Lett. 14, 6361 (2014)
B. Li, D.C. Higgins, Q. Xiao, D. Yang, C. Zhng, M. Cai, Z. Chen, J. Ma, Appl. Catal. B Environ. 162, 133 (2015)
Y. Furuya, T. Mashio, A. Ohma, N. Dale, K. Oshihara, G. Jerkiewicz, J. Chem. Phys. 141, 164705 (2014)
B.E. Conway, B. Barnett, H. Angerstein-Kozlowska, B.V. Tilak, J. Chem. Phys. 93, 8361 (1990)
S.G. Rinaldo, J. Stumper, M. Eikerling, J. Phys. Chem. C 114, 5773 (2010)
S.G. Rinaldo, W. Lee, J. Stumper, M. Eikerling, Phys. Rev. E 86, 041601 (2012)
L. Xing, M.A. Hossain, M. Tian, D. Beauchemin, K.T. Adjemian, G. Jerkiewicz, Electrocatalysis 5, 96 (2014)
S.G. Rinaldo, P. Urchaga, J. Hu, W. Lee, J. Stumper, C. Rice, M. Eikerling, Phys. Chem. Chem. Phys. 16, 26876 (2014)
M. Wakisaka, S. Asizawa, H. Uchida, M. Watanabe, Phys. Chem. Chem. Phys. 12, 4184 (2010)
A. Sun, J. Franc, D.D. Macdonald, J. Electrochem. Soc. 153, B260 (2006)
R.R. Sasaki, K. Marinković, N. Isaacs, H.S. Adzic, ACS Catal. 6, 69 (2016)
A.M. Gómez-Marín, J.M. Feliu, Electrochim. Acta 82, 558 (2012)
A. Björling, E. Ahlberg, J.M. Feliu, Electrochem. Commun. 12, 359 (2010)
H. You, D.J. Zurawski, Z. Nagy, R.M. Yonco, J. Chem. Phys. 100, 4699 (1994)
S.P. Devarajan, J.A. Hinojosa, J.F. Weaver, Surf. Sci. 602, 3116 (2008)
R.M. Darling, J.P. Meyers, J. Electrochem. Soc. 150, A1523 (2003)
S.G. Rinaldo, W. Lee, J. Stumper, M. Eikerling, Electrocatalysis 5, 262 (2014)
A.D. Fantauzzi, J.E. Mueller, L. Sabo, A.C.T. van Duin, T. Jacob, ChemPhysChem 16, 2797 (2015)
P.B. Balbuena, R. Callejas-Tovar, P. Hirunsit, J.M. Martínez De La Hoz, Y. Ma, G.E. Ramírez-Caballero, Top. Catal. 55, 322 (2012)
J.M. Hawkins, J.F. Weaver, A. Asthagiri, Phys. Rev. B 79, 125413 (2009)
P.B. de la Hoz, J.M.M. León-Quintero, D.F. Hirunsit, P. Balbuena, Chem. Phys. Lett. 498, 328 (2010)
Z.H. Gu, P.B. Balbuena, J. Phys. Chem. C 111, 17388 (2007)
E.F. Holby, J. Greeley, D. Morgan, J. Phys. Chem. C 116, 9942 (2012)
G. Kresse, J. Furthmüller, Phys. Rev. B 54, 11169 (1996)
P.E. Blöchl, Phys. Rev. B 50, 17953 (1994)
J. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996)
S. Schnur, A. Groß, New J. Phys. 11, 125003 (2009)
K. Tonigold, A. Groß, J. Comput. Chem. 33, 695 (2012)
J.D. Pack, H.J. Monkhorst, Phys. Rev. B 16, 1748 (1977)
M. Neugebauer, J. Scheffler, Phys. Rev. B 46, 16067 (1992)
J.S. Filhol, M.L. Doublet, Catal. Today 202, 87 (2013)
M.J. Eslamibidgoli, M.H. Eikerling, ACS Catal. 5, 6090 (2015)
A. Michaelides, P. Hu, J. Am. Chem. Soc. 122, 9866 (2000)
A. Michaelides, P. Hu, A. Alavi, J. Chem. Phys. 111, 1343 (1999)
L. Yu, X. Pan, X. Cao, P. Hu, X. Bao, J. Catal. 282, 183 (2011)
Z.P. Liu, P. Hu, J. Am. Chem. Soc. 125, 1958 (2003)
G. Henkelman, A. Arnaldsson, H. Jónsson, Comput. Mater. Sci. 36, 354 (2006)
L. Li, F. Abild-Pedersen, J. Greeley, J.K. Nørskov, J. Phys. Chem. Lett. 6, 3797 (2015)
B. Hammer, J.K. Nørskov, Surf. Sci. 343, 211 (1995)
A. Eichler, F. Mittendorfer, J. Hafner, Phys. Rev. B 62, 4744 (2000)
J. Tymoczko, F. Calle-Vallejo, V. Čolić, W. Schuhmann, A.S. Bandarenka, Electrochim. Acta 179, 469 (2015)
Acknowledgments
This research was conducted as a part of the Catalysis Research for Polymer Electrolyte Fuel Cell (CaRPE-FC) Network, administered by Simon Fraser University and funded by Automotive Partnership Canada grant No. APCPJ 417858 – 11, through the National Sciences and Engineering Research Council of Canada (NSERC)). Computational studies were performed with resources provided by WestGrid and Compute Canada.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 940 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Eslamibidgoli, M.J., Eikerling, M.H. Atomistic Mechanism of Pt Extraction at Oxidized Surfaces: Insights from DFT. Electrocatalysis 7, 345–354 (2016). https://doi.org/10.1007/s12678-016-0313-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-016-0313-2