
Abstract
We studied the genetic resistance to Campylobacter coli and Campylobacter jejuni in wild boar using both STR analysis and genome-wide association studies (GWAS). A total of 60 wild boars hunted in Tuscany (Italy) during the 2018/2019 hunting season were analyzed and genotyped. During postmortem operations, fecal swabs, liver samples and kidneys were collected. Two groups of animals were considered for the statistical analysis: 28 Campylobacter positive (22 for C. coli and 6 for C. jejuni) and 32 Campylobacter negative. Regarding STR analysis, 15 markers belonging to a marker panel validated by the International Society of Animal Genetics (ISAG) for swine were used: for each marker, alleles and genotype frequencies between the two groups of animals were compared using the Chi-square test and Fisher’s exact tests. To analyze the genetic variability within groups, the following parameters were computed: molecular coancestry coefficients (fij), kinship distance (Dk), inbreeding coefficient (Fi), and genetic similarities (GS). The internal relatedness (IR) was also calculated, and ANOVA was used to verify the relationships between IR and Campylobacter groups. For GWAS, the Geneseek Genomic Profiler Porcine HD (70 k), containing 62,330 SNPs, was used. No differences in the internal relatedness (IR) were observed between the two groups (F = 5.64, P = 0.065) and no significant association between STRs and SNPs and Campylobacter positivity was observed. Although genetic resistance to bacterial diseases is often regulated by multiple genes controlling different processes of the host–pathogen interaction, in our studies no candidate genes that could be directly or indirectly involved in the development of the disease were identified.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Wild boar populations in Europe have significantly increased since the 1980s and they are the second most abundant wild ungulate in Europe with close to 4 million animals (Massei et al. 2015; Iacolina et al. 2018). In Italy, wild boar population is largely diffused, reaching high density level in some specific areas (Pittiglio et al. 2018) such as Tuscany region, which is an area particularly suitable for wild boar, as suggested by the high number of animals hunted in this zone (Carnevali et al. 2009; Pittiglio et al. 2018). The geographical range of this omnivore species reflects its great adaptive potential to live in various climatic zones, as well as in those environments subject to major anthropogenic impact, coupled with its high reproductive rates. The high density of wild boars in a determinate area represents a serious problem for the agriculture, causing extensive damage to croplands (Lombardini et al. 2017), particularly maize, wheat, potatoes, rye and grassland; yearly compensation payments in Europe amount to several million euros (Johann et al. 2020). Wild boar populations are identified as one of the “100 of the World’s Worst Invasive Alien Species” (McClure et al. 2018; Risch et al. 2021); indeed, many studies show that wild boars can have a negative impact on biodiversity due to the predation upon invertebrates, mainly earthworm, small vertebrates and eggs of ground-nesting birds (Mori et al. 2020; Cappa et al. 2021) threatening 672 taxa in 54 different countries across the globe (Risch et al. 2021). In addition to their direct impacts on both wildlife and plant communities, they are generally known to disturb ecosystem structure due to their unique rooting and digging behavior (Risch et al. 2021).
Furthermore, this species could represent a severe hazard for both human health and animals, especially domestic pigs which belong to the same species and share most of their pathogens (Boadella et al. 2012; Malmsten et al. 2018). In fact, it is known that wild boar can be the host for different etiological agents, thus contributing to maintaining and/or disseminating important zoonotic diseases (Blanchong et al. 2016) caused by viral, bacterial and parasitic pathogens, such as hepatitis E, tuberculosis, brucellosis, salmonellosis, yersiniosis, toxoplasmosis and trichinellosis (Rugna et al. 2014; Fredriksson-Ahomaa 2019), besides animal diseases, such as African Swine fever, transmissible to farm animals (Gavier-Widén et al. 2015) with great economic impact.
Among the pathogens transmissible by wild boars, Campylobacter spp. is one of the major human foodborne pathogens around the world. Among zoonoses, Campylobacter is the most reported cause of gastrointestinal infection in Europe (almost 220,700 cases), with the annual number of cases exceeding those caused by the second and the third cause, Salmonella (about 88,000 cases) and Shiga toxin-producing (STEC) Escherichia coli (about 8000 cases) (EFSA and ECDC 2021). Among the species in the Campylobacter genus, Campylobacter jejuni and Campylobacter coli are the two species mostly involved in human campylobacteriosis (EFSA and ECDC 2021).
Bloody diarrhea, fever, stomach cramps, abdominal pain, nausea and vomiting are the main symptoms of campylobacteriosis in humans. The infection is usually self-limiting and resolves after few days; however, especially in patients with a compromised immune system, it can lead to severe and persistent bacteremia (Fernández-Cruz et al. 2010). Campylobacter infection can also lead to chronic gastrointestinal pathologies (Riddle et al. 2012), as well as severe extraintestinal pathologies, such as Guillan–Barré syndrome and reactive arthritis (Callahan et al. 2021).
Antimicrobial resistance against antimicrobials of common use in therapy (fluoroquinolones, tetracyclines, aminoglycosides and macrolides, with different percentages) is well known in Campylobacter strains either of human or animal origin. Antibiotic resistance represents a major challenge to preserve public health and it is important to address the problem with a “One Health” strategy, recognized by the European Union (https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52017DC0339&from=IT) and aimed at protecting in a closely interconnected way human health, animal health and welfare, and environmental salubrity, through monitoring, surveillance and contrast actions in both human and veterinary sectors,
Campylobacteriosis is mainly a foodborne disease in which foods of animal origin, such as poultry meat and raw milk, play a primary role, followed by meats of various animal species, above all pork, lamb and beef (Walker et al. 2019).
Wild boars can harbor different species of Campylobacter, including resistant and multi-resistant strains which may be a concern for public health (Carbonero et al. 2014). C. coli, C. jejuni, C. lari and C. lanienae are the most commonly reported Campylobacter species in wild boars (Carbonero et al. 2014; Navarro-Gonzalez et al. 2014; Hulánková et al. 2019).
Moreover, there is a potential risk of a spillover of Campylobacter from wild boars, especially to free-ranging livestock (Navarro-Gonzalez et al. 2014). This spillover could impact particularly free-ranging and extensive pig farming, as is the case for some valuable autochthonous pig breeds, such as the Cinta Senese in Tuscany.
For these reasons, wild boar populations harboring Campylobacter infections may represent a potential food hazard which requires to be monitored, together with the different aspects related to the infection in this reservoir. Among these, the mechanisms of genetic resistance can play a role.
The wild boar population studied in this paper has never been genetically analyzed before, but the evolutionary history and genetics of wild boar in Italy and in other countries have received much attention. Previous studies have revealed the complex genetic structure of wild boar populations in Eurasia, including multiple domestication events and the gene flow between wild boar and domestic pig breeds (Šprem et al. 2014; Khalilzadeh et al. 2016). Results of different studies (Lattuada et al. 2009; Vilaça et al. 2014; Maselli et al. 2016) revealed two major clades of mitochondrial DNA types in Europe (the Italian and the European) and one in Asia. The European clade is the most widespread throughout Europe (Kusza et al. 2014), while wild boar in east Asia has a higher gradient of genetic variation than its European counterparts as shown by mitochondrial, Y-chromosome and microsatellite diversity (Scandura et al. 2011). The European and Asia groups include wild boars as well domestic pig breeds. In Italy the wild boar population seems to have a mixed origin, both clades and interbreeding between domesticated strains have probably occurred. The Italian clade is present in the Castelporziano Reserve and Maremma Regional Park and in Croatia (Lattuada et al. 2009; Kusza et al. 2014). Most southern Italian wild boar populations seem to belong to the European clade, with a smaller proportion belonging to the other haplogroups (Maselli et al. 2016).
Genetic resistance to microbial diseases has only been partially explored. The genetic determinants for resistance to Campylobacter infections in poultry have been the subject of various studies, especially regarding resistance to C. jejuni (Calenge and Beaumont 2012; Psifidi et al. 2021; Russell et al. 2021). Regarding suids, microbial disease resistance genes have been studied in pigs focusing on Gram-negative bacilli (Zhao et al. 2012); however to the best of our knowledge, our study is one of the first attempts to identify genomic regions associated with infection by C. coli and C. jejuni in wild boars.
2 Materials and methods
2.1 Hunting information and sample collection
Wild boars hunted in Tuscany during the 2018–2019 and 2019–2020 hunting seasons (from November to January) were sampled. The study was part of a project investigating the role of wild boar in the epidemiology of infectious diseases of relevance for livestock and humans (Bertelloni et al. 2020; Cilia et al. 2020; Pacini et al. 2020). Animals were hunted according to regional hunting laws in various areas of the provinces of Pisa, Siena, Grosseto and Livorno, characterized by an abundance of wild boar and other wild ungulates (Carnevali et al. 2009). The hunting method was that in the “braccata” mode, characterized by a large number of hunters assisted by dogs (Monaco et al. 2006). Hunting of wild boar has a long tradition in Italy both to obtain meat and other resources and as an instrument for reducing human–wild boar conflicts. Hunting in Tuscany is regulated by some regional regulations (https://www.regione.toscana.it/caccia). The texts of the regional laws and regulations and of the acts of the regional bodies were first published by regional authorities of Tuscany in 1971.The first classification of the different types of hunting to which ungulates have historically been subjected is distinguished between (i) scheduled hunting (i.e., culling) and selective collection and (ii) collective hunts and individual hunts. To hunt ungulates, there has to be a sampling plan (or slaughter plan), which takes into account the structure and dynamics of the population (density, birth rate, mortality etc.) and the management objectives. Scheduled hunting presupposes the definition of a theoretical game bag determined by the number of animals that can be killed by each hunter for each hunting day and by the number of days when hunting is permitted. On the other hand, selective culling is subject to the prior definition of both the quantity of animals to be culled and their division into sex and age thus reaching or maintaining pre-established population densities and structures.
The forms of hunting that make it possible to respect the fundamental principle of selection (discrimination and preventive choice of the animal to be killed) are hunting by aspect or by stalking (fixed or temporary) and the hunting technique (Pirsch method) which consists in looking for the assigned animals, along pre-established transepts, within the assigned hunting territory. Hunters are instructed with regard to personal protective equipment and the procedures for the correct handling of killed wild ungulates to avoid any risk of contact with infected animals. In particular, after the death of the animal has been verified, disposable latex gloves must be worn for handling the carcasses and anti-cut gloves (steel mesh) for the protection of the hands during evisceration, skinning and sectioning, which must all be carried out carefully and in safe conditions.
Hunting is the rational exploitation of a renewable resource; it is therefore essential (for both ethical and economic reasons) to recover as many injured animals as possible. To be able to carry out the recovery promptly and correctly, a sufficient number of research units must be set up in each management area, consisting of qualified dog–handler pairs, which are an indispensable complement to the activity of hunters. These groups must always be ready to respond to calls from hunters, who in turn must be made aware that they should request such interventions (https://www.regione.toscana.it/documents/10180/13846198/Modulo_cacciatore_di_ungulati.pdf/5d8d4d0d-fcfa-4963-bcf1-f81ea80c7d1b).
A total of 60 wild boars (29 females and 31 males) homogenously divided into Campylobacter-positive and -negative animal groups were included in the study. All sampling operations were performed by experienced veterinarians using appropriate personal protective equipment to ensure proper sampling and to ensure the safety of operators during the contact with potentially infected animals. At postmortem, samples presented no relevant lesions related to infectious diseases. During necroscopy, fecal swabs, liver and kidney samples were collected.
2.2 Campylobacter isolation and identification
On fecal and liver samples, Campylobacter isolation and preliminary identification at the genus level were carried out after enrichment in Bolton Selective Enrichment Broth (Oxoid, Basingstoke, UK) following the ISO 10272-1:2017 method with a supplemental filtration phase of the incubated enrichment broth, as described in Pedonese et al. (2017).
The isolates were cultured on Columbia blood agar plates at 42 °C for 48 h in microaerobic atmosphere and DNA was extracted using Maxwell instrument (Promega Corporation, Madison, WI, USA) and quantified using a Nanodrop Spectrophotometer (Nanodrop Technologies, Celbio Srl., Milan, Italy). Isolates were identified at the species level using a multiplex PCR (Wang et al. 2002) for the five most important Campylobacter species from a clinical point of view and also a simplex PCR (Di Giannatale et al. 2016) for C. jejuni.
Among all the sampled animals (193), 60 animals were chosen for the analysis of possible genetic resistance to Campylobacter and divided in two groups: Campylobacter negative (32 animals) and Campylobacter positive (28 animals). Of the 28 Campylobacter-positive wild boars, 22 were positive for C. coli and 6 for C. jejuni). In the positive animals, Campylobacter was isolated from the feces (78.57%), the liver (14.29%) and from both feces and liver (7.14%).
2.3 Wild boar DNA extraction
Total DNA extraction for each sample of kidney was performed, starting with about 100 µl of homogenized tissue, according to the salting out procedure proposed by Armani et al. (2011, 2014). The final DNA concentration and purity were assessed with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, US) by two subsequent measurements of the absorbance value at 260 nm and by calculating the A260/A280 and of A260/230 ratios. 260/280 and 260/230 values ≥ 2 were considered indicative of nucleic acid purity according to the manufacturer's indications (https://tools.thermofisher.com/content/sfs/brochures/TN52646-E-0215M-NucleicAcid.pdf).
2.4 Genotyping analysis
A first approach involved the genotyping of the 60 samples using 15 STR markers (S0005, S0090, S0101, S0155, SW24, SW240, SW857, SW72, SW936, SW911, S0227, S0228, S0386, S0355, SW951) according to the guidelines of the International Society of Animal Genetics–Food and Agriculture Organization (ISAG–FAO) Advisory Committee for parentage control and linkage mapping in swine. The STR analyses were performed at a genetics laboratory in Migliaro (Italy). Table 1 gives detailed information on these markers. The 60 samples were then genotyped using the “Geneseek Genomic Profiler Porcine HD (70 k)”, containing 62,330 SNPs. SNP genotyping was outsourced at the Science and Technology Park of Sardinia.
2.5 Statistical analysis
For each STR marker, and for each group (Campylobacter-positive group and Campylobacter-negative group), the following parameters were computed using Molkin v. 2.0 (Gutièrrez et al. 2005) to analyze within and between-group genetic variability: number of alleles, allelic frequencies, effective allele size, observed heterozygosity and polymorphism information content (PIC), molecular coancestry coefficients (fij), kinship distance (Dk), and inbreeding coefficient (Fi). Wright’s (1978) FST was also calculated.
Genetic similarities among all animals and between groups were investigated by comparing the individual multilocus genotype of each individual. Genetic similarity is defined as P = A/2L, where P is the proportion of common alleles (A) in relation to the 2L possibilities (L = number of considered loci). To investigate possible genetic influences of wild boar on the susceptibility or resistance to Campylobacter for each marker, allele frequencies between the Campylobacter-positive group and Campylobacter-negative group were compared using the Chi-square test and Fisher’s exact tests. The internal relatedness (IR) (Amos et al. 2001) was also calculated using ‘IRMACRON’, an MS Excel macro written in visual basic. Relationships between IR and Campylobacter groups were analyzed using ANOVA (SAS, JMP 2007).
Regarding the GWAS as a preliminary analysis, the SNP quality control (QC) was performed with PLINK v.1.07 (http://zzz.bwh.harvard.edu/plink/) and only autosomal SNPs with a call rate higher than 95%, a minor allele frequency (MAF) > 1% and with no extreme deviation from Hardy–Weinberg equilibrium (P value > 0.00001), were included in the analysis. After QC, 42,431 SNPs mapped on the 18 porcine autosomes and 60 individuals were retained. The number of SNPs per chromosome is reported in Table 2.
The association analysis was performed with the R package GenABEL (Aulchenko et al. 2007), which performs a simple linear regression marker–phenotype analysis. Firstly, the genomic relationship matrix was calculated with the function ibs (https://rdrr.io/cran/GenABEL/man/ibs.html), where for a given pair of individuals i and j, the identical by state coefficients (fi, j) is calculated as follows:
where N is the number of markers used, xi, k is the genotype of the ith individual at the kth SNP (coded as 0, ½ and 1), pk is the frequency of the “ + ” allele and k = 1, …, N.
Then, the additive polygenic model described above was applied:
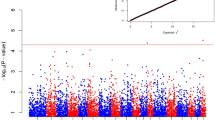
Negative/positive (0/1) phenotypes were considered; β was a vector with the fixed sex effect, and X was the incidence matrix that associated each observation to factor levels in β. The random effects in the model were the animal and the residual, which were assumed to be normally distributed as \(\alpha \sim N\left( {0,G\sigma_{g}^{2} } \right)\) and \(e \sim N\left( {0,I\sigma_{e}^{2} } \right)\), where G is the genomic relationship matrix, I is an identity matrix, and \(\sigma_{g}^{2}\) and \(\sigma_{e}^{2}\) are the additive genomic and residual variances, respectively. Regression was performed using the GenABEL function mmscore and the associations between marker and phenotype with a P value ≤ 0.00001 were considered as significant (Cecchi et al. 2019). A Manhattan plot was produced using the R software (R Core Team, R 2013).
3 Results and discussion
Wildlife can act as a reservoir for different infections, among which is Campylobacter; together with wild birds, small rodents and insects, wild boar may play this role, being a possible source of Campylobacter for farmed animals and humans (Marotta et al. 2020). The percentage of wild boars from which it was possible to isolate different species of Campylobacter in feces or organs and tissues was variable in different geographical areas and in different researches (Atanassova et al. 2008; Díaz-Sánchez et al. 2013; Carbonero et al. 2014; Navarro-Gonzalez et al. 2014; Stella et al. 2018), but the results confirm the role played by this wild mammal as carrier of the pathogen.
The aim of the present study was to investigate for the first time whether genetic variation influences susceptibility to Campylobacter in wild boar. We tested for possible associations between C. coli and C. jejuni positivity and molecular markers. Our initial analysis involved the use of 15 STR markers, and as preliminary analysis the genetic variability within and between groups was analyzed.
The number of alleles and private alleles, the effective number of alleles, observed heterozygosity, and the PIC for each marker in the two groups are reported in Table 3. Two markers (S0355 and SW951) were monomorphic and were thus not used for the subsequent statistical analysis. The other 13 microsatellite markers were found to be polymorphic in the whole sample and in each group. The most polymorphic loci were: S0005 (17 alleles in both groups) and SW24 (8 alleles in the Campylobacter-positive group and 9 alleles in the Campylobacter-negative group).
As reported in the literature, the most commonly used marker is S0005 due to its high degree of polymorphism. In fact, a minimum number of eight alleles was highlighted by Hajji and Zachos (2011) for the wild Sus scrofa in Tunisia, and a maximum of 33 alleles in an East Asian population by Choi et al. (2014). In contrast, the least polymorphic loci were: S0228 (three alleles in both groups), SW857 and S0155 (three alleles in the Campylobacter-positive group).
A total of 74 alleles were found in the Campylobacter-positive group and 81 in the Campylobacter-negative group, with the number of alleles (Na) ranging from 3 to 17 in both groups (mean value 5.69 ± 3.66 in Campylobacter-positive and 6.23 ± 3.63 in Campylobacter-negative group). Although comparisons with other populations can be biased due to the different marker sets used by different authors, the values observed for the average number of alleles were similar to those reported by Choi et al. (2014), in a study that included wild boar samples from ten regions across East Asia. Our values were higher than those reported by Costa et al. (2012) in Hungarian wild boars. Conversely, higher average allele numbers were found by Veličković et al. (2016) in a large sample set from 13 countries across Europe.
The effective number of alleles ranged from 1.29 in the Campylobacter-positive group and 1.46 in the Campylobacter-negative group for the S0228 marker, to 8.48 in the Campylobacter-positive group and 8.64 in the Campylobacter-negative group for the S0005 marker. The average within-population Hobs values found in our study varied from 0.569 in the Campylobacter-positive, to 0.656 in the Campylobacter-negative group. In both groups, the PIC per locus showed two markers with values under 50% (SW857 and S0228), while S00005 and S0101 and SW24 microsatellites appeared to be the most informative.
We found small differences in alleles and genotype frequencies between the animals belonging to the Campylobacter-positive and Campylobacter-negative groups. A total of 13 group-specific alleles were observed (3 in the Campylobacter-positive group and 10 in the Campylobacter-negative group), but all at a frequency lower than 10%. Table 4 reports the percentage of each of the most frequent alleles (> 10%) for each marker in the two groups. There were many genotypes observed at the individual loci (100 in the Campylobacter-positive group and 149 in the Campylobacter-negative group; data not reported), ranging from 3 (S0228 and SW857) to 17 (locus S0005) in the Campylobacter-positive group (7.7 ± 3.71 as the average value per locus), and from 5 (locus S0228) to 24 (locus S0005) in the Campylobacter-negative group (10.8 ± 6.28 as the average value per locus).
All the parameters concerning the genetic diversity between the two groups are shown in Table 5. Molecular coancestry ranged from 0.367 (Campylobacter positive) to 0.381 (Campylobacter negative), while kinship distances ranged from 0.304 (Campylobacter negative) to 0.334 (Campylobacter positive). The highest within-population inbreeding level was observed in the Campylobacter-positive group (F = 0.431). Molecular coancestry and kinship distances were similar to those reported in pigs (Guastella et al. 2010). However, the genetic similarity values, for which we have no comparisons in the literature on swine species, were higher than those reported for other species such as sheep (D’Angelo et al. 2009), and similar to those observed in some Italian endangered donkey breeds (Ciampolini et al. 2007; Matassino et al. 2014), highlighting a low genetic variability above all in the Campylobacter-positive group. The overall FST value (0.008) showed less differentiation between groups. After these preliminary analyses, we explored the differences in the internal relatedness and carried out gene association analyses.
Genetic heterozygosity in wild, unmanaged animal populations is often associated with protection against infectious diseases; however in our study no differences in the internal relatedness (IR) were observed between the two groups (F = 5.64, P = 0.065).
Many studies on genetic heterozygosity and disease resistance have focused on natural populations of animals, with a high genetic heterozygosity generally associated with a reduced risk of infectious diseases (Acevedo-Whitehouse et al. 2005; Rijks et al. 2008). Animals that have a relatively homozygous panel of microsatellite markers tend to be at greater risk of infection and disease, as reported by Acevedo-Whitehouse et al. (2005) in wild boar, together with a greater mortality risk (Rijks et al. 2008). The correlation between individual multilocus heterozygosity (MLH) at microsatellite loci and fitness‐related traits, also called the heterozygosity–fitness correlation, has been studied and discussed for over four decades (Smith et al. 2012).
Despite the lower genetic variability and higher inbreeding of Campylobacter-positive animals, the internal relatedness (IR) was not different from that observed in negative animals. This is probably due to the fact that IR weights each genotype by the frequencies of the alleles involved, which in our case were very similar between the two groups. It is also well known that heterozygosity estimates based on a limited number of loci are expected to correlate only weakly with inbreeding coefficients (Slate et al. 2004). For these reasons an effect due to the inbreeding depression cannot be hypothesized.
Association studies performed for each marker and comparing allele frequencies between the Campylobacter-positive group and Campylobacter-negative group and using the Chi-square test and Fisher’s exact tests showed no significant differences between groups. Gene association studies in pig and in wild boar are limited; however in the literature, some STR markers associated with traits of interest in pigs have been highlighted. The only study to our knowledge on this topic is the association of alleles of the marker S0386 on SSC11 with the inverted teat defect in German Landrace and Large White pig breeds (Jonas et al. 2008). Despite this, some genes have been found in the region near the SW240 marker. For example, Okamura et al. (2010) mapped quantitative trait loci (QTLs) for mycoplasmal pneumonia in swine on SSC2 between SW1650 marker and SW240 marker in Landrace Purebred Swine. Kadowaki et al. (2012) mapped QTLs for respiratory disease and immune capacity traits in pigs and identified a significant QTL for swine enzootic pneumonia between microsatellite markers SW1650 and SW240 on SSC2.
GWAS also showed no significant results: no significant association between SNPs and Campylobacter infection were observed as reported in Fig. 1 which shows the Manhattan plot of the test values obtained for each marker.

Manhattan plot of the test values obtained, for each marker for Campylobacter. The horizontal red line separates the significant markers (p value < 0.00005)
Resistance to Campylobacter intestinal colonization has been studied in poultry, with differences reported in response to Campylobacter infection according to the chicken line tested (Stern et al. 1990; Boyd et al. 2005). In fact, the genetic control of Campylobacter infection has been shown in commercial broiler lines and inbred layer lines (Psifidi et al. 2021).
Reiner (2009) reported that there is an abundant variability in resistance/susceptibility of pig populations and breeds to a number of pathogens, including several viruses, bacteria, and parasites, but resistance to Campylobacter has not been reported. Although genetic resistance to bacterial diseases is often regulated by multiple genes controlling different processes of the host–pathogen interaction, the genetics of natural resistance is increasingly being revealed by the identification and characterization of candidate genes, microsatellite markers and comparative gene mapping. Both types of markers are commonly used in association studies. The development of high-density single nucleotide polymorphism (SNPs) arrays has improved genome-wide screening and, therefore, the detection of disease-associated genes. For example, in wild Sus scrofa, Queirós et al. (2018) found candidate genes for the host genetic susceptibility to tuberculosis, while Fabbri et al. (2021) identified candidate genes involved in pseudorabies, Brucella and Leptospira infections in wild boar.
4 Conclusions
Campylobacter is a Gram-negative pathogen living in the host intestine, which can be silently carried by the host and cause foodborne disease in humans, and thus has a great impact on food safety. Campylobacter infections can be treated with antibacterial drugs; however, antibiotic resistance is not rare (Yang et al. 2019). It is thus of particular interest to highlight genes in the host which may be directly or indirectly involved in the development of infection. Unfortunately, in this study the use of the two types of markers led to the same result and no candidate genes were identified in the animals studied. Further studies on larger numbers of samples are needed to try to clarify the mechanisms of genetic resistance to this microorganism in wild boar.
More generally, the application of operational strategies aimed at the wild boar populations control in the environment (González-Crespo et al. 2018; Wong 2021) and the implementation of a constant epidemiological observatory against the most important zoonoses at the time of hunting remains of fundamental importance to ensure the consumer safety and to check the health status of animals. Indeed, at the time of hunting, and in particular during the evisceration phase, it is possible to detect the presence of anatomical–pathological lesions referable to diseases transmissible to humans and animals. As a contribution to the knowledge concerning the infection dynamics of transmissible diseases to humans and animals in wild boar, in this research we presented for the first time the results of the genetic variability analysis of 60 adult wild boar hunted in four provinces of Tuscany (Italy), using a set of 15 swine-specific microsatellite markers; further studies are needed to deepen the research including domestic pigs as a reference population to study the effect of hybridization and using also a high-density genome-wide SNP analysis that is more affordable in the identification of private wild and domestic polymorphisms.
References
Acevedo-Whitehouse K, Vicente J, Gortazar C, Hofle U, Fernandez-De-Mera IG, Amos W (2005) Genetic resistance to bovine tuberculosis in the Iberian wild boar. Mol Ecol 14:3209–3217
Amos W, Wilmer WJ, Fullard KT, Burg M, Croxall JP, Bloch D, Coulson T (2001) The influence of parental relatedness on reproductive success. Proc Royal Soc London Ser B, Biol Ser 268:2021–2027
Armani A, Castigliego L, Tinacci L, Gianfaldoni D, Guidi A (2011) Molecular characterization of icefish, (Salangidae family), using direct sequencing of mitochondrial cytochrome b gene. Food Control 22(6):888–895
Armani A, Tinacci L, Xiong X, Titarenko E, Guidi A, Castigliego L (2014) Development of a simple and cost-effective bead-milling method for DNA extraction from fish muscles. Food Anal Methods 7(4):946–955
Atanassova V, Apelt J, Reich F, Klein G (2008) Microbiological quality of freshly shot game in Germany. Meat Sci 78(4):414–419
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM (2007) GenABEL: an R library for genome-wide association analysis. Bioinforma Oxf Engl 23:1294–1296. https://doi.org/10.1093/bioinformatics/btm108
Bertelloni F, Mazzei M, Cilia G, Forzan M, Felicioli A, Sagona S, Bandecchi P, Turchi B, Cerri D, Fratini F (2020) Serological survey on bacterial and viral pathogens in wild boars hunted in Tuscany. EcoHealth 17:85–93
Blanchong JA, Robinson SJ, Samuel MD, Foster JT (2016) Application of genetics and genomics to wildlife epidemiology. J Wildl Manag 80:593–608
Boadella M, Ruiz-Fons JF, Vicente J, Martín M, Segalés J, Gortazar C (2012) Seroprevalence evolution of selected pathogens in Iberian wild boar. Transbound Emerg Dis 59(5):395–404
Boyd Y, Herbert E, Marston K, Jones M, Barrow P (2005) Host genes affect intestinal colonisation of newly hatched chickens by Campylobacter jejuni. Immunogenetics 57:248–253
Calenge F, Beaumont C (2012) Toward integrative genomics study of genetic resistance to Salmonella and Campylobacter intestinal colonization in fowl. Front Genet 3:261
Callahan SM, Dolislager CG, Johnson JG (2021) The host cellular immune response to infection by Campylobacter spp. and its role in disease. Infect Immun IAI 89(8):e00116–21
Cappa F, Bani L, Meriggi A (2021) Factors affecting the crop damage by wild boar (Sus scrofa) and effects of population control in the Ticino and Lake Maggiore Park (North-western Italy). Mamm Biol 101:451–463
Carbonero A, Paniagua J, Torralbo A, Arenas-Montes A, Borge C, Garcia Bocanera I (2014) Campylobacter infection in wild artiodactyl species from southern Spain: occurrence, risk factors and antimicrobial susceptibility. Comp Immunol Microbiol Infect Dis 37(2):115–121
Carnevali L, Pedrotti L, Riga F, Toso S (2009) Banca Dati Ungulati: status, distribuzione, consistenza, gestione e prelievo venatorio delle popolazioni di Ungulati in Italia Rapporto 2001–2005. Biologia e Conservazione Della Fauna 117:77–92
Cecchi F, Dadousis C, Bozzi R, Fratini F, Russo C, Bandecchi P et al (2019) Genome scan for the possibility of identifying candidate resistance genes for goat lentiviral infections in the Italian Garfagnina goat breed. Trop Anim Health Prod 51:729–733
Choi SK, Lee JE, Kim YJ, Min MS, Volishina I, Myslenkov A, Oh JG, Kim TH, Markov N, Seryodkin I et al (2014) Genetic structure of wild boar (Sus scrofa) populations from east Asia based on microsatellite loci analysis. BMC Genet 15:85
Ciampolini R, Cecchi F, Mazzanti E, Ciani E, Tancredi M, De Sanctis B (2007) The genetic variability analysis of the Amiata donkey breed by molecular data. Ital J Anim Sci 6(1):78–80
Cilia G, Bertelloni F, Mignone W, Spina S, Berio E, Razzuoli E, Vencia W, Franco V, Cecchi F, Bogi S, Turchi B, Cerri D, Fratini F (2020) Molecular detection of Leptospira spp in wild boar (Sus scrofa) hunted in Liguria region (Italy). Comp Immunol Microb 68:101410
Costa V, Pérez-González J, Santos P, Fernández-Llario P, Carranza J, Zsolnai A, Anton I, Buzgó J, Varga G, Monteiro N et al (2012) Microsatellite markers for identification and parentage analysis in the European wild boar (Sus scrofa). BMC Res Notes 5:479
D’Angelo F, Albenzio M, Sevi A, Ciampolini R, Cecchi F, Ciani E, Muscio A (2009) Genetic variability of the Gentile di Puglia sheep breed based on microsatellite polimorphism. J Anim Sci 87(4):1205–1209
Di Giannatale E, Garofolo G, Alessiani A, Di Donato G, Candeloro L, Vencia W, Decastelli L, Marotta F (2016) Tracing back clinical Campylobacter jejuni in the Northwest of Italy and assessing their potential source. Front Microbiol 7:887
Díaz-Sánchez S, Sánchez S, Herrera-León S, Porrero C, Blanco J, Dahbi G, Blanco JE, Mora A, Mateo R, Hanning I, Vidal D (2013) Prevalence of Shiga toxin-producing Escherichia coli, Salmonella spp. and Campylobacter spp. large game animals intended for consumption: relationship with management practices and livestock influence. Vet Microbiol 163(3–4):274–281
EFSA (European Food Safety Authority) and ECDC (European Centre for Disease Prevention and Control) (2021) The European Union One Health 2019 Zoonoses Report. EFSA J 19:286
Fabbri MC, Crovetti A, Tinacci L, Bertelloni F, Armani A, Mazzei M, Fratini F, Bozzi R, Cecchi F (2021) Identification of candidate genes associated with bacterial and viral infections in wild boars hunted in Tuscany (Italy). Sci Rep. https://doi.org/10.21203/rs.3.rs-1046070/v1
Fernández-Cruz MP, Mohedano R, Maricela V, Mercedes M, Alcalá L, Rodriguez-Créixems M, Cercenado E, Bouza E (2010) Campylobacter bacteremia: clinical characteristics, incidence, and outcome over 23 years. Medicine 89(5):319–330
Fredriksson-Ahomaa M (2019) Wild boar: a reservoir of foodborne zoonoses. Foodborne Pathog Dis 16(3):153–165
Gavier-Widén D, Ståhl K, Neimanis AS, Segerstad CH, Gortázar C, Rossi S et al (2015) African swine fever in wild boar in Europe: a notable challenge. Vet Rec 176:199–200
González-Crespo C, Serrano E, Cahill S, Castillo-Contreras R, Cabañeros L, López-Martín JM, Roldán J, Lavín S, López-Olvera JR (2018) Stochastic assessment of management strategies for a Mediterranean peri-urban wild boar population. PLoS ONE 13(8):e0202289
Guastella AM, Criscione A, Marletta D, Zuccaro A, Chies L, Bordonaro S (2010) Molecular characterization and genetic structure of the Nero Siciliano pig breed. Mol Cell Biol 33:650–656
Gutièrrez JP, Royo LJ, Alvarez I, Goyache F (2005) MolKin v20: a computer program for genetic analysis of populations using molecular coancestry information. J Hered 96:718–721
Hajji GEM, Zachos FE (2011) Mitochondrial and nuclear DNA analyses reveal pronounced genetic structuring in Tunisian wild boar Sus scrofa. Eur J Res 57:449–456
Hulánková R., Bořilová G., Plhal R (2019) Occurrence and characterisation of selected bacterial pathogens in the intestinal tract of wild boars hunted in the Czech Republic. In: Conference proceedings of the 12th International Symposium on Wild Boar and other suids—4–7 September 2018, Lázně Bělohrad, Czech Republic, p 50
Iacolina L, Pertoldi C, Amills M, Kusza S, Megens HJ, Bâlteanu VA, Bakan J, Cubric-Curik V, Oja R, Saarma U, Scandura M, Šprem N, Stronen AV (2018) Hotspots of recent hybridization between pigs and wild boars in Europe. Sci Rep 8:17372
ISO 10272-1:2017. Microbiology of food and animal feeding stuffs—horizontal method for detection and enumeration of Campylobacter spp.— part 1: detection method
Johann F, Handschuh M, Linderoth P, Dormann CF, Arnold J (2020) Adaptation of wild boar (Sus scrofa) activity in a human-dominated landscape. BMC Ecol 20:4
Jonas E, Schreinemachers HJ, Kleinwächter T, Un C, Oltmanns I, Tetzlaff S, Jennen D, Tesfaye D, Ponsuksili S, MuraniE JH, Tholen E, Schellander K, Wimmers K (2008) QTL for the heritable inverted teat defect in pigs. Mamm Genome 19:127–138
Kadowaki H, Suzuki E, Kojima-Shibata C, Suzuki K, Okamura T, Onodera W, Shibata T, Kano H (2012) Selection for resistance to swine mycoplasmal pneumonia over 5 generations in Landrace pigs. Livest Sci 147:20–26
Khalilzadeh P, Rezaei HR, Fadakar D, Serati M, Aliabadian M, Haile J, Goshtasb H (2016) Contact zone of Asian and European wild boar at north west of Iran. PLoS ONE 11(7):e0159499
Kusza S, Podgórski T, Scandura M, Borowik T, Jávor A, Sidorovich VE, Bunevich AN, Kolesnikov M, Jędrzejewska B (2014) Contemporary genetic structure, phylogeography and past demographic processes of wild boar Sus scrofa population in central and eastern Europe. PLoS ONE 9(3):e91401
Lattuada L, Quaglia F, Iannelli F, Gissi C, Mantecca P, Bacchetta R, Polli M (2009) Mitochondrial DNA sequence variations in some Italian wild boar populations. J Anim Breed Genet 26:154–216
Lombardini M, Meriggi A, Fozzi A (2017) Factors influencing wild boar damage to agricultural crops in Sardinia (Italy). Curr Zool 63:507–514
Malmsten A, Magnusson U, Ruiz-Fons F, González-Barrio D, Dalin AM (2018) A serologic survey of pathogens in wild boar (Sus scrofa) in Sweden. J Wildl Dis 54(2):229–237
Marotta F, Di Marcantonio L, Janowicz A, Pedonese F, Di Donato G, Ardelean A, Nuvoloni R, Di Giannatale E, Garofolo G (2020) Genotyping and antibiotic resistance traits in Campylobacter jejuni and coli from pigs and wild Boars in Italy. Front Cell Infect Microbiol 10:592512. https://doi.org/10.3389/fcimb.2020.592512
Maselli V, Rippa D, De Luca A, Larson G, Wilkens B, Linderholm A, Masseti M, Fulgione D (2016) Southern Italian wild boar population, hotspot of genetic diversity. Hystrix, It J Mamm 27:11489
Massei G, Kindberg J, Licoppe A, Gačić D, Šprem N et al (2015) Wild boar populations up, numbers of hunters down? A review of trends and implications for Europe. Pest Manag Sci 71(4):492–500
Matassino D, Cecchi F, Ciani F, Incoronato C, Occidente M, Santoro L, Ciampolini R (2014) Genetic diversity and variability in two Italian autochthonous donkey genetic types assessed by microsatellite markers. It J Anim Sci 13:53–60
McClure ML, Burdett CL, Farnsworth ML et al (2018) A globally-distributed alien invasive species poses risks to United States imperiled species. Sci Rep 8:5331
Monaco A, Carnevali L, Riga F, Toso S (2006) Il cinghiale sull’arco alpino: status e gestione delle popolazioni. Rep Centro Ecol Alpina 38:5–23
Mori E, Ferretti F, Lagrotteria A, La Greca L, Solano E, Fattorini N (2020) Impact of wild boar rooting on small forest-dwelling rodents. Ecol Res 35(4):675–681
Navarro-Gonzalez N, Ugarte-Ruiz M, Porrero MC, Zamora L, Mentaberre G, Serrano E et al (2014) Campylobacter shared between free-ranging cattle and sympatric wild ungulates in a natural environment (NE Spain). EcoHealth 11(3):333–342
Okamura T, Kadowaki H, Shibata C, Suzuki E, Uenishi H, Hayashi T, Mikawa S, Awata T, Okumura H, Matsumoto T, Kanaya N, Mikawa A, Toki D, Suzuki K (2010) Genome-wide scan and fine-mapping of QTL for respiratory disease in Landrace purebred swine. In: Proceeding of the 9th World Congress of Genetics applied to Livestock Production, Leipzig, Germany August 1–6
Pacini MI, Forzan M, Cilia G, Bernardini L, Marzoli F, Pedonese F, Bandecchi P, Fratini F, Mazzei M (2020) Detection of pseudorabies virus in wild boar foetus. Animals 10(2):366
Pedonese F, Nuvoloni TB, Torracca B, Di Giannatale E, Marotta F, Cerri D (2017) Prevalence, phenotypic and genetic diversity of Campylobacter in poultry fresh meat and poultry products on retail sale in Tuscany (Italy). Vet Ital 53(1):29–37
Pittiglio C, Khomenko S, Beltran-Alcrudo D (2018) Wild boar mapping using population-density statistics: From polygons to high resolution raster maps. PLoS ONE 13:e0193295
Psifidi A, Kranis A, Rothwell LM, Bremner A, Russell K, Robledo D, Bush SJ, Fife M, Hocking PM, Banos G, Hume DA, Kaufman J, Bailey RA, Avendano S, Watson KA, Kaiser P, Stevens MP (2021) Quantitative trait loci and transcriptome signatures associated with avian heritable resistance to Campylobacter. Sci Rep 11(1):1–18
Queirós J, Alves PC, Vicente J, Gortázar C, de la Fuente J (2018) Genome-wide associations identify novel candidate loci associated with genetic susceptibility to tuberculosis in wild boar. Sci Rep 8:1980
R Core Team. R: a language and environment for statistical computing. 2013. R: The R Project for Statistical Computing Available online: https://www.r-project.org/ (Accessed on 31 January 2021).
Reiner G (2009) Investigations on genetic disease resistance in swine: a contribution to the reduction of pain, suffering and damage in farm animals. Appl Anim Behav Sci 118:217–221
Riddle MS, Gutierrez RL, Verdu EF, Porter CK (2012) The chronic gastrointestinal consequences associated with Campylobacter. Curr Gastroenterol Rep 14(5):395–405
Rijks JM, Hoffman JI, Kuiken T, Osterhaus AD, Amos W (2008) Heterozygosity and lungworm burden in harbour seals (Phoca vitulina). Hered 100:587–593
Risch DR, Ringma J, Price MR (2021) The global impact of wild pigs (Sus scrofa) on terrestrial biodiversity. Sci Rep 11:13256
Rugna G, Bonilauri P, Garbarino C, Licata E, Tamba M, Merialdi G (2014) Monitoring zoonotic diseases in the wild boar (Sus scrofa) population of the Emilia-Romagna Region (Northern Italy). In: Bauer A, Smulders FJM, Paulsen P (eds) Trends in game meat hygiene. From forest to fork. Wageningen Academic Publishers, The Netherlands
Russell KM, Smith J, Bremner A, Chintoan-Uta C, Vervelde L, Psifidi A, Stevens MP (2021) Transcriptomic analysis of caecal tissue in inbred chicken lines that exhibit heritable differences in resistance to Campylobacter jejuni. BMC Genomics 22(1):1–15
SAS, Jmp (2007) User’s guide, ver 70 SAS INST Cary. NC, USA
Scandura M, Iacolina L, Appolonio M (2011) Genetic diversity in the European wild boar Sus scrofa: phylogeography, population structure and wild x domestic hybridization. Mammal Rev 41:125–137
Slate J, David P, Dodds KG, Veenvliet BA, Glass BC, Broad TE, McEwan JC (2004) Understanding the relationship between the inbreeding coefficient and multilocus heterozygosity: theoretical expectations and empirical data. Hered 93:255–265
Smith EM, Hoffman JI, Green LE, Amos W (2012) Preliminary association of microsatellite heterozygosity with footrot in domestic sheep. Livest Sci 143:293–299
Šprem N, Salajpal K, Safner T, Đikić D, Jurić J, Curik I, Đikić M, Cubric-Curik V (2014) Genetic analysis of hybridization between domesticated endangered pig breeds and wild boar. Liv Sci 162:1–4
Stella S, Tirloni E, Castelli E, Colombo F, Bernardi C (2018) Microbiological evaluation of carcasses of wild boar hunted in a hill area of Northern Italy. J Food Prot 81(9):1519–1525
Stern NJ, Meinersmann RJ, Dickerson HW (1990) Influence of antibody treatment of Campylobacter jejuni on the dose required to colonize chicks. Avian Dis 34:595–601
Veličković N, Ferreira E, Djan M, Ernst M, Vidaković DO, Monaco A, Fonseca C (2016) Demographic history, current expansion and future management challenges of wild boar populations in the Balkans and Europe. Hered 117:348–357
Vilaça ST, Biosa D, Zachos F, Iacolina L, Kirschning J, Alves PC et al (2014) Mitochondrial phylogeography of the European Wild Boar: the effect of climate on genetic diversity and spatial lineage sorting across. Europe J Biogeor 41:987–998
Walker LJ, Wallace RL, Smith JJ, Graham T, Saputra T, Symes S, Stylianopoulos A, Polkinghorne BG, Kirk MD, Glass K (2019) Prevalence of Campylobacter coli and Campylobacter jejuni in retail chicken, beef, lamb, and pork products in three Australian states. J Food Prot 82(12):2126–2134
Wang G, Clark CG, Taylor TM, Pucknell C, Barton C, Price L et al (2002) Colony multiplex PCR assay for identification and differentiation of Campylobacter jejuni, C. coli, C. lari, C. upsaliensis, and C. fetus subsp. fetus. J Clin Microbiol 40(12):4744–4747
Wong A (2021) Developing population control strategies for wild boar management in Canada. Theses and Dissertations (Comprehensive). 2366. https://scholars.wlu.ca/etd/2366.
Wright S (1978) Evolution and the genetics of populations. Variability within and among natural populations, vol 4. University of Chicago Press, Chicago
Yang Y, Feye KM, Shi Z, Pavlidis HO, Kogut MJ, Ashworth A, Ricke SC (2019) A historical review on antibiotic resistance of foodborne Campylobacter. Front Microbiol 10:1509
Zhao S, Zhu M, Chen H (2012) Immunogenomics for identification of disease resistance genes in pigs: a review focusing on Gram-negative bacilli. J Anim Sci Biotechnol 3(1):1–13
Acknowledgements
The authors thank Mr. Mauro Vignozzi, Voluntary Hunting Guard (authorized to control wildlife in the Tuscan region pursuant to Art. 37 of the L.R. 3/1994 since 2001) for information regarding hunting.
Funding
This work is supported by a grant of the University of Pisa (PRA2018_56—Prof. Filippo Fratini; Ateneo Funding 2019—Prof. Francesca Cecchi) and Istituto Zooprofilattico Sperimentale dell’Abruzzo e del Molise “G. Caporale”, Teramo, Italy (IZS AM 03/20 RC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors report that they have no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Animal studies statement
All animal procedures used in this study were in agreement with the ethical and animal welfare concerns of the Committee on the Ethics of Animal Experiments of Minimally Invasive Surgery Centre and fully complied with recommendations outlined by the Italian laws. Animals were hunted following regional hunting laws (Regolamento di attuazione della legge regionale 12 gennaio 1994 n 3 DPGR 48/R/2017).
Informed consent
All authors have approved this manuscript, agree to the order in which their names are listed, declare that no conflict of interest exist and deny any commercial affiliation.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cecchi, F., Fabbri, M.C., Tinacci, L. et al. Genetic resistance to Campylobacter coli and Campylobacter jejuni in wild boar (Sus scrofa L.). Rend. Fis. Acc. Lincei 33, 407–417 (2022). https://doi.org/10.1007/s12210-022-01052-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12210-022-01052-1