
Abstract
Recently an early onset lethal encephalopathy has been described in relation to mutations of NFU1, one of the genes involved in iron-sulfur cluster metabolism. We report a new NFU1 mutated patient presenting with a milder phenotype characterized by a later onset, a slowly progressive spastic paraparesis with relapsing-remitting episodes, mild cognitive impairment and a long survival. The early white matter abnormalities observed on MRI was combined with a mixed sensory-motor neuropathy in the third decade. Our case clearly suggests the importance of considering NFU1 mutation in slowly evolving leukoencephalopathy with high glycine concentration.
Similar content being viewed by others
Dear editor,
Mutations of NFU1, one of the genes involved in iron-sulfur cluster metabolism have been recently reported in 14 patients with an early onset and rapidly fatal encephalopathy and/or severe pulmonary hypertension [1-4]. The patients reported died before the age of 15 months [1-3] except for one who was still alive, but severely affected, at the age of 2.5 years [4]. The MRI performed only in this last patient shown white matter abnormalities with cystic degeneration in the periventricular region and in corpus callosum [4]. The brain autopsy analysis performed in 7 patients, demonstrated also white matter demyelination, vacuolization and astrogliosis. We report a new NFU1 mutated patient with a significantly milder phenotype than previously reported.
Clinical data
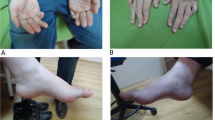
The patient was the second born child of unrelated healthy parents. Pregnancy and delivery were uneventful. Psychomotor development was initially normal. At the age of 18 months, concomitantly to an intercurrent viral illness, he lost the ability to walk, stand, sit, grip, he partially lost head control and spastic tetraparesis appeared. After this acute episode he partially recovered: one year later he was able to sit again and to stand alone but he never regained the ability to walk. He was first evaluated in our department at 17 years of age. Neurological examination showed a spastic paraparesis with mild cognitive impairment. Electrophysiological studies performed at 18 years of age (BAEP, VEP, ERG, SEP, EMG, NCV, EEG) were normal. Progressive scoliosis required spine surgery at 18 years of age. At the age of 24 years he subacutely developed flaccid paraplegia with superficial and deep hypoesthesia, EMG and NCV revealed the presence of a severe mixed motor-sensory neuropathy on lower limbs. Lipoic acid treatment (200 mg/day) has been introduced at the age of 28, without significant improvement. The patient now aged 30 years remains stable. However, during the last year, he developed an acute oedema of the lower limbs related to severe bladder dysfunctions requiring a urinary catheter.
Radiological findings
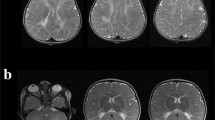
MRI performed at 3, 7, 16, 19, 24, 28 years of age revealed the presence of white matter abnormalities involving the periventricular regions and the posterior part of the corpus callosum which was also severely atrophic. Single voxel proton MR spectroscopy of the abnormal lobar white matter showed a severe reduction of the N-acetyl aspartate (NAA) peak, an increase in the choline (Cho) peak and a mild double inverted peak consistent with lactate (Figure 1). MRI abnormalities remained globally stable during the long radiological follow-up.

Radiological findings. MRI (A-D) and H-MRS (E) of patient 1 at 28 years of age. FLAIR (A-C) and T1 (D) sequences. MRI revealed the presence of white matter abnormalities hyperintense on FLAIR sequences, involving the periventricular regions (A-C) and the posterior part of corpus callosum (A) which was also severely atrophic (D). H-MRS (TE = 136 milliseconds) of the abnormal lobar white matter showed a severe reduction of the N-acetyl aspartate (NAA) peak, an increase of the choline (Cho) peak and a mild doublet inverted peak (white arrow) consistent with elevated lactate (lac).
Laboratory investigations
No documentation concerning laboratory investigations performed before referring to our Institute is still available. Aminoacid chromatographies on urine, plasma and CSF performed at 27 years of age revealed high concentration of glycine (Table 1)
. Redox status evaluations at 22 and 27 years of age showed a mild elevation of lactic and pyruvic acids after meal (Table 1). Activities of the respiratory chain complexes (mainly complex II) were low in muscle and fibroblasts as well as the pyruvate dehydrogenase complex and the α-ketoglutarate dehydrogenase in fibroblasts (Table 1).
Molecular analysis of NFU1 revealed the presence of one novel mutation (c.146delC, p.Pro49LeufsX8) predicted to be damaging (Align DGVD, Polyphen-2, SIFT, MutationTaster) inherited from the mother and a previously reported missense mutation (c.565G > A, p.Gly189Arg) [4] inherited from the father.
Discussion
Iron-sulfur clusters (Fe-S) are essential components of many mitochondrial, nuclear and cytosolic proteins [5]. We described a new patient affected by a disorder of iron-sulfur cluster metabolism due to heterozygous mutations of the NFU1 gene. All patients so far described presented a clinical picture dominated by an early onset severe encephalopathy and/or a severe pulmonary hypertension that lead in both cases to early death [1-4]. In sharp contrast, our patient presented a milder progressive leucoencephalopathy with a later age of onset (18 months of age) characterized by stress induced deterioration followed by partial recover, as usually observed in mitochondrial diseases [6]. No signs of pulmonary hypertension were ever found. The patient reached an age of 30 years with relatively stable neurological conditions. A subacute demyelinating mixed motor sensory neuropathy, never yet reported in NFU1 mutations, appeared in the third decade. Peripheral neuropathy is a typical feature in PDHA1 deficiency [7]. On our patient, NFU1 mutations result in a severe PDH defect demonstrated in fibroblasts. Moreover, differently to what is reported to date, our patient’s lactate and pyruvate levels were only high postprandially and the lactate/pyruvate ratio was normal. These data suggest a PDH deficiency more than an oxidative phosphorylation deficiency [1-4]. For this reason we suggest the possibility that the neuropathy in this patient may be related to the severe PDH deficiency.
The reason for the clinical and biological differences between our patient and those previously described is not clear. We can observe that patients reported to date were all homozygous either for the same null mutation or for the same missense mutation with only two patient heterozygotes for two different missense mutations [3,4]. Our patient was found to be compound heterozygote for a null and a missense mutation, one never reported so far. We could speculate that this genetic situation and probably some unknown epigenetic factors could differently modulate the final phenotype.
Our case clearly suggests the importance of considering NFU1 mutation not only in early onset leukoencephalopathy or in presence of a severe pulmonary hypertension but also in a more slowly evolving and later onset leukoencephalopathy with sometimes acute phases of deterioration. High concentration of glycine in body fluids is a key diagnostic marker.
Abbreviations
- BAEP:
-
Brainstem Auditory Evoked Potentials
- VEP:
-
Visual Evoked Potentials
- ERG:
-
Electroretinogram
- SEP:
-
Somatosensorial Evoked Potentials
- EMG:
-
Electromyography
- NCV:
-
Nerve Conduction Velocity
- EEG:
-
Electroencephalogram
- MRI:
-
Magnetic Resonance Imaging
- NAA:
-
N-Acetyl-Aspartate
- CSF:
-
Cerebrospinal fluid
References
Cameron JM, Janer A, Levandovskiy V, Mackay N, Rouault TA, Tong WH, et al. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet. 2011;89:486–95.
Seyda A, Newbold RF, Hudson TJ, Verner A, MacKay N, Winter S, et al. A novel syndrome affecting multiple mitochondrial functions, located by microcell-mediated transfer to chromosome 2p14-2p13. Am J Hum Genet. 2001;68:386–96.
Navarro-Sastre A, Tort F, Stehling O, Uzarska MA, Arranz JA, Del Toro M, et al. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet. 2011;89:656–67.
Nizon M, Boutron A, Boddaert N, Slama A, Delpech H, Sardet C, et al. Leukoencephalopathy with cysts and hyperglycinemia may result from NFU1 deficiency. Mitochondrion. 2014;15:59–64.
Lill R, Mühlenhoff U. Maturation of iron-sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu Rev Biochem. 2008;77:669–700.
Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. 2002;59:1406–11.
Imbard A, Boutron A, Vequaud C, Zater M, de Lonlay P, de Baulny HO, et al. Molecular characterization of 82 patients with pyruvate dehydrogenase complex deficiency. Structural implications of novel amino acid substitutions in E1 protein. Mol Genet Metab. 2011;104:507–16.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
All authors disclaim any financial or commercial involvement or other conflicts of interest. All authors take full responsibility for the data, the analyses and interpretation, and the conduct of the research; all authors have read and agreed to the content of the manuscript and all authors are included on the author list. All authors reports no disclosures.
Authors’ contributions
DT: study design, data analysis, manuscript and figure preparation. ID: study design, genetic analysis, manuscript preparation. AI: metabolic and genetic analysis, AS: respiratory chain analysis, manuscript review and correction. AB: PDH activity analysis, manuscript review and correction. SP: data collection, manuscript review and corrections. ME: neuroimaging evaluation, manuscript review and corrections. LV: data collection, manuscript review and corrections. JFB: metabolic and genetic analysis, manuscript review and corrections. HO: study design, data collection and analysis, manuscript review and corrections. OBT: study design, data collection and analysis, manuscript review and corrections. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Tonduti, D., Dorboz, I., Imbard, A. et al. New spastic paraplegia phenotype associated to mutation of NFU1 . Orphanet J Rare Dis 10, 13 (2015). https://doi.org/10.1186/s13023-015-0237-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-015-0237-6