
Abstract
Background
Multiple Mitochondrial Dysfunctions Syndrome 4 (MMDS4) is manifested as a result of ISCA2 mutations. ISCA2 is a vital component of 4Fe-4S clusters assembly machine. Therefore, in MMDS4 patients, deficient mitochondrial respiratory chain complexes I and II, Aconitase and Succinate dehydrogenase of Kerbs cycle and Lipoic Acid Synthetase in the biosynthesis of lipoic acid are expected.
Case presentations
A 7 months boy in an Iranian consanguineous family with progressive neurodegenerative problems was referred to us. Primarily, general laboratory tests, Abdomen ultrasonography and brain magnetic resonance imaging were performed. In order to find out the genetic problem in this case Whole Exome Sequencing (WES) following by Sanger sequencing was carried out. A novel variant (c.355G > A, p.Ala119Thr) in ISCA2 gene was identified by WES in the proband. Confirmation and segregation in the family for this variant was performed by Sanger sequencing. In-Silico prediction of the ISCA2 secondary structure showed that a helix motif in the Fe-S biosynthesis domain of ISCA2 protein will be eliminated as a result of this variant.
Conclusions
We reported the first patient with ISCA2 variant in Iranian population and the third one in the world reported for ISCA2 gene, so far associated with early-onset mitochondrial neurodegeneration. However further functional studies on this variant or finding it in other patients with similar clinical problems are needed to confirm the pathogenicity of this variant.
Similar content being viewed by others


Background
Mitochondria contain about 1500 functional proteins but only 13 of them are coded by the mitochondrial genome (mtDNA), and the rest are coded by the nuclear genome (nDNA). This fact justifies the inheritance patterns of mitochondrial diseases [1, 2]. mtDNA mutations are responsible for approximately 80% of adult mitochondrial diseases and only 20–25% of childhood cases. Accordingly, most cases of childhood mitochondrial diseases are due to mutations in nDNA [3]. Mitochondrial Iron-Sulfur Clusters (ISCs) are the necessary cofactors for pivotal life processes, such as electron transport in energy production as their main role, gene expression regulation, DNA maintenance, sulfur delivery in the process of lipoic acid synthesis, and the antiviral responses [4].
The production of ISCs in the mitochondria requires lots of proteins in stages like assembly, maturation and delivery to the targets. Defects in any of these proteins in this pathway play a major role in reducing the de novo production of ISCs. Up to now, Studies in yeast and human cell lines have shown the function of at least 17 protein components in the mitochondrial ISC assembly system, which mutation in any of them can cause severe disorders in humans [5,6,7,8,9].
A class of mitochondrial diseases that defect energy production and also affect the white matter of brain are MMDSs (Multiple Mitochondrial Dysfunctions Syndrome), of which four types have been introduced. NFU1 (MIM #608100) [10, 11], BOLA3 (MIM #613183) [10, 12], IBA57 (MIM #615316) [13] and ISCA2 (MIM #615317) [9] genes are responsible for 4 types of MMDSs, respectively. Recently, it has been argued whether ISCA1 mutations could be considered as the cause of another type of MMDSs or not [14, 15]. Common features of all of these dysfunctions are neurodevelopmental delay, defected lipoic acid biosynthesis, seizures, lactic acidosis, weakness, leukodystrophy, hypotonia, dystonia, and autosomal recessive mode of inheritance [6, 9, 10, 12,13,14,15].
In this report, we present a novel variant in the ISCA2 gene which is the first variant in this gene will be reported in Iranian population and the third variant in the world reported for ISCA2 gene so far.
Case presentation
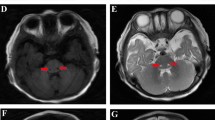
The proband (Fig. 1a), a boy who is the first child of an Iranian consanguineous family, was born after an uneventful pregnancy, but seven month after birth he developed some problems such as, malaise, insomnia, irritability, muscle stiffness and hypotonia. All general tests of the patient, including Complete Blood Count, Calcium, Phosphorous, Alkaline phosphatase, Total Vitamin D and Urine Analysis were normal. The patient’s abdomen sonography showed a normal size of kidney and spleen for his age. Brain magnetic resonance imaging showed nearly symmetrical abnormal changes in the areas of the periventricular white matter that was distributed in both centrum semiovale and has involvement of both middle cerebellar peduncles (Fig. 1b, c, d). Additionally, magnetic resonance spectroscopy which measures the amount of brain metabolites, indicated an increase in the lactate levels and a relative increase in the choline with respect to N-acetyl aspartate (NAA) in the periventricular regions of the patient’s brain, which is mostly reported in mitochondrial diseases (Fig. 1e).

a Pedigree of the family. Chromatograms represent homozygous and heterozygous state of c.355G > A variant in ISCA2 in the patient and his parents respectively. b, c Nearly symmetrical involvement of white matter in the axial view d Sagittal T2 view that shows extensive white matter signal change e Magnetic Resonance Spectroscopy that shows an increase in lactate levels and a relative increase in the choline with respect to N-acetyl aspartate (NAA)
Sample collection and DNA extraction
DNA was extracted from the peripheral blood of the patient, his parents, and one of his aunts using the Roche DNA Extraction Kit (Product No. 11814770001). The quality and quantity of the extracted DNAs were examined by Nanodrop and running on the gel.
Whole exome sequencing
Whole Exome Sequencing (WES) analysis was performed for the proband, where approximately 37 Mb (214,405 exons) of the Consensus Coding Sequences were enriched from fragmented genomic DNA by > 340,000 probes designed against the human genome (Nextera Rapid Capture Exome). The generated library was sequenced on the Illumina Hiseq2500 platform to an average coverage depth of 175X. All in all, 95.9, 98.2 and 99.8% of exons were covered with at least 20, 10 and one reads respectively (Additional file 1: Table S1).
In-silico bioinformatics analysis
An end to end in-house bioinformatics pipeline including base calling, adapters trimming, FASTQ file quality controls, primary filtering of low quality reads and probable artefacts were applied. Subsequently, reads alignment to reference human genome (hg 19), variant calling, recalibration of quality scores, annotation and filtration of variants were performed by HISAT2 [16], Genome Analysis Toolkit (GATK) [17], Annovar tool [18] and MySQL 8.0, respectively. It should be noted that dbSNP (build 150), 1000 Genome Project (1000 GP) and Exome Aggregation Consortium (ExAC) [19] databases were used for filtering purposes. The clinical significances of variants were taken from ClinVar (September 5, 2017), list of patient’s phenotype-related genes was also extracted from CentoMD database [20]. The probable effects of prioritized variants were predicted using PolyPhen-2 [21], SIFT [22], PANTHER [23], Mutation Taster [24], PMut [25], Human Splicing Finder, SNAP [26] and PROVEAN [27] algorithms. Finally, filtering of Iranian population variants were performed using Iranome database (http://www.iranome.ir). Protein residue conservations were also examined using the ConSurf server (http://consurf.tau.ac.il/2016/) [28]. Secondary structures of Wild-type and Mutant ISCA2 proteins were predicted by GORIV [29].
Sanger sequencing
Sanger sequencing was used to validate the identified variant in ISCA2 in the patient, his parents, and one of his aunts (Applied Biosystems). Primer sequences and PCR conditions are available upon request.
Results
After quality control and alignment, single nucleotide variants and small indel variants were called by GATK (74353 variants). Subsequently, GATK recommended filters for quality score recalibrations were applied and rest of the variants were annotated by Annovar tool (67205 variants). Because only one affected existed in this consanguineous family, both of heterozygous (42628 variants) and homozygous (24577 variants) variants, was considered.
In the filtration process, by excluding the variants with allele frequency greater than 1% in 1000GP, ExAC, dbSNP databases only 2194 homozygous and 6645 heterozygous variants remained. Then, variants that located in the exonic and splicing regions were chosen and Synonymous variants were excluded which ended up with 318 homozygous and 956 heterozygous variants. After that, we chose only variants located in the patient’s phenotype-related genes (20 homozygous and 36 heterozygous variants). The details of this filtration steps are presented in Fig. 2.

Flowchart of variants filtration process
Finally heterozygous variants were prioritized according to the expected inheritance mode of mutated genes, clinical significances and in-silico effect predictions (Additional file 1: Table S2 and Table S3). In other word, heterozygous variants identified in the genes which only homozygous variants with pathogenic effect for it have been reported in the literature, have not been considered. Otherwise, heterozygous variants were judged based on clinical significances and in-silico effect predictions, respectively. The homozygous variants were filtered only based on clinical significances and in-silico predictions. Also some variants were ruled out based on ethnic matched allele frequency database.
We concluded that the homozygous variant [c.355G > A, p.Ala119Thr] in ISCA2 gene (NM_194279.3), which affects a highly conserved domain of ISCA2 (encoded by exon 4) leads to the severe phenotypes observed in the patient (Fig. 3). The PROVEAN algorithm predicted that this variant is Neutral but other algorithms such as Mutation Taster, PANTHER, PolyPhen-2, SIFT, SNAP, PMut suggested that it is, in fact, detrimental to protein function. Sanger Sequencing results showed co-segregation of the c.355G > A variant with the disease in the family. Altogether these evidences strongly suggest the causality of this variant in the pathogenicity of infantile leukoencephalopathy in this family.

Gene structure of ISCA2 and position of the identified pathogenic variant. (Introns are not drawn in scale)
Discussion and conclusions
Each person has ~ 400–500 protein-modifying variants in the coding regions of his or her genome which makes it highly challenging in diagnostic to choose the pathogenic culprit variants among the others. In the current report, due to lack of functional analysis, we cannot be completely sure about the diagnosis, but the evidences provided here highly suggest that the c.355G > A variant in ISCA2 is a causative variant in this family.
The [4Fe-4S] clusters are prosthetic groups of a wide spectrum of proteins, such as Mitochondrial Respiratory Chain (MRC) complexes I and II, Aconitase and Succinate dehydrogenase of Kerbs cycle and Lipoic Acid Synthetase in the biosynthesis of lipoic acid. Also, lipoic acid is a cofactor of 5 multimeric enzymes, which are taking part in energy metabolism and catabolism of Lysine and Glycine, generally [30, 31]. Considering the cascading events, it can be inferred that deficiency in the ISCA2 disrupts the energy metabolism and Glycine catabolism. Hyperglycinaemia causes Glycine encephalopathy, which is one of the fatal features of patients with defected ISCA2.
Furthermore, a functional study in HeLa cells have stated that in the ISCA2 depleted cells by RNA interfering technology, the function of [4Fe-4S] clusters-free proteins may also be affected, like MRC complex IV, which is probably due to a pleiotropic effect of deficiency in assembling of MRC complexes I and II and their supercomplexes [32]. An example of this pleiotropic effect has been reported in an ISCA2 mutated patient [33].
Al-Hassnan et al. for the first time reported a d.G74961032A; c.G229A; p.G77S mutation in ISCA2 gene in 6 patients of 5 consanguineous families with following phenotypes: failure to thrive, spasticity, optic atrophy, severe leukodystrophy, and neurological regression. The age of onset in these patients was 3–7 months and the maximum lifespan was 5 years. The uniformity of mutation in all the families in their report indicated the existence of a founder effect. Later Al-Fadhel et al., in a retrospective review, reported 10 additional patients with the same mutation in Saudi Arabia [34].
The second mutation in the ISCA2 has been reported by Toldo et al. in an Italian family, recently. The case was a girl with compound heterozygote state for a nonsense and a missense mutation in ISCA2 gene. Interestingly, their biochemical analyses showed that the [4Fe-4S] clusters-free MRC complex IV function has also been affected [33]. Here we report the third mutation in the ISCA2 gene.
Based on the location of mutations, these three reports neither include nor exclude the possibility of a hotspot for mutations in this gene. However, Multiple Sequence Alignment by ConSurf, indicates that ISCA2 C-Terminal residues are highly conserved, and also [Fe-S] clusters synthesis domain is coded by C-Terminal. Therefore, more reports of mutations affecting this region of ISCA2 in the future can be expected (Fig. 4a, b). Additionally, prediction of the Wild-Type and Mutant ISCA2 protein secondary structures showed that the c.355G > A mutation eliminates a helix motif in the Fe-S biosynthesis domain and therefore could strongly affect on its function (Fig. 4b).

a The amino acid residues of ISCA2 colored based on conservation scores produced by ConSurf database and presentation of all mutations that have been reported to date. (p.Gly77Ser) reported by Al-Hassnan et al. as the first mutation in ISCA2 (Grey Box). (p.Phe99Leufs*18/p.Ser112Gly) reported by Toldo et al. as the second mutation (Black Boxes). (p.Ala119Thr) found by our investigation in an Iranian family (White Box). b Secondary structure prediction and comparison of Wild-Type and Mutant ISCA2 protein and presentation of Pfam domain which is involved in Fe-S clusters biosynthesis. In-silico prediction shows that the (p.Ala119Thr) mutation eliminates Helix motif in this area
Based on clinical observations and neuroradiological findings, genotype-phenotype correlation of ISCA2 deficiency is clinically overlapped with a wide range of other diseases such as Leukoencephalopathies with Brain Stem and Spinal Cord involvement and Lactate elevation, infantile Metachromatic Leukodystrophy (MLD) and MLD caused by Saposin B deficiency, Krabbe disease, infantile Vanishing White Matter disease, Canavan disease, and Alexander disease [33, 34]. Accordingly, efforts have been made to enable differential diagnosis, but no diagnosis is more accurate and faster than the genetic test.
As stated, impairment in the ISCA2 function disrupts crucial life pathways, so there is no definitive treatment available so far, other than supportive cares, and early demise occurs, usually.
Overall, we have discovered an Iranian family with the diagnosis of MMDS4 as a result of c.355G > A variant in ISCA2 gene. Since neurological disease lesions bring lots of psychological issues to the families and also an economic burden to the health system of each country, our report and the two previously mutation reports in ISCA2 gene suggest to be place this gene in targeted sequencing panels for Prenatal Testing and Preimplantation Genetic Diagnosis in families who are at the risk of infantile leukoencephalopathies.
Availability of data and materials
The datasets generated and/or analysed during the current study are not publicly available but are available from the corresponding author on reasonable request.
Abbreviations
- GATK:
-
Genome Analysis Toolkit
- ISCs:
-
Iron-Sulfur Clusters
- MLD:
-
Metachromatic Leukodystrophy
- MMDS:
-
Multiple Mitochondrial Dysfunctions Syndrome
- MRC:
-
Mitochondrial Respiratory Chain
- NAA:
-
N-Acetyl Aspartate
- WES:
-
Whole-Exome Sequencing
References
Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet. 2010;11:25–44.
Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong S-E, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134(1):112–23.
Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, et al. Mitochondrial diseases. Nature Reviews Disease Primers. 2016;2:16080.
Braymer JJ, Lill R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J Biol Chem. 2017. https://doi.org/10.1074/jbc.R117.787101.
Stehling O, Wilbrecht C, Lill R. Mitochondrial iron–sulfur protein biogenesis and human disease. Biochimie. 2014;100:61–77.
Ahting U, Mayr JA, Vanlander AV, Hardy SA, Santra S, Makowski C, et al. Clinical, biochemical, and genetic spectrum of seven patients with NFU1 deficiency. Front Genet. 2015;13(6):123.
Rouault TA, Tong WH. Iron–sulfur cluster biogenesis and human disease. Trends Genet. 2008;24(8):398–407.
Sheftel A, Stehling O, Lill R. Iron–sulfur proteins in health and disease. Trends in Endocrinology & Metabolism. 2010;21(5):302–14.
Al-Hassnan ZN, Al-Dosary M, Alfadhel M, Faqeih EA, Alsagob M, Kenana R, et al. ISCA2 mutation causes infantile neurodegenerative mitochondrial disorder. J Med Genet. 2015;52(3):186–94.
Cameron JM, Janer A, Levandovskiy V, MacKay N, Rouault TA, Tong W-H, et al. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet. 2011;89(4):486–95.
Seyda A, Newbold RF, Hudson TJ, Verner A, MacKay N, Winter S, et al. A novel syndrome affecting multiple mitochondrial functions, located by microcell-mediated transfer to chromosome 2p14-2p13. Am J Hum Genet. 2001;68(2):386–96.
Baker PR, Friederich MW, Swanson MA, Shaikh T, Bhattacharya K, Scharer GH, et al. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain. 2013;137(2):366–79.
Ajit Bolar N, Vanlander AV, Wilbrecht C, Van der Aa N, Smet J, De Paepe B, et al. Mutation of the iron-sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy. Hum Mol Genet. 2013;22(13):2590–602.
Finsterer J, Zarrouk-Mahjoub S. A commentary on homozygous p.(Glu87Lys) variant in ISCA1 is associated with a multiple mitochondrial dysfunctions syndrome. J Hum Genet. 2017;62(9):865.
Shukla A, Hebbar M, Srivastava A, Kadavigere R, Upadhyai P, Kanthi A, et al. Homozygous p.(Glu87Lys) variant in ISCA1 is associated with a multiple mitochondrial dysfunctions syndrome. J Hum Genet. 2017;62(7):723–27.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164–e.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285.
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D7. https://doi.org/10.1093/nar/gkx1153.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4.
Mi H, Guo N, Kejariwal A, Thomas PD. PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways. Nucleic Acids Res. 2006;35(suppl_1):D247–D52.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–2.
López-Ferrando V, Gazzo A, de la Cruz X, Orozco M, Gelpí JL. PMut: a web-based tool for the annotation of pathological variants on proteins, 2017 update. Nucleic Acids Res. 2017.
Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35(11):3823–35.
Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745–7.
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44(W1):W344–W50.
Garnier J. GOR secondary structure prediction method version IV. Meth Enzym, RF Doolittle Ed. 1998;266:540–53.
Tort F, Ferrer-Cortes X, Ribes A. Differential diagnosis of lipoic acid synthesis defects. J Inherit Metab Dis. 2016;39(6):781–93.
Berg J, Tymoczko J, Stryer L. Chapter 17: The citric acid cycle. In: Biochemistry. New York: WH Freeman and Company; 2002.
Sheftel AD, Wilbrecht C, Stehling O, Niggemeyer B, Elsässer H-P, Mühlenhoff U, et al. The human mitochondrial ISCA1, ISCA2, and IBA57 proteins are required for [4Fe-4S] protein maturation. Mol Biol Cell. 2012;23(7):1157–66.
Toldo I, Nosadini M, Boscardin C, Talenti G, Manara R, Lamantea E, et al. Neonatal mitochondrial leukoencephalopathy with brain and spinal involvement and high lactate: expanding the phenotype of ISCA2 gene mutations. Metab Brain Dis. 2018;33(3):805–12.
Alfadhel M, Nashabat M, Alrifai MT, Alshaalan H, Al Mutairi F, Al-Shahrani SA, et al. Further delineation of the phenotypic spectrum of ISCA2 defect: a report of ten new cases. Eur J Paediatr Neurol. 2018;22(1):46–55.
Acknowledgements
We would like to thank the patient’s family for their participation that made this work possible. We sincerely appreciate Dr. Mohammad Mehran Pursina, Tehran Islamic Azad University of Medical Sciences, Tehran, Iran for his neuroscientific supports and additionally grateful to the staffs of DeNA laboratory for helping us in this research. This work was supported by the Medical Genetics Department of Tarbiat Modares University of Iran. The undeniable fact is that equipmental and financial limitations prevented us from further exploration in this study.
Funding
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
MG conceived and designed the experiments. MG, ME conducted the experiments. ME analysed the data. ME, MG wrote the paper. ME designed all the figures. All co-authors have seen and agree with the contents of the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethics committee of Tarbiat Modares University. Patient’s clinical information and medical histories of the family were collected at the Department of Medical Genetics, DeNA Laboratory, Tehran, Iran. Written informed consent was obtained from the patient’s guardian for blood sampling and genetic tests.
Consent for publication
Written informed consent for publication of this case was obtain from legal guardians.
Competing interests
The report is void of any biases and the result hasn’t been manipulated.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Whole Exome Sequencing statistical analysis. Table S2. Reminder heterozygous variants after exclusion phenotype unrelated variants. Table S3. Reminder homozygous variants after exclusion phenotype unrelated variants. (DOCX 32 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Eidi, M., Garshasbi, M. A novel ISCA2 variant responsible for an early-onset neurodegenerative mitochondrial disorder: a case report of multiple mitochondrial dysfunctions syndrome 4. BMC Neurol 19, 153 (2019). https://doi.org/10.1186/s12883-019-1387-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-019-1387-2