
Abstract
Background
Campylobacter is the leading cause of foodborne diarrhoeal illness in humans and is mostly acquired from consumption or handling of contaminated poultry meat. In the absence of effective licensed vaccines and inhibitors, selection for chickens with increased resistance to Campylobacter could potentially reduce its subsequent entry into the food chain. Campylobacter intestinal colonisation levels are influenced by the host genetics of the chicken. In the present study, two chicken populations were used to investigate the genetic architecture of avian resistance to colonisation: (i) a back-cross of two White Leghorn derived inbred lines [(61 x N) x N] known to differ in resistance to Campylobacter colonisation and (ii) a 9th generation advanced intercross (61 x N) line.
Results
The level of colonisation with Campylobacter jejuni following experimental infection was found to be a quantitative trait. A back-cross experiment using 1,243 fully informative single nucleotide polymorphism (SNP) markers revealed quantitative trait loci (QTL) on chromosomes 7, 11 and 14. In the advanced intercross line study, the location of the QTL on chromosome 14 was confirmed and refined and two new QTLs were identified located on chromosomes 4 and 16. Pathway and re-sequencing data analysis of the genes located in the QTL candidate regions identified potential pathways, networks and candidate resistance genes. Finally, gene expression analyses were performed for some of the candidate resistance genes to support the results.
Conclusion
Campylobacter resistance in chickens is a complex trait, possibly involving the Major Histocompatibility Complex, innate and adaptive immune responses, cadherins and other factors. Two of the QTLs for Campylobacter resistance are co-located with Salmonella resistance loci, indicating that it may be possible to breed simultaneously for enhanced resistance to both zoonoses.
Similar content being viewed by others

Background
Campylobacter is the leading cause of foodborne acute enteritis in humans in the developed world. The condition is usually self-limiting and symptoms last for 5–7 days, but in some cases the infection may be complicated by severe sequelae [1–3]. Epidemiology unequivocally implicates poultry as a key reservoir of human infection and up to 80 % of human cases may be attributable to the avian reservoir as a whole [4]. A recent year-long survey found Campylobacter in 73 % of chicken on retail sale in the United Kingdom [5]. There were 66,575 laboratory-confirmed human infections (mostly due to Campylobacter jejuni) and an estimated total of 685,000 cases in the UK in 2013 [6]. The number of Campylobacter jejuni (C. jejuni) in the caeca of chickens can exceed 108 colony-forming units (CFU)/g and escape of gut contents and cross-contamination at slaughter is difficult to avoid. Quantitative risk assessments predict that even a relatively modest 2 log10 reduction in the number of Campylobacter in broiler carcasses could reduce the incidence of human disease due to infected chicken by up to 30-fold [7]. Therefore, a pressing need exists for strategies to reduce the entry of Campylobacter into the food chain. In the absence of effective licensed vaccines and inhibitors, selection for chickens with increased resistance to Campylobacter intestinal colonisation provides a sustainable complimentary control strategy.
There is a widely–held perception that Campylobacter is an inert commensal of birds. However, experimental infection of chickens with Campylobacter induces a rapid influx of heterophils (the avian functional equivalent of the mammalian neutrophil) into the gut and the production of pro-inflammatory cytokines and chemokines in the intestinal epithelium [8]. Maternal C. jejuni-specific antibodies protect chicks against experimental infection [9, 10] and are associated with the delayed incursion of Campylobacter into flocks [10, 11]. In some breeds of chicken, C. jejuni elicits prolonged inflammatory responses, damage to the intestinal mucosa, diarrhoea and failure to thrive [12, 13]. Conversely, both innate and acquired immune responses have been associated with differential resistance to Campylobacter intestinal colonisation [14–16].
The innate immune response to pathogen challenge and disease resistance varies between birds in inbred lines and outbred populations [12, 17–21]. The ability of C. jejuni to colonise the intestines differs amongst White Leghorn chicken inbred lines, with lines 61 and N being at the extremes of phenotype [22]. An initial reciprocal backcross experiment between inbred lines 61 and N revealed that the difference in bacterial numbers was heritable [22], but the host genetic mechanism of resistance to Campylobacter colonisation is not known. One published genome-wide association study (GWAS) of C. jejuni intestinal colonisation status (phenotypes analysed as a binary trait) in a novel dual-purpose chicken breed revealed one candidate locus on chromosome 11 near the CDH13 gene [23]. There are also several studies of caecal gene expression analysis in chicken lines with different susceptibility to Campylobacter colonisation showing variation in transcription of genes influencing immune response [14, 15, 24].
The aim of the present study was to extend a previous investigation of inbred lines of chickens to determine the genetic architecture of resistance to C. jejuni colonisation using a focussed genotyping platform. This initially involved challenge of a back-cross population (n = 288) of White Leghorn chicken inbred lines 61 and N with C. jejuni and genotyping of the birds for 1,243 fully informative single nucleotide polymorphism (SNP) markers. An independent replication study was then performed by challenging a 9th generation advanced intercross line (AIL) population (n = 218) from a cross of the same two inbred lines with the same C. jejuni strain and genotyping with a 580 K SNP high density whole genome DNA array (Affymetrix® Axiom® HD) [25] to refine and identify new quantitative trait loci (QTLs). SNP markers significantly associated with Campylobacter intestinal colonisation resistance were detected on chromosomes 4, 7, 11, 14 and 16. We also performed pathway analysis and examined gene expression and re-sequencing data to identify candidate genes within the relevant genomic intervals.
Results
Phenotypes for parental lines, back-cross and AIL birds
Mean values and standard deviations of log-transformed caecal C. jejuni levels following experimental inoculation with strain 11168H for line 61 and N parental birds, the [(61 x N) x N] back-cross and the 9th generation AIL (61 x N) birds are listed in Additional file 1: Table S1. With a single exception of low counts on third day post-infection (dpi), no C. jejuni colonies were detected by direct plating of homogenates in any bird of resistant line 61. In contrast, significant levels of C. jejuni colonisation were identified in susceptible line N birds, with the number of birds showing colonisation rising over time after infection. The results are consistent with the original report [22]. Resistance was semi-dominant in that levels of C. jejuni in the backcross and AIL population, measured five dpi, were intermediate between the levels seen in the two parental lines.
Interval mapping and GWAS analysis of the back-cross experiment
The back-cross genotypes were analysed both using interval mapping (linkage analysis) and GWAS analysis (linkage disequilibrium (LD) analysis). In contrast to earlier studies [22, 23] we found with both analyses that levels of bacteria measured in challenged birds behaved as a quantitative trait, and mapped genetically to multiple loci. Two QTLs were detected on chromosomes 7 and 14 that were significantly associated with the log-transformed number of C. jejuni in the caeca at 5 dpi by the interval mapping analysis. The QTL on chromosome 7 was located at 26 Mb (Fig. 1) with a 1-LOD interval of 19.3 to 27.12 Mb. This QTL was significant at the chromosome-wide level (P-value <0.01) and, with an F value of 12.79, was close to genome-wide significance (5 % F-statistic threshold = 14.73). The QTL on chromosome 14 at 7 Mb (1 LOD interval 2.46 to 13.25 Mb) was significant at the chromosome-wide level (P-value <0.05) with an F value of 7.59 (Fig. 1).

F-statistic scores obtained from least-squares interval mapping analysis in the back-cross experiment. F-statistic score of log-transformed number of C. jejuni per gram of caecal contents is plotted against location for chromosome 7 (above) and chromosome 14 (below)
GWAS analysis, using the limited informative marker set, identified both significant associations on chromosomes 7 and 14 as in the interval mapping analysis (Table 1). In addition, one SNP on chromosome 11 was also significantly associated at chromosome-wide level with the log-transformed caecal C. jejuni load (Table 1). The significant SNPs located on chromosome 7 were in high LD with each other (r 2 = 0.89–0.98) and the same was the case for the SNPs on chromosome 14 (r 2 = 0.97). Additional file 2: Figure S1 shows the Manhattan plot and the Q-Q plot displaying the GWAS results.
The SNPs in all three regions were confirmed to have a significant effect in the mixed model analysis (P-value <0.05). The additive genetic effects of the SNPs located on chromosome 7, 14 and 11 were log10 cfu/g 0.72 (P-value = 0.007), 0.71 (P-value = 0.006) and 0.68 (P-value = 0.009), respectively, and the phenotypic variance explained by these SNPs was 4.5 %, 4.3 % and 4.0 %, respectively.
GWAS analyses of the AIL experiment
Multidimensional scaling analysis (MSA) revealed five substructure clusters in the AIL population, which were subsequently included in the GWAS model to correct results for population stratification.
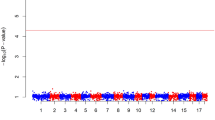
GWAS analysis identified two SNPs significantly associated with the log-transformed number of C. jejuni in the caeca at 5 dpi on chromosome 14, located within the 1 LOD interval of the chromosome 14 QTL identified in the back-cross experiment (Table 2). Thus, the QTL on chromosome 14 was confirmed. Additionally, two SNPs crossing the suggestive genome-wide significant threshold were identified on chromosomes 4 and one SNP reaching the chromosome-wide significant threshold on chromosome 16 (Table 2). The Manhattan plot and the Q-Q plot for the GWAS results are displayed in Fig. 2.

Manhattan plot and Q-Q plot displaying the GWAS results from the AIL experiment (continuous phenotypes). Genomic location is plotted against -log10(P) in the Manhattan plot (above). Genome-wide (P < 0.05) and suggestive genome-wide thresholds are shown as dashed lines. Q–Q plot (below) of observed P-values against the expected P-values for Campylobacter gut colonisation (log-transformed number of C. jejuni per gram of caecal contents)
The estimated additive effects for SNP markers on chromosomes 14, 4 and 16 were log10cfu/g 1.2 (P-value = 0.01), 1.1 (P-value = 0.04) and 2.6 (P-value = 0.0001), respectively; proportions of the total phenotypic variance explained were 10, 9 and 6 %, respectively. Collectively, these three loci explained 25 % of the phenotypic variance.
The GWAS data was also reanalysed as a binary trait. This approach identified both the chromosome 4 and 16 significant associations with Campylobacter colonisation status (Table 2) and the significant SNP on chromosome 4 crossed the genome-wide significance threshold (Table 2). The Manhattan plot and the Q-Q plot for the GWAS results from the case-control analysis are displayed in Additional file 3: Figure S2.
Annotation of QTL regions identified from the back-cross and AIL experiments
The large region encompassed by the QTL on chromosome 7 contains a relatively small number of genes, a total of 124 genes and 12 microRNA, inside the 1 LOD interval region (Additional file 4: Table S2).
The significant SNP on chromosome 11 is located in an intergenic region between two cadherin genes, cadherin 11 precursor (CDH11) and cadherin 8 (CDH8). Very close to this SNP a third cadherin gene, cadherin 5 (CDH5) was located (Additional file 4: Table S2).
The two significant SNPs identified on chromosome 14 in the AIL experiment were in strong LD and belonged to the same small LD block (154 bp) (Additional file 5: Figure S3). The two significant SNPs were in high LD with the other SNP markers located in regions 0.2 Mb upstream and downstream (Additional file 5: Figure S3). In this 0.4 Mb region, 22 genes and two microRNAs are located (Additional file 4: Table S2). The two significant markers were located in the intronic region of an undescribed gene in the chicken genome. Further investigation in the Ensembl database suggested that this was the orthologue of the N-acetyltransferase 15 (NAT15) gene in humans.
The significant SNPs on chromosome 4 were in high LD and located in a very small LD block (Additional file 6: Figure S4). These SNPs were also in high LD with SNP markers located in regions 0.2 Mb upstream and downstream (Additional file 6: Figure S4). In this 0.4 Mb region, five annotated genes and two microRNAs are located (Additional file 4: Table S2); all significant SNPs were located in the intronic region of the Ephrin receptor A5 (EPHA5) gene.
The significant SNP on chromosome 16 was located inside a single LD block with a length of 224 Kb and was in high LD with the other SNPs located there (Additional file 7: Figure S5). This region contains 29 annotated genes most of which are related in the Major Histocompatibility Complex (MHC) (Additional file 4: Table S2).
Re-sequencing data analysis of back-cross and AIL results
To identify possible protein-coding genes associated with the detected QTLs, the genomic sequences of lines 61 and N birds in the regions of interest were compared. The focus was on the identification of exonic single nucleotide variants (SNVs) with high importance (i.e., nonsense (stop-gain) and missense (non-synonymous) exonic and splicing), since these can affect the function of the gene leading to different isoforms of the transcribed proteins. Genomic regions located within 1 kb upstream of the respective genes were also analysed to identify SNVs with a potential regulatory effect. Due to the lack of regulatory element annotation in chickens, we developed our own pipeline for identifying possible regulatory site mutations. The 1 kb upstream genomic regions were scanned for putative TATA box and CpG island motifs. TATA boxes help facilitate transcription factor binding [26] thus TATA box mutations can effect transcription rates. CpG islands play an important role in methylation regulatory pathways [27]. CpG islands are characterised by regions which have a high density of CpG sites that can be methylated to down-regulate gene expression. Thus mutations in CpG sites can alter transcription regulation.
Summary statistics of all the SNVs identified in the candidate regions for Campylobacter colonisation resistance and all the SNVs detected, the Variant Effect Predictor annotation and the SIFT predictions are presented in Additional file 8: Figure S6 and Additional file 9: Table S3, respectively. In total, 20,125 variants were identified. The SNVs located in exonic regions were in total less than 5 % while the rest of the SNVs (95 %) were located in intronic, upstream and downstream regions. A few genes with SNVs that potentially could lead to non-functional transcripts were detected. More specifically, two genes AXIN 1 located on chromosome 14 and BG1 on chromosome 16 were found to contain a stop-gain SNV, a sequence variant whereby at least one base of a codon is changed, resulting in a premature stop codon, leading to a shortened transcript; three genes, BG1 and ENSGALG00000028367 located on chromosome 16 and CCDC108 on chromosome 7 contained a splice acceptor variant, a splice variant that changes the 2 base region at the 3' end of an intron and might lead to splicing changes; two genes, RACGAP1 and SPEG on chromosome 7 contained a splice donor variant, a splice variant that changes the 2 base region at the 5' end of an intron and can lead to splicing changes, as well. Furthermore, seventeen genes contained missense and according to SIFT prediction deleterious SNVs that might create partially or completely non-functional proteins. More specifically, C16orf96 gene on chromosome 14, C4, BFIV21, B-BTN2, TAP2 and IL4I1 on chromosome 16, IFIH1, LY75, SLC11A1, SLC38A11, SPEG, ZNF142, CCDC108, TTC21B, OBSL1, PTPRN, GLB1L on chromosome 7 had missense deleterious SNVs. Several other genes contained SNVs with moderate impact. Details of genes containing splicing, 5′ UTR, both missense and UTR SNVs are presented in Additional file 10: Table S4.
TATA box motifs were identified in the upstream region of some genes but no variation was detected there. On the other hand, CpG island motifs were detected in many of the genes studied and some had SNVs. However, only in few cases the SNVs occur in CpG sites. Details of the genes containing SNVs in CpG sites are presented in the Additional file 10: Table S4.
The gene transcript from each experiment with the highest rate of non-synonymous coding SNVs (i.e., number of non-synonymous SNVs divided by the length of the coding DNA sequence (CDS) of the transcript, dN/L), rate of non-synonymous to synonymous SNVs (dN/dS), rate of exonic SNVs (number of exonic SNVs divided by exonic length of the transcript), rate of intronic SNVs (number of intronic SNVs divided by intronic length of the transcript) are presented in Table 3. These rates were considered to pertain to transcripts that differed between the two parental lines and an indication of positive selection that might result in the creation of different alleles responsible for functional differences in immune responses affecting disease resistance in the two lines.
Ingenuity pathway analysis of back-cross and AIL results
To identify potential canonical pathways and networks underlying the QTLs detected, we performed pathway analysis using the genes located in these regions. Pathways involved in innate and adaptive immune response, inflammatory response, response to infectious diseases, cell signalling and adhesion, and metabolism constituted the majority of the pathways highlighted for both back-cross and AIL results (Fig. 3). Moreover, two networks of molecular interactions related to immune response were constructed using the list of candidate genes for AIL (Additional file 11: Figure S7).

Pathway analysis using the IPA software. The most highly represented canonical pathways of genes located at the candidate regions for Campylobacter colonisation resistance derived from the back-cross (above) and the advance intercross line (below) experiments. The solid yellow line represents the significance threshold. The line with squares represents the ratio of the genes represented within each pathway to the total number of genes in the pathway
Gene expression analysis
Many quantitative traits are associated with altered gene expression rather than coding variation. For example, variable expression of the satiety signal receptor, CCKAR, is associated with appetite control in chickens [28]. Two chemokine genes, CXCLi1 and CXCLi2, lie in close proximity with the significant markers identified on chromosome 4. Both chemokines are induced after Campylobacter infection in chickens [8, 12, 29] and might be involved in effective host immune response. We therefore examined their expression in caecal tonsils of challenged birds. The data are shown in Additional file 12: Figure S8. Data are expressed as the fold change in mRNA levels when samples from infected birds were compared to non-infected birds of the same age from each line. In C. jejuni infected birds from both lines CXCLi1 and CXCLi2 were down-regulated. Nevertheless, the level of chemokine CXCLi2 was decreased in line N significantly (P-value <0.05) more than in line 61, while CXCLi1 levels did not significantly differ between the lines.
Selection of candidate genes
A total of 20 genes were selected amongst all genes located in the regions of interest identified from the analysis of the back-cross and AIL populations as good candidate genes for avian resistance to Campylobacter colonisation (Additional file 13: Table S5). Gene selection was based on their biological function, proximity to significant markers, sequencing differences, mRNA expression (tested for CXCLi1 and CXCLi2) after Campylobacter infection, their involvement in immune response pathways and networks, and any previously known involvement in other infectious diseases in poultry (Additional file 9: Table S4).
Discussion
The present study indicates that the precise level of Campylobacter intestinal colonisation is a heritable complex quantitative trait under the genetic control of multiple loci, genes and linked sequence variants. In the back-cross analysis we identified candidate QTLs in three genomic locations on chromosomes 7, 11 and 14. Using a 9th generation AIL population we refined the location of the QTL identified on chromosome 14 (from a 12 Mb region to a 0.4 Mb region) and detected two additional QTLs located on chromosomes 4 and 16. The initial candidate regions identified in the back-cross experiment have limited resolution and the detected QTLs may reflect the effect of many linked variants which are separated when the LD blocks break after many generations of recombination [30]. The identification of new QTLs in the AIL analysis could be attributed to genotyping based on many more markers (approximately 300,000 compared to 1,300 in the back-cross experiment), which considerably increased the power of QTL detection.
Two good candidate genes, TRAP 1 and AXIN 1 were identified in the refined QTL location on chromosome 14; this exemplifies the common findings from the two experimental designs (back-cross and AIL). TRAP1 is coding for a mitochondrial heat shock protein with antioxidant and anti-apoptotic functions [31, 32]. Our re-sequencing data revealed that TRAP1 is divergent between the two lines. It has a high SNV/L and dN/dS rate that may affect its function and also has 3′/5′ UTR SNV that may impact on gene expression. AXIN 1, is coding for a scaffolding protein controlling the levels of β-catenin, which in turn regulates NF-kB activity. This gene is also very divergent in the two lines with a stop/coding SNV, a high dN/L and dN/dS rate as well as UTR SNV.
The two parental lines, 61 and N, have a different MHC haplotype, B2 and B21, respectively. Therefore, the involvement of MHC, a genomic region that encodes molecules that provide the context in which T cells recognize foreign antigens, in Campylobacter colonisation resistance, is not unexpected. This genomic region is the focus of considerable interest because of the strong, reproducible infectious disease associations found with specific MHC haplotypes [33–36]. However, the highly polymorphic nature of the genes, the strong LD that exists within this region [37–39], and the limited number of SNP markers on chromosome 16 present obstacles to associating individual genes with disease responses. The previous study of the inbred lines suggested that MHC was not involved [22], but this was based on only 41 back-cross (between lines 61 and N) birds and the genotyping was performed using only one microsatellite marker located close to the MHC. The study of Boyd et al. inferred autosomal dominance at a single locus, but that conclusion was based upon the failure to detect the differential colonisation between the parent line 61 and crossed birds [22].
As noted in the introduction, the only previous GWAS study of Campylobacter intestinal colonisation resistance in chickens [23], identified a risk locus on chromosome 11 associated with the T-cadherin (CDH13) gene. This finding might be linked to the cadherin genes (CDH5, CDH11, CDH8) identified in our back-cross analysis. Cadherins are a super-family of calcium-dependent proteins, with a significant role in cell-cell adhesion and the maintenance of structural and functional tissue integrity [40]. The cadherins might interact with C. jejuni and consequentially affect resistance to colonisation either by facilitating internalisation or inciting a protective host response [41–44].
Connell et al. examined gene expression of caeca tissue from high colonised and nil colonised birds of the same breed challenged with Campylobacter, and found evidence of more rapid innate immune response to the infection in nil colonised birds [14]. Among the candidates genes identified in the current study, there are two genes associated with two major pro-inflammatory chemokines (CXCLi1 and CXCLi2) that are reportedly induced during Campylobacter infection both in chickens and humans [45, 46]. Stimulated heterophils produce CXCLi1 and CXCLi2 to induce leukocyte (primarily heterophil) chemotaxis [47, 48]. Re-sequencing data analysis of the two parental inbred lines (61 and N) revealed no protein-coding variation, but did identify an SNV in the 3′UTR of CXCLi2. Although SNV located in the regulatory regions of individual genes are of high importance, relevant knowledge in chickens is very limited and literature virtually non-existent. Whether this particular variant or more distal control elements are responsible, direct measurement of mRNA levels in caecal tonsils of infected birds confirmed differential expression of CXCLi2 between the two lines. Similarly, Connell et al. detected a differential expression only for CXCLi2 [14]. According to these results CXCLi2 may plausibly contribute to resistance to Campylobacter colonisation in chickens.
A previous study of SNP associations with innate and adaptive immune responses in laying hens identified a QTL on chromosome 7 at the same region where the peak of our QTL on the same chromosome is located, associated with complement activity [49]. Several other innate immune genes, very different at sequence level between the two inbred lines, lie under the peak of our identified QTL within the 1 LOD-drop confidence interval. The interferon-induced helicase C domain-containing protein 1 (IFIH1) gene with a predicted splicing SNV is located at 20.5 Mb, the lymphocyte antigen 75 precursor (LY75) gene, also known as CD205, which encodes a receptor on dendritic cells, is located at 21.5 Mb. The natural resistance-associated macrophage protein 1 (NRAMP1) gene, also known as SLC11A1, located at 22 Mb, is another potentially exciting candidate gene to explain this QTL. NRAMP1 regulates intracellular pathogen proliferation and macrophage inflammatory responses, by influencing the phagolysosomal function of macrophages. Variants in this gene have been associated with resistance to Salmonella infection in mice and poultry [50, 51]. Interestingly, host resistance to C. jejuni infection in the mouse is also NRAMP1-dependent [52].
Inbred lines 61 and N are resistant and susceptible respectively not only to Campylobacter gut colonisation, but also to Salmonella gut colonisation, supporting speculation that the genetic control of colonisation could be at least partly common for these bacteria [53]. Extensive QTL mapping studies have taken place to identify loci for resistance to S. enterica serovar Typhimurium and Enteritidis colonisation over the last 20 years using these two inbred chicken lines [2, 20, 51, 54, 55]. Interestingly, two QTLs for resistance to enteric carriage of Salmonella have been identified at the same regions on chromosome 14 [54] and 16 [51, 55] as in our study. Both TRAP1 and AXIN 1, the two putative candidate genes on chromosome 14 for Campylobacter colonisation resistance, have been also associated with Salmonella infection in previous studies [56, 57].
Line 61 is also resistant to infectious bursal disease virus (IBDV) and Marek’s disease virus (MDV) while line N is resistant to infectious bronchitis virus (IBV) and MDV. Whole-genome gene expression analyses comparing resistant line 61 with susceptible inbred line 7 for MDV and inbred Brown Leghorn [Brl] line for IBDV have been conducted to investigate the host response to these infections [58–63]. Likewise, similar analysis comparing resistant line N with susceptible inbred line 15I for IBV has been conducted. Among the genes highlighted as candidate genes for IBV, IBDV or MDV resistance, with different expression during the host response and differential expression between the resistant and susceptible lines, were many of the genes identified in our study as good candidates for Campylobacter intestinal colonisation resistance (Additional file 13: Table S5). These genes may play a central role in an overall effective host immune response.
Conclusion
The multiple QTLs identified as well as the many immune response and inflammation pathways and the predicted involvement of MHC attest to a complex trait controlled by many candidate genes each with a moderate or weak effect. However, the magnitude of the additive effect size of the significant markers and the large proportion of the phenotypic variance explained by them is encouraging for informing breeding strategies for enhanced Campylobacter colonisation resistance in broiler chicken. A study by Gormley et al. revealed that levels of contamination generated by natural exposure varied by at least two orders of magnitude in a wide range of commercial broiler genotypes [64]. As our findings pertain to inbred lines and linkage disequilibrium will likely be different in outbred broiler populations future research is now warranted to explore the genetic basis for resistance in commercial birds and extent of variation at the QTLs identified. Our study highlights the utility of using inbred lines as a resource to map resistance-associated loci, gaining increased power to detect QTLs with modest effects, and informs the design of selective breeding strategies for control of a major zoonosis.
Methods
Ethical statement
All animal experiments were conducted in accordance with the Animals (Scientific Procedures) Act 1986, with the approval of the Ethical Review Committee of The Pirbright Institute (under project licence PPL 30/2462) and the Animal Welfare and Ethical Review Body of The Roslin Institute (under PPL 60/4420).
Animals
Chicken inbred lines 61 (resistant to Campylobacter colonisation) and N (susceptible to Campylobacter colonisation) were originally derived from White Leghorn flocks at the USDA-ARS Avian Disease and Oncology Laboratory in East Lansing, MI, USA. The lines have been maintained by random mating within the specified-pathogen-free (SPF) flocks at the Pirbright Institute in the UK since 1972 (line 61) and 1982 (line N). To generate the back-cross (n = 288 for the present study), F1 progeny (61 x N) were crossed with the susceptible line N.
Colonies of lines 61 and N were given to INRA Tours by the Pirbright Institute over a decade ago. A 9th generation AIL between lines 61 and N was generated there. At each generation, animals from distinct families were crossed to minimise inbreeding and increase the number of recombinations as suggested by Darvasi and Soller [65]. In total, 218 AIL birds were generated for the present study.
Bacterial challenge
In the parent lines, back-cross and AIL, day-old birds were orally inoculated with 0.1 ml of an overnight Mueller-Hinton broth culture of Campylobacter-free gut flora originally taken from the caecal contents of an adult SPF chicken. Birds were orally challenged at three weeks of age with 108 CFU C. jejuni strain 11168H, a hypermotile variant of the sequenced strain NCTC11168 that readily colonises chickens [66] or with control Mueller-Hinton medium only. To confirm resistance or susceptibility to Campylobacter colonisation, control birds (n = 4–5 per line) and infected birds (n = 8–10 per line) were killed at 2, 3, 4 and 5 dpi and a sample of caecal contents was obtained. In addition, caecal tonsil samples of the birds killed 5 dpi were removed to RNAlater for RNA extraction, and gene expression analysis, as described below. Three separate challenge experiments were performed for logistical reasons on the back-cross and AIL populations to assess the level of Campylobacter colonisation in which birds were killed at 5 dpi and caecal contents were removed to measure the Campylobacter load and blood samples were taken for DNA extraction and genotyping, as described below.
Phenotyping
The level of bacterial colonisation in the caecal contents of infected birds was determined in all birds from the back-cross and AIL populations as well as the respective parents as described previously [67]. Bacterial counts for both the back-cross and AIL population were skewed and were accordingly log-transformed in order to normalise their distribution. Thus, phenotypes were expressed on a continuous scale as log-transformed bacterial counts. Moreover, in the case of AIL the phenotypes were also expressed on a binary (0/1) scale indicating absence or minimal colonisation (i.e., no colonies detected by direct plating of 0.1 g homogenate; thus corresponding to birds with Campylobacter load ≤ 100 CFU/g of content)/presence of colonisation.
Genotyping and QTL detection
This part of the study was conducted as two separate experiments described below.
Back-cross experiment
QTL mapping analysis
Over 18,000 genome-wide SNPs were screened in the parent lines (61 and N) to identify fully informative markers for the back-cross mapping study. All SNPs were available through Ensembl (www.ensembl.org) and were analysed using existing panels of chicken SNPs. Markers were selected in the parent lines on the basis of their fixation for the alternate allele at each position, providing maximum information content in the back-cross population. A total of 1,385 SNPs were selected from the 18,000 SNPs panel for analysis in the present study. Genotyping and quality control were performed as described before [20]. A full list of the SNPs (1,243) used in the final analysis is displayed in Additional file 14: Table S6. Positions of SNP markers were obtained using the Gal-gal4 assembly in Ensembl Genome Browser (www.ensembl.org).
QTL analysis was performed by regression interval mapping [68] using the QTL Express software [69] available through GRIDQTL (http://gridqt1.cap.ed.ac.uk) as described previously [20].
GWAS analysis
The back-cross genotypes were also analysed in a GWAS in order to compare with and validate interval mapping results. The following thresholds were used for quality control: minor allele frequency (MAF) < 0.05 and call rate > 95 %. Deviation from Hardy-Weinberg equilibrium was not considered as a criterion for excluding SNPs since this was a back-cross population. After the quality control, 1,212 SNP markers remained for further analysis. The software GEMMA [70] was used to run the GWAS analysis using a standard univariate linear mixed model in which sex (male, female) and experiment number (1, 2, 3) were fitted as fixed effects and the genomic relationship matrix among individuals was included as a random effect. A Bonferroni correction was applied for multiple testing [71]. After Bonferroni correction, significance thresholds were P ≤ 4.12 × 10−5 and P ≤ 8.24 × 10−4 for genome-wide (P ≤ 0.05) and suggestive (namely one false positive per genome scan) levels, corresponding to -log10(P) of 4.38 and 3.08, respectively. Searches for significant SNPs were performed also at chromosome-wide level (P ≤ 0.05).
SNP validation
Individual markers found to be significant in GWAS were further verified in an association analysis where each SNP was fitted as a fixed effect simultaneously with other fixed effects previously fitted (in the GWAS analysis) in a mixed model. The analysis was carried out with the software ASREML [72]. This analysis yielded estimates of the magnitude of the additive SNP effect, as the difference in value between the homozygous (line N-AA) and the heterozygous (AB) genotypes. Solutions were the predicted trait values for each genotype class.
Finally, to evaluate the extent of LD and identify potential regions of causal mutations for Campylobacter colonisation resistance, LD among SNPs was calculated as an r-square value using the software Plink [73]. Furthermore, LD blocks in the regions where significant SNPs were found with GWAS were visualised using the software Haploview [74].
Advanced intercross line experiment
GWAS
AIL birds were genotyped using a 580 K SNP high density whole genome SNP array (Affymetrix® Axiom® HD) [74]. The SNP genotype data were subjected to quality control measures using the following thresholds: MAF < 0.02 and call rate < 95 %. Deviation from Hardy-Weinberg equilibrium was not considered as a method for excluding SNPs since this was an AIL population. After quality control, 286,432 SNP markers remained for further analysis. Positions of SNP markers were obtained using the Gal-gal4 assembly in Ensembl Genome Browser (www.ensembl.org).
The phenotypes were treated both as continuous and as binary data (based on a case-control model). In the case of the binary analysis, 133 birds with no or minimal colonisation where considered as controls and the remaining 85 birds were considered as cases.
The AIL data was analysed using GWAS. To investigate any population stratification present in the population, a genomic relationship matrix was generated from all individuals. This genomic relationship matrix was converted to a distance matrix that was used to carry out classical MSA. All analyses of population stratification were performed using GenABEL software [75]. The same software and the mixed model used in the back-cross was also used for the AIL analyses. In addition to sex and experiment, population cluster (1–5) was fitted as a fixed effect to account for population structure. A Bonferroni correction for multiple testing was applied. After Bonferroni correction significance thresholds were P ≤ 1.75 × 10−7 and P ≤ 3.50 × 10−6 for genome-wide (P ≤ 0.05) and suggestive levels, corresponding to -log10(P) of 6.75 and 5.45, respectively. In addition, a search for SNPs significant at the chromosome-wide level was performed.
SNP validation
Individual SNP markers that were significant at the genome- and chromosome-wide level in GWAS were further verified in an association analysis as described for the back-cross. This model also yielded estimates of the magnitude of the significant SNPs effects as described previously by [76].
Finally, the extent of LD among the significant SNP markers was calculated and LD blocks were built as described for the back-cross.
Further analyses pertaining to both experiments
Annotation of the genes in the QTL candidate regions
The Biomart data mining tool within the Ensembl database (http://www.ensembl.org/biomart/martview/) and the Gal-gal4 assembly were used to locate genes in the candidate regions for Campylobacter colonisation resistance identified by the QTL mapping analysis and GWAS for the back-cross and AIL populations. In the case of GWAS, the limits of genomic candidate regions were determined by the LD block structure and the LD present among the significant SNP markers and the neighbouring markers.
The Variant Effect Predictor tool within the Ensembl database (http://www.ensembl.org/Tools/VEP) and the Gal-gal4 assembly were used to determine the location of the significant SNPs identified in the GWAS analyses on the reference genome. The effect of SNPs located in exonic regions on the protein sequence was determined with a SIFT prediction [77] of the protein sequence changes.
Re-sequencing data analysis
The two parental lines (61 and N) were fully re-sequenced to 15–20 fold coverage, using pools of 10 individuals per line. Sequencing was performed on Illumina GAIIx platform using a paired end protocol [74]. Re-sequencing data of the candidate regions for Campylobacter colonisation resistance, derived from both the back-cross and AIL analysis results, was extracted. More specifically, the extracted sequence included the gene in question and 1 kb upstream and 1 kb downstream of the gene. The SNVs between the two parental lines and the reference genome in these regions were detected using the Mpileup tool for SNP calling (SAMtools v0.1.7). Detected SNVs were annotated using the VEP tool within the Ensembl database (http://www.ensembl.org/Tools/VEP). Information for all SNVs present in the regions of interest was collated. SNV that were in common in the two parental lines but different from the reference genome were filtered out. We separated SNVs into categories based on their relationship to genes. These categories were exonic, intronic, and 1000 bp upstream and downstream. Within the exonic category, there were additional subcategories of UTR and CDS. For SNV’s occurring in CDS regions we also predicted SNV effect as being synonymous, missense, nonsense (stop gain), or splicing. SIFT predictions of the effects of CDS SNVs (tolerated or deleterious) were also acquired through VEP. While mutations in transcribed regions can lead to altered transcriptional products, mutations in regulatory regions can affect transcription rates which ultimately alters phenotype. However, due to the lack of regulatory element annotation in chickens, we developed our own pipeline for predicting TATA boxes and CpG islands. Tata boxes are short DNA regions which act as Transcription Factor Binding sites. CGIs are interspersed DNA sequences that deviate significantly from the average genomic pattern by being GC-rich, and predominantly non-methylated. TATA boxes were predicted by looking for TATA box motifs identified in humans (Computational modeling of oligonucleotide positional densities for human promoter prediction) within the region 20–40 bp upstream of each gene. CpG islands were predicted by using 100 bp windows and scanning across the 1000 bp upstream region with 1 bp intervals to find >200 bp regions with >50 % GC content and an Observed/Expected ratio greater than 0.6 (CpG Islands in Vertebrate Genomes, http://www.bioinformatics.org/sms2/cpg_islands.html). By intersecting our SNV data with our predicted TATA boxes and CpG islands we were able to analyse putative regulatory region mutations.
All the genes, including micro RNAs, were ranked on the rate of non-synonymous coding SNV (dN/L), rate of non-synonymous to synonymous SNV (dN/dS), rate of exonic SNV, rate of intronic SNV.
Pathway analysis
The gene lists in the QTL candidate regions for Campylobacter colonisation resistance were analysed using the IPA programme (www.ingenuity.com) in order to identify canonical pathways and gene networks constructed by the products of the genes located there. IPA constructs many possible upstream regulators, pathways and networks serving as hypotheses for the biological mechanism underlying the data based on a large-scale causal network derived from the Ingenuity Knowledge Base. Then, IPA infers the most suitable pathways and networks based on their statistical significance, thus a threshold above which the pathways are significant is derived.
Gene expression analysis
Genes whose expression was studied included CXCLi1 and CXCLi2. Total RNA from a caecal tonsil was isolated from four replicate control birds and eight replicate infected birds per line (61 and N) using the RNeasy Mini Kit (Qiagen West Sussex, United Kingdom) following the manufacturer’s instructions. For each gene, mRNA levels were quantified using TaqMan quantitative RT-PCR. Primers and probes for the 28S rRNA, CXCLi1 and CXCLi2 genes were as described previously [78, 79]. TaqMan assays were performed as described by Sutton et al. [80]. Data was expressed as cycle threshold (Ct) value, which was normalised using the formula; Ct + (N′t-C′t)*(S/S′) where N′t is the mean Ct value for 28S RNA among all samples, C′t is the mean value for 28S RNA in the sample and the S and S′ are the slopes of regression of the standard plots for the specific cytokine or chemokine mRNA and the 28S RNA, respectively. Expression data was calculated as fold changes compared with the mean of the control mock-infected samples. Results are expressed as the mean of the fold changes between replicate samples and error bars represent the standard error of this mean. Differences between control and infected samples were examined with an analysis of variance.
Ethics
All animal experiments were conducted in accordance with the Animals (Scientific Procedures) Act 1986, with the approval of the Ethical Review Committee of The Pirbright Institute (under project licence PPL 30/2462) and the Animal Welfare and Ethical Review Body of The Roslin Institute (under PPL 60/4420).
Availability of supporting data
The SNP data used in this study is available in NCBI dbSNP at the following web page: http://www.ncbi.nlm.nih.gov/SNP/snp_viewBatch.cgi?sbid=1062063. The same data is also available at the National Avian Research Facility website through the link: http://www.narf.ac.uk/VariationDatabase.
Abbreviations
- AIL:
-
advanced intercross line
- B-BTN2 :
-
Butyrophilin subfamily 2 member A2
- BFIV21 :
-
MHC class I alpha chain II
- BG1 :
-
MHC class IV antigen (B-G)
- BLEC-2 :
-
B-cell CLL/Lymphoma 2
- C. jejuni :
-
Campylobacter jejuni
- C16orf96 :
-
chromosome 16 open reading frame 96
- C4 :
-
complement 4 precursor
- CCDC108 :
-
coiled-coil domain containing 108
- CDH11 :
-
cadherin 11 precursor gene
- CDH13 :
-
T-cadherin
- CDH5 :
-
cadherin 5-type 2
- CDH8 :
-
cadherin 8-type 2
- CFU:
-
colony-forming units
- dN/dS:
-
rate of non-synonymous to synonymous SNV
- dN/L:
-
number of non-synonymous SNV divided by the length of the coding region of the gene
- EPHA5 :
-
ephrin receptor A5
- GLB1L :
-
galactosidase, beta 1-like
- GWAS:
-
genome-wide association study
- IBDV:
-
infectious bursal disease virus
- IBV:
-
infectious bronchitis virus
- IFIH1 :
-
interferon-induced helicase C domain-containing protein 1
- IL4I1 :
-
interleukin 4 Induced 1
- LD:
-
linkage disequilibrium
- LY75 :
-
lymphocyte antigen 75 precursor
- MAF:
-
minor allele frequency
- MDV:
-
Marek’s disease virus
- MHC:
-
major histocompatibility complex
- MSA:
-
multidimensional scaling analysis
- NAT15 :
-
N-acetyltransferase 15
- NRAMP1 (SLC11A1) :
-
natural resistance-associated macrophage protein 1
- OBSL1 :
-
Obscurin-like 1
- PTPRN :
-
protein tyrosine phosphatase, receptor type, N
- QTL:
-
quantitative trait loci
- RACGAP1 :
-
Rac GTPase-activating protein 1
- SLC38A11 :
-
solute carrier family 38, member 11
- SNP:
-
single nucleotide polymorphism
- SNV:
-
single nucleotide variant
- SPEG :
-
SPEG complex locus
- SPF:
-
specified pathogen-free
- TAP2 :
-
antigen peptide transporter 2
- TRAP :
-
Tumour necrosis factor receptor-associated protein 1
- TRIM 27 :
-
Tripartite motif–containing 27
- TTC21B :
-
Tetratricopeptide repeat domain 21B
- UTR:
-
untranslated region
- ZNF142 :
-
Zinc finger protein 142
References
Tam CC, O’Brien SJ, Petersen I, Islam A, Hayward A, Rodrigues LC. Guillain-Barre syndrome and preceding infection with campylobacter, influenza and Epstein-Barr virus in the general practice research database. PLoS One. 2007;2(4):e344.
Townes JM, Deodhar AA, Laine ES, Smith K, Krug HE, Barkhuizen A, Thompson ME, Cieslak PR, Sobel J. Reactive arthritis following culture-confirmed infections with bacterial enteric pathogens in Minnesota and Oregon: a population-based study. Ann Rheum Dis. 2008;67(12):1689–96.
Koga M, Takahashi M, Masuda M, Hirata K, Yuki N. Campylobacter gene polymorphism as a determinant of clinical features of Guillain-Barre syndrome. Neurology. 2005;65(9):1376–81.
EFSA. Scientific Opinion on Campylobacter in broiler meat production: control options and performance objectives and/or targets at different stages of the food chain. EFSA J. 2011;9(4):2105, 141pp.
Food Standards Agency. Cumulative results: first 12 months of retail survey on levels of Campylobacter on chicken. 2015. https://www.food.gov.uk/sites/default/files/full-campy-survey-report.pdf.
Department for the Environment, Food & Rural Affairs. Zoonoses Report 2013. 2015. https://www.gov.uk/government/publications/zoonoses-report-uk-2013.
Rosenquist H, Nielsen NL, Sommer HM, Norrung B, Christensen BB. Quantitative risk assessment of human campylobacteriosis associated with thermophilic Campylobacter species in chickens. Int J Food Microbiol. 2003;83(1):87–103.
Smith CK, Abuoun M, Cawthraw SA, Humphrey TJ, Rothwell L, Kaiser P, Barrow PA, Jones MA. Campylobacter colonization of the chicken induces a proinflammatory response in mucosal tissues. FEMS Immunol Med Microbiol. 2008;54(1):114–21.
Sahin O, Luo N, Huang S, Zhang Q. Effect of Campylobacter-specific maternal antibodies on Campylobacter jejuni colonization in young chickens. Appl Environ Microbiol. 2003;69(9):5372–9.
Cawthraw SA, Newell DG. Investigation of the presence and protective effects of maternal antibodies against Campylobacter jejuni in chickens. Avian Dis. 2010;54(1):86–93.
Sahin O, Zhang Q, Meitzler JC, Harr BS, Morishita TY, Mohan R. Prevalence, antigenic specificity, and bactericidal activity of poultry anti-Campylobacter maternal antibodies. Appl Environ Microbiol. 2001;67(9):3951–7.
Humphrey S, Chaloner G, Kemmett K, Davidson N, Williams N, Kipar A, Humphrey T, Wigley P. Campylobacter jejuni is not merely a commensal in commercial broiler chickens and affects bird welfare. MBio. 2014;5(4):e01364–01314.
Awad WA, Aschenbach JR, Ghareeb K, Khayal B, Hess C, Hess M. Campylobacter jejuni influences the expression of nutrient transporter genes in the intestine of chickens. Vet Microbiol. 2014;172(1–2):195–201.
Connell S, Meade KG, Allan B, Lloyd AT, Kenny E, Cormican P, Morris DW, Bradley DG, O’Farrelly C. Avian resistance to Campylobacter jejuni colonization is associated with an intestinal immunogene expression signature identified by mRNA sequencing. PLoS One. 2012;7(8):e40409.
Li X, Swaggerty CL, Kogut MH, Chiang HI, Wang Y, Genovese KJ, He H, Zhou H. Gene expression profiling of the local cecal response of genetic chicken lines that differ in their susceptibility to Campylobacter jejuni colonization. PLoS One. 2010;5(7):e11827.
Li XY, Swaggerty CL, Kogut MH, Chiang HI, Wang Y, Genovese KJ, He H, Pevzner IY, Zhou HJ. Caecal transcriptome analysis of colonized and non-colonized chickens within two genetic lines that differ in caecal colonization by Campylobacter jejuni. Anim Genet. 2011;42(5):491–500.
Barrow PA, Bumstead N, Marston K, Lovell MA, Wigley P. Faecal shedding and intestinal colonization of Salmonella enterica in in-bred chickens: the effect of host-genetic background. Epidemiol Infect. 2004;132(1):117–26.
Kaiser P, Howell J, Fife M, Sadeyen JR, Salmon N, Rothwell L, Young J, van Diemen P, Stevens M, Poh TY, et al. Integrated immunogenomics in the chicken: deciphering the immune response to identify disease resistance genes. Dev Biol (Basel). 2008;132:57–66.
Fife MS, Salmon N, Hocking PM, Kaiser P. Fine mapping of the chicken salmonellosis resistance locus (SAL1). Anim Genet. 2009;40(6):871–7.
Fife MS, Howell JS, Salmon N, Hocking PM, van Diemen PM, Jones MA, Stevens MP, Kaiser P. Genome-wide SNP analysis identifies major QTL for Salmonella colonization in the chicken. Anim Genet. 2011;42(2):134–40.
Li X, Swaggerty CL, Kogut MH, Chiang H, Wang Y, Genovese KJ, He H, Stern NJ, Pevzner IY, Zhou H. The paternal effect of Campylobacter jejuni colonization in ceca in broilers. Poult Sci. 2008;87(9):1742–7.
Boyd Y, Herbert EG, Marston KL, Jones MA, Barrow PA. Host genes affect intestinal colonisation of newly hatched chickens by Campylobacter jejuni. Immunogenetics. 2005;57(3–4):248–53.
Connell S, Meade KG, Allan B, Lloyd AT, Downing T, O’Farrelly C, Bradley DG. Genome-wide association analysis of avian resistance to Campylobacter jejuni colonization identifies risk locus spanning the CDH13 gene. G3 (Bethesda). 2013;3(5):881–90.
Li X, Swaggerty CL, Kogut MH, Chiang HI, Wang Y, Genovese KJ, He H, McCarthy FM, Burgess SC, Pevzner IY, et al. Systemic response to Campylobacter jejuni infection by profiling gene transcription in the spleens of two genetic lines of chickens. Immunogenetics. 2012;64(1):59–69.
Kranis A, Gheyas AA, Boschiero C, Turner F, Yu L, Smith S, Talbot R, Pirani A, Brew F, Kaiser P, et al. Development of a high density 600K SNP genotyping array for chicken. BMC Genomics. 2013;14:59.
Wang Y, Jensen RC, Stumph WE. Role of TATA box sequence and orientation in determining RNA polymerase II/III transcription specificity. Nucleic Acids Res. 1996;24(15):3100–6.
Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010–22.
Dunn IC, Meddle SL, Wilson PW, Wardle CA, Law AS, Bishop VR, Hindar C, Robertson GW, Burt DW, Ellison SJ, et al. Decreased expression of the satiety signal receptor CCKAR is responsible for increased growth and body weight during the domestication of chickens. Am J Physiol Endocrinol Metab. 2013;304(9):E909–921.
Borrmann E, Berndt A, Hanel I, Kohler H. Campylobacter-induced interleukin-8 responses in human intestinal epithelial cells and primary intestinal chick cells. Vet Microbiol. 2007;124(1–2):115–24.
Besnier F, Wahlberg P, Ronnegard L, Ek W, Andersson L, Siegel PB, Carlborg O. Fine mapping and replication of QTL in outbred chicken advanced intercross lines. Genet Sel Evol. 2011;43(1):3.
Felts SJ, Owen BA, Nguyen P, Trepel J, Donner DB, Toft DO. The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J Biol Chem. 2000;275(5):3305–12.
Montesano Gesualdi N, Chirico G, Pirozzi G, Costantino E, Landriscina M, Esposito F. Tumor necrosis factor-associated protein 1 (TRAP-1) protects cells from oxidative stress and apoptosis. Stress. 2007;10(4):342–50.
Schierman LW, Collins WM. Influence of the major histocompatibility complex on tumor regression and immunity in chickens. Poult Sci. 1987;66(5):812–8.
Lamont SJ, Warner CM, Nordskog AW. Molecular analysis of the chicken major histocompatibility complex gene and gene products. Poult Sci. 1987;66(5):819–24.
Lillehoj HS, Ruff MD, Bacon LD, Lamont SJ, Jeffers TK. Genetic control of immunity to Eimeria tenella. Interaction of MHC genes and non-MHC linked genes influences levels of disease susceptibility in chickens. Vet Immunol Immunopathol. 1989;20(2):135–48.
Taylor Jr RL. Major histocompatibility (B) complex control of responses against Rous sarcomas. Poult Sci. 2004;83(4):638–49.
Simonsen M, Crone M, Koch C, Hala K. The MHC haplotypes of the chicken. Immunogenetics. 1982;16(6):513–32.
Hala K, Chausse AM, Bourlet Y, Lassila O, Hasler V, Auffray C. Attempt to detect recombination between B-F and B-L genes within the chicken B complex by serological typing, in vitro MLR, and RFLP analyses. Immunogenetics. 1988;28(6):433–8.
Kaufman J, Volk H, Wallny HJ. A “minimal essential Mhc” and an “unrecognized Mhc”: two extremes in selection for polymorphism. Immunol Rev. 1995;143:63–88.
Halbleib JM, Nelson WJ. Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes Dev. 2006;20(23):3199–214.
Hoy B, Geppert T, Boehm M, Reisen F, Plattner P, Gadermaier G, Sewald N, Ferreira F, Briza P, Schneider G, et al. Distinct roles of secreted HtrA proteases from gram-negative pathogens in cleaving the junctional protein and tumor suppressor E-cadherin. J Biol Chem. 2012;287(13):10115–20.
Boehm M, Hoy B, Rohde M, Tegtmeyer N, Baek KT, Oyarzabal OA, Brondsted L, Wessler S, Backert S. Rapid paracellular transmigration of Campylobacter jejuni across polarized epithelial cells without affecting TER: role of proteolytic-active HtrA cleaving E-cadherin but not fibronectin. Gut pathogens. 2012;4(1):3.
Boehm M, Lind J, Backert S, Tegtmeyer N. Campylobacter jejuni serine protease HtrA plays an important role in heat tolerance, oxygen resistance, host cell adhesion, invasion, and transmigration. Eur J Microbiol Immunol (Bp). 2015;5(1):68–80.
Elmi A, Nasher F, Jagatia H, Gundogdu O, Bajaj-Elliott M, Wren BW, Dorrell N. Campylobacter jejuni outer membrane vesicle-associated proteolytic activity promotes bacterial invasion by mediating cleavage of intestinal epithelial cell E-cadherin and occludin. Cell Microbiol. 2015;18(4):561-72.
Zheng J, Meng J, Zhao S, Singh R, Song W. Campylobacter-induced interleukin-8 secretion in polarized human intestinal epithelial cells requires Campylobacter-secreted cytolethal distending toxin- and Toll-like receptor-mediated activation of NF-kappaB. Infect Immun. 2008;76(10):4498–508.
MacCallum AJ, Harris D, Haddock G, Everest PH. Campylobacter jejuni-infected human epithelial cell lines vary in their ability to secrete interleukin-8 compared to in vitro-infected primary human intestinal tissue. Microbiology. 2006;152(Pt 12):3661–5.
Swaggerty CL, Ferro PJ, Pevzner IY, Kogut MH. Heterophils are associated with resistance to systemic Salmonella enteritidis infections in genetically distinct chicken lines. FEMS Immunol Med Microbiol. 2005;43(2):149–54.
Tareen AM, Dasti JI, Zautner AE, Gross U, Lugert R. Campylobacter jejuni proteins Cj0952c and Cj0951c affect chemotactic behaviour towards formic acid and are important for invasion of host cells. Microbiology. 2010;156(Pt 10):3123–35.
Biscarini F, Bovenhuis H, van Arendonk JA, Parmentier HK, Jungerius AP, van der Poel JJ. Across-line SNP association study of innate and adaptive immune response in laying hens. Anim Genet. 2010;41(1):26–38.
Lamont SJ, Kaiser MG, Liu W. Candidate genes for resistance to Salmonella enteritidis colonization in chickens as detected in a novel genetic cross. Vet Immunol Immunopathol. 2002;87(3–4):423–8.
Calenge F, Lecerf F, Demars J, Feve K, Vignoles F, Pitel F, Vignal A, Velge P, Sellier N, Beaumont C. QTL for resistance to Salmonella carrier state confirmed in both experimental and commercial chicken lines. Anim Genet. 2009;40(5):590–7.
Champion OL, Valdez Y, Thorson L, Guttman JA, Menendez A, Gaynor EC, Finlay BB. A murine intraperitoneal infection model reveals that host resistance to Campylobacter jejuni is Nramp1 dependent. Microbes Infect. 2008;10(8):922–7.
Calenge F, Beaumont C. Toward integrative genomics study of genetic resistance to Salmonella and Campylobacter intestinal colonization in fowl. Front Genet. 2012;3:261.
Calenge F, Vignal A, Demars J, Feve K, Menanteau P, Velge P, Beaumont C. New QTL for resistance to Salmonella carrier-state identified on fowl microchromosomes. Mol Genet Genomics. 2011;285(3):237–43.
Tilquin P, Barrow PA, Marly J, Pitel F, Plisson-Petit F, Velge P, Vignal A, Baret PV, Bumstead N, Beaumont C. A genome scan for quantitative trait loci affecting the Salmonella carrier-state in the chicken. Genet Sel Evol. 2005;37(5):539–61.
Silva-Garcia O, Valdez-Alarcon JJ, Baizabal-Aguirre VM. The Wnt/beta-catenin signaling pathway controls the inflammatory response in infections caused by pathogenic bacteria. Mediators Inflammation. 2014;2014:310183.
Chiang H-I, Swaggerty CL, Kogut MH, Dowd SE, Li X, Pevzner IY, Zhou H. Gene expression profiling in chicken heterophils with Salmonella enteritidis stimulation using a chicken 44 K Agilent microarray. BMC Genomics. 2008;9(1):1–11.
Smith J, Sadeyen JR, Cavanagh D, Kaiser P, Burt DW. The early immune response to infection of chickens with Infectious Bronchitis Virus (IBV) in susceptible and resistant birds. BMC Vet Res. 2015;11(1):256.
Smith J, Sadeyen JR, Paton IR, Hocking PM, Salmon N, Fife M, Nair V, Burt DW, Kaiser P. Systems analysis of immune responses in Marek’s disease virus-infected chickens identifies a gene involved in susceptibility and highlights a possible novel pathogenicity mechanism. J Virol. 2011;85(21):11146–58.
Smith J, Smith N, Yu L, Paton IR, Gutowska MW, Forrest HL, Danner AF, Seiler JP, Digard P, Webster RG, et al. A comparative analysis of host responses to avian influenza infection in ducks and chickens highlights a role for the interferon-induced transmembrane proteins in viral resistance. BMC Genomics. 2015;16:574.
Haunshi S, Cheng HH. Differential expression of Toll-like receptor pathway genes in chicken embryo fibroblasts from chickens resistant and susceptible to Marek’s disease. Poult Sci. 2014;93(3):550–5.
Subramaniam S, Preeyanon L, Cheng HH. Transcriptional profiling of mEq-dependent genes in Marek’s disease resistant and susceptible inbred chicken lines. PLoS One. 2013;8(10):e78171.
Maceachern S, Muir WM, Crosby SD, Cheng HH. Genome-wide identification and quantification of cis- and trans-regulated genes responding to Marek’s disease virus infection via analysis of allele-specific expression. Front Genet. 2011;2:113.
Gormley FJ, Bailey RA, Watson KA, McAdam J, Avendano S, Stanley WA, Koerhuis AN. Campylobacter colonization and proliferation in the broiler chicken upon natural field challenge is not affected by the bird growth rate or breed. Appl Environ Microbiol. 2014;80(21):6733–8.
Darvasi A, Soller M. Advanced intercross lines, an experimental population for fine genetic mapping. Genetics. 1995;141(3):1199–207.
Karlyshev AV, Linton D, Gregson NA, Wren BW. A novel paralogous gene family involved in phase-variable flagella-mediated motility in Campylobacter jejuni. Microbiology. 2002;148(Pt 2):473–80.
Chintoan-Uta C, Cassady-Cain RL, Al-Haideri H, Watson E, Kelly DJ, Smith DG, Sparks NH, Kaiser P, Stevens MP. Superoxide dismutase SodB is a protective antigen against Campylobacter jejuni colonisation in chickens. Vaccine. 2015;33:6206–11.
Haley CS, Knott SA. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity (Edinb). 1992;69(4):315–24.
Seaton G, Haley CS, Knott SA, Kearsey M, Visscher PM. QTL Express: mapping quantitative trait loci in simple and complex pedigrees. Bioinformatics (Oxford, England). 2002;18(2):339–40.
Zhou X, Stephens M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat Methods. 2014;11(4):407–9.
Dunn OJ. Multiple comparisons among means. J Am Stat Assoc. 1961;56(293):52–64.
Gilmour AR, Cullis BR, Thompson R. ASREML User Guide, Release 3.0. Australia: NSW Department of Primary Industries; 2009.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics (Oxford, England). 2005;21(2):263–5.
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics (Oxford, England). 2007;23(10):1294–6.
Riggio V, Matika O, Pong-Wong R, Stear MJ, Bishop SC. Genome-wide association and regional heritability mapping to identify loci underlying variation in nematode resistance and body weight in Scottish Blackface lambs. Heredity (Edinb). 2013;110(5):420–9.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Swaggerty CL, Kogut MH, Ferro PJ, Rothwell L, Pevzner IY, Kaiser P. Differential cytokine mRNA expression in heterophils isolated from Salmonella-resistant and -susceptible chickens. Immunology. 2004;113(1):139–48.
Kogut MH, Swaggerty C, He H, Pevzner I, Kaiser P. Toll-like receptor agonists stimulate differential functional activation and cytokine and chemokine gene expression in heterophils isolated from chickens with differential innate responses. Microbes Infect. 2006;8(7):1866–74.
Sutton KM, Hu T, Wu Z, Siklodi B, Vervelde L, Kaiser P. The functions of the avian receptor activator of NF-kappaB ligand (RANKL) and its receptors, RANK and osteoprotegerin, are evolutionarily conserved. Dev Comp Immunol. 2015;51(1):170–84.
Acknowledgements
The authors thank all members of the Chicken SNP Consortium for their contribution to the validation and characterization of a high-density chicken SNP map, including the US Department of Agriculture Agricultural Research Service (USDA-ARS); the USDA Cooperative State Research, Education and Extension Service, National Research Initiative Competitive Grants Program (USDA CSREES NRICGP); and Hans Cheng, William Muir, Gane Wong, Martien Groenen, and Huanmin Zhang. The authors also thank Cathrerine Beaumont and Michael Riou from INRA Institute (Tours) for the performance of the AIL experiment. Finally, authors would like to thank Steve Bishop for his advice on the interpretation of the interval mapping and GWAS analysis results.
Funding
This work was supported by the Department for the Environment, Food and Rural Affairs (LINK grant no. LK0665), the European Union (SABRE - FOOD-CT-2006-01625), EADGENE, the Biotechnology and Biological Sciences Research Council (Core Strategic Grants to The Pirbright Institute and The Roslin Institute and BBSRC-Aviagen LINK grant BB/J006815/1) and funding from the Scottish Government via the Strategic Partnership for Animal Science Excellence initiative.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PK, MS, MJ, NS conceived and designed the backcross study and secured substantial funding; PK, MS, MF, conceived and designed the AIL study and secured substantial funding; MS and MJ developed the infection models used in this study. JH, NS, MF and PMD performed the experimental challenge, the phenotyping of the back-cross experiment and the cytokine expression work; JH, NS, MF created the SNP set and genotyped the backcross DNA; MF, PMH, NS and AP collated, edited and performed the genetic analysis of the backcross data; AP, GB, OM collated, edited and performed the genetic analysis of the AIL data; RK annotated the sequence data, AP, RK and GB analysed the re-sequencing data; AP and JS performed the pathway analyses; JS analysed the expression of candidate genes in other viral infections; PK, DH, and NS helped with the interpretation of the results. AP wrote the manuscript. All other co-authors provided manuscript editing and feedback. All authors read and approved the final manuscript.
Additional files
Additional file 1: Table S1
Levels of C. jejuni colonisation in the caeca of line 61, line N, back-cross and advanced intercross line birds. The measurements took place one, two, three, four and five days after challenge at 3 weeks of age. Data are presented as mean log10 (CFU/g) ± standard deviation of the mean. (XLSX 12 kb)
Additional file 2: Figure S1
Manhattan plot and Q-Q plot displaying the GWAS results from the back-cross experiment. Genomic location is plotted against -log10(P) in the Manhattan plot (above). Suggestive genome-wide threshold is shown as a horizontal line. Q–Q plot (below) of observed P-values against the expected P-values for Campylobacter gut colonisation (log-transformed number of C. jejuni per gram of caecal contents). (PPTX 1422 kb)
Additional file 3: Figure S2
Manhattan plot and Q-Q plot displaying the GWAS results from the AIL experiment (binary (0/1) phenotypes). Genomic location is plotted against -log10(P). Genome-wide (P < 0.05) and suggestive genome-wide thresholds are shown as dashed lines. Q–Q plot (below) of observed P-values against the expected P-values for Campylobacter gut colonisation level. (PPTX 1524 kb)
Additional file 4: Table S2
Genes located in the QTL candidate regions for Campylobacter colonisation resistance. (XLSX 27 kb)
Additional file 5: Figure S3
Linkage disequilibrium (LD) pattern for significant SNPs on chromosome 14. LD between SNPs in the 0.4 Mb on chromosome 14 region. LD blocks are marked with triangles. The significant markers are illustrated with a blue arrow. Strongest LD signals are in red and weakest in white. (PPTX 2065 kb)
Additional file 6: Figure S4
Linkage disequilibrium (LD) pattern for significant SNPs on chromosome 4. LD between SNPs in the 0.4 Mb on chromosome 4 region. LD blocks are marked with triangles. The significant marker is illustrated with a blue arrow. Strongest LD signals are in red and weakest in white. (PPTX 1646 kb)
Additional file 7: Figure S5
Linkage disequilibrium (LD) block containing the significant SNP identified on chromosome 16. (PPTX 1681 kb)
Additional file 8: Figure S6
Summary statistics of the single nucleotide variants identified in the candidate regions for Campylobacter colonisation resistance. (PNG 41 kb)
Additional file 9: Table S3
List of single nucleotide variants located in the candidate regions for Campylobacter colonisation resistance. Variant Effect Predictor annotation results, SIFT predictions and allelic frequencies per each parental line are also presented. (XLSX 1242 kb)
Additional file 10: Table S4
Genes located in the QTL candidate regions for Campylobacter resistance containing splicing, 5′UTR, both missense and UTR, CpG sites SNVs. (XLSX 14 kb)
Additional file 11: Figure S7
Network analysis using IPA software. The two networks (A related to immunological disease and hereditary disorder) and (B related to cell cycle, cell death and survival, connective tissue development and function) illustrate molecular interactions between products of candidate genes selected from QTL regions from the AIL. Arrows with solid lines represent direct interactions, and arrows with broken lines represent indirect interactions. The white colour indicates gene products added to the IPA analysis because of their interaction with the target gene products. (PPTX 1586 kb)
Additional file 12: Figure S8
Chemokine gene expression. Quantitation of CXCLi1 and CXCLi2 mRNA expression in caecal tonsils of line 61 and line N Campylobacter jejuni infected birds at 5 days post infection. Data are expressed as the fold change in mRNA levels when samples from infected birds were compared to non-infected birds of the same age from each line. Error bars show ± S.E.M. Asterisks indicate significant differences (P < 0.05) between line 61 and line N. (PPTX 204 kb)
Additional file 13: Table S5
List of selected candidate genes for Campylobacter colonisation resistance detected with the back-cross and AIL experiment. Expression of the candidate genes for Campylobacter resistance identified in the present study using data from previously published studies of other poultry infections. (XLSX 15 kb)
Additional file 14: Table S6
List of SNP markers used in the back-cross analysis. (XLSX 48 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Psifidi, A., Fife, M., Howell, J. et al. The genomic architecture of resistance to Campylobacter jejuni intestinal colonisation in chickens. BMC Genomics 17, 293 (2016). https://doi.org/10.1186/s12864-016-2612-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-016-2612-7