
Abstract
Multiple mitochondrial dysfunctions syndrome (MMDS) is an autosomal recessive disorder of systemic energy metabolism. This study is to present the diagnosis of two MMDS Chinese sufferers. Physical and auxiliary examination was performed. Next generation sequencing (NGS) was conducted to identify candidate causal genes and Sanger sequencing was adopted to validate the variants detected. Fluorescence quantitative polymerase chain reaction (FQ-PCR) amplification was carried out to testify allelic loss existence. Structural investigation was performed to study the possibility of the candidate variants for disease onset. Physical examination showed that the children were with neurological impairment. Auxiliary examination demonstrated energy metabolism disturbance and abnormal brain signals. NGS found that the probands had homozygous mutation of c.545 + 5G > A and compound heterozygous variants of exon 4 deletion and c.721G > T in NFU1, respectively. NFU1 was considered as candidate molecular etiology and indicating that the kids were with MMDS. Sanger sequencing confirmed the variants. FQ-PCR amplification characterized that patient 1 had a de novo allele mutation while patient 2 inherited from his parents. Structural investigation demonstrated that the variants were possible for MMDS occurrence. This is the first report of patients diagnosed as MMDS with novel mutation types from the Asia-Pacific region.
Similar content being viewed by others

Introduction
Iron-sulfur (Fe-S) clusters (ISCs) are one of the most ubiquitous and functionally versatile inorganic protein cofactors involved in a variety of biological processes (Rouault 2012; Netz et al. 2014). The most common types of ISCs are [2Fe-2S] and [4Fe-4S] (Lill 2009; Crack et al. 2012). Notwithstanding the simple composition, the biosynthesis of ISCs is rather complex. For example, [4Fe–4S] cluster is included in a complex metabolic pathway possessing iron–sulfur cluster scaffold homolog (ISCU, NFU1), bolA family member 3 (BOLA3), and iron-sulfur cluster assembly homolog (IBA57) (Tong et al. 2000; Tong et al. 2003; Zhou et al. 2008; Ajit Bolar et al. 2013). Genetic or functional deficiency of ISCs is responsible for the onset of some human energy metabolism diseases (Ajit Bolar et al. 2013; Cameron et al. 2011; Al-Hassnan et al. 2015).
Among ISCs associated diseases, there is a series of severe autosomal recessive diseases characterized by systemic dysfunction of energy metabolism, which are entitled multiple mitochondrial dysfunction syndrome (MMDS). MMDS is associated with defective lipoate synthesis and defects in respiratory complexes I, II, and III (Melber et al. 2016). The symptoms of MMDS included depression, dyspnea, pulmonary hypertension, hyperlactacidemia, hyperglycinemia, ketonuria, agenesis of the corpus callosum, brain white matter hyperintensity, and early death (Seyda et al. 2001; Navarro-Sastre et al. 2011; Nizon et al. 2014; Baker et al. 2014).
To date, four genes are found to be linked to MMDS, namely NFU1 (causing MMDS1), BOLA3 (causing MMDS2), IBA57 (causing MMDS3), and iron-sulfur cluster assembly 2 (ISCA2, causing MMDS4) (Ajit Bolar et al. 2013; Cameron et al., 2011; Al-Hassnan et al. 2015; Navarro-Sastre et al. 2011). NFU1 is considered as a late targeting factor of ISCs, playing roles in proper assembly of [4F2-4S] proteins (Nizon et al. 2014). Hitherto, reported cases of MMDS caused NFU1 all are from Europe and America (Seyda et al. 2001; Navarro-Sastre et al. 2011; Nizon et al. 2014; Invernizzi et al. 2014; Tonduti et al. 2015). In this study, we present two cases of children diagnosed as MMDS form China. To our knowledge, this is the first study of MMDS from Asia-Pacific region.
Materials and Methods
Patients’ Data
Patient 1, a boy of 4 months and 15 days old, was hospitalized because of having fever for 2 days and epileptic seizure for once. His temperature continued at 38~39 °C, and the twitch presented as generalized tonic-clonic seizure. He was the second child with a healthy elder brother from a non-consanguineous Chinese family. The boy could support his head and be amused at the age of 3 months, however, developmental regression started at 4 months of age. Patient 2 was a boy of 6 months and 1 days old. He was hospitalized because of emesia, cyanosis, and drowsiness for a half day. Feeding difficulty and development retrogression appeared after he was 3 months old. This study was approved by the ethics committee of Anhui Provincial Children’s Hospital and Hunan Provincial People’s Hospital. Informed consents were received from the parents prior to the study.
Physical and Auxiliary Examination
Once admitted, physical and auxiliary examination was performed. Venous blood was collected for blood routine and biochemical test. Cerebral spinal fluid examination and blood gas analysis were undertaken. Gas chromatography/mass spectrometry (GC/MS) was applied for urine metabolites detection. Pathological changes of the heart and lung were detected by chocardiography. Head computed tomography (CT) and magnetic resonance imaging (MRI) scanning were carried out to identify intracranial changes.
Next Generation Sequencing
NGS was performed using Illumina Hiseq2500 (Illumina, Santiago, USA). Briefly, genomic DNA was extracted, hybridized, and enriched for exome sequencing (ES) and whole genome sequencing (WGS). Raw image files were processed and the sequencing reads were aligned to the human reference genome (hg19) using BWA. GATK (McKenna et al. 2010) and Annovar (Wang et al. 2010) were used to analyze the candidate variants. A 0.5% frequency cut-off of 1000 genomes project database was applied. Non-synonymous, loss-of-function, indel, duplication, splice site variants, and copy number variants (CNVs) were taken for candidate variants identification. Protein biological function was predicted using Provean (Choi et al., 2015) and PolyPhen-2 (Adzhubei et al. 2010).
Sanger Sequencing
Polymerase chain reaction (PCR) was used for NFU1 amplification and the primers were listed in Table. 1. Amplification was performed in a 25 μL system with an annealing temperature of 60 °C. The PCR products were sequenced with ABI 3730XL (Thermo Fisher Scientific Inc., Waltham, USA) and analyzed by DNASTAR 5.0 software (DNASTAR, Inc., Madison, USA).
Fluorescence Quantitative Polymerase Chain Reaction Amplification
FQ-PCR amplification was performed using whole genome DNA from the probands, their family members, and healthy controls. The prixmers were illustrated in Table. 2 and the program was as follows: 95 °C for 1 min, followed by 40 cycles of 95 °C for 15 s, and 55 °C for 40 s. ABL gene was taken as reference gene. The amount of the amplified PCR product was calculated by comparing with that of the ABL.
Sequential Analysis and Structural Investigation
BioEdit sequence alignment editor was used for sequence study (http://www.mbio.ncsu.edu/bioedit/bioedit.html). Nuclear magnetic structure of N and C terminal domains of Homo sapiens NFU1 (PDB: 2ltm, 2m5o), N-terminal domain of Saccharomyces cerevisiae Nfu1 (PDB: 2ltl), and C-terminal domain of Mus musculus Nfu1 (PDB:1veh) were searched from PDB (http://www.rcsb.org/pdb/home/home.do). The structures were visualized using PyMOL software (http://www.pymol.org/).
Results and Discussion
Clinical Finding
To gain insight into the condition of the patients, physical examination was performed once they were hospitalized. The general status for patient 1 was as follows: weight 7.6 kg, T 39.3 °C, HR 190 /min, RR 5 /min, and Bp 56/35 mmHg. The boy was with facial cyanosis and irregular respiratory rhythm. He was in a deep coma with Glasgow outcome score of 5 (language 1, eyes open 1, movement 3). His pupils were with equal diameter (4 cm) and papillary light reflex was insensitive. Neither neck resistance nor Rales lung sounds were detected. The liver, with moderate hardness, was touched 6 cm under the right subcostal margin. The ascites was positive. Neurologic examination showed muscular hypertonia of the limbs and active bilateral knee jerk reflexes. Klinefelter syndrome, Brandt syndrome, and bilateral Babinski sign were negative.
For patient 2, the general condition was T 37.3 °C, P 128 /min, R 30 /min, WT 5 kg, and Bp 58/22 mmHg. The boy showed growth retardation, poor nutrition, and lethargy. The transcutaneous oxygen saturation decreased to 87%. His anterior fontanelle was 2 cm × 2 cm, uplifted, and with increased tension. Pupil diameter was 3 mm and light reflex was insensitive. The kid was with facial cyanosis, regular respiratory rhythm, and decreased breath sounds. His abdomen was flat and the liver was 2.5 cm under subcostal margin. He was with lower muscle strength and normal Achilles tendon reflex. Klinefelter syndrome, Brandt syndrome, and bilateral Babinski sign were negative.
In terms of the clinical symptoms, we could conclude that the children were with neurological impairment. After active rescue including cardiopulmonary resuscitation, tracheal intubation, ventilator assisted breathing, and fluid resuscitation, their heart rate recovered to normal range. They continued in deep coma, and as spontaneous breathing failed, they discharged a few days after treatment. Follow-up showed that they died soon after they left the hospital, and therefore, we had no chance to perform further examination.
Auxiliary Examination Indicating Energy Metabolism Disturbance
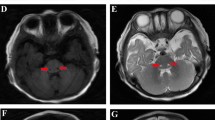
In order to make a positive diagnosis, auxiliary examination was applied. Blood gas analysis of patient 1 was pH 7.29, PaCO2 27 mmHg, Pa02 179.5 mmHg (Fi02 40%), HC03−12.8 mmol/L, and BE-15.5. Anion gap (AG) was 32.3, suggesting a high degree of AG metabolic acidosis. Routine blood test, C-reactive protein (CRP), and cerebrospinal fluid lactic acid were normal, excluding the existence of central nervous system infection. Biochemical result was as follows: alanine aminotransferase 88 U/L, albumin 27.5 g/l, urea nitrogen 17 mmol/L, uric acid 882.2 umol/L, blood lactate 5.2 mmol/L (normal 1.0 ~ 1.7 mmol/L), and troponin 0.26 ng/m L. Blood ammonia was 96 mmol/ L, argued the possibility of hyperammonemia. Echocardiography showed right heart enlargement and pulmonary hypertension. Thoracic and abdominal plain X-ray and CT scanning demonstrated pulmonary hypertension and ascites. Urinary organic acid analysis displayed mild phenylketonuria combined with mild methylmalonic aciduria. Skull CT demonstrated decreased symmetry density of bilateral white matter, indicating the presence of leukoencephalopathy Fig. 1a. Cranial MRI revealed extensive signal abnormalities in the subcortical white matter of both cerebral hemispheres, with bilateral internal capsule, external capsule, corpus callosum, and medulla involved. MR diffusion-weighted imaging (DWI) sequences showed limited value of diffusion, illustrating active pathological process period (Fig. 1b).

Extensive signal abnormalities (a, b patient 1; c, d patient 2) illustrated by imaging examination. Symmetrical low density (a) and T2WI high signal (b) are found in bilateral periventricular region with corpus callosum and semioval center affected. Head MR (c) and DWI (d) demonstrated abnormal signals across medulla, midbrain, thalamus, lateral ventricle, and centrum ovale
Patient 2 presented similar clinical manifestations. Routine blood test, CRP, and cerebrospinal fluid lactic acid were generally normal. Blood ammonia was 23.5 umol/L, eliminating the existence of hyperammonemia. Cerebrospinal fluid virus detection was negative therefore central nervous system infectious diseases were excluded. Urinary organic acid analysis showed increase of pyruvic acid, lactic acid, and 3-hydroxy butyric acid, suggesting ketonuria. CT illustrated abnormal signal in frontotemporal portion, excluded eripheral hydrocephalus and brain growth retardation. Head MR and DWI demonstrated abnormal signals across medulla, midbrain, thalamus, lateral ventricle, and centrum ovale (Fig.1c, d). In line of the results, the sufferers were with energy metabolism disturbance and suspected as metachromatic leukodystrophy.
NGS Identified NFU1 as the Candidate Pathogenic Gene
To unravel genetic causes of the patients, NGS was performed. For ES, identified Mendelian inheritance disease-related genes were sequenced. Finally, homozygous mutation of c.545 + 5G > A (RefSeq, NM_001002755.2) and heterozygosis variants of exon 4 deletion and c.721G > T in NFU1 were identified as the candidate variants. Seyda. A et al. showed that NFU1 which playing a critical role in ISCs biogenesis is the pathogenic gene of MMDS1 (Cameron et al., 2011; Melber et al. 2016; Stehling et al., 2013). Navarro-Sastre A and Xènia Ferrer-Cortès all demonstrate compound heterozygous mutations of c.545 + 5G > A and c.622G > T in NFU1 are responsible for MMDS1 (Navarro-Sastre et al. 2011; Ferrer-Cortes et al. 2016). In line with this, the identified mutations were possible to be the pathogenic mutations.
Validation of Candidate Causative Variant
In order to validate the mutations identified by NGS, Sanger sequencing was performed in the family trios. As illustrated in Fig. 2a, the c.545 + 5G > A variant was found to be homozygous in patient 1 and heterozygous in his father, whereas the mother was with wild type gene. As NFU1 is autosomal recessive inheritance for MMDS, we could not rule out the existence of allelic loss in the mother. To further characterize the allele in them, FQ-PCR was carried out. Compared with the control, the expression levels were both higher in the proband and his mother (Fig. 2b), indicating no allelic loss in them. Accordingly, it is possible to assume that the patient had a do nove and a paternal inherited c.545 + 5G > A mutation.

Sanger sequencing and fluorescence quantitative polymerase chain reaction (FQ-PCR) amplification results. Sanger sequencing argued that the sufferers were with homozygous mutation (a) and heterozygous mutation (b) in NFU1 gene. FQ-PCR illustrated that the expression of NFU1 in the patient 1 and his mother was normal (c), and patient 2 and his father were with heterozygous NFU1 exon 4 deletion while his mother was normal (d). Diagrammatic sketch of nucleic acid and protein of the identified NFU1 variants (E, c.545 + 5G > A; F, exon 4 deletion)
NGS showed patient 2 had compound heterozygous mutation of exon 4 deletion and c.721G > T. To our knowledge, these two sites are novel candidate mutation types of NFU1 gene for the occurrence of MMDS1. Sanger sequencing argued that c.721G > T mutation is passed from the mother (Fig. 2c). As Sanger sequencing could not identify the existence of heterozygous deletion, FQ-PCR was performed to better validate the inheritance of this deletion. As shown in Fig. 2d, FQ-PCR result demonstrated that the patient and his father were heterozygous deletion of exon 4, indicating that the compound heterozygous mutation identified in patient 2 was taken from his parents.
Structural Investigation Demonstrated that the Variants are Possible to Cause the Disease
In order to examine the hypothesis that the patients were caused by variants identified, we sought to assess convincing evidence theoretically. NFU1 is mainly composed by an N-terminal domain and a C-terminal domain. Sequence alignment shows that both domains are highly conserved and the C terminus of NFU1 contains a “CXXC” motif involved in Fe-S cluster binding (Cai et al. 2016), therefore, structural integrity of this C-terminal domain is vital to the function of the protein.
Hannie and Cheng et al. consider that changes in splicing region could cause exon skipping (Zandberg et al. 1995; Cheng et al. 2007). Navarro-Sastre A et al. demonstrate that c.545 + 5G > A variant could cause NFU1 exon 6 skipping (Navarro-Sastre et al. 2011). We studied the transcript this point mutation induces. It brings about a protein with frameshift starts from site 162 and premature stops at position 172 (Fig. 2e). As shown in Fig. 3a, this could almost cause whole C-terminal domain deletion. We then performed structural alignment and the root mean square (RMS) of the N-terminal domain of Homo sapiens NFU1 to Saccharomyces cerevisiae Nfu1and C-terminal domain of Homo sapiens NFU1 to Mus musculus Nfu1 are 1.376 and 2.038, respectively. The results argued that the structures are quite similar, indicating highly structural conservation of NFU1 (Fig. 3b, c). A protein with high sequential and structural conservation among different species usually demonstrates it is susceptible to mutation. To sum up, it is possible to assume that c.545 + 5G > A variant could induce a functional deficiency protein, which is responsible for the occurrence of MMDS in patient 1.

Structure schematic diagrams of NFU1 domains. (a) Diagram of NFU1 C domain (green + cyan) and the c.545 + 5G > A mutation induced truncation (green). Structural alignment of the N-terminal domain of Homo sapiens NFU1 to Saccharomyces cerevisiae Nfu1 and b C-terminal domain of Homo sapiens NFU1 to Mus musculus Nfu1 (c) showed that NFU1 domains are highly conservative. d Exon 4 deletion could lead to a truncated N domain (green + red) with the C terminus (cyan) lost. This will lost two β-sheets which play important roles in structural stability of the N domain. The peptides in red are caused by frameshift. e The V241 forms polar bond with L204 of β-sheet 2, which might affect the function of C domain
Patient 2 had two novel variants and as illustrated in Fig. 2f, deletion of exon 4 could lead to frameshift of NFU1 and induce a truncated protein with frameshift initiates from position 101 and stops at site 111. From Fig. 3d, it is obvious that most of the amino acids (cyan) of the N domain are lost. There are five β-sheets in the N terminus of NFU1, which, form a large number of main chain and side chain hydrogen bonds playing important roles in the stability of the structure. For the truncation, two β-sheets in the center are lost, which will disrupt the structural stability and in turn induce dysfunction of NFU1. The mutation of c.721G > T will incur V241F variant. As shown in Fig. 3e, this site locates at the C terminus of NFU1 C domain. Notwithstanding this site is on a loop, it is in close proximity to the C-terminal β-sheet and forms polar bond with L204 in co-rotating parallel β-sheet of the C-terminal β-sheet and might play certain role for the C domain structural stability. We could not directly infer that this mutation could lead to functional breakdown of NFU1, however, it is quite possible. Based on the structural analysis, it is possible to assume that the compound heterozygous mutations identified together could lead to the onset of patient 2.
In conclusion, in the present study, we reported two Chinese children diagnosed as MMDS1 in two non-consanguineous families using NGS. To our knowledge, they are the first cases of MMDS1 from Asia-Pacific region with specific mutation types never have been reported.
References
Adzhubei IA et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. doi:10.1038/nmeth0410-248
Ajit Bolar N et al (2013) Mutation of the iron-sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy. Hum Mol Genet 22:2590–2602. doi:10.1093/hmg/ddt107
Al-Hassnan ZN et al (2015) ISCA2 mutation causes infantile neurodegenerative mitochondrial disorder. J Med Genet 52:186–194. doi:10.1136/jmedgenet-2014-102592
Baker PR et al (2014) Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain : a journal of neurology 137:366–379. doi:10.1093/brain/awt328
Cai K et al (2016) Structural/functional properties of human NFU1, an intermediate [4Fe-4S] carrier in human mitochondrial iron-sulfur cluster biogenesis. Structure 24:2080–2091. doi:10.1016/j.str.2016.08.020
Cameron JM et al (2011) Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet 89:486–495. doi:10.1016/j.ajhg.2011.08.011
Cheng J et al (2007) A novel DFNA5 mutation, IVS8+4 A>G, in the splice donor site of intron 8 causes late-onset non-syndromic hearing loss in a Chinese family. Clin Genet 72:471–477. doi:10.1111/j.1399-0004.2007.00889.x
Choi Y et al (2015) PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31:2745–2747. doi:10.1093/bioinformatics/btv195
Crack JC et al (2012) Iron-sulfur cluster sensor-regulators. Curr Opin Chem Biol 16:35–44. doi:10.1016/j.cbpa.2012.02.009
Ferrer-Cortes X et al (2016) A leaky splicing mutation in NFU1 is associated with a particular biochemical phenotype. Consequences for the diagnosis Mitochondrion 26:72–80. doi:10.1016/j.mito.2015.12.004
Invernizzi F et al (2014) Cavitating leukoencephalopathy with multiple mitochondrial dysfunction syndrome and NFU1 mutations. Front Genet 5:412. doi:10.3389/fgene.2014.00412
Lill R (2009) Function and biogenesis of iron-sulphur proteins. Nature 460:831–838. doi:10.1038/nature08301
McKenna A et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi:10.1101/gr.107524.110
Melber A et al. (2016) Role of Nfu1 and Bol3 in iron-sulfur cluster transfer to mitochondrial clients. Elife 5 doi:10.7554/eLife.15991
Navarro-Sastre A et al (2011) A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet 89:656–667. doi:10.1016/j.ajhg.2011.10.005
Netz DJ et al (2014) Maturation of cytosolic and nuclear iron-sulfur proteins. Trends Cell Biol 24:303–312. doi:10.1016/j.tcb.2013.11.005
Nizon M et al (2014) Leukoencephalopathy with cysts and hyperglycinemia may result from NFU1 deficiency. Mitochondrion 15:59–64. doi:10.1016/j.mito.2014.01.003
Rouault TA (2012) Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis Model Mech 5:155–164. doi:10.1242/dmm.009019
Seyda A et al (2001) A novel syndrome affecting multiple mitochondrial functions, located by microcell-mediated transfer to chromosome 2p14-2p13. Am J Hum Genet 68:386–396. doi:10.1086/318196
Stehling O et al (2013) The role of mitochondria in cellular iron-sulfur protein biogenesis: mechanisms, connected processes, and diseases. Cold Spring Harb Perspect Biol 5:a011312. doi:10.1101/cshperspect.a011312
Tonduti D et al (2015) New spastic paraplegia phenotype associated to mutation of NFU1. Orphanet journal of rare diseases 10:13. doi:10.1186/s13023-015-0237-6
Tong WH et al (2003) Subcellular compartmentalization of human Nfu, an iron-sulfur cluster scaffold protein, and its ability to assemble a [4Fe-4S] cluster. Proc Natl Acad Sci U S A 100:9762–9767. doi:10.1073/pnas.1732541100
Tong WH et al (2000) Distinct iron-sulfur cluster assembly complexes exist in the cytosol and mitochondria of human cells. EMBO J 19:5692–5700. doi:10.1093/emboj/19.21.5692
Wang K et al (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164. doi:10.1093/nar/gkq603
Zandberg H et al (1995) Cooperation of 5′ and 3′ processing sites as well as intron and exon sequences in calcitonin exon recognition. Nucleic Acids Res 23:248–255
Zhou YB et al (2008) hBolA, novel non-classical secreted proteins, belonging to different BolA family with functional divergence. Mol Cell Biochem 317:61–68. doi:10.1007/s11010-008-9809-2
Acknowledgements
We greatly appreciate the understanding and cooperation of family members.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study was approved by the ethics committee of Anhui Provincial Children’s Hospital and Hunan Provincial People’s Hospital. Informed consents were received from the parents prior to the study.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
New and Noteworthy
Two novel mutation types resulting in MMDS were identified in this study and to our knowledge, this is the first report of patients diagnosed with MMDS from the Asia-Pacific region.
Rights and permissions
About this article

Cite this article
Jin, D., Yu, T., Zhang, L. et al. Novel NFU1 Variants Induced MMDS Behaved as Special Leukodystrophy in Chinese Sufferers. J Mol Neurosci 62, 255–261 (2017). https://doi.org/10.1007/s12031-017-0927-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-017-0927-8