
Abstract
Over the past years, infectious disease has caused enormous economic loss in pig industry. Among the pathogens, gram negative bacteria not only cause inflammation, but also cause different diseases and make the pigs more susceptible to virus infection. Vaccination, medication and elimination of sick pigs are major strategies of controlling disease. Genetic methods, such as selection of disease resistance in the pig, have not been widely used. Recently, the completion of the porcine whole genome sequencing has provided powerful tools to identify the genome regions that harboring genes controlling disease or immunity. Immunogenomics, which combines DNA variations, transcriptome, immune response, and QTL mapping data to illustrate the interactions between pathogen and host immune system, will be an effective genomics tool for identification of disease resistance genes in pigs. These genes will be potential targets for disease resistance in breeding programs. This paper reviewed the progress of disease resistance study in the pig focusing on Gram-negative bacilli. Major porcine Gram-negative bacilli and diseases, suggested candidate genes/pathways against porcine Gram-negative bacilli, and distributions of QTLs for immune capacity on pig chromosomes were summarized. Some tools for immunogenomics research were described. We conclude that integration of sequencing, whole genome associations, functional genomics studies, and immune response information is necessary to illustrate molecular mechanisms and key genes in disease resistance.
Similar content being viewed by others


Introduction
Infectious disease, caused by bacteria or virus, has always been a big barrier of effective pig production worldwide. The economics loss caused by infectious disease estimated to be RMB 40 billion annually in China. Gram negative bacteria, such as Escherichia coli, Salmonella, Haemophilus parasuis etc., has been a significant problem in pig industry. Pathogens generated by these bacteria not only cause inflammation, but also cause different disease and make the pigs more susceptible to virus infection. Different strains of E. Coli can cause piglet diarrhoea and enterotoxemia, which result in decreased weight gain or even sudden death [1]. Salmonella enterica can cause both pneumonia and diarrhea in the pig, as well as human food-borne gastroenteritis [2, 3]. Another important harmful bacterium, Haemophilus parasuis (HPS), is a pathogen that can causes fibrinous polyserositis, meningitis and arthritis, called Glässer's disease [4].
Traditionally, producers use vaccination, medication and elimination of sick pigs as major strategies of controlling disease [1, 2]. Genetic methods such as selection of disease resistance in the pig has not been widely used due to several reasons: First, selection of meat production traits ignored the improvement of disease resistance traits; Second, due to the complexity of infectious disease caused by multiple pathogens, selection hardly become effective to control all type of diseases; Third, basic research has not identified enough genes/pathways that can be used in disease resistant breeding.
Recently, the completion of the porcine whole genome sequencing [5], has provided powerful tools to dissect the genome that harboring genes controlling immunity. Over the past few years, large amount of data was generated through QTL mapping, expression profiling by microarray or expressed sequence tags, RNA-sequencing, and SNP chips in the pig. Immunogenomics, which combines DNA variations, transcriptome, immune response data, and QTL mapping to illustrate the interactions between pathogen and host immune system, will be an effective genomics tool for identification of disease resistance genes in pigs. These genes will be potential targets for disease resistance in breeding programs. This paper reviewed the progress of disease resistance study in the pig focusing on Gram-negative bacilli.
General consideration of disease resistance
Genetics of disease resistance
Understanding the genetics of disease resistance is a key issue in improving animal health through traditional or molecular breeding approach. Early studies demonstrated that resistance to infection of certain pathogen is heritable. Lundeheim [6] showed that the heritability for disease resistance to Enzootic pneumonia, Pleuritis, and Atrophic Rhinitis in the pig were 0.12, 0.13 and 0.16, respectively [6]. Occurrence of Enteric, respiratory, and chronic Pleuritis diseases was significantly different among Landrace, Yorkshire, Hampshire, and Duroc breeds [7]. Henryon et al. [8] found that heritabilities of respiratory diseases and diarrhoea are 0.24 and 0.30. These studies revealed additive genetic variation of resistance to certain pathogen does exist in different pig breeds, suggesting that selection in breeding for disease resistance could be efficient. However, there are many factors affect heritability estimation, e.g. different genetic backgrounds, environmental conditions can cause inaccuracy of heritability estimation, which could induce errors in breeding value estimation in using these parameters. Bishop et al. [9] showed that factors including incomplete exposure to infection, or imperfect diagnostic test sensitivity and specificity may reduce the estimable heritabilities, which reduces the power of datasets, but are not critical for illustration of host genetic differences in resistance.
Direct selection based on recovery after infection is not a practical way of disease resistance breeding. Moreover, focusing on one bacteria or virus cannot improve the disease resistance to multiple pathogens. Indicator traits, such as lymphocyte counts and proportions of various leucocyte subsets, innate immune response parameters, and adaptive immune response parameters, are high rank traits to improve the health status and disease resistance ability of pig populations. Thus, understanding the genetic basis of general resistance to multiple infectious diseases, and identification of indicator traits that can be used in breeding program are very important in future pig industry. Edfors-Lilja et al. [10] showed that heritability estimates of responses to E. coli antigens were ranging from 0.29 to 0.45. Mallard et al. [11] showed that heritability estimates in a Yorkshire population were 0.25, 0.23, 0.08 and zero for secondary antibody response to Hen Egg White Lysozyme, blastogenic response to Con A, serum IgG, and monocyte function(uptake and killing of S. typhimurium), respectively. Clapperton et al. [12] demonstrated that significant differences existed between Meishan and Large White pigs for a number of innate immune traits, e.g. total white cell counts were similar between the pig breeds but the numbers of lymphocytes, neutrophils and monocytes differed significantly in that Meishans having higher neutrophil and monocyte counts and lower lymphocyte counts. Juul-Madsen et al. [13] showed that serum porcine mannan-binding lectin A (pMBL-A) concentration is significantly higher in Landrace breed than that in Duroc breed, and the heritability of pMBL-A level is high in the Landrace (h2=0.8) but not in the Duroc breed (h2=0.15). pMBL-A is an innate immune collectin binding to microbial carbohydrates, higher concentration enhance pathogen killing and clearance. Flori et al. [14] scored and analyzed heritabilities of a number of immunity parameters three weeks after vaccination against Mycoplasma hyopneumoniae in a French Large White population, the results showed that 42 of the 54 measured parameters showed moderate to high heritabilities (≥0.2). However, relationship between improved levels of these parameters and disease resistance has not been illustrated clearly, e.g. how to define a pig is resistant, yet deciding of the levels of the innate immune factors that can keep the pigs healthier is of great challenging.
Genetic parameter estimation and application based on phenotypic records remains a big challenge in selection for disease resistance in the pig. Moderate to high heritability of some traits indicates there are genes/markers controlling these traits in the genome remain identified. Genomics tools, including genetic and physical maps, sequencing and expression studies offered powerful tools to discover genetic markers and genes that can be used in improvement of disease resistance. More genes have been identified in mice comparing to farm animals due to the advance of genomic tools and application of inbred strains. In the pig, few genes have been identified to control disease. A typical example is the presence or absence of the receptor of K88, a cell-surface antigen on some Escherichia coli, can cause diarrhoea in the pig [15, 16]. Alleles in mucin genes have been shown to have strong association with susceptibility to enterotoxigenic Escherichia coli F4ab/ac in the pig [17, 18]. Unfortunately, not many pathogens have an inheritance pattern as the Escherichia coli F4, there are more genes and complicated pathways involved in controlling their infections. It is urgent to use immunogenomics tools to uncover the useful genes and pathways to specific pathogen resistance and enhance the innate immunity in different pig breeds.
Strategies for revealing genes and molecular mechanisms of disease resistance
There are different ways to identify genes and molecular mechanisms in disease resistance. However, no single approach can effectively identify genes and the controlling pathways. Integration of sequencing, whole genome associations, functional genomics studies, and immune response information is necessary to illustrate molecular mechanisms and key genes in disease resistance.
Candidate gene approach
In a previous review, we have described the progress and challenges of candidate gene approach [19]. As mentioned above, many diseases have genetic basis, identifying the major genes that play roles in controlling these diseases is of great interest to researchers. Candidate genes are genes with known biological function that directly or indirectly regulating the developing processes of the investigated disease. With the modern developed sequencing techniques today, these studies are relatively cheap and quick to perform. Moreover, the selected genes have been shown to be related to disease on the biology function from previous studies, thus it is more likely to find associations with the target disease traits. Typically, case/control studies from susceptible or resistant animals, or different livestock breeds were carried out to identify mutations in the candidate genes. The basic idea is to analyze the mutations in susceptible / resistant animals, or different breeds with different susceptibility to a certain infection. Alleles based on the mutations find in these genes may be useful markers for disease resistance breeding. In the pig, a number of studies have been focused on immune response and disease resistance traits (See 3.3).
Genome wide scan approach
Many traits, including disease resistance and immune response traits, are quantitative traits that usually determined by multiple genes. Candidate gene approach is largely limited in identifying multiple genes simultaneously that contribute to a phenotype. Thus, genome wide scans to identify chromosome regions and genes have been applied widely. Many QTL studies have been conducted in the pig [20–23]. A number of QTLs associated with a single trait were identified. (http://www.animalgenome.org/cgi-bin/QTLdb/SS/summary?summ=type&qtl=6433&pub=283&trait=594). A total of 40 QTLs were identified for Samonella count in liver and spleen [24]. Six QTLs were identified for resistance to E. coli[25]. No studies were attempted to identify QTLs for other gram negative bacteria. This may be due to the difficulty of creation of QTL mapping populations. QTL studies offered good clue for further cloning of the genes that controlling a trait. However, for the identified QTLs in livestock, a large number of them were located in relatively large chromosome regions,which brings difficulty of positional cloning of the key genes or QTNs for a certain trait. Also, the rather low accuracy of QTL locations is a big challenge of using them in breeding programs. Further, the interactions of the QTLs remain largely unclear, which leads to uncertain results when using them in selection.
Along with the completion swine genome sequencing and development of the high throughput SNP chip, genome wide association study (GWAS) tools became available in identification of key genes associated with disease resistance traits. The analytical tools of GWAS provided us powerful means to identify the possible variants underlying the target traits at the whole-genome scale. In the past years, the applications of GWAS in identification of susceptibility/resistance loci for a range of diseases and resistance-related traits have achieved great progress. Despite of obvious merits, GWAS currently has some limitations in its applications for pig disease studies, which includes high costs, common hypothesis-dependence and other ones that traditional candidate gene studies meet. Especially, one of the disadvantages of GWAS is that the association results are rarely supported by studies on the identification of causal variants or on the functional characterizations of putative SNPs. In addition, with development of genome techniques and accumulation of porcine genome resources, systems biology approach provides a new strategy for identification of disease resistance loci in pigs. In the near future, it is anticipated that systems biological approaches would encourage and accelerate the steps of pig disease resistance investigations.
Genetic control of porcine resistance to Gram-negative bacilli infections
A brief introduction of porcine Gram-negative bacilli
Although it is somewhat ambiguous, we normally describe bacteria as Gram-positive and Gram-negative. Gram-negative bacteria do not retain crystal violet dye after adding a counterstain, such as safranin, in the Gram staining protocol, thus are colored with a red or pink color. Lipopolysaccharide (LPS), also known as endotoxin, is an essential constituent of the outer Gram-negative cell wall. Lipid components such as Lipid A and polysaccharide are closely associated with the toxicity and antigenicity of Gram-negative bacteria, respectively [26]. Pathogenic Gram-negative bacteria, e.g. Salmonella, Escherichia coli, have Pathogenicity Islands (PAI) on the bacterial chromosome, which play a pivotal role in the virulence [27]. PAIs are found mainly in Gram-negatives, but have been reported in a few Gram-positive bacteria. For more details, readers can reference Schmidt and Hensel (2004) [28]. Based on cell shape, Gram-negative bacteria comprise three major subdivisions: coccobacilli, cocci, and baciili. In pigs, there are many Gram-negative bacilli of clinical significance (Table 1) besides the species mentioned above. These include species of the family Enterobacteriaceae (e.g. Salmonella enterica, Escherichia coli, Klebsiella pneumoniae, and Yersinia species). Infection with these members may or may not lead to high mortality, but affects pig industry world-wild and leads to significant economic losses. Additionally, they are causative agent (especially the salmonella and Escherichia coli) of food borne illness in humans, which has become a severe public health problem. Another bacillus of public health significance is Campylobacter coli, which belongs to the family of Campylobacteraceae and causes enteritis in pigs. Infection with Lawsonia intracellularis causes proliferative hemorrhagic enteropathy (PHE), which has become one of the most economically important diseases in pigs [29]. Production of PHE result in bloody diarrhea and sudden death in adult pigs; but in growing pigs, Lawsonia intracellularis infection causes chronic proliferative enteropathy (PE) (reviewed in [30]). Other important Gram-negative bacilli include Actinobacillus pleuropneumoniae and Haemophilus parasuis, two bacilli of the family of Pasteurellaceae. Actinobacillus pleuropneumoniae is the causative agent of porcine pleuropneumonia and fibrinous pleuritis [31, 32]. Haemophilus parasuis infection also leads to respiratory diseases such as acute pneumonia, but the major clinical manifestation is Glässer's disease [33]. In brief, Gram-negative bacilli that have great impacts on pig industry are mainly classified into two categories: 1) the enteric agents that are closely associate with diarrhea; 2) the members of Pasteurellaceae family that cause pneumonia and other severe inflammations.
Resistance indicator for Gram-negative bacilli
Complete resistance to Gram-negative bacilli would be expected in pigs that have capability of 100% elimination, similar to the resistance to other pathogenic invaders e.g. virus, parasites, and Gram-positives. In order to fight infection, vertebrates including the pig own three lines of defense. The first line of defense such as the skin and mucosae provides physical and chemical barriers. In most instances, these barriers unfortunately do not work very well. Severe respiratory diseases and intestine problems caused by Gram-negative bacilli (Table 1) are obvious examples for this. The failed guarding of the first line results in colonization and deeper host tissue invasion, which is mediated by several complex mechanisms [46].
In fact, resistance has happened once host-pathogen interactions begin. Gram-negative bacilli-resistant pigs have strong capability of elimination, due mainly to the effectiveness of the host immune response (IR) mechanisms. The earliest resistance indicators are developed during innate immunity, which is the second line of defense. Gram-negative cell wall components (LPS, porins in the outer membrane, and peptidoglycan monomers etc.), which are collectively called pathogen-associated molecular patterns (PAMPs), play critical roles in host initial IRs [47]. Generally, bindings of PAMPs to pattern-recognition receptors (PRRs) on a variety of host defense cells (e.g. macrophages, neutrophils, and epithelia) result in secretions of many cytokines (e.g. tumor necrosis factor-alpha, interleukin-1, and interleukin-8), which in turn lead to protective inflammatory processes, phagocytosis, and activation of the complement classical pathways and the coagulation pathway [47]. Uthe et al. [48] showed that innate immunity and the inflammatory T helper 1 (Th1) response were observed during both Salmonella enterica serovar Choleraesuis infection and serovar Typhimurium infection. Our previous results suggested that Haemophilus parasuis infection also engaged immune-inflammatory mechanisms [4, 49]. These early resistance indicators belong to nonspecific immune responses but are critical for detecting non-selves and clearing infection. On the other hand, some bacilli infections such as E. coli and Y. pseudotuberculosis can inhibit complement, attenuate host inflammatory response [50], and reduce the recruitment of professional phagocytes [51], indicating these host resistance indicators are main targets in immune evasion mechanisms of Gram-negative bacilli. To better fight against invaders, pigs have to use the last line of defense which is called adaptive immunity. Antibodies produced by plasmacytes are major mediators of adaptive immunity; they are involved in a wild range of host protective IRs including opsonophagocytosis, MAC cytolysis, neutralization, and ADCC (antibody-dependent cell mediated cytotoxicity), so it is the most effective defense by host. For example, this has been proved by the fact that maternal antibodies play important roles in protection against H. parasuis infection [52, 53]. Protective antibody responses has also been observed during A. pleuropneumoniae infection [54, 55]. Guedes and Gebhart [56] showed that cell-mediated immune response was weak during L. intracellularis infection but specific local intestinal humoral IR mediated by IgG was observed.
Candidate gene identification by genetics and omics approaches
A critical goal of porcine genome research is the development of genomic-based tools to select for disease resistance/susceptibility and improved health traits. Although much consideration is needed, good news is that variations in resistance to many porcine pathogens including Gram-negatives do exist [57–60]. For breeding programs such as MAS, the first task is to attempt to identify host candidate genes/genetic markers. With the development of genomics, both traditional methods (e.g. QTL mapping) and state of the art approaches (e.g. GWAS) have been pursued to understand genetic control of host resistance to various causing agents. To date, majority studies have been focused on Gram-negative bacilli infections such as E. coli, S. enterica, A. pleuropneumoniae, and H. parasuis, whereas few have been done on other kind of bacilli (Table 2). This may due to the importance of some bacilli in pig industry.
S. Choleraesuis and S. Typhimurium are the most commonly isolated serovars that effecting performances in pigs [61]. NF-kappaB pathway, antigen-presentation, ERK1/2 activation, and apoptosis involved in both innate and adaptive IRs are associated with infections [2, 35, 62, 63]. Interestingly, in vivo and in vitro studies from different groups showed different or even contradictory results as indicated in expression patterns of PAMP receptors and porcine β-defensins (PBD-1, PBD-2) [64, 65]. Additionally, IR patterns were different due to different subspecies infections and samplings (Table 2); differences in gene expressions exist even in different intestine regions [64]. Results above implied that host IRs are complex and co-works are usually necessary in resistance to S. enterica infections. This is consistent with the theory that most disease resistance traits are quantitative traits. Based on bioinformatic SNP predictions, Uthe et al. [3] revealed that SNPs in HP, NCF2, PGD were associated with Salmonella shedding. In our lab, Ma et al. [66] identified SNPs in GBP family genes that differentially expressed under Salmonella infection, and showed the SNPs were associated with blood parameter traits.
Thanks to candidate gene identification, associations between SNPs and E. coli F4ab/F4ac susceptibility have been extensively studied. Previous studies revealed that the loci controlling F4ab/F4ac receptor is located on SSC13q41 (reviewed in [70]). Mutations in functional genes including HEG1, ITGB5[65], MUC4[71, 72], FUT1[73, 74], B3GNT5[75, 76], MUC20[77], and MUC13[18] were found to be associated with porcine susceptibility. Jacobsen et al. [91] characterized 34 SNPs in five candidate genes within the ETEC F4ab/ac candidate region, but no obvious causative mutations were identified for E. coli F4ab/F4ac susceptibility. These associations may be helpful for genetic improvement of porcine disease resistance to this bacterium.
Several groups have focused on candidate gene identifications following A. pleuropneumoniae and/or H. parasuis infections, but few mutations have not been revealed (Table 2). Daniłowicz et al. [83] identified 62 polymorphisms in TF genome sequence on a panel of 10 different pig breeds, but only found one possible association of the severity of A. pleuropneumoniae infection with TF genotypes. Cav1 was differentially expressed under H. parasuis infection, SNPs in this gene were found associated with blood parameter traits [89].
As for candidate gene identification, much consideration should be given in the future. First, identification of candidate genes needs to be strengthened by employing high throughput approaches, bioinformatics, and functional genomics. In response to pathogenic agents, different kinds of host IRs would be induced or inhibited simultaneously. It is a challenge to attempt to interpret tens of thousands data and then identify the right genes against diseases. Our previous work on H. parasuis infection revealed systematic changes of gene pathways/networks during the process. In combining with gene function analysis, one selective way is to try to identify the main actors that closely related to clinical signs (Figure 1), but this may not easy to do especially when lacking of powerful tools. Some approaches such as GWAS can help us to identify genetic markers directly but the following analysis on gene functions (if it is not available) is usually necessary. This is not only let us to know “what” but also “how” the candidate genes play roles in porcine resistance/susceptibility. Second, candidate genes resistant to multiple pathogens are more useful. In most cases, multiple pathogens play roles simultaneously in porcine infectious diseases. For example, Actinobacillus pleuropneumoniae, Pasteurella multocida and Staphylococcus aureus in porcine lung infections [81]; and, presence of pathogenic porcine reproductive and respiratory syndrome virus (PRRSV) accelerates H. parasuis infection [92]. Third, animal infection models are mainly but not limited in pigs. Since some Gram-negative bacilli have many hosts including human and mouse, host functional genes in other species might also be good references for pig research. An example for this is that Oh et al. [93] has revealed cell cycle and cell differentiation genes in mouse intestinal crypt epithelial cells following L. intracellularis infection. In brief, the candidate gene approach has been widely used to identify genes, but very limited mutations have been found to be powerful in breeding schemes. A number of mutations need to be tested in large field data set. Moreover, only a few genes and a small number of mutations have been preliminarily studied. High throughput strategies (e.g. SNPchip and deep sequencing) and bioinformatic meta-analysis enable researchers to identify genes in a more efficient way.

Gene-interaction network analysis of porcine S100A8 and S100A9 in H. parasuis infected spleen. Depicted are the results of the functional analysis of networks A, B in which porcine S100A8 and S100A9 are involved. The intensity of the node color indicates the degree of up- (red) or down- (green) regulation. Nodes are displayed using various shapes that represent the functional class of the gene product as indicated in the list in B [49].
Database resources for livestock immunomic research
One of the characteristics of contemporary life science is that most of the research results have been digitalized and deposited in public or specific databases, and are able to be traced. Up to date, there are a series of different types of digital repositories for disease resistance or immunity-related resources, which vary from phenotype to DNA, to mRNA, or to protein. These resources can be easily retrieved and widely applied to accelerate our own researches on livestock immunogenomics.
AnimalQTL database
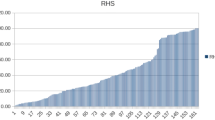
The AnimalQTL database, led by Iowa State University under the NAGRP Bioinformatics Coordination Program, has gathered the published QTLs identification results from the species of pig, cattle, chicken, sheep and rainbow trout. There are totally 640 health-related QTLs deposited in the division of PigQTLdb, in which containing 174 QTLs for immune capacity (e.g., CD4-positive leukocyte number, interferon-gamma level, eosinophil number, monocyte number, and lymphocyte percentage), 107 for disease susceptibility (e.g., chronic pleuritis, melanoma susceptibility, pseudorabies susceptibility, and PRRSV antibody titer), 43 for pathogen (e.g., parasite load and Salmonella counts), and some for blood parameters (e.g., mean corpuscular volume, hematocrit, haptoglobin concentration, and red cell distribution width). It can be found that the QTLs of health traits have located across all porcine chromosomes, some of which are locally clustered. Figure 2 displayed the distributions of QTLs for immune capacity on pig chromosomes. The database resources of disease or immunity-related QTLs provide us useful reference information when mining of resistance alleles.

Distributions of QTLs for immune capacity on pig chromosomes. The red lines represent for significant QTLs and the blue ones for suggestive statistical evidence (This figure is generated from AnimalGenome.ORG with kind permission).
KEGG database
KEGG (Kyoto Encyclopedia of Genes and Genomes) is a widely-used digital resource that contains the regulatory relationship between molecules/genes based on the network/pathway forms (KEGG pathway maps, BRITE functional hierarchies, and KEGG modules). KEGG provides several entries to extract the disease or immunity-related resources. In KEGG DISEASE, the molecular networks of different types of diseases, including single-gene (monogenic) diseases, multifactorial diseases such as cancers, immune system diseases, neurodegenerative diseases, cardiovascular diseases, metabolic diseases, and infectious diseases, can be searched. The KEGG PATHWAYDatabase provides a collection of manually drawn pathway maps according to the functional classifications such as metabolism, genetic information processing, cellular processes, organismal systems, and diseases. Immune system under the organismal systems contains a series of immune signaling pathways, in which including Hematopoietic cell lineage, Complement and coagulation cascades, Toll-like receptor signaling pathway, NOD-like receptor signaling pathway, RIG-I-like receptor signaling pathway, Cytosolic DNA-sensing pathway, Natural killer cell mediated cytotoxicity, Antigen processing and presentation, T cell receptor signaling pathway, B cell receptor signaling pathway, Fc epsilon RI signaling pathway, Fc gamma R-mediated phagocytosis, Leukocyte transendothelial migration, Intestinal immune network for IgA production, and Chemokine signaling pathway. Most of the immune signaling pathways are mainly organized by species (including human, chimpanzee, rat, pig, dog, chicken, mouse, cow, zebrafish, and so on). KEGG provides a reference knowledge base for integration and interpretation of large-scale data produced from genome sequencing and other high-throughput experiments [94], which can be also applied to provide the molecular network-based views of diseases resistance.
Interactome databases
There are many interactome databases such as APID, BioGRID database, Bioverse database, ConsensusPathDB, MIPS database, PSIMAP database, InterPare database,Biomolecular Interaction Network Database (BIND), Online Predicted Human Interaction Database (OPHID), Human Protein Reference Database (HPRD), Human Protein Interaction Database (HPID), Molecular INTeraction database (MINT), Proteins Interacting in the Nucleus Database (PINdb), Molecular Interaction Database (IntAct), VirHostNet, Interactome Databases at CCSB,TRANSFAC, and PSIbase Database. Most of these interactome databases are not independent, the website of Pathguide has displayed the relationships between some databases (Figure 3). These databases contain a large volume of molecules involving disease or immunity, especially the interactions between protein molecules, which also provide references for interpretating and understanding the molecular bases of disease resistance.

The links among databases in Pathguide. (This figure is generated from the Pathguide website).
Microarray databases
Functional genomics is one important aspect of livestock immunogenomics. Microarray experimental techniques have largely and historically, and now still, contributed to the studies on livestock immunogenomics, which produced a large amount of microarray resources on Internet, also including exons arrays and RNA-seq data. There are many microarray databases including the most known public database, the Gene Expression Omnibus (GEO) from NCBI or ArrayExpress from EBI. In addition, some specific or curated databases also host microarray data repositories for disease or immunity such as the Immunological Genome Project, caArray at NCI, ImmGen database, and Stanford Microarray Database. Obviously, functional information or molecular bases of disease resistances can be mined or re-mined from these microarray databases.
Prospects on livestock disease resistance research
Different from other economic traits, it is difficult to make direct measurements of the traits or indicators of disease resistances that are ordinarily not measurable or unknowable on most occasions. Exactly speaking, most of the disease resistances, usually belonging to threshold or quantitative traits under polygenic, genetically heterogeneous control with a small proportion of additive genetic effects, are essentially revealed by death rate or mortality rate when exposed to disease pathogens. But, in practice, it is high cost and difficult to operate the direct measurement of disease resistance and thus disease resistances are alternatively measured by immunity-related traits. Thus, the immune traits are closely associated but not directly with the measurements of disease resistance. Nevertheless, there are so many immune traits, and it is still unknown which immune trait is the best one for measuring the disease resistance. There is an urgent need to find an immune trait or a restructured indicator to pinpoint or be possible mostly near the disease resistance. Currently, although microarray techniques have been widely used to investigate the molecular basis of disease resistance, other genomic approaches such as proteomics and metabonomics/metabolomics are rarely involved in. Furthermore, because of the complexity, high cost, lack of available data, and diversity of host-pathogen interaction, computational aspects of disease genomics are still in challenge. In this field, new and innovative research approaches such as novel genomic and systems biological techniques will be heavily dependent and applied more widely in future. In our opinion, one important aspect of the future studies should focus on Chinese indigenous pig breeds that deposit a large complex of gene resources for disease resistance, including isolation, cloning, and identification of specific resistance alleles accompanying with large-scale explorations of their biological functions. Besides mining gene materials for anti-disease breeding, we should also eye on the application-oriented issues such as molecular techniques for gene diagnosis and testing reagent boxes, and molecular selection approaches matching the characteristics of pig breeding system.
Conclusions
Currently, immunotherapies, such as inoculation or immunization, and medicine administration are the main control strategies on pig diseases. The recently emerged strategy of disease resistance breeding is still immature, which is far from the actual applications. Compared with other economic traits, little breakthrough in disease resistance studies has been achieved. The limited progress and fragmentary results in this field cannot underpin an efficient application of genetic improvement programs to disease resistance. Aiding with novel genomic and systems biological techniques, such as high throughput sequencing, GWAS, and gene function analysis, will help to uncover the disease resistance genes and strengthen the studies of pig disease resistance. It is believed that disease resistance breeding will benefit the future pig industry.
References
Fairbrother JM, Nadeau E, Gyles CL: Escherichia coli in postweaning diarrhea in pigs: an update on bacterial types, pathogenesis, and prevention strategies. Anim Health Res Rev. 2005, 6: 17-39. 10.1079/AHR2005105.
Zhao SH, Kuhar D, Lunney JK, Dawson H, Guidry C, Uthe JJ, Bearson SM, Recknor J, Nettleton D, Tuggle CK: Gene expression profiling in Salmonella Choleraesuis-infected porcine lung using a long oligonucleotide microarray. Mamm Genome. 2006, 17: 777-789. 10.1007/s00335-005-0155-3.
Uthe JJ, Qu L, Couture O, Bearson SM, O'Connor AM, McKean JD, Torres YR, Dekkers JC, Nettleton D, Tuggle CK: Use of bioinformatic SNP predictions in differentially expressed genes to find SNPs associated with Salmonella colonization in swine. J Anim Breed Genet. 2011, 128: 354-365. 10.1111/j.1439-0388.2011.00935.x.
Chen H, Li C, Fang M, Zhu M, Li X, Zhou R, Li K, Zhao S: Understanding Haemophilus parasuis infection in porcine spleen through a transcriptomics approach. BMC Genomics. 2009, 10: 64-10.1186/1471-2164-10-64.
Archibald AL, Bolund L, Churcher C, Fredholm M, Groenen MA, Harlizius B, Lee KT, Milan D, Rogers J, Rothschild MF: Pig genome sequence–analysis and publication strategy. BMC Genomics. 2010, 11: 438-10.1186/1471-2164-11-438.
Lundeheim N: Genetic analysis of respiratory diseases in pigs. Acta Agriculturae Scandinavica. 1979, 29: 209-215. 10.1080/00015127909435230.
Lundeheim N: Health disorders and growth performance at a swedish pigprogeny testing station. Acta Agriculturae Scandinavica. 1988, 38: 77-88. 10.1080/00015128809436590.
Henryon MBP, Jensen J, Andersen S: Genetic variation for resistance to clinical and subclinical diseases exists in growing pigs. Anim Sci. 2001, 73: 375-387.
Bishop SC, Woolliams JA: On the genetic interpretation of disease data. PLoS One. 2010, 5: e8940-10.1371/journal.pone.0008940.
Edfors-Lilja I, Wattrang E, Marklund L, Moller M, Andersson-Eklund L, Andersson L, Fossum C: Mapping quantitative trait loci for immune capacity in the pig. J Immunol. 1998, 161: 829-835.
Mallard BA WB, Kennedy BW, Quinton M: Use of estimated breeding values in a selection index to breed Yorkshire pigs for high and low immune and innate resistance factors. Anim Biotechnol. 1992, 3: 257-280. 10.1080/10495399209525776.
Clapperton M, Bishop SC, Glass EJ: Innate immune traits differ between Meishan and large white pigs. Vet Immunol Immunopathol. 2005, 104: 131-144. 10.1016/j.vetimm.2004.10.009.
Juul-Madsen HR, Krogh-Meibom T, Henryon M, Palaniyar N, Heegaard PM, Purup S, Willis AC, Tornoe I, Ingvartsen KL, Hansen S: Identification and characterization of porcine mannan-binding lectin A (pMBL-A), and determination of serum concentration heritability. Immunogenetics. 2006, 58: 129-137. 10.1007/s00251-006-0092-7.
Flori L, Gao Y, Laloe D, Lemonnier G, Leplat JJ, Teillaud A, Cossalter AM, Laffitte J, Pinton P, de Vaureix C: Immunity traits in pigs: substantial genetic variation and limited covariation. PLoS One. 2011, 6: e22717-10.1371/journal.pone.0022717.
Gibbons RA, Jones GW, Sellwood R: An attempt to identify the intestinal receptor for the K88 adhesin by means of a haemagglutination inhibition test using glycoproteins and fractions from sow colostrum. J Gen Microbiol. 1975, 86: 228-240.
Moon HW, Hoffman LJ, Cornick NA, Booher SL, Bosworth BT: Prevalences of some virulence genes among Escherichia coli isolates from swine presented to a diagnostic laboratory in Iowa. J Vet Diagn Invest. 1999, 11: 557-560. 10.1177/104063879901100617.
Peng QL, Ren J, Yan XM, Huang X, Tang H, Wang YZ, Zhang B, Huang LS: The g.243A>G mutation in intron 17 of MUC4 is significantly associated with susceptibility/resistance to ETEC F4ab/ac infection in pigs. Anim Genet. 2007, 38: 397-400. 10.1111/j.1365-2052.2007.01608.x.
Zhang B, Ren J, Yan X, Huang X, Ji H, Peng Q, Zhang Z, Huang L: Investigation of the porcine MUC13 gene: isolation, expression, polymorphisms and strong association with susceptibility to enterotoxigenic Escherichia coli F4ab/ac. Anim Genet. 2008, 39: 258-266. 10.1111/j.1365-2052.2008.01721.x.
Zhu M, Zhao S: Candidate gene identification approach: progress and challenges. Int J Biol Sci. 2007, 3: 420-427.
Rothschild MF, Hu ZL, Jiang Z: Advances in QTL mapping in pigs. Int J Biol Sci. 2007, 3: 192-197.
Hu ZL, Fritz ER, Reecy JM: AnimalQTLdb: a livestock QTL database tool set for positional QTL information mining and beyond. Nucleic Acids Res. 2007, 35: D604-609. 10.1093/nar/gkl946.
Hu ZL, Ramos AM, Humphray SJ, Rogers J, Reecy JM, Rothschild MF: Use of genome sequence information for meat quality trait QTL mining for causal genes and mutations on pig chromosome 17. Front Genet. 2011, 2: 43.
Hu ZL, Reecy JM: Animal QTLdb: beyond a repository. A public platform for QTL comparisons and integration with diverse types of structural genomic information. Mamm Genome. 2007, 18: 1-4. 10.1007/s00335-006-0105-8.
Galina-Pantoja L, Siggens K, van Schriek MG, Heuven HC: Mapping markers linked to porcine salmonellosis susceptibility. Anim Genet. 2009, 40: 795-803. 10.1111/j.1365-2052.2009.01916.x.
Yang B, Huang X, Yan X, Ren J, Yang S, Zou Z, Zeng W, Ou Y, Huang W, Huang L: Detection of quantitative trait loci for porcine susceptibility to enterotoxigenic Escherichia coli F41 in a White Duroc x Chinese Erhualian resource population. Animal. 2009, 3: 946-950. 10.1017/S1751731109004509.
Wang X, Quinn PJ: Endotoxins: lipopolysaccharides of gram-negative bacteria. Subcell Biochem. 2010, 53: 3-25. 10.1007/978-90-481-9078-2_1.
Hacker J, Blum-Oehler G, Muhldorfer I, Tschape H: Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol Microbiol. 1997, 23: 1089-1097. 10.1046/j.1365-2958.1997.3101672.x.
Schmidt H, Hensel M: Pathogenicity islands in bacterial pathogenesis. Clin Microbiol Rev. 2004, 17: 14-56. 10.1128/CMR.17.1.14-56.2004.
Lawson GH, Gebhart CJ: Proliferative enteropathy. J Comp Pathol. 2000, 122: 77-100. 10.1053/jcpa.1999.0347.
Pedersen KS, Holyoake P, Stege H, Nielsen JP: Diagnostic performance of different fecal Lawsonia intracellularis-specific polymerase chain reaction assays as diagnostic tests for proliferative enteropathy in pigs: a review. J Vet Diagn Invest. 2010, 22: 487-494. 10.1177/104063871002200401.
Macinnes JI, Rosendal S: Prevention and Control of Actinobacillus (Haemophilus) pleuropneumoniae Infection in Swine: A review. Can Vet J. 1988, 29: 572-574.
Chiers K, De Waele T, Pasmans F, Ducatelle R, Haesebrouck F: Virulence factors of Actinobacillus pleuropneumoniae involved in colonization, persistence and induction of lesions in its porcine host. Vet Res. 2010, 41: 65-10.1051/vetres/2010037.
Oliveira S, Pijoan C: Haemophilus parasuis: new trends on diagnosis, epidemiology and control. Vet Microbiol. 2004, 99: 1-12. 10.1016/j.vetmic.2003.12.001.
Wang Y, Couture OP, Qu L, Uthe JJ, Bearson SM, Kuhar D, Lunney JK, Nettleton D, Dekkers JC, Tuggle CK: Analysis of porcine transcriptional response to Salmonella enterica serovar Choleraesuis suggests novel targets of NFkappaB are activated in the mesenteric lymph node. BMC Genomics. 2008, 9: 437-10.1186/1471-2164-9-437.
Hur J, Choi YY, Park JH, Jeon BW, Lee HS, Kim AR, Lee JH: Antimicrobial resistance, virulence-associated genes, and pulsed-field gel electrophoresis profiles of Salmonella enterica subsp. enterica serovar Typhimurium isolated from piglets with diarrhea in Korea. Can J Vet Res. 2011, 75: 49-56.
Nataro JP, Kaper JB: Diarrheagenic escherichia coli. Clin Microbiol Rev. 1998, 11: 142-201.
Li Y, Qiu X, Li H, Zhang Q: Adhesive patterns of escherichia coli F4 in piglets of three breeds. J Genet Genomics. 2007, 34: 591-599. 10.1016/S1673-8527(07)60067-8.
Wilcock BP: Experimental Klebsiella and Salmonella infection in neonatal swine. Can J Comp Med. 1979, 43: 200-206.
SEPTICAEMIA: APEGK.http://vla.defra.gov.uk/science/docs/sci_klebsiella.pdf.
Slee KJ, Button C: Enteritis in sheep, goats and pigs due to Yersinia pseudotuberculosis infection. Aust Vet J. 1990, 67: 320-322. 10.1111/j.1751-0813.1990.tb07814.x.
Harper PA, Hornitzky MA, Rayward DG: Enterocolitis in pigs associated with Yersinia pseudotuberculosis infection. Aust Vet J. 1990, 67: 418-419. 10.1111/j.1751-0813.1990.tb03039.x.
Bottone EJ: Yersinia enterocolitica: the charisma continues. Clin Microbiol Rev. 1997, 10: 257-276.
McOrist S, Gebhart CJ, Boid R, Barns SM: Characterization of Lawsonia intracellularis gen. nov., sp. nov., the obligately intracellular bacterium of porcine proliferative enteropathy. Int J Syst Bacteriol. 1995, 45: 820-825. 10.1099/00207713-45-4-820.
Jacobson M, Hard af Segerstad C, Gunnarsson A, Fellstrom C, de Verdier Klingenberg K, Wallgren P, Jensen-Waern M: Diarrhoea in the growing pig - a comparison of clinical, morphological and microbial findings between animals from good and poor performance herds. Res Vet Sci. 2003, 74: 163-169. 10.1016/S0034-5288(02)00187-X.
Olubunmi PA, Taylor DJ: Production of enteritis in pigs by the oral inoculation of pure cultures of Campylobacter coli. Vet Rec. 1982, 111: 197-202. 10.1136/vr.111.10.197.
Katsikogianni M, Missirlis YF: Concise review of mechanisms of bacterial adhesion to biomaterials and of techniques used in estimating bacteria-material interactions. Eur Cell Mater. 2004, 8: 37-57.
Heumann D, Roger T: Initial responses to endotoxins and Gram-negative bacteria. Clin Chim Acta. 2002, 323: 59-72. 10.1016/S0009-8981(02)00180-8.
Uthe JJ, Royaee A, Lunney JK, Stabel TJ, Zhao SH, Tuggle CK, Bearson SM: Porcine differential gene expression in response to Salmonella enterica serovars Choleraesuis and Typhimurium. Mol Immunol. 2007, 44: 2900-2914. 10.1016/j.molimm.2007.01.016.
Chen H, Lunney JK, Cheng L, Li X, Cao J, Zhu M, Zhao S: Porcine S100A8 and S100A9: molecular characterizations and crucial functions in response to Haemophilus parasuis infection. Dev Comp Immunol. 2011, 35: 490-500. 10.1016/j.dci.2010.11.017.
Thorgersen EB, Pharo A, Haverson K, Axelsen AK, Gaustad P, Kotwal GJ, Sfyroera G, Mollnes TE: Inhibition of complement and CD14 attenuates the Escherichia coli-induced inflammatory response in porcine whole blood. Infect Immun. 2009, 77: 725-732. 10.1128/IAI.01305-08.
Pisano F, Kochut A, Uliczka F, Geyer R, Stolz T, Thiermann T, Rohde M, Dersch P: In vivo-induced InvA-like autotransporters Ifp and InvC of Yersinia pseudotuberculosis promote interactions with intestinal epithelial cells and contribute to virulence. Infect Immun. 2012, 80: 1050-1064. 10.1128/IAI.05715-11.
Blanco I, Galina-Pantoja L, Oliveira S, Pijoan C, Sanchez C, Canals A: Comparison between Haemophilus parasuis infection in colostrums-deprived and sow-reared piglets. Vet Microbiol. 2004, 103: 21-27. 10.1016/j.vetmic.2004.06.011.
Pomorska-Mol M, Markowska-Daniel I, Rachubik J, Pejsak Z: Effect of maternal antibodies and pig age on the antibody response after vaccination against Glassers disease. Vet Res Commun. 2011, 35: 337-343. 10.1007/s11259-011-9478-6.
Bosse JT, Johnson RP, Nemec M, Rosendal S: Protective local and systemic antibody responses of swine exposed to an aerosol of Actinobacillus pleuropneumoniae serotype 1. Infect Immun. 1992, 60: 479-484.
Crawley AM, Mallard B, Wilkie BN: Genetic selection for high and low immune response in pigs: effects on immunoglobulin isotype expression. Vet Immunol Immunopathol. 2005, 108: 71-76. 10.1016/j.vetimm.2005.07.006.
Guedes RM, Gebhart CJ: Evidence of cell-mediated immune response and specific local mucosal immunoglobulin (Ig) A production against Lawsonia intracellularis in experimentally infected swine. Can J Vet Res. 2010, 74: 97-101.
Jeyasingham MD, Butty P, King TP, Begbie R, Kelly D: Escherichia coli K88 receptor expression in intestine of disease-susceptible weaned pigs. Vet Microbiol. 1999, 68: 219-234. 10.1016/S0378-1135(99)00052-8.
Opriessnig T, Fenaux M, Thomas P, Hoogland MJ, Rothschild MF, Meng XJ, Halbur PG: Evidence of breed-dependent differences in susceptibility to porcine circovirus type-2-associated disease and lesions. Vet Pathol. 2006, 43: 281-293. 10.1354/vp.43-3-281.
Blanco I, Canals A, Evans G, Mellencamp MA, Cia C, Deeb N, Wang L, Galina-Pantoja L: Differences in susceptibility to Haemophilus parasuis infection in pigs. Can J Vet Res. 2008, 72: 228-235.
Lunney JK, Steibel JP, Reecy JM, Fritz E, Rothschild MF, Kerrigan M, Trible B, Rowland RR: Probing genetic control of swine responses to PRRSV infection: current progress of the PRRS host genetics consortium. BMC Proc. 2011, 5 (Suppl 4): S30-10.1186/1753-6561-5-S4-S30.
Veldhuizen EJ, Koomen I, Ultee T, van Dijk A, Haagsman HP: Salmonella serovar specific upregulation of porcine defensins 1 and 2 in a jejunal epithelial cell line. Vet Microbiol. 2009, 136: 69-75. 10.1016/j.vetmic.2008.09.072.
Wang Y, Qu L, Uthe JJ, Bearson SM, Kuhar D, Lunney JK, Couture OP, Nettleton D, Dekkers JC, Tuggle CK: Global transcriptional response of porcine mesenteric lymph nodes to Salmonella enterica serovar Typhimurium. Genomics. 2007, 90: 72-84. 10.1016/j.ygeno.2007.03.018.
Vandenbroucke V, Croubels S, Verbrugghe E, Boyen F, De Backer P, Ducatelle R, Rychlik I, Haesebrouck F, Pasmans F: The mycotoxin deoxynivalenol promotes uptake of Salmonella Typhimurium in porcine macrophages, associated with ERK1/2 induced cytoskeleton reorganization. Vet Res. 2009, 40: 64-10.1051/vetres/2009045.
Collado-Romero M, Arce C, Ramirez-Boo M, Carvajal A, Garrido JJ: Quantitative analysis of the immune response upon Salmonella typhimurium infection along the porcine intestinal gut. Vet Res. 2010, 41: 23-10.1051/vetres/2009072.
Meurens F, Berri M, Auray G, Melo S, Levast B, Virlogeux-Payant I, Chevaleyre C, Gerdts V, Salmon H: Early immune response following Salmonella enterica subspecies enterica serovar Typhimurium infection in porcine jejunal gut loops. Vet Res. 2009, 40: 5-10.1051/vetres:2008043.
Ma G, Huang J, Sun N, Liu X, Zhu M, Wu Z, Zhao S: Molecular characterization of the porcine GBP1 and GBP2 genes. Mol Immunol. 2008, 45: 2797-2807. 10.1016/j.molimm.2008.02.007.
Veldhuizen EJ, Hendriks HG, Hogenkamp A, van Dijk A, Gaastra W, Tooten PC, Haagsman HP: Differential regulation of porcine beta-defensins 1 and 2 upon Salmonella infection in the intestinal epithelial cell line IPI-2I. Vet Immunol Immunopathol. 2006, 114: 94-102. 10.1016/j.vetimm.2006.07.012.
Veldhuizen EJ, Rijnders M, Claassen EA, van Dijk A, Haagsman HP: Porcine beta-defensin 2 displays broad antimicrobial activity against pathogenic intestinal bacteria. Mol Immunol. 2008, 45: 386-394. 10.1016/j.molimm.2007.06.001.
Uthe JJ, Wang Y, Qu L, Nettleton D, Tuggle CK, Bearson SM: Correlating blood immune parameters and a CCT7 genetic variant with the shedding of Salmonella enterica serovar Typhimurium in swine. Vet Microbiol. 2009, 135: 384-388. 10.1016/j.vetmic.2008.09.074.
Fu WX, Liu Y, Lu X, Niu XY, Ding XD, Liu JF, Zhang Q: A genome-wide association study identifies two novel promising candidate genes affecting escherichia coli F4ab/F4ac susceptibility in swine. PLoS ONE. 2012, 7: e32127-10.1371/journal.pone.0032127.
Erickson AK, Baker DR, Bosworth BT, Casey TA, Benfield DA, Francis DH: Characterization of porcine intestinal receptors for the K88ac fimbrial adhesin of Escherichia coli as mucin-type sialoglycoproteins. Infect Immun. 1994, 62: 5404-5410.
Joller D, Jorgensen CB, Bertschinger HU, Python P, Edfors I, Cirera S, Archibald AL, Burgi E, Karlskov-Mortensen P, Andersson L: Refined localization of the Escherichia coli F4ab/F4ac receptor locus on pig chromosome 13. Anim Genet. 2009, 40: 749-752. 10.1111/j.1365-2052.2009.01881.x.
Bao WB, Ye L, Zi C, Su XM, Pan ZY, Zhu J, Zhu GQ, Huang XG, Wu SL: Study on the age-dependent tissue expression of FUT1 gene in porcine and its relationship to E. coli F18 receptor. Gene. 2012, 497: 336-339. 10.1016/j.gene.2012.01.035.
Bao WB, Ye L, Pan ZY, Zhu J, Du ZD, Zhu GQ, Huang XG, Wu SL: The effect of mutation at M307 in FUT1 gene on susceptibility of Escherichia coli F18 and gene expression in Sutai piglets. Mol Biol Rep. 2012, 39: 3131-3136. 10.1007/s11033-011-1078-6.
Python P, Jorg H, Neuenschwander S, Asai-Coakwell M, Hagger C, Burgi E, Bertschinger HU, Stranzinger G, Vogeli P: Inheritance of the F4ab, F4ac and F4ad E. coli receptors in swine and examination of four candidate genes for F4acR. J Anim Breed Genet. 2005, 122 (Suppl 1): 5-14.
Ouyang J, Zeng W, Ren J, Yan X, Zhang Z, Yang M, Han P, Huang X, Ai H, Huang L: Association of B3GNT5 polymorphisms with susceptibility to ETEC F4ab/ac in the white Duroc x Erhualian intercross and 15 outbred pig breeds. Biochem Genet. 2012, 50: 19-33. 10.1007/s10528-011-9454-4.
Ji H, Ren J, Yan X, Huang X, Zhang B, Zhang Z, Huang L: The porcine MUC20 gene: molecular characterization and its association with susceptibility to enterotoxigenic Escherichia coli F4ab/ac. Mol Biol Rep. 2011, 38: 1593-1601. 10.1007/s11033-010-0268-y.
Wang Y, Ren J, Lan L, Yan X, Huang X, Peng Q, Tang H, Zhang B, Ji H, Huang L: Characterization of polymorphisms of transferrin receptor and their association with susceptibility to ETEC F4ab/ac in pigs. J Anim Breed Genet. 2007, 124: 225-229. 10.1111/j.1439-0388.2007.00664.x.
Bao WB, Ye L, Pan ZY, Zhu J, Du ZD, Cai JJ, Huang XG, Zhu GQ, Wu SL: [cDNA microarray on differently expressed genes in duodenum in porcine sensitive or resistant to Escherichia coli F18]. Yi Chuan. 2011, 33: 60-66. 10.3724/SP.J.1005.2011.00060.
Niewold TA, van der Meulen J, Kerstens HH, Smits MA, Hulst MM: Transcriptomics of enterotoxigenic Escherichia coli infection. Individual variation in intestinal gene expression correlates with intestinal function. Vet Microbiol. 2010, 141: 110-114. 10.1016/j.vetmic.2009.08.014.
Bruun CS, Leifsson PS, Johansen LK, Jensen HE, Nielsen J, Fredholm M: Expression of matrix metalloproteinase-9 and −12 in porcine lung infections. J Comp Pathol. 2012, 146: 253-257. 10.1016/j.jcpa.2011.05.005.
Skovgaard K, Mortensen S, Boye M, Hedegaard J, Heegaard PM: Hepatic gene expression changes in pigs experimentally infected with the lung pathogen Actinobacillus pleuropneumoniae as analysed with an innate immunity focused microarray. Innate Immun. 2010, 16: 343-353. 10.1177/1753425909342730.
Danilowicz E, Martinez-Arias R, Dolf G, Singh M, Probst I, Tummler B, Holtig D, Waldmann KH, Gerlach GF, Stanke F: Characterization of the porcine transferrin gene (TF) and its association with disease severity following an experimental Actinobacillus pleuropneumoniae infection. Anim Genet. 2010, 41: 424-427.
Moser RJ, Reverter A, Lehnert SA: Gene expression profiling of porcine peripheral blood leukocytes after infection with Actinobacillus pleuropneumoniae. Vet Immunol Immunopathol. 2008, 121: 260-274. 10.1016/j.vetimm.2007.10.007.
Hedegaard J, Skovgaard K, Mortensen S, Sorensen P, Jensen TK, Hornshoj H, Bendixen C, Heegaard PM: Molecular characterisation of the early response in pigs to experimental infection with Actinobacillus pleuropneumoniae using cDNA microarrays. Acta Vet Scand. 2007, 49: 11-10.1186/1751-0147-49-11.
Wang Y, Liu C, Fang Y, Liu X, Li W, Liu S, Liu Y, Charreyre C, Audonnet JC, Chen P: Transcription analysis on response of porcine alveolar macrophages to Haemophilus parasuis. BMC Genomics. 2012, 13: 68-10.1186/1471-2164-13-68.
Wang SJ, Liu WJ, Yang LG, Sargent CA, Liu HB, Wang C, Liu XD, Zhao SH, Affara NA, Liang AX: Effects of FUT1 gene mutation on resistance to infectious disease. Mol Biol Rep. 2012, 39: 2805-2810. 10.1007/s11033-011-1039-0.
Wilkinson JM, Sargent CA, Galina-Pantoja L, Tucker AW: Gene expression profiling in the lungs of pigs with different susceptibilities to Glasser's disease. BMC Genomics. 2010, 11: 455-10.1186/1471-2164-11-455.
Liu XD, Chen HB, Tong Q, Li XY, Zhu MJ, Wu ZF, Zhou R, Zhao SH: Molecular characterization of caveolin-1 in pigs infected with Haemophilus parasuis. J Immunol. 2011, 186: 3031-3046. 10.4049/jimmunol.0902687.
Jacobson M, Andersson M, Lindberg R, Fossum C, Jensen-Waern M: Microarray and cytokine analyses of field cases of pigs with diarrhoea. Vet Microbiol. 2011, 153: 307-314. 10.1016/j.vetmic.2011.06.003.
Jacobsen M, Cirera S, Joller D, Esteso G, Kracht SS, Edfors I, Bendixen C, Archibald AL, Vogeli P, Neuenschwander S: Characterisation of five candidate genes within the ETEC F4ab/ac candidate region in pigs. BMC Res Notes. 2011, 4: 225-10.1186/1756-0500-4-225.
Yu J, Wu J, Zhang Y, Guo L, Cong X, Du Y, Li J, Sun W, Shi J, Peng J: Concurrent highly pathogenic porcine reproductive and respiratory syndrome virus infection accelerates Haemophilus parasuis infection in conventional pigs. Vet Microbiol. 2012, 158: 316-321. 10.1016/j.vetmic.2012.03.001.
Oh YS, Lee JB, McOrist S: Microarray analysis of differential expression of cell cycle and cell differentiation genes in cells infected with Lawsonia intracellularis. Vet J. 2010, 184: 340-345. 10.1016/j.tvjl.2009.03.018.
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M: KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40: D109-114. 10.1093/nar/gkr988.
Acknowledgements
This work was supported by National Natural Science Foundation of China (30901021), 863, and the Key Programs for Science and Technology Development of Hubei Province.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SHZ completed the introduction and the first part of the paper, MJZ drafted the second part, HBC completed the third part of the paper. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhao, S., Zhu, M. & Chen, H. Immunogenomics for identification of disease resistance genes in pigs: a review focusing on Gram-negative bacilli. J Animal Sci Biotechnol 3, 34 (2012). https://doi.org/10.1186/2049-1891-3-34
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2049-1891-3-34