
Abstract
Purpose of Review
This comprehensive review discusses the complex relationship between Alzheimer’s disease (AD) and osteoporosis, two conditions that are prevalent in the aging population and result in adverse complications on quality of life. The purpose of this review is to succinctly elucidate the many commonalities between the two conditions, including shared pathways, inflammatory and oxidative mechanisms, and hormonal deficiencies.
Recent Findings
AD and osteoporosis share many aspects of their respective disease-defining pathophysiology. These commonalities include amyloid beta deposition, the Wnt/β-catenin signaling pathway, and estrogen deficiency. The shared mechanisms and risk factors associated with AD and osteoporosis result in a large percentage of patients that develop both diseases. Previous literature has established that the progression of AD increases the risk of sustaining a fracture. Recent findings demonstrate that the reverse may also be true, suggesting that a fracture early in the life course can predispose one to developing AD due to the activation of these shared mechanisms. The discovery of these commonalities further guides the development of novel therapeutics in which both conditions are targeted.
Summary
This detailed review delves into the commonalities between AD and osteoporosis to uncover the shared players that bring these two seemingly unrelated conditions together. The discussion throughout this review ultimately posits that the occurrence of fractures and the mechanism behind fracture healing can predispose one to developing AD later on in life, similar to how AD patients are at an increased risk of developing fractures. By focusing on the shared mechanisms between AD and osteoporosis, one can better understand the conditions individually and as a unit, thus informing therapeutic approaches and further research. This review article is part of a series of multiple manuscripts designed to determine the utility of using artificial intelligence for writing scientific reviews.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
This is one of many articles evaluating the utility of using AI to write scientific review articles on musculoskeletal topics [1]. The first draft of this review was written entirely by humans. Refer to this edition’s Comment paper for more information [2]. Alzheimer’s disease and related dementias (AD/ADRD) and osteoporosis are two diseases that are prevalent in our aging population, and they unfortunately have a deleterious impact on quality of life [3]. Individuals living with AD typically experience a progressive loss of cognition, while those with osteoporosis are at increased risk of developing fractures. Patients diagnosed with both AD and osteoporosis may experience decreased cognitive agility, decreased mobility, and decreased ability to take care of themselves. Research has shown that these two disease processes are more intertwined than previously thought—in fact, they share many of the same molecular pathways and risk factors, such as old age, lifestyle, and fractures [4, 5]. While not the focus of this review, it is important to note that bone-brain cross-talk may be important in this process. Indeed, Yuan et al. recently reviewed the role of bone-derived modulators and AD progression. They describe that bone-derived cells and secreted proteins interact with multiple organ systems including the central nervous system, and such cross-talk between systems is important in the progression of AD [6]. In the current review, the commonalities between AD and osteoporosis will be elucidated, and a discussion of AD and fractures will seek to uncover whether each of the diseases affects the onset and progression of the other. Uncovering the complex relationship between these conditions could have important implications for improving prognosis and quality of life for those afflicted, which will be especially significant for our aging population.
Background on Alzheimer’s Disease and Related Dementias (AD/ADRD)
AD is the most common cause of dementia in the elderly and affects 6.7 million people in the USA, equaling roughly 1 in 9 individuals over the age of 65 [7]. Alzheimer’s disease and other dementias cost the USA $345B every year, with an additional estimated $339.5B in unpaid care, such as that provided by family and friends in the home [7]. AD is a deadly disease: deaths resulting from AD complications have doubled since 2000, and the 10-year survival rate for 70-year-old AD patients is half that of those without AD. AD is a multifactorial disease with many associated risk factors including advanced age, sex, genetic markers (e.g., apolipoproteinE4 (ApoE4) allele), traumatic head injuries, and environmental factors. Patients with AD present with multiple impairments including declines in cognition and memory and behavioral changes.
AD/ADRD is characterized by extracellular amyloid plaque deposition and intracellular neurofibrillary tangles in the medial temporal lobe of the brain, as well as widespread cerebral atrophy [8]. These pathological abnormalities result in many neurological changes in AD, which can be divided into two categories: positive lesions and negative lesions. Positive lesions are characterized by accumulations of abnormal deposits in the brain, such as amyloid plaques, and neurofibrillary tangles and negative lesions involve neuronal and synaptic loss. Abnormal deposition of beta-sheets has a strong correlation with dementia; beta-sheets provide the composition of fibrils, which aggregate to form amyloid plaques [9]. The transmembrane amyloid precursor protein (APP) is cleaved by proteolytic enzymes, yielding several varieties of amyloid beta (Aβ) monomers, including large and insoluble amyloid fibrils [10,11,12]. APP has been identified as a cause of early-onset AD when mutated [13]. The amyloid hypothesis posits that the degradation of the Aβ plaques is decreased with advanced age, thus leading to the aggregation of amyloid plaques. These amyloid plaques accumulate in chains of 39–43 amino acid Aβ peptides [14] throughout the brain causing neurotoxicity and inhibiting neural function, which can lead to cognitive impairment [8, 9, 12, 15]. Neurofibrillary tangles are hyperphosphorylated tau proteins and consist of accumulations of paired helical filaments that are characteristic of intracellular changes in AD [16]. Normally, tau acts as a scaffolding protein in microtubules to enrich axonal connections. Tau can undergo many post-translational modifications, such as monomethylation, acetylation, phosphorylation, and ubiquitination [17]. When tau protein becomes hyperphosphorylated, it begins to aggregate and loses its specificity for microtubules, impairing axonal function and causing neurodegeneration. The observed cerebral atrophy of negative lesions in AD is due to the loss of neurons and synapses throughout the brain, which may be more pronounced in the hippocampus and amygdala [8].
Despite the increasing prevalence of the disease, there is no cure for AD. Although the few drugs that have been approved by the FDA for AD are useful in temporarily alleviating symptoms, there has been little success in slowing or halting the progression of AD [17]. In July 2023, a new FDA-approved drug, lecanemab, showed a modest slowing of AD progression and reduced Aβ; however, debate is ongoing over the benefits in light of the undesirable side effects such as infusion-related reactions [18•].
Background on Osteoporosis
Osteoporosis is a prevalent skeletal condition associated with bone fragility due to low bone mass and compromised bone structure. It is estimated that there are over 10 million individuals living with osteoporosis in the USA [19]. The disruption of bone architecture seen in osteoporosis is a result of greater rates of bone loss than bone formation, which reduces bone strength and leads to an increased risk of fractures [20]. Bone remodeling is a continuous process of replacing older bone material with new bone material, helping to repair microfractures and preventing the onset of macrofractures. However, this process becomes impaired with aging-induced increases in bone resorption and reductions in bone formation. With this impaired balance, the architectural structure of bone becomes weakened due, in part, to a significantly reduced mass combined with deleterious changes in bone structure such as cortical thinning, leading to an increased incidence of fractures and subsequent decline in daily functioning.
Osteoporosis typically goes undiagnosed until a fracture occurs. A fracture of the hip or vertebrae without any severe trauma is diagnostic of the disease. Osteoporosis can also be diagnosed using a metric known as the T-score. This scoring involves measuring the patient’s bone mineral density (BMD) with a dual X-ray absorptiometry (DXA) scan and comparing this measurement to the mean BMD of young healthy people aged 20–29. BMD accounts for 70% of bone strength, with an additional 20% coming from bone quality, which is currently unmeasurable [20].
Overview of Alzheimer’s and Osteoporosis
While AD and osteoporosis seemingly affect very different organ systems, there are many commonalities between them. In fact, multiple AD mouse models have been shown to express an osteoporotic phenotype [21••]. In the current section, we will look at the underlying risk factors and pathways shared by AD and osteoporosis. It has been observed that osteoporosis and bone fracture occur at roughly twice the rate in AD patients compared to non-AD patients of comparable age [22]. Indeed, a previous cross-sectional study reported that individuals with AD were more likely to have sustained a hip fracture during their lifetime, have concurrent osteoporosis, and have fallen, as compared to individuals with no diagnosis of AD [23]. The risk factors between AD and osteoporosis that are shared include advanced age of the patients, poor nutrition, poor gait, impaired metabolism due to underlying co-morbidities, and sex-based differences in physiology.
Aβ has been implicated in the damage of bone tissue, as it has been shown that Aβ directly interacts with bone cells to increase bone resorption by osteoclasts and inhibit differentiation of osteoblasts, thus compromising bone architecture. APP, a transmembrane protein mentioned earlier as a precursor to amyloid plaques in the brain, is also expressed in osteoblasts and osteoclasts, two cell types important for bone remodeling. When certain mutations in APP occur, osteoblast differentiation is suppressed, preventing new bone growth and laying the foundation for osteoporosis [13].
Many pathways have been identified as having commonalities in both AD and osteoporosis. Studies have shown that patients with osteoporosis have an increased risk of developing AD compared to those without osteoporosis [24]. Furthermore, osteoporosis typically precedes a diagnosis of AD. This could indicate a pathophysiological link, which is not yet well understood.
While many observational studies have established an association between AD and osteoporosis, a recent two-sample Mendelian randomization study found that there was no distinct causal genetic link between the two conditions [25]. These researchers isolated potentially pleiotropic single nucleotide polymorphisms and found that the removal of such genes did not confer the development of osteoporosis or AD directly. It is worth mentioning that in addition to genetic links, environmental and physiological causes can be determinants of diseases, especially diseases associated with aging. As mentioned earlier and discussed in detail in the sections to follow, AD and osteoporosis share similar pathways and pathogenesis, which continue to require further investigation.
Shared Pathways Between Alzheimer’s Disease and Osteoporosis
The Wnt/β-catenin signal transduction pathway regulates many cellular processes in the body, including cell survival [26]. In the brain, this pathway works to increase neuronal survival, promote neurogenesis, and regulate synaptic plasticity [26]. The Wnt/β-catenin signaling pathway has been linked to AD, as its normal activation serves to inhibit Aβ production and tau phosphorylation (p-tau) in the brain. In aging brains, Wnt/β-catenin signaling is downregulated, and this suppression is even greater in AD brains [27]. Loss of function of the Wnt co-receptor LRP6 has been shown to downregulate the Wnt/β-catenin signaling pathway and is associated with an increased risk of developing AD [28, 29] while contributing to the synaptic dysfunction and Aβ accumulation seen in AD [30].
The Wnt/β-catenin signaling pathway is also a critical player in the facilitation of bone formation. The loss of Wnt/β-catenin signaling in osteocytes, specifically β-catenin gene deletion, causes an elevation of both the number and activity of osteoclasts, leading to substantial bone loss [31]. Furthermore, osteoclasts stimulate osteoblast differentiation through the secretion of Wnt ligands and chemoattractants to aid in skeletal remodeling [32]. Osteoblastic cells in turn impact osteoclastogenesis through the expression of RANKL and OPG, which work to differentiate osteoclasts [33, 34]. Thus, the interplay between bone regeneration and remodeling involves cytokine signaling, including Wnt/β-catenin, RANKL, and OPG which at least the former has also been implicated in AD as described in more detail below.
Studies using mouse models of AD, rat neurons in vitro cultures, and samples from human Alzheimer’s patients have identified deficits in the Wnt/β-catenin signaling pathway that accounts for both the Aβ and tau pathogenesis seen in AD, as well as the characteristic bone loss of osteoporosis. The Wnt/β-catenin signaling pathway has been shown to facilitate bone formation and promote synapse formation in the brain [22], and the disruption of this pathway has been implicated in both the onset of osteoporosis and AD. In relation to AD, the inhibition of the pathway allows for the unregulated production of p-tau and Aβ, leading to the accumulation and deposition of these proteins [22, 35]. Upon accumulation of p-tau and Aβ, inflammatory pathways are activated, further inhibiting the Wnt/β-catenin signaling pathway and contributing to a vicious cycle of p-tau and Aβ deposition. Disruptions in Wnt/β-catenin signaling are typically seen prior to the onset of AD [22, 36]. Dengler-Crish and Elefteriou in 2019 hypothesized that a disruption in the Wnt/β-catenin signaling pathway causes peripheral accumulation of Aβ initially and that the positive-feedback loop of further accumulation leads to deposition in the central nervous system, contributing to the pathogenesis of AD [22].
Angiogenesis
Angiogenesis is the process of forming new blood vessels from existing vasculature [37]. Angiogenesis is vital for proper bone repair, as it is involved in the development of new bone tissue and bone remodeling [38,39,40,41]. Angiogenesis is also a highly relevant process in AD, as the accumulation of amyloid plaques damages the cerebrovasculature.
A study using a transgenic mouse model (Tg APPsw) of amyloidosis found that the overexpression of APP may oppose angiogenesis, leading to decreased functional vasculature in the brain [14]. The impaired angiogenesis seen in AD patients leads to decreased capillary diameter, thinning of the capillary basement membrane, and atrophy of the cerebrovascular smooth muscle [14, 42,43,44]. Furthermore, Aβ peptides are also powerful inhibitors of angiogenesis, in both in vitro and in vivo studies [45]. The capillary network in cerebral cortices has demonstrated severe amyloid plaque accumulation and deposition, compromising the cerebrovasculature with a loss of small cortical arterioles and capillaries [14, 46]. Previous studies have shown that AD patients have increased levels of VEGF in the brain, a potent angiogenic factor necessary for the growth of vascular endothelial cells. This increase of VEGF suggests a compensatory mechanism in response to damaged cerebral structure, even though this mechanism ultimately fails to yield proper angiogenesis [14, 47, 48].
Bone repair following fracture constitutes an interplay between angiogenic and osteogenic pathways. Angiogenesis is necessary for fracture healing to occur and to prevent the onset of osteoporosis. Angiogenesis in actively regenerating calluses supplies the nutrients, oxygen, cytokines, and growth factors necessary for the formation of osteoblasts and osteoclasts, ultimately leading to bone formation [49]. Studies have shown that disruption in angiogenesis precedes the onset of osteoporosis, as inadequate blood flow is linked to impaired bone remodeling and subsequent low bone mass [40]. By a similar mechanism, angiogenesis also precedes osteogenesis. The endothelial cells are arguably the most important components of the vasculature, as they maintain a permeable barrier and allow for the recruitment of hematopoietic cells to the bone site to maintain bone homeostasis and facilitate fracture repair [40, 50,51,52].
Bone Mineral Density (BMD)
Lower BMD is associated with an increased risk of developing AD [53]. On the other hand, studies have shown that AD patients have reduced hip BMD and are at twice the risk of developing hip fractures [54, 55]. Large prospective studies have demonstrated an association between reduced BMD and an increased incidence of AD in the elderly [5, 53, 56,57,58]. Supporting this idea, a recent meta-analysis of three longitudinal studies found that a higher baseline BMD has a significant protective association with incident dementia (new cases of dementia); however, prior bone loss was not found to be associated with incident dementia [59].
A study in a Chinese population examined the potential role of low BMD on the transition from mild cognitive impairment to AD and found a positive relationship between osteoporosis and the decline in cognitive function observed in AD. Subjects in the lowest quartile for BMD were at twice the risk for AD compared to controls. Furthermore, the study revealed that individuals who were identified to have mild cognitive impairment at study onset were more likely to develop AD if they had a low baseline BMD. The study also showed that severe low BMD at baseline was associated with an increased risk of developing AD; this association was seen in both men and women [57]. Together, these studies imply a link between bone density loss and Alzheimer’s that requires exploration of common risk factors to identify a potential root cause.
The Protective Effect of Estrogen
Dementias have a variety of risk factors, including sex. Depending on the type of dementia, the epidemiology of male-to-female prevalence varies. AD is the most common form of dementia, with females constituting roughly 2/3 of all affected individuals [60]. The increased prevalence of AD in females as compared to men is likely due to the loss of the protective effect of estrogen in females with menopause. Indeed, in the normal brain, estrogen works in the nucleus basalis of Meynert to maintain normal cognitive function [61]. Furthermore, a cross-sectional study conducted in the Netherlands reported that women in the highest quintile of estradiol or estrone were 40% less likely to experience cognitive impairment compared to those in the lowest quintile [62], suggesting a protective effect of estrogen. Studies indicate that estrogen deprivation plays a vital role in the onset of cognitive decline and increased risk for AD in both men and women [61].
A study by Hoskin et al. found that levels of sex hormone–binding globulin (SHBG) were 20% higher in AD patients and that levels of estradiol were significantly reduced, compared to controls [63]. An estimated 37% of estradiol in elderly women circulates in the body bound to SHBG, the form that is postulated to be unable to cross the blood–brain barrier and thus cannot exert effects on the CNS. In other words, roughly 37% of elderly women’s estradiol is unable to be used for the protective effect of estrogen on the brain. Similarly, several observational studies have found an association between increased levels of SHBG and AD [61, 64, 65].
While these studies are promising for elucidating the negative effects of low estrogen levels on increased risk of developing AD in elderly women, there are conflicting studies that suggest a lack of association. The Rancho Bernardo study did not find a significant effect on cognitive test outcome due to bioavailable estradiol [66], and the Rotterdam study found that women with greater bioavailable estradiol levels demonstrated significantly poorer cognitive function [67]. However, these conflicting results may be due to variations in hormone measurement procedures [61].
Estrogen receptors are heavily expressed in osteoblasts, osteoclasts, and osteocytes, making their interaction with estrogen an important factor in the success of bone remodeling throughout the lifetime. Osteoporosis in post-menopausal women is directly related to estrogen deficiency. A deficiency in estrogen leads to increased bone resorption and a negative balance between bone resorption and formation [68]. Estrogen binds to estrogen receptors to inhibit osteoclast formation via the expression of osteoprotegerin. Estrogen can also activate the Wnt/β-catenin signaling pathway to increase osteogenesis. Thus, a lack of estrogen will alter the expression of target genes such as interleukin-1 (IL-1), IL-6, tumor necrosis factor alpha (TNFα), insulin-like growth factor (IGF), and transforming growth factor beta (TGFβ), decreasing osteogenesis. In females, the primary treatment for estrogen deficiency–related osteoporosis is estrogen supplements [68], primarily in the form of transdermal estradiol [69].
When estrogen binds to its receptors, it can also regulate the expression of gene-encoding proteins such as IL-1, IGF, and TGFβ [70]. Estrogen works to upregulate bone morphogenetic protein (BMP) signaling, which promotes mesenchymal stem cell differentiation from pre-osteoblasts to osteoblasts, enhancing bone formation in the remodeling process [68]. Estrogen also suppresses the action of receptor activator of nuclear factor kβ ligand (RANKL) to inhibit osteoclast activity. RANK is expressed on osteoclast precursors, and binding by RANKL promotes osteoclast formation and subsequent resorption. When estrogen binds to osteoclast-expressed estrogen receptors, RANK activity is suppressed [71]. Furthermore, estrogen inhibits the differentiation of osteoclasts and promotes osteoclast apoptosis through the increase of TGFβ production. Thus, estrogen serves to regulate the bone resorption rate.
Osteocytes are the foundational bone material and serve to control bone remodeling and mineralization [68]. The decline in estrogen levels in menopausal women has been associated with bone loss [72, 73] characterized by an increase in both osteoblasts and osteoclasts [74]. In men, low androgen levels result in bone loss and increased bone remodeling [75, 76], in part due to lower levels of estrogen [77]. A study found that in the absence of estrogen receptors, osteocytes were not able to provide an adequate response to received mechanical strain, thus representing a deficiency of osteocyte mechanosensory ability in the absence of estrogen [78].
Estrogen deficiency has major contributions to the pathophysiology of both AD and osteoporosis, affecting both the risk and progression of both diseases. While there has been some conflicting evidence about the role of estrogen in AD, it is reasonable to identify the deficiency of this hormone as a common risk factor between the two conditions, and estrogen may play a role in a shared disease mechanism. Identifying and unraveling the complex relationship between sex hormones and AD progression as well as the shared commonalities of pathways in bone disorders may help in developing potential therapies to improve bone mass while slowing the progression of AD, especially in post-menopausal women.
Benefits of FSH Blockade
Follicle-stimulating hormone (FSH) is an important regulator in the reproductive systems of men and women, and its blockade is shown to have beneficial effects on inhibiting the hallmarks of AD such as Aβ deposition and p-tau [79••]. Previous studies in mice have shown that FSH works to increase bone mass and enhance thermogenesis, two factors which are dysregulated in AD [80,81,82]. A study by Xiong et al. demonstrates that FSH accelerates Aβ and tau deposition in the hippocampus and cortical neurons, thus impairing cognition in 3xTg-AD mice. The study shows that blocking the action of FSH in 3xTg-AD mice inhibits the formation of plaque and neurofibrillary tangles, thus alleviating these adverse cognitive symptoms [79••]. Furthermore, recent results indicate that anti-FSH antibody is useful in increasing the bone formation of the femur and spine in mice [83•].
Neuroinflammation
Current evidence suggests that the progression and severity of AD can be attributed to the immunological mechanisms that occur in the brain [84]. For example, expression of immune receptors, such as triggering receptor expressed on myeloid cells 2 (TREM2) [85] and CD33 [86, 87], has been found to be associated with AD, suggesting that neuroinflammation contributes to the onset and progression of AD [84]. TREM2 is expressed in the microglia of the brain, and the variant R47H has been found to present a significantly higher risk of late-onset AD development [88]. Furthermore, the TREM2 variant Y38C in the brain disrupts the normal functionality of TREM2, causing changes in the microglia morphology and impairing the synaptic plasticity in the hippocampus. The downstream effects of the dysfunction of TREM2 provide an explanation of the events leading to AD and dementia [88]. These downstream effects are discussed in detail by Lee-Gosselin et al., who found in the brains of TREM2−/− mice injected with human tau extract that there was a significant decrease in microglial density compared to controls, as well as diminished tau pathology. This suggests that the experimental mice may not demonstrate a sufficient activated inflammatory response in the presence of tau pathologies, such as aggregation [89]. The observations from Lee-Gosselin et al. suggest that deletion of TREM2 may be beneficial in improving certain hallmarks of AD.
Additionally, it is hypothesized that the formation of neurofibrillary tangles is due to the neurotoxicity seen in neuroinflammation [16]. Furthermore, activated microglia and astrocytes seen in the inflammatory process surround the amyloid plaque depositions, resulting in higher levels of inflammatory mediators than observed in non-AD brains [90]. Reactive astrogliosis has been shown to occur in many neurodegenerative tauopathies, such as AD [91]. Taken together, these observations implicate neuroinflammation and the glial response as contributors to the damage of neurons and ultimately AD [92, 93].
Limited work has been conducted looking at the link between neuroinflammation and bone. TREM2 is expressed on osteoclasts, regulating the rate of osteoclastogenesis, and a study by Otero et al. reports that TREM2−/− mice exhibit osteopenic phenotype resembling the Nasu-Hakola disease [94, 95]. Furthermore, the TREM2 R47H variant has been implicated in low bone mass and skeletal muscle strength seen in TREM2R47H/+ mutant female mice, independent of central nervous system pathology [96].
Oxidative Stress
Reactive oxygen species (ROS) are free radicals that regulate cellular homeostasis and can be formed from both endogenous and exogenous sources. Endogenous sources of ROS include the mitochondrial respiratory chain and various enzymatic reactions, while exogenous sources are various stressors such as ionizing radiation and oxidizing chemicals [97]. Normally, ROS are important messengers in cell signaling, but at high concentrations, they can cause damage to cells leading to necrosis and apoptosis [97].
Oxidative stress is a major contributor to the progression of AD [97], with ROS being a critical player in the pathology of AD [98]. Oxidative stress has been shown to expedite aging and accelerate the onset of AD. The progressive cell loss due to oxidative stress can lead to the onset of neurodegenerative diseases; in AD, this causes abnormal aggregation of amyloid proteins [97]. In patients with AD, there is significant oxidative damage to brain tissue [99, 100], which leads to the upregulation of Aβ and p-tau formation [97, 99]. Double bond peroxidation in polyunsaturated neuronal lipid products forms molecules that stimulate p-tau [98, 101,102,103,104,105].
Oxidative stress has also been implicated as a causative factor in the diminished BMD in osteoporosis [106]. Kimball et al. cite four avenues through which oxidative stress affects the pathway of bone metabolism: (1) upregulation of osteoclastogenesis, (2) decreased osteoprogenitor differentiation, (3) decreased osteoblast activity, and (4) increased osteoblast and osteocyte apoptosis [106]. Oxidative stress causes increased osteoclastogenesis through the upregulation of RANKL and downregulation of osteoprotegerin; these two factors are an osteoclast activator and inhibitor, respectively [107,108,109], and occur via the Wnt/β-catenin pathway [107]. A study has shown that hydrogen peroxide–induced oxidative stress decreases osteoblast differentiation, thus inhibiting the formation of new bone [110]. Decreased osteoblast differentiation leads to decreased osteoblast activity and thus decreased osteoprotegrin production [106], ultimately ceasing regulation of osteoclast activity. Osteoblast and osteocyte apoptosis increase with oxidative stress, further inhibiting osteogenesis [106], while stimulating osteoclastogenesis via decreased osteoblastic cytokine activity [33, 111,112,113,114,115].
Therapies
There are currently three classes of FDA-approved drugs to treat AD: cholinesterase inhibitors, NMDA antagonists [8], and monoclonal antibodies. Acetylcholinesterase inhibitors function to block the breakdown of acetylcholine, thereby increasing the levels of acetylcholine in the synaptic cleft [116,117,118]. This medication helps to reduce the effects of the reduced cholinergic transmission throughout the brain due to the destruction of acetylcholine-producing cells in AD [8]. NMDA antagonists work to prevent cell death and synaptic dysfunction caused by excitotoxic overactivation of the NMDA receptor and subsequent increased levels of calcium [119, 120]. These two drugs are effective in managing the symptoms of AD but do not cure the disease [8, 17]. Due to the lack of disease-modifying therapies, research has focused on prevention or risk reduction of AD [121]. Studies have shown that lifestyle modifications such as physical activity, diet, and cognitive training can increase or maintain cognitive function and reduce new cases of AD in the elderly [8, 122]. Monoclonal antibodies have shown some promise in slowing the progression of AD. Trials using the monoclonal antibody aducanumab reported that high doses of the drug had the potential to slow the cognitive decline seen in AD, and the drug was given conditional FDA approval in 2022 [123]. Lecanemab, a humanized IgG1 monoclonal antibody [18•] which received FDA approval in July 2023, was found to reduce the markers of amyloid plaques in early-onset AD and led to less cognitive decline after 18 months of use, when compared to placebo. While this is the first drug that demonstrates slowing of AD progression to receive full FDA approval, ongoing studies are being conducted to determine the overall safety of the drug [18•].
In contrast, lifestyle modifications are the first-line treatment for the prevention or treatment of osteoporosis [124]. These modifications include eating a healthy and varied diet with calcium-rich and vitamin-rich foods [125], as well as reducing alcohol consumption and avoiding smoking [126]. However, these lifestyle modifications may not be enough for some patients, and thus, there are a variety of pharmaceutical options. There are two main treatment categories: anabolic treatments, which activate osteoblasts [127], and inhibitors of catabolism, which inhibit osteoclast-mediated resorption [128]. The most commonly prescribed anabolic treatments are parathyroid hormone (PTH) derivatives [124], although few patients receive full-length PTH and administration of PTH derivatives is given intermittently. PTH is a hormone known to promote bone resorption when administered continuously and promote bone regeneration when administered intermittently, regulating endochondral bone development [124], while maintaining higher BMD [129]. The most prescribed anti-catabolic treatments for osteoporosis are bisphosphonates. Bisphosphonates inhibit osteoclast activity and induce osteoclast apoptosis, thereby blocking bone resorption and stopping bone loss [124]. Thus, these two treatment categories serve to target osteoporotic pathologies at the mechanistic level to slow progression.
Increased Risk of Fractures Following Alzheimer’s Disease Diagnosis
It is well known that fracture is the most common sequela of osteoporosis, but it has been found to be a complication in AD as well. Research has shown that individuals with AD are more than twice as likely to sustain fractures at disease onset, despite having comparable risk to controls prior to the onset of AD symptoms [130•]. This increased risk occurs as soon as the first year of disease onset [131, 132]. The main risk factors for the increased incidence of hip fractures in AD patients are low BMD [4, 133], low concentrations of serum ionized calcium, and low concentrations of 25-hydroxyvitamin with compensatory hyperparathyroidism [134, 135]. Following a hip fracture, functional recovery is poor in AD patients [134, 136, 137], with individuals having a significantly lower ambulatory level [138] and greater risk of immobilization [139] compared to controls. Furthermore, AD patients have a higher risk of post-fracture mortality [140]. Another study found that individuals with dementia were at an even higher risk of developing a hip fracture if they also had diagnosed osteoporosis [54]. This idea is supported by the fact that individuals with AD have an increased risk of falling and subsequent fracture, with co-occurring osteoporosis being one of the strongest predictors of hip fractures [4, 54, 141, 142].
Increased Risk of Developing Dementia/AD Following Fracture Incidence
A 2020 observational study found that an incidence of distal radius, hip, and spine fractures increased the risk of developing dementia in individuals greater than 60 years of age [143]. A retrospective study found that, after a 12-year follow-up period, the overall incidence rate of dementia following fracture was 41% higher than in individuals who did not experience a fracture incidence [3]. Interestingly, the degree of increased risk varies depending on the fracture site, with hip fractures being the greatest at 60% higher risk of developing dementia. Comparatively, those with vertebral fractures had a 47% higher risk, those with thigh/leg/ankle fractures had a 35% higher risk, and those with an upper limb fracture exhibited a 29% increased risk [3].
There are several factors, both during the fracture incidence and healing process, that have been proposed to predispose or increase the risk of one developing dementia. It is hypothesized that fractures can predispose individuals to developing dementia due to the inflammatory process and reactive oxidative stress associated with fracture healing [143], as well as impaired balance [144,145,146] and vestibular asymmetry [147,148,149]. Following a fracture incidence, the inflammatory cytokines TNFα and IL-6 are elevated [150] in both the cerebrospinal fluid and peripheral blood [151, 152], and these two factors have been implicated in dementia [153]. Furthermore, ROS levels increase during fracture healing [154], which may lead to oxidative brain injury, thus increasing the risk of dementia [155].
Following a fracture, the complications of recovery may increase the risk of dementia via decreased physical activity and postoperative delirium [143]. Observed functional mobility declined in patients following a fracture [156], and a retrospective study found that roughly 32% of hip fracture patients who experienced postoperative delirium were later diagnosed with dementia [157].
Even though there are some observational clinical studies showing an association between the incidence of fractures and an increased risk of AD, to date, no studies exist in either humans or animal models of AD showing a link between fractures and AD progression. Due to a lack of these studies, it is imperative that this less understood link be explored further to uncover the intertwined pathways between fractures, bone health, inflammation, and AD.
Conclusion
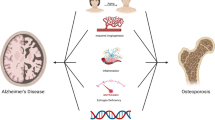
As illustrated in Fig. 1, Alzheimer’s disease and osteoporosis share many of the same disease mechanisms, such as altered angiogenesis, low BMD, a decrease in estrogen levels, neuroinflammation, and increased oxidative stress. The diseases both involve some of the same signaling pathways, and some of the characteristic molecular hallmarks of AD, such as Aβ and APP, have been shown to play a role in osteoporosis as well. One disease often predisposes an individual to the other, and many elderly patients concurrently have both AD and osteoporosis. The co-occurrence of these two degenerative diseases provides a great negative impact on the individual, namely, an increase in fractures and subsequent decreased mobility. Fractures have been shown to occur at greater rates in AD and osteoporosis patients compared to controls, largely due to the low BMD seen in both diseases. Interestingly, recent evidence suggests that an incidence of fracture also predisposes one to developing dementia, suggesting further commonalities between these two common geriatric diseases. Further exploration on fracture incidence causing AD onset is warranted, as this could uncover additional mechanistic commonalities and provide more insight into the pathogenesis of AD. Osteoporosis, AD, and fracture are debilitating ailments with recently uncovered similarities in pathophysiology. It is important to note that mouse models exploring such links have a variety of limitations, largely due to incomplete replication of the remodeling patterns seen in humans. As a result, there is a need for more human studies. That said, a limitation of human studies is the difficulty of drawing inferences related to pathways and mechanisms involved. Thus, both preclinical and clinical studies are essential to tackle these debilitating diseases. Finally, understanding the complex relationship between osteoporosis, AD, and fracture healing will be crucial to the development of therapies to improve the lives of people everywhere, especially the elderly.

Alzheimer’s disease and osteoporosis share many commonalities in their disease processes, such as inflammation, oxidative stress, estrogen deficiency, and, therefore, potential for shared therapeutics
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Kacena MA, Plotkin LI, Fehrenbacher JC. The use of artificial intelligence in writing scientific review articles. Curr Osteoporos Rep. 2024. https://doi.org/10.1007/s11914-023-00852-0.
Margetts TJ, Karnik SJ, Wang HS, et al. Use of AI language engine ChatGPT 4.0 to write a scientific review article examining the intersection of alzheimer’s disease and bone. Curr Osteoporos Rep. 2024. https://doi.org/10.1007/s11914-023-00853-z.
Tsai CH, et al. Fracture as an independent risk factor of dementia: a nationwide population-based cohort study. Medicine (Baltimore). 2014;93(26): e188.
Friedman SM, et al. Dementia and hip fractures: development of a pathogenic framework for understanding and studying risk. Geriatr Orthop Surg Rehabil. 2010;1(2):52–62.
Lui LY, et al. Bone loss predicts subsequent cognitive decline in older women: the study of osteoporotic fractures. J Am Geriatr Soc. 2003;51(1):38–43.
Yuan J, et al. The potential influence of bone-derived modulators on the progression of Alzheimer’s disease. J Alzheimers Dis. 2019;69(1):59–70.
2023 Alzheimer’s disease facts and figures. Alzheimers Dement. 2023;19(4):1598–695. https://doi.org/10.1002/alz.13016.
Breijyeh Z, Karaman R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules. 2020;25(24):5789. https://doi.org/10.3390/molecules25245789.
Chen GF, et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin. 2017;38(9):1205–35.
Cras P, et al. Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc Natl Acad Sci U S A. 1991;88(17):7552–6.
Perl DP. Neuropathology of Alzheimer’s disease. Mt Sinai J Med. 2010;77(1):32–42.
Armstrong RA. The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer’s disease. Folia Neuropathol. 2009;47(4):289–99.
Xia WF, et al. Swedish mutant APP suppresses osteoblast differentiation and causes osteoporotic deficit, which are ameliorated by N-acetyl-L-cysteine. J Bone Miner Res. 2013;28(10):2122–35.
Paris D, et al. Impaired angiogenesis in a transgenic mouse model of cerebral amyloidosis. Neurosci Lett. 2004;366(1):80–5.
Tabaton M, Piccini A. Role of water-soluble amyloid-beta in the pathogenesis of Alzheimer’s disease. Int J Exp Pathol. 2005;86(3):139–45.
Metcalfe MJ, Figueiredo-Pereira ME. Relationship between tau pathology and neuroinflammation in Alzheimer’s disease. Mt Sinai J Med. 2010;77(1):50–8.
Du X, Wang X, Geng M. Alzheimer’s disease hypothesis and related therapies. Transl Neurodegener. 2018;7:2.
• van Dyck CH, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388(1):9–21. This study reported a reduction in cognitive and functional decline in AD patients treated with lecanemab, a new monoclonal antibody that targets amyloid beta-soluble protofibrils.
Sarafrazi N, Wambogo EA, Shepherd JA. Osteoporosis or low bone mass in older adults: United States, 2017–2018. NCHS Data Brief. 2021;405:1–8.
Sozen T, Ozisik L, Basaran NC. An overview and management of osteoporosis. Eur J Rheumatol. 2017;4(1):46–56.
•• JE LL, et al. Degradation of bone quality in a transgenic mouse model of Alzheimer’s disease. J Bone Miner Res. 2022;37(12):2548–65. https://doi.org/10.1002/jbmr.4723. This primary research study utilized a 5xFAD transgenic model of AD to establish a relationship between elevated Aβ levels and impairment of bone health in mice.
Dengler-Crish CM, Elefteriou F. Shared mechanisms: osteoporosis and Alzheimer’s disease? Aging (Albany NY). 2019;11(5):1317–8.
Weller II. The relation between hip fracture and Alzheimer’s disease in the canadian national population health survey health institutions data, 1994–1995 A cross-sectional study. Ann Epidemiol. 2000;10(7):461.
Chen YH, Lo RY. Alzheimer’s disease and osteoporosis. Ci Ji Yi Xue Za Zhi. 2017;29(3):138–42.
Hu H, et al. No genetic causal association between Alzheimer’s disease and osteoporosis: a bidirectional two-sample Mendelian randomization study. Front Aging Neurosci. 2023;15:1090223.
Jia L, Pina-Crespo J, Li Y. Restoring Wnt/beta-catenin signaling is a promising therapeutic strategy for Alzheimer’s disease. Mol Brain. 2019;12(1):104.
Garcia-Velazquez L, Arias C. The emerging role of Wnt signaling dysregulation in the understanding and modification of age-associated diseases. Ageing Res Rev. 2017;37:135–45.
De Ferrari GV, et al. Common genetic variation within the low-density lipoprotein receptor-related protein 6 and late-onset Alzheimer’s disease. Proc Natl Acad Sci U S A. 2007;104(22):9434–9.
Alarcon MA, et al. A novel functional low-density lipoprotein receptor-related protein 6 gene alternative splice variant is associated with Alzheimer’s disease. Neurobiol Aging. 2013;34(6):1709.e9-18.
Liu CC, et al. Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer’s disease. Neuron. 2014;84(1):63–77.
Kramer I, et al. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol Cell Biol. 2010;30(12):3071–85.
Pederson L, et al. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc Natl Acad Sci U S A. 2008;105(52):20764–9.
Henriksen K, et al. Local communication on and within bone controls bone remodeling. Bone. 2009;44(6):1026–33.
Martin T, Gooi JH, Sims NA. Molecular mechanisms in coupling of bone formation to resorption. Crit Rev Eukaryot Gene Expr. 2009;19(1):73–88.
Inestrosa NC, Varela-Nallar L. Wnt signaling in the nervous system and in Alzheimer’s disease. J Mol Cell Biol. 2014;6(1):64–74.
Dengler-Crish CM, et al. Evidence of Wnt/beta-catenin alterations in brain and bone of a tauopathy mouse model of Alzheimer’s disease. Neurobiol Aging. 2018;67:148–58.
Adair TH, Montani JP. Angiogenesis. San Rafael (CA). Integrated Systems Physiology: from Molecule to Function to Disease. 2010. https://doi.org/10.4199/C00017ED1V01Y201009ISP009.
Colnot CI, Helms JA. A molecular analysis of matrix remodeling and angiogenesis during long bone development. Mech Dev. 2001;100(2):245–50.
Gerstenfeld LC, et al. Fracture healing as a post-natal developmental process: molecular, spatial, and temporal aspects of its regulation. J Cell Biochem. 2003;88(5):873–84.
Saran U, Gemini Piperni S, Chatterjee S. Role of angiogenesis in bone repair. Arch Biochem Biophys. 2014;561:109–17.
Bhatti FUR, et al. The effects of high fat diet, bone healing, and BMP-2 treatment on endothelial cell growth and function. Bone. 2021;146:115883.
Claudio L. Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer’s disease patients. Acta Neuropathol. 1996;91(1):6–14.
Kalaria RN, Hedera P. Differential degeneration of the cerebral microvasculature in Alzheimer’s disease. NeuroReport. 1995;6(3):477–80.
Mancardi GL, et al. Thickening of the basement membrane of cortical capillaries in Alzheimer’s disease. Acta Neuropathol. 1980;49(1):79–83.
Paris D, et al. Inhibition of angiogenesis by Abeta peptides. Angiogenesis. 2004;7(1):75–85.
Suter OC, et al. Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke. 2002;33(8):1986–92.
Kalaria RN, et al. Vascular endothelial growth factor in Alzheimer’s disease and experimental cerebral ischemia. Brain Res Mol Brain Res. 1998;62(1):101–5.
Tarkowski E, et al. Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol Aging. 2002;23(2):237–43.
Horner A, et al. Immunolocalisation of vascular endothelial growth factor (VEGF) in human neonatal growth plate cartilage. J Anat. 1999;194(Pt 4):519–24.
Imai K, et al. Selective transendothelial migration of hematopoietic progenitor cells: a role in homing of progenitor cells. Blood. 1999;93(1):149–56.
Imai K, et al. Selective secretion of chemoattractants for haemopoietic progenitor cells by bone marrow endothelial cells: a possible role in homing of haemopoietic progenitor cells to bone marrow. Br J Haematol. 1999;106(4):905–11.
Peled A, et al. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood. 2000;95(11):3289–96.
Tan ZS, et al. Bone mineral density and the risk of Alzheimer disease. Arch Neurol. 2005;62(1):107–11.
Wang HK, et al. Increased risk of hip fractures in patients with dementia: a nationwide population-based study. BMC Neurol. 2014;14:175.
Zhao Y, Shen L, Ji HF. Alzheimer’s disease and risk of hip fracture: a meta-analysis study. Sci World J. 2012;2012:872173.
Yaffe K, et al. Association between bone mineral density and cognitive decline in older women. J Am Geriatr Soc. 1999;47(10):1176–82.
Zhou R, et al. Bone loss and osteoporosis are associated with conversion from mild cognitive impairment to Alzheimer’s disease. Curr Alzheimer Res. 2014;11(7):706–13.
Brownbill RA, Ilich JZ. Cognitive function in relation with bone mass and nutrition: cross-sectional association in postmenopausal women. BMC Womens Health. 2004;4(1):2.
Lary CW, et al. Bone mineral density and the risk of incident dementia: a meta-analysis. J Am Geriatr Soc. 2023.
Castro-Aldrete L, et al. Sex and gender considerations in Alzheimer’s disease: the Women’s Brain Project contribution. Front Aging Neurosci. 2023;15:1105620.
Janicki SC, Schupf N. Hormonal influences on cognition and risk for Alzheimer’s disease. Curr Neurol Neurosci Rep. 2010;10(5):359–66.
Lebrun CE, et al. Endogenous oestrogens are related to cognition in healthy elderly women. Clin Endocrinol (Oxf). 2005;63(1):50–5.
Hoskin EK, et al. Elevated sex-hormone binding globulin in elderly women with Alzheimer’s disease. Neurobiol Aging. 2004;25(2):141–7.
Yaffe K, et al. Cognitive decline in women in relation to non-protein-bound oestradiol concentrations. Lancet. 2000;356(9231):708–12.
Schupf N, et al. Onset of dementia is associated with age at menopause in women with Down’s syndrome. Ann Neurol. 2003;54(4):433–8.
Barrett-Connor E, et al. Endogenous levels of dehydroepiandrosterone sulfate, but not other sex hormones, are associated with depressed mood in older women: the Rancho Bernardo Study. J Am Geriatr Soc. 1999;47(6):685–91.
Geerlings MI, et al. Endogenous estradiol and risk of dementia in women and men: the Rotterdam Study. Ann Neurol. 2003;53(5):607–15.
Cheng CH, Chen LR, Chen KH. Osteoporosis due to hormone imbalance: an overview of the effects of estrogen deficiency and glucocorticoid overuse on bone turnover. Int J Mol Sci. 2022;23(3):1376. https://doi.org/10.3390/ijms23031376.
Henderson VW. Estrogens, episodic memory, and Alzheimer’s disease: a critical update. Semin Reprod Med. 2009;27(3):283–93.
Eastell R, et al. Postmenopausal osteoporosis. Nat Rev Dis Primers. 2016;2:16069.
Abu-Amer Y. NF-kappaB signaling and bone resorption. Osteoporos Int. 2013;24(9):2377–86.
Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21(2):115–37.
Riggs BL, Khosla S, Melton LJ 3rd. A unitary model for involutional osteoporosis: estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men. J Bone Miner Res. 1998;13(5):763–73.
Manolagas SC, O’Brien CA, Almeida M. The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol. 2013;9(12):699–712.
Katznelson L, et al. Increase in bone density and lean body mass during testosterone administration in men with acquired hypogonadism. J Clin Endocrinol Metab. 1996;81(12):4358–65.
Daniell HW. Osteoporosis after orchiectomy for prostate cancer. J Urol. 1997;157(2):439–44.
Falahati-Nini A, et al. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men. J Clin Invest. 2000;106(12):1553–60.
Lee K, et al. Endocrinology: bone adaptation requires oestrogen receptor-alpha. Nature. 2003;424(6947):389.
•• Xiong J, et al. FSH blockade improves cognition in mice with Alzheimer’s disease. Nature 2022;603(7901):470–476. This study shows the potential of FSH blocking as a new treatment for AD, as this treatment in mice led to a reduction of AD phenotype.
Geng W, et al. Immunization with FSHbeta fusion protein antigen prevents bone loss in a rat ovariectomy-induced osteoporosis model. Biochem Biophys Res Commun. 2013;434(2):280–6.
Ji Y, et al. Epitope-specific monoclonal antibodies to FSHbeta increase bone mass. Proc Natl Acad Sci U S A. 2018;115(9):2192–7.
Liu P, et al. Blocking FSH induces thermogenic adipose tissue and reduces body fat. Nature. 2017;546(7656):107–12.
• Gera S, et al. FSH-blocking therapeutic for osteoporosis. Elife. 2022;11:e78022. https://doi.org/10.7554/eLife.78022. This study discussed the efficacy of FSH blockade in osteoporosis treatment in mice and introduced a new FSH-blocking antibody to be used in human trials.
Heneka MT, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405.
Guerreiro R, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–27.
Bradshaw EM, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013;16(7):848–50.
Griciuc A, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78(4):631–43.
Jadhav VS, et al. Trem2 Y38C mutation and loss of Trem2 impairs neuronal synapses in adult mice. Mol Neurodegener. 2020;15(1):62.
Lee-Gosselin A, et al. TREM2-deficient microglia attenuate tau spreading in vivo. Cells. 2023;12(12):1597. https://doi.org/10.3390/cells12121597.
Olabarria M, et al. Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: mechanism for deficient glutamatergic transmission? Mol Neurodegener. 2011;6:55.
Kovacs GG, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. 2016;131(1):87–102.
McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. 1995;21(2):195–218.
McGeer PL, Rogers J. Anti-inflammatory agents as a therapeutic approach to Alzheimer’s disease. Neurology. 1992;42(2):447–9.
Otero K, et al. TREM2 and beta-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J Immunol. 2012;188(6):2612–21.
Humphrey MB, et al. TREM2, a DAP12-associated receptor, regulates osteoclast differentiation and function. J Bone Miner Res. 2006;21(2):237–45.
Essex AL, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) R47H variant causes distinct age- and sex-dependent musculoskeletal alterations in mice. J Bone Miner Res. 2022;37(7):1366–81.
Chen X, Guo C, Kong J. Oxidative stress in neurodegenerative diseases. Neural Regen Res. 2012;7(5):376–85.
Cassidy L, et al. Oxidative stress in Alzheimer’s disease: a review on emergent natural polyphenolic therapeutics. Complement Ther Med. 2020;49:102294.
Gella A, Durany N. Oxidative stress in Alzheimer disease. Cell Adh Migr. 2009;3(1):88–93.
Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12(10):1161–208.
Mark RJ, et al. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 1997;68(1):255–64.
Bradley-Whitman MA, Lovell MA. Biomarkers of lipid peroxidation in Alzheimer disease (AD): an update. Arch Toxicol. 2015;89(7):1035–44.
Keller JN, et al. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: role of the lipid peroxidation product 4-hydroxynonenal. J Neurochem. 1997;69(1):273–84.
Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium. 2003;34(4–5):385–97.
Tamagno E, et al. H2O2 and 4-hydroxynonenal mediate amyloid beta-induced neuronal apoptosis by activating JNKs and p38MAPK. Exp Neurol. 2003;180(2):144–55.
Kimball JS, Johnson JP, Carlson DA. Oxidative stress and osteoporosis. J Bone Joint Surg Am. 2021;103(15):1451–61.
Fontani F, et al. Glutathione, N-acetylcysteine and lipoic acid down-regulate starvation-induced apoptosis, RANKL/OPG ratio and sclerostin in osteocytes: involvement of JNK and ERK1/2 signalling. Calcif Tissue Int. 2015;96(4):335–46.
Filaire E, Toumi H. Reactive oxygen species and exercise on bone metabolism: friend or enemy? Joint Bone Spine. 2012;79(4):341–6.
Romagnoli C, et al. Role of GSH/GSSG redox couple in osteogenic activity and osteoclastogenic markers of human osteoblast-like SaOS-2 cells. FEBS J. 2013;280(3):867–79.
Bai XC, et al. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem Biophys Res Commun. 2004;314(1):197–207.
Bellido T. Osteocyte-driven bone remodeling. Calcif Tissue Int. 2014;94(1):25–34.
Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26(2):229–38.
Jilka RL, Noble B, Weinstein RS. Osteocyte apoptosis. Bone. 2013;54(2):264–71.
Al-Dujaili SA, et al. Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J Cell Biochem. 2011;112(9):2412–23.
Mulcahy LE, et al. RANKL and OPG activity is regulated by injury size in networks of osteocyte-like cells. Bone. 2011;48(2):182–8.
Singh R, Sadiq NM. Cholinesterase inhibitors. In: StatPearls. Treasure Island (FL); 2023.https://www.ncbi.nlm.nih.gov/books/NBK544336/. Accessed 19 Sept 2023.
Eldufani J, Blaise G. The role of acetylcholinesterase inhibitors such as neostigmine and rivastigmine on chronic pain and cognitive function in aging: a review of recent clinical applications. Alzheimers Dement (N Y). 2019;5:175–83.
Sharma K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol Med Rep. 2019;20(2):1479–87.
Wang R, Reddy PH. Role of glutamate and NMDA receptors in Alzheimer’s disease. J Alzheimers Dis. 2017;57(4):1041–8.
Kuns B, Rosani A, Varghese D. Memantine. In: StatPearls. Treasure Island (FL); 2023. https://www.ncbi.nlm.nih.gov/books/NBK500025/. Accessed 19 Sept 2023.
Passeri E, et al. Alzheimer’s disease: treatment strategies and their limitations. Int J Mol Sci. 2022;23(22):13954. https://doi.org/10.3390/ijms232213954.
Crous-Bou M, et al. Alzheimer’s disease prevention: from risk factors to early intervention. Alzheimers Res Ther. 2017;9(1):71.
Beshir SA, et al. Aducanumab therapy to treat Alzheimer’s disease: a narrative review. Int J Alzheimers Dis. 2022;2022:9343514.
Tonk CH, et al. Therapeutic treatments for osteoporosis-which combination of pills is the best among the bad? Int J Mol Sci. 2022;23(3):1393. https://doi.org/10.3390/ijms23031393.
Nicholls AR, Holt RI. Growth hormone and insulin-like growth factor-1. Front Horm Res. 2016;47:101–14.
de Vernejoul MC, et al. Evidence for defective osteoblastic function. A role for alcohol and tobacco consumption in osteoporosis in middle-aged men. Clin Orthop Relat Res. 1983;179:107–15.
Russow G, et al. Anabolic therapies in osteoporosis and bone regeneration. Int J Mol Sci. 2018;20(1):83. https://doi.org/10.3390/ijms20010083.
Bi H, et al. Key triggers of osteoclast-related diseases and available strategies for targeted therapies: a review. Front Med (Lausanne). 2017;4:234.
Makino A, et al. Abaloparatide exerts bone anabolic effects with less stimulation of bone resorption-related factors: a comparison with teriparatide. Calcif Tissue Int. 2018;103(3):289–97.
• Zhang M, Hu S, Sun X. Alzheimer’s disease and impaired bone microarchitecture, regeneration and potential genetic links. Life (Basel). 2023;13(2):373. https://doi.org/10.3390/life13020373. This is a comprehensive review article that explores mechanisms and genetic influences shared between AD and osteoporosis.
Melton LJ 3rd, et al. Fracture risk in patients with Alzheimer’s disease. J Am Geriatr Soc. 1994;42(6):614–9.
Tolppanen AM, et al. Incident hip fractures among community dwelling persons with Alzheimer’s disease in a Finnish nationwide register-based cohort. PLoS ONE. 2013;8(3):e59124.
Amboni M, Barone P, Hausdorff JM. Cognitive contributions to gait and falls: evidence and implications. Mov Disord. 2013;28(11):1520–33.
Sato Y, et al. Risk factors for hip fracture among elderly patients with Alzheimer’s disease. J Neurol Sci. 2004;223(2):107–12.
Kipen E, et al. Bone density, vitamin D nutrition, and parathyroid hormone levels in women with dementia. J Am Geriatr Soc. 1995;43(10):1088–91.
Morrison RS, Siu AL. Mortality from pneumonia and hip fractures in patients with advanced dementia. JAMA. 2000;284(19):2447–8.
Holmes JD, House AO. Psychiatric illness in hip fracture. Age Ageing. 2000;29(6):537–46.
Matsueda M, Ishii Y. The relationship between dementia score and ambulatory level after hip fracture in the elderly. Am J Orthop (Belle Mead NJ). 2000;29(9):691–3.
Tanaka S, et al. The Fracture and Immobilization Score (FRISC) for risk assessment of osteoporotic fracture and immobilization in postmenopausal women–a joint analysis of the Nagano, Miyama, and Taiji Cohorts. Bone. 2010;47(6):1064–70.
Baker NL, et al. Hip fracture risk and subsequent mortality among Alzheimer’s disease patients in the United Kingdom, 1988–2007. Age Ageing. 2011;40(1):49–54.
Bukata SV, Kates SL, O’Keefe RJ. Short-term and long-term orthopaedic issues in patients with fragility fractures. Clin Orthop Relat Res. 2011;469(8):2225–36.
Gleason LJ, et al. Diagnosis and treatment of osteoporosis in high-risk patients prior to hip fracture. Geriatr Orthop Surg Rehabil. 2012;3(2):79–83.
Kim SY, et al. Increased risk of dementia after distal radius, hip, and spine fractures. Medicine (Baltimore). 2020;99(10):e19048.
Goto S, et al. Relationship between cognitive function and balance in a community-dwelling population in Japan. Acta Otolaryngol. 2018;138(5):471–4.
Deschamps T, et al. Postural control and cognitive decline in older adults: position versus velocity implicit motor strategy. Gait Posture. 2014;39(1):628–30.
Bahureksa L, et al. The impact of mild cognitive impairment on gait and balance: a systematic review and meta-analysis of studies using instrumented assessment. Gerontology. 2017;63(1):67–83.
Kristinsdottir EK, Jarnlo GB, Magnusson M. Asymmetric vestibular function in the elderly might be a significant contributor to hip fractures. Scand J Rehabil Med. 2000;32(2):56–60.
Kristinsdottir EK, et al. Observation of vestibular asymmetry in a majority of patients over 50 years with fall-related wrist fractures. Acta Otolaryngol. 2001;121(4):481–5.
Ekvall Hansson E, Dahlberg LE, Magnusson M. Vestibular rehabilitation affects vestibular asymmetry among patients with fall-related wrist fractures - a randomized controlled trial. Gerontology. 2015;61(4):310–8.
Yu MD, Su BH, Zhang XX. Morphologic and molecular alteration during tibia fracture healing in rat. Eur Rev Med Pharmacol Sci. 2018;22(5):1233–40.
Hall RJ, et al. Cerebrospinal fluid levels of neopterin are elevated in delirium after hip fracture. J Neuroinflammation. 2016;13(1):170.
Neerland BE, et al. Associations between delirium and preoperative cerebrospinal fluid C-reactive protein, interleukin-6, and interleukin-6 receptor in individuals with acute hip fracture. J Am Geriatr Soc. 2016;64(7):1456–63.
Darweesh SKL, et al. Inflammatory markers and the risk of dementia and Alzheimer’s disease: a meta-analysis. Alzheimers Dement. 2018;14(11):1450–9.
Yeler H, Tahtabas F, Candan F. Investigation of oxidative stress during fracture healing in the rats. Cell Biochem Funct. 2005;23(2):137–9.
Mecocci P, et al. A long journey into aging, brain aging, and Alzheimer’s disease following the oxidative stress tracks. J Alzheimers Dis. 2018;62(3):1319–35.
Piirtola M, et al. Fractures as an independent predictor of functional decline in older people: a population-based study with an 8-year follow-up. Gerontology. 2012;58(4):296–304.
Olofsson B, et al. Development of dementia in patients with femoral neck fracture who experience postoperative delirium-a three-year follow-up study. Int J Geriatr Psychiatry. 2018;33(4):623–32.
Funding
The funding for these studies was provided in part by the NIH (AG060621-05S1/-05S2 (MAK), U54AG054345 (ALO), AG078861/AG078861-S1 (LIP), AG-064003 (AM), K02AG-068595 (AM), and T35HL110854 (HSW)). This work was also supported in part by the Indiana University School of Medicine, the Indiana Clinical and Translational Sciences Institute (funded in part by NIH UM1TR004402), the Indiana Center for Musculoskeletal Health, the Stark Neuroscience Research Institute, and the Department of Orthopaedic Surgery. This material is also the result of work supported with resources and the use of facilities at the Richard L. Roudebush VA Medical Center, Indianapolis, IN: VA Merit I01BX006399 (MAK) and I01RX003552 (MAK) and I01BX005154 (LIP). The presented contents are solely the responsibility of the authors and do not necessarily represent the official views of any of the aforementioned agencies.
Author information
Authors and Affiliations
Contributions
This review article was conceived by MAK, JCF, LIP, AM, and ALO. HSW performed the initial literature search and wrote the first draft of the manuscript. All authors revised the manuscript critically for important intellectual content, take responsibility for all aspects of the work, and approve of the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Dr. Kacena is Editor-in-Chief for Current Osteoporosis Reports. Drs. Fehrenbacher and Plotkin are Section Editors for Current Osteoporosis Reports.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, H.S., Karnik, S.J., Margetts, T.J. et al. Mind Gaps and Bone Snaps: Exploring the Connection Between Alzheimer’s Disease and Osteoporosis. Curr Osteoporos Rep (2024). https://doi.org/10.1007/s11914-023-00851-1
Accepted:
Published:
DOI: https://doi.org/10.1007/s11914-023-00851-1