
Abstract
Purpose of Review
This review examines the linked pathophysiology of Alzheimer’s disease/related dementia (AD/ADRD) and bone disorders like osteoporosis. The emphasis is on “inflammaging”—a low-level inflammation common to both, and its implications in an aging population.
Recent Findings
Aging intensifies both ADRD and bone deterioration. Notably, ADRD patients have a heightened fracture risk, impacting morbidity and mortality, though it is uncertain if fractures worsen ADRD. Therapeutically, agents targeting inflammation pathways, especially Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) and TNF-α, appear beneficial for both conditions. Additionally, treatments like Sirtuin 1 (SIRT-1), known for anti-inflammatory and neuroprotective properties, are gaining attention.
Summary
The interconnectedness of AD/ADRD and bone health necessitates a unified treatment approach. By addressing shared mechanisms, we can potentially transform therapeutic strategies, enriching our understanding and refining care in our aging society. This review article is part of a series of multiple manuscripts designed to determine the utility of using artificial intelligence for writing scientific reviews.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
This is one of many articles evaluating the assistance of using AI to write scientific review articles on musculoskeletal topics [1]. The first draft of this review was written by ChatGPT 4.0 but was edited and carefully checked for accuracy resulting in a final manuscript which was significantly different from the original draft. Refer to this edition’s Comment paper for more information [2]. Alzheimer’s disease and related dementias (ADRD) and osteoporosis represent two prominent health issues with global implications as their prevalence is soaring in tandem with an increasingly aging demographic. Historically perceived as independent ailments, there is mounting evidence of a complex entanglement between these diseases, signifying a nuanced interplay between neuronal and skeletal health [3,4,5,6,7,8,9]. ADRDs, which account for the largest proportion of dementia cases, are characterized by progressive cognitive decline. ADRDs presently affect approximately 53 million individuals globally, a number projected to triple by 2050 [10,11,12]. A notable feature of ADRD epidemiology is the significant gender bias, with women constituting nearly two-thirds of the diagnosed cases [13]. This discrepancy was thought to be due to women’s longer lifespan; however, latest research shows a more complex picture with hormonal interplay post-menopause, differences in brain structure and physiology in different sexes, socio-economic factors instead of longevity, and other factors yet to be well understood [14,15,16,17,18].
Concurrently, osteoporosis is a systemic skeletal condition characterized by diminished bone mass and compromised bone tissue microarchitecture. This alteration predisposes affected individuals to an increased fracture risk, rendering osteoporosis a substantial public health challenge [19,20,21,22]. Like ADRD, osteoporosis prevalence amplifies with age and is more prevalent in women, especially post-menopausal women, partially due to the discontinuation of protective estrogen levels [23,24,25]. Fractures, especially hip fractures, contribute to significant morbidity, functional decline, and heightened mortality, further complicating the clinical trajectory for those concurrently dealing with ADRD [26,27,28,29].
The interconnection of ADRD and bone health, which encompasses conditions such as osteoporosis and subsequent fractures, has sparked considerable scientific interest. Recent studies indicate a network of shared risk factors, including advanced age, gender, genetic susceptibility, and lifestyle factors, which all play a key role in mediating the pathophysiology of both ADRD and bone-related disorders [5, 8, 9, 13, 30, 31, 32•]. Additionally, the molecular hallmarks intrinsic to ADRD, such as chronicinflammation, oxidative stress, and the accumulation of amyloid-beta (Aβ) and tau proteins, have been linked with dysregulated bone metabolism and elevated fracture risk [33, 34, 35, 36, 37, 38, 39, 40, 41]. A study in experimental AD-like mouse model (APP/PS1) found Aβ to be expressed in bones and potentially involved in the pathogenesis of osteoporosis in this mouse model of AD [41]. Amyloid precursor protein (APP) and Aβ42 were found to be elevated in the osteoporotic tissues collected from bone biopsies of female patients with osteoporosis compared to healthy controls [4]. Mouse models of AD have shown low bone mineral density (BMD) and altered osteogenesis and osteoclastogenesis [42••, 43, 44, 45, 46, 47•]. Alternatively, impaired bone health may expedite AD progression, instigating a vicious cycle of disease advancement and deteriorating quality of life [5, 6, 30, 48]. Neuroinflammation within the brain is one of the hallmarks of ADRD and leads to progression of the and in turn, ADRD can increase the inflammatory response leading to a positive feedback loop resulting in the worsening of the condition [49]. Low-grade systemic inflammation due to aging or bone disorders can also lead to progression of ADRD [7, 38, 50]. Since inflammation is a common theme in ADRD, bone disorders, fracture healing, and aging; we discuss it in detail and in context of these linked and intertwined conditions in the following sections.
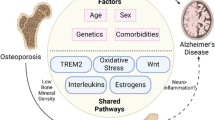
This review aims to coalesce our present understanding of the reciprocal relationship between ADRD and poor bone health, delving into common risk factors, overlapping molecular pathways, and implications for fracture risk and osteoporosis (Fig. 1). Further, we highlight potential therapeutic strategies that might target both conditions simultaneously. Deciphering this intricate interface could significantly enhance clinical management and prognosis for millions of patients globally living with ADRD and bone disorders.

Overlapping factors between aging, inflammation, bone disorders, and ADRD. Potential therapies can be developed to target both ADRD and bone disorders such as osteoporosis that reduce inflammation and/or target ROS
Sexual Dimorphism in Alzheimer’s Disease Incidence: A Complex Confluence of Factors
The epidemiology of ADRD demonstrates a marked sex-based disparity, with women shouldering a higher burden of disease incidence compared to men [13]. In the USA, for instance, women constitute nearly two-thirds of the total ADRD patient population [13]. This disproportion cannot be wholly relegated to the well-documented longevity advantage women have over men, inviting a closer scrutiny of multifactorial contributors including hormonal dynamics, sex, genetics, and physiological differences [14, 17, 32, 51, 52].
Sex hormones, particularly estrogen, have been suggested to exert a crucial modulatory influence on ADRD risk [17, 53,54,55,56,57]. Estrogen exerts several neuroprotective effects, such as mitigating oxidative stress, stimulating neuronal growth, and fine-tuning synaptic plasticity [15, 57,58,59,60,61]. Estrogen is known to decrease inflammation in the brain [62]. Studies on the effect of estrogen on inflammatory factors show complex interactions and sometimes opposing effects between inflammation and estrogen [63,64,65,66,67,68]. In neuroinflammation, pro-inflammatory factors, such as cyclooxygenases 1 and 2 (COX1 and 2), are found to be elevated [69, 70]. Immune cells located in the brain (microglia and astrocytes) are also involved in increasing the expression of pro-inflammatory factors. COX 1 and 2 generate prostaglandins such as PGE2 [71]. Estrogen is known to have a protective effect by inhibiting or counteracting the inflammatory factors such as COX 1 and 2, prostaglandins, inducible nitric oxide, and aromatases [72,73,74].
Intriguingly, estrogen is also implicated in inhibiting the aggregation of beta-amyloid plaques, a defining neuropathological feature of AD, thereby potentially attenuating disease risk [54, 60]. Menopause results in a pronounced reduction in estrogen levels in women, which may, in turn, amplify their susceptibility to ADRD [16]. A study in female 3xTg mice (a transgenic mouse model for AD) demonstrated the roles female sex hormones play in AD pathology [55]. In 2007, Carroll et al. [55] showed that administering estrogen to ovariectomized female 3xTg mice had beneficial effects, reducing Aβ accumulation and improving working memory-based performance in a Y-maze test [55]. Progesterone on the other hand blocked estrogen’s beneficial action on Aβ accumulation, but not cognitive performance, when administered together with estrogen. Progesterone, however, was beneficial in tau pathology by reducing phosphorylation of tau, when either administered alone or in combination with estrogen [55]. This study highlights the importance and synergistic action of female sex hormones, estrogen and progesterone, in 3xTg mice. However, menopausal women do not completely lack ovaries and the secretion of sex hormones is only reduced and not eliminated as is the case in ovariectomized female mice. Hence, caution needs to be exercised in interpreting the results in animal models and translating them to humans. Another study involving 3xTg mice showed that blocking follicle stimulating hormone (FSH), by FSH binding antibody (FSH-Ab), slowed the progression of AD in 3xTg mice [75••]. FSH blockade resulted in a reduction in Aβ and Tau levels in ovariectomized 3xTg mice. The study also showed that FSH acts on the hippocampal and cortical neurons to accelerate Aβ accumulation. Studies done in another AD mouse model, 5xFAD, also shows beneficial effects of estrogen [57] and membrane-associated estrogen receptor, G protein-coupled receptor 30 (GPR30) [76]. FSH has also been explored as a potential target for different diseases including AD and osteoporosis. It was demonstrated by Gera et al. that blocking FSH in mice using a humanized antibody against FSH helped stimulate new bone formation [77, 78]. More studies are needed in different female mouse models for AD that study the effects of estrogen, progesterone, FSH, and luteinizing hormone (LH) synergistically. These nuances will likely impact the translatability of animal studies to humans, especially when considering potential therapies.
Emerging evidence also points toward a potential role of the female sex chromosome gene expression in shaping ADRD risk [31]. Certain genetic variants, such as the apolipoprotein E (APOE) ε4 allele, a well-documented genetic risk factor for AD, appear to confer a heightened risk in women compared to men [79]. Moreover, physiological differences between sexes, encompassing differences in brain structure and functionality, may contribute to sex-dependent incidence of ADRD. Women are reported to exhibit a higher prevalence of brain alterations such as pronounced atrophy in regions prone to ADRD, potentially augmenting disease risk [80]. However, whether atrophy in the regions of brain is a cause or an effect of ADRD is yet to be fully understood. Furthermore, the cause of brain atrophy and its relationship to sex is not fully understood. Due to the critical role that APOE isoforms play in ADRD pathology, mice expressing human APOE (h-APOE) isoforms have been developed and used for studying ADRD pathology [81]. These mice show human isoform-specific differences in lipid physiology and synaptic function which are hallmarks of ADRD pathogenesis [81], suggesting that the allele ε4 in human APOE shows highest genetic risk for ADRD compounded by female sex [79, 82].
A comprehensive understanding of sex-based differences in ADRD incidence could offer critical insights into the pathophysiological underpinnings of the disease, possibly informing the development of sex-specific therapeutic strategies. Further research is warranted to decode the mechanistic basis of this gender disparity in ADRD, as well as potential intersections with bone health and fracture risk.
Convergence of Central Nervous System and Peripheral Inflammation: Roles in ADRD and Bone Health
The physiological process of aging is also accompanied by “inflammaging,” a chronic, low-grade inflammatory state believed to be a significant contributor to both neurodegenerative and skeletal diseases [83]. This inflammatory response in aging, ADRD, and osteoporosis shares common molecular pathways. These include the activation of the Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) signaling pathway, an increase in the production of reactive oxygen species (ROS), and the emergence of cellular senescence [84,85,86].
In 2016, DiSabato et al. [87] defined neuroinflammation as an inflammatory response in the brain and spinal cord, mediated by inflammatory cytokines, chemokines, and ROS. The intricate pathogenesis of ADRD prominently features an augmented state of neuroinflammation, a phenomenon increasingly acknowledged as a crucial accelerator of the disease trajectory [88,89,90,91,92]. This persistent inflammation within the brain is primarily orchestrated by microglia and astrocytes, the resident immune cells and support cells of the central nervous system (CNS), respectively, which exhibit a heightened levels of activity within an ADRD environment [93,94,95,96,97].
In a physiologically normal state, microglia enact an essential role in preserving brain homeostasis. They function as innate immune cells, facilitating the clearance of cellular debris and aberrant proteins, inclusive of Aβ peptides [98,99,100]. However, as ADRD advances, these microglial cells undergo a significant phenotypic transformation, instigating a potent neuroinflammatory response marked by the production of pro-inflammatory cytokines, ROS, and additional neurotoxic entities such as Aβ and tau tangles [89, 94, 95, 98]. This phenotypic switch in microglial behavior is further exacerbated by the progressive accumulation of AD hallmark structural pathological features, namely Aβ plaques and neurofibrillary tangles composed of hyperphosphorylated tau proteins [101].
Astrocytes, another cell type implicated in the genesis of neuroinflammation, contribute to the inflammatory cascade within the ADRD-afflicted brain [96, 97, 102, 103]. Under pathological circumstances, these cells transition into a hyperactive state, upregulating the expression of inflammatory mediators while simultaneously forfeiting their capacity to support neuronal health and synaptic functionality [104,105,106].
The neuroinflammatory response not only augments neurodegeneration but also appears to underpin other pathological facets of ADRD, such as synaptic dysfunction and disruption of neuronal networks [87, 107, 108]. Moreover, mounting evidence proposes that neuroinflammation may arise early in the ADRD continuum, potentially even preceding the formation of Aβ plaques and tau tangles. This places neuroinflammation squarely in the crosshairs as a viable therapeutic target [109,110,111].
Persistent neuroinflammation in ADRD implies a plausible link to altered bone metabolism and heightened fracture risk, though the precise molecular mediators bridging these processes remain to be unraveled [3, 4, 6, 9, 30, 48, 112]. While neuroinflammation primarily affects the central nervous system (CNS), it can have systemic effects as well. Studies have shown that neuroinflammation can communicate with the peripheral immune system, leading to the release of cytokines and other immune mediators into the bloodstream. This process is known as the “neuroimmune interface.” The communication between the CNS and the peripheral immune system is bidirectional. Peripheral cytokines can cross the blood-brain barrier and enter the brain, contributing to neuroinflammation. Conversely, pro-inflammatory molecules released by activated immune cells within the brain can signal to peripheral immune cells, triggering an inflammatory response in the periphery. Therefore, in the context of brain neuroinflammation, it is possible to observe an increase in peripheral cytokines. The specific cytokines released can vary depending on the underlying neuroinflammatory condition. For example, in conditions like multiple sclerosis or stroke, the peripheral levels of cytokines such as interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and interleukin-1 beta (IL-1β) have been found to be elevated.
It is important to note that the relationship between brain neuroinflammation and peripheral cytokines is complex and can be influenced by various factors. Additionally, the exact mechanisms and consequences of this bidirectional communication are still being studied. As we refine our understanding of neuroinflammation’s role in ADRD, it becomes imperative to further scrutinize its association with bone health. Such investigations may uncover novel therapeutic strategies, shedding new light on our approach to this formidable disease.
The importance of inflammation is equally apparent in the context of bone health. Pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, have been implicated in osteoclastogenesis, the formation of bone resorbing osteoclasts [113, 114]. Upregulation of osteoclastogenesis can precipitate bone loss and subsequent osteoporosis [115, 116]. Additionally, chronic inflammation can thwart bone formation by inhibiting the function and differentiation of bone forming osteoblasts, thereby tipping the balance of bone homeostasis towards resorption [117, 118].
Emerging evidence suggests a potential bidirectional interplay between ADRD and bone health, with inflammation acting as a critical mediator. Neuroinflammation in ADRD may indirectly influence bone health via dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis, which could lead to elevated cortisol production and, consequently, bone loss [119,120,121]. Conversely, systemic inflammation ensuing from osteoporosis and fractures could augment neuroinflammation and further propagate ADRD pathology [7, 39, 86, 122,123,124].
Observational studies in humans following surgeries showed an increase in cognitive decline and is called postoperative cognitive dysfunction (POCD) [125,126,127,128]. Several studies have suggested that abdominal and cardiac surgery can induce neuroinflammatory responses [125, 129, 130]. For example, research in animal models has shown that surgical trauma can result in increased levels of pro-inflammatory cytokines within the brain [129]. Additionally, clinical studies have found evidence of neuroinflammatory markers in the cerebrospinal fluid of patients undergoing abdominal and cardiothoracic surgeries [131,132,133]. It is worth noting that the extent and duration of neuroinflammation following abdominal or cardiothoracic surgery may vary among individuals and depend on factors such as the type of surgery, the individual’s overall health, and the presence of pre-existing conditions. The consequences of neuroinflammation after surgery are still being studied, and more research is needed to fully understand its implications.
Therefore, targeting the shared inflammatory pathways could hold therapeutic potential for both ADRD and bone disorders. Unraveling these intertwined pathways could reveal innovative therapeutic targets and strategies that concurrently address ADRD and compromised bone health, offering a more holistic and effective approach to improving patient prognosis.
Alzheimer’s Disease and Fractures: A Connection Worthy of Investigation
In the current section, we will explore the link between ADRD and the propensity for fractures in ADRD patients. We will also explore the potential link between fractures leading to ADRD progression and cognitive decline. Understanding the connection between fractures and cognitive decline is imperative as this connection is not well understood and very few studies exist to date.
Emerging evidence suggests a compelling link between ADRD and increased susceptibility to fractures, a common manifestation of deteriorating bone health. Fractures, particularly in the elderly, can significantly compromise quality of life and escalate morbidity and mortality rates, underlining the necessity for further exploration of this relationship [134,135,136].
Clinical studies indicate that individuals with ADRD are at a substantially higher risk of sustaining fractures, primarily attributable to increased falls and decreased BMD [3, 134, 137, 138]. Indeed, altered lifestyle factors associated with ADRD, including decreased physical activity and poor nutrition, can lead to compromised BMD and overall poor bone health [8, 138], further increasing the propensity for fractures [29, 139]. Additionally, cognitive impairment in ADRD can lead to a higher risk of falls and subsequent fractures due to improper gait [29].
One of the intriguing aspects of this relationship is whether fractures can contribute to the progression of ADRD. Even though there are no animal studies that suggest that systemic inflammation resulting from a fracture could exacerbate pre-existing ADRD pathology, several clinical observational studies suggest that there might be an association between fractures and ADRD progression [140, 141•]. However, human studies present a more complex picture. While no direct association between fracture and the rate of cognitive decline in ADRD patients has been established, fractures can indirectly impact cognitive and functional outcomes in these patients, possibly due to complications such as immobility, pain, and the use of sedative medications post-fracture [29, 142]. As discussed earlier, surgeries can lead to POCD with neuroinflammation playing a main role [125,126,127,128]. Hence, it would not be surprising to see similarities in human patients and cognitive decline following fracture surgeries. Strategies to minimize fracture risk, such as fall prevention measures and maintaining bone health through adequate nutrition and exercise, become crucial components of patient management [143,144,145,146].
In summary, a complex interplay exists between ADRD and fractures. While individuals with ADRD appear more susceptible to fractures, the potential impact of fractures on ADRD progression is yet to be definitively established. Given the significant implications of these findings, further comprehensive studies are warranted to fully understand this bidirectional link, which could potentially uncover novel avenues for patient management and therapeutic interventions.
Potential Therapeutic Interventions for Alzheimer’s Disease: A Focus on Neuroinflammation and Overlapping Pathways with Bone Health Disorders
Emerging evidence highlighting the intersection between ADRD and bone health—specifically through shared inflammatory pathways—provides new opportunities for the development of therapeutic strategies that could concurrently address both conditions [6,7,8,9]. Given the pivotal role that inflammation plays in both ADRD and osteoporosis, anti-inflammatory agents are a key focus of current research. However, due to the complex nature and intertwined pathways shared between ADRD and bone health, the approaches taken should not be limited only to anti-inflammatory agents but should also include bioactive molecules targeting multiple pathways.
Non-steroidal anti-inflammatory drugs (NSAIDs), conventionally used for pain and inflammation management, have been examined for their potential role in ADRD progression. Though research has produced mixed results, some epidemiological studies involving long-term NSAID usage suggest a decreased ADRD incidence, thereby indicating potential therapeutic benefits [147,148,149,150]. The putative mechanism of action is the mitigation of inflammation, thereby decelerating the disease progression. Correspondingly, NSAIDs could potentially offer protection against bone loss by inhibiting prostaglandin synthesis, a process known to stimulate osteoclast activity and subsequent bone resorption [151, 152].
Another therapeutic target of interest is the NF-kB signaling pathway, a central regulator of inflammation and immune responses, implicated in both ADRD and osteoporosis [153, 154]. Drugs inhibiting NF-kB have demonstrated promising results in preclinical models by reducing neuroinflammation and improving cognitive function in ADRD, and by attenuating bone loss in osteoporosis [155,156,157,158]. Denosumab is a human monoclonal antibody that binds to receptor activator of NF-κB ligand (RANKL) approved by the FDA for the treatment of post-menopausal osteoporosis [159, 160]. RANKL is indispensable for osteoclast (OC) differentiation and activation [160]. OCs resorb bone and an increase in OCs leads to higher bone resorption and osteoporosis. Denosumab, by blocking RANKL, inhibits OC differentiation and activation. RANKL has also been found to be expressed in normal rodent brain [161]; however, its role in neuronal functions and neuroprotection is not completely understood. Given the intermingled pathways involved in both bone health and disorders as well as ADRD progression [7], Denosumab as a potential candidate for treating neuroinflammation and osteoporosis is intriguing and might need to be explored as a potential dual target osteoporosis and ADRD therapy.
Romosozumab, a humanized monoclonal antibody used to treat osteoporosis, inhibits sclerostin, a molecule expressed by osteocytes and involved with inhibition of osteogenesis via Wnt/β-catenin pathway [162, 163]. In a study conducted to compare the performance of Denosumab and Romosozumab in treating osteoporotic patients, the authors observed a significant increase in BMD of the lumbar spine, total hip, and femoral neck after 12 months of starting the treatments [164]. Wnt signaling is also involved in synaptic plasticity and amyloid pathology in AD [165]. Activation of the Wnt signaling pathway has also been involved in microglial survival [166]. Intracerebrovascular injections of sclerostin in ICR mice reduced the dendritic complexity of pyramidal neurons in the hippocampus showing that there might be an overlap of pathways such as Wnt signaling via sclerostin and neuronal dendritic growth [167]. Since inhibition of sclerostin would activate Wnt signaling, and this might benefit bone formation as well as potentially benefit neurons, synaptic plasticity, and microglia, Romosozumab might also be a good candidate for both osteoporosis and ADRD therapy.
Biological therapies, including monoclonal antibodies that target pro-inflammatory cytokines (for example, TNF-α and IL-1β), may also offer potential benefits for ADRD and bone disorders [168,169,170,171,172,173,174,175]. Preliminary studies in rodents suggest these therapies could reduce neuroinflammation and slow cognitive decline in ADRD, while concurrently inhibiting OC activity and reducing bone loss in osteoporosis [168,169,170,171].
Despite the potential of the monoclonal antibodies as dual target treatments for both osteoporosis and ADRD, what needs to be kept in mind is that under normal conditions, monoclonal antibodies do not cross the blood brain barrier (BBB) and therefore would not be able to neutralize locally produced proteins of interest in the brain [176]. To make these biological molecules a viable option to treat ADRD, these need to be delivered in a way that can cross the BBB [176, 177]. However, recently there have been some studies that show that modifying large molecules such as monoclonal antibodies as an IgG fusion may help deliver them to the target proteins past the BBB [178]. The BBB transport system involves two modalities to deliver biological molecules, namely receptor-mediated transcytosis (RMT) for large molecules and carrier-mediated transport (CMT) for small molecules. Exploiting the biology and chemistry of these endogenous modalities of BBB transport can lead to effective and efficient delivery of monoclonal antibodies and other drugs across BBB, making dual target treatment for ADRD and osteoporosis a reality in the future [179].
Emerging research has underscored the potential therapeutic role of hormones, such as estrogen, which have shown protective effects on both brain and bone health. Estrogen replacement therapy following menopause may reduce ADRD risk and slow disease progression, while concurrently preventing postmenopausal bone loss [180,181,182,183]. FSH is also involved in regulating bone mass and has been described as a key hormone, along with estrogen, that modifies the risk of developing osteoporosis in post-menopausal women [184]. As mentioned earlier, blocking FSH in the 3xTg AD mouse model slowed the progression of AD [75••]. The findings from AD mouse models for FSH blockade as well as beneficial effect of FSH blockade on bone mass show a promise for exploring FSH either as a standalone therapy for targeting both osteoporosis and ADRD or in combination with estrogen replacement therapy in post-menopausal women.
Similarly, calcitonin gene-related peptide (CGRP) has shown promise as a target in preclinical models for its neuroprotective effects and for its traditional application in treating osteoporosis [185,186,187]. The flip side to CGRP treatment is that peripheral CGRP is also involved in the pathophysiology of migraine. CGRP levels were elevated in the plasma, cerebrospinal fluid, and tear samples from migraine patients [188,189,190] and intravenous administration of CGRP to migraineurs can trigger headache [191]. Currently, there are FDA-approved drugs used to treat migraine that target either CGRP or its receptor in patients [192]. This is the major clinical conundrum that needs to be considered while exploring CGRP or CGRP receptor as potential targets for treating osteoporosis and ADRD.
One family of molecules, Sirtuins, has been studied and explored for their involvement in aging and longevity. Sirtuins are a class of deacetylases that are NAD+ dependent [193,194,195,196]. Among the seven Sirtuins, Sirtuin 1 (SIRT1) has been most extensively studied for its role as an anti-inflammatory molecule; in addition, it protects against ROS, is anti-apoptotic, and is involved in energy metabolism in brain and bone health [195, 197,198,199,200,201,202,203]. SRT1720 (a SIRT1 activator) is known to enhance endothelial cell function and promote angiogenesis in 20–22-month mice which may be beneficial for fracture healing [204]. Since SIRT1 not only acts on one axis, such as inflammation or ROS, for its protective role, it may be a good candidate for developing therapies for ADRD as well as bone disorders. Even though there are numerous studies on mouse models of AD and SIRT1 as a neuroprotective agent [205,206,207,208,209,210,211] and the role of SIRT1 in preventing bone loss [203, 212, 213], there are no studies to date that explored the effect of SIRT1 as having a potential dual protection in reducing AD while preventing bone loss.
However, as promising as these therapeutic strategies may be, it is crucial to evaluate the potential side effects and risks associated with long-term use. Continued research is needed to substantiate these therapies’ effectiveness and safety through well-designed, large-scale clinical trials. Harnessing the shared pathological mechanisms between ADRD and bone health may potentially unlock innovative and integrated treatment approaches that could significantly improve the quality of life for those dealing with ADRD and/or osteoporosis.
Conclusion
In conclusion, the intricate relationship between ADRD, bone disorders such as osteoporosis, and inflammation has emerged as a promising research frontier. An increased susceptibility to fractures, largely due to falls and reduced bone density, is observed in individuals with ADRD, potentially exacerbating morbidity and mortality in these patients. Interestingly, many studies on mouse models indicate that osteoporosis or low bone turnover states lead to exacerbation of hallmarks of ADRD. Therefore, it would not be surprising to find that fractures might also potentially intensify ADRD progression based on clinical follow-up studies on POCD after other types of surgeries [214]. As shown in Fig. 1, the overlap between aging, inflammation, ADRD, and osteoporosis is significant. Through this lens, we may uncover novel therapeutic targets, leading to integrated care strategies aimed at reducing fracture risk in ADRD patients and limiting the potential impact of fractures on ADRD progression. As we unravel the shared pathophysiological pathways in ADRD, bone health, and inflammation, we are not merely enriching our academic knowledge but taking crucial strides towards improving patient care in our aging population.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Kacena MA, Plotkin LI, Fehrenbacher JC. The use of artificial intelligence in writing scientific review articles. Curr Osteoporos Rep. 2024;1–7. https://doi.org/10.1007/s11914-023-00852-0.
Margetts TJ, Karnik SJ, Wang HS, et al. Use of AI language engine ChatGPT 4.0 to write a scientific review article examining the intersection of alzheimer’s disease and bone. Curr Osteoporos Rep. 2024;1–5. https://doi.org/10.1007/s11914-023-00853-z.
Melton LJ 3rd, et al. Fracture risk in patients with Alzheimer’s disease. J Am Geriatr Soc. 1994;42(6):614–9.
Li S, et al. Amyloid beta peptide is elevated in osteoporotic bone tissues and enhances osteoclast function. Bone. 2014;61:164–75.
Zhou R, et al. Bone loss and osteoporosis are associated with conversion from mild cognitive impairment to Alzheimer’s disease. Curr Alzheimer Res. 2014;11(7):706–13.
Frame G, Bretland KA, Dengler-Crish CM. Mechanistic complexities of bone loss in Alzheimer’s disease: a review. Connect Tissue Res. 2020;61(1):4–18.
Culibrk RA, Hahn MS. The role of chronic inflammatory bone and joint disorders in the pathogenesis and progression of Alzheimer’s disease. Front Aging Neurosci. 2020;12: 583884.
Kumar S, et al. Alzheimer’s disease and its association with bone health: a case-control study. Cureus. 2021;13(3): e13772.
Chen YH, Lo RY. Alzheimer’s disease and osteoporosis. Ci Ji Yi Xue Za Zhi. 2017;29(3):138–42.
Prince M, et al. Recent global trends in the prevalence and incidence of dementia, and survival with dementia. Alzheimers Res Ther. 2016;8(1):23.
Alzheimer’s Disease International [ADI]. Improving healthcare for people living with dementia: Coverage, quality, and costs now and in the future. World Alzheimer report 2016. Alzheimer’s Disease International, London. 2016. Retrieved from https://www.alz.co.uk/research/files/WorldAlzheimer-Report2016.pdf.
Brightfocus.org. Alzheimer’s disease: Facts & Figures. 2022. (www.brightfocus.org/alzheimers/article/alzheimers-disease-facts-figures).
Alzheimer’s Association. Women at risk. 2023. (www.alz.org/alzheimers-dementia/facts-figures#:~:text=An%20estimated%206.7%20million%20Americans,Americans%20with%20Alzheimer's%20are%20women).
Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin Epidemiol. 2014;6:37–48.
Henderson VW. Estrogens, episodic memory, and Alzheimer’s disease: a critical update. Semin Reprod Med. 2009;27(3):283–93.
Iqbal J, Zaidi M. Understanding estrogen action during menopause. Endocrinology. 2009;150(8):3443–5.
Rahman A, et al. Sex and gender driven modifiers of Alzheimer’s: the role for estrogenic control across age, race, medical, and lifestyle risks. Front Aging Neurosci. 2019;11:315.
Rosende-Roca M, et al. The role of sex and gender in the selection of Alzheimer patients for clinical trial pre-screening. Alzheimers Res Ther. 2021;13(1):95.
Kanis JA, et al. Intervention thresholds and the diagnosis of osteoporosis. J Bone Miner Res. 2015;30(10):1747–53.
Dalle Carbonare L, Giannini S. Bone microarchitecture as an important determinant of bone strength. J Endocrinol Invest. 2004;27(1): 99–105.
Brandi ML. Microarchitecture, the key to bone quality. Rheumatology (Oxford). 2009;48 Suppl 4:iv3–8.
Kanis JA, et al. European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos Int. 2013;24(1):23–57.
Sozen T, Ozisik L, Basaran NC. An overview and management of osteoporosis. Eur J Rheumatol. 2017;4(1):46–56.
Ji MX, Yu Q. Primary osteoporosis in postmenopausal women. Chronic Dis Transl Med. 2015;1(1):9–13.
Cheng CH, Chen LR, Chen KH. Osteoporosis due to hormone imbalance: an overview of the effects of estrogen deficiency and glucocorticoid overuse on bone turnover. Int J Mol Sci. 2022;23(3):1376.
Haentjens P, et al. Meta-analysis: excess mortality after hip fracture among older women and men. Ann Intern Med. 2010;152(6):380–90.
Bai J, et al. Association between dementia and mortality in the elderly patients undergoing hip fracture surgery: a meta-analysis. J Orthop Surg Res. 2018;13(1):298.
Ha YC, et al. Effect of dementia on postoperative mortality in elderly patients with hip fracture. J Korean Med Sci. 2021;36(38): e238.
Friedman SM, et al. Dementia and hip fractures: development of a pathogenic framework for understanding and studying risk. Geriatr Orthop Surg Rehabil. 2010;1(2):52–62.
Zhou R, et al. Association between bone mineral density and the risk of Alzheimer’s disease. J Alzheimers Dis. 2011;24(1):101–8.
Dumitrescu L, et al. Genetic variants and functional pathways associated with resilience to Alzheimer’s disease. Brain. 2020;143(8):2561–75.
• Castro-Aldrete L, et al. Sex and gender considerations in Alzheimer’s disease: The Women’s Brain Project contribution. Front Aging Neurosci. 2023;15:1105620. The Women’s Brain Project is a significant endeavor that focuses on understanding gender-specific vulnerabilities in various neurological disorders, including AD. This publication seems to shed light on the role of gender in the context of bone health and AD.
Cassidy L, et al. Oxidative stress in Alzheimer’s disease: a review on emergent natural polyphenolic therapeutics. Complement Ther Med. 2020;49: 102294.
Domazetovic V, et al. Oxidative stress in bone remodeling: role of antioxidants. Clin Cases Miner Bone Metab. 2017;14(2):209–16.
Kimball JS, Johnson JP, Carlson DA. Oxidative stress and osteoporosis. J Bone Joint Surg Am. 2021;103(15):1451–61.
Gella A, Durany N. Oxidative stress in Alzheimer disease. Cell Adh Migr. 2009;3(1):88–93.
Kinney JW, et al. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y). 2018;4:575–90.
Xie J, Van Hoecke L, Vandenbroucke RE. The impact of systemic inflammation on Alzheimer’s disease pathology. Front Immunol. 2021;12: 796867.
Ginaldi L, Di Benedetto MC, De Martinis M. Osteoporosis, inflammation and ageing. Immun Ageing. 2005;2:14.
Zhang P, et al. Potential association of bone mineral density loss with cognitive impairment and central and peripheral amyloid-beta changes: a cross-sectional study. BMC Musculoskelet Disord. 2022;23(1):626.
Wang TH, Jiang Y, Xiao LP. Expression of amyloid beta-protein in bone tissue of APP/PS1 transgenic mouse. Zhonghua Yi Xue Za Zhi. 2013;93(1):65–8.
•• Je LL, et al. Degradation of bone quality in a transgenic mouse model of Alzheimer’s disease. J Bone Miner Res. 2022;37 Suppl 12:2548–2565. Using a transgenic mouse model, this article offers a direct investigation into the degradation of bone quality in the context of AD. The use of a transgenic model adds weight to the findings, as it provides mechanistic insights into the AD-bone health relationship.
Dengler-Crish CM, et al. Evidence of Wnt/beta-catenin alterations in brain and bone of a tauopathy mouse model of Alzheimer’s disease. Neurobiol Aging. 2018;67:148–58.
Dengler-Crish CM, Smith MA, Wilson GN. Early evidence of low bone density and decreased serotonergic synthesis in the dorsal raphe of a tauopathy model of Alzheimer’s disease. J Alzheimers Dis. 2017;55(4):1605–19.
Cui S, et al. APPswe/Abeta regulation of osteoclast activation and RAGE expression in an age-dependent manner. J Bone Miner Res. 2011;26(5):1084–98.
Xia WF, et al. Swedish mutant APP suppresses osteoblast differentiation and causes osteoporotic deficit, which are ameliorated by N-acetyl-L-cysteine. J Bone Miner Res. 2013;28(10):2122–35.
• Zhang M, Hu S, Sun X. Alzheimer’s disease and impaired bone microarchitecture, regeneration and potential genetic links. Life (Basel). 2023;13(2). This article is particularly important as it not only delves into the link between AD and bone health, but it also explores potential genetic links and the impairment of bone microarchitecture and regeneration.
Cui Z, et al. Schizophrenia, bipolar disorder, and Alzheimer’s disease are not causal factors of bone mineral density: a Mendelian randomization analysis. Calcif Tissue Int. 2020;106(2):131–46.
Doig AJ. Positive feedback loops in Alzheimer’s disease: the Alzheimer’s feedback hypothesis. J Alzheimers Dis. 2018;66(1):25–36.
Kosyreva AM, et al. Alzheimer’s disease and inflammaging. Brain Sci. 2022;12(9):1237.
Fisher DW, Bennett DA, Dong H. Sexual dimorphism in predisposition to Alzheimer’s disease. Neurobiol Aging. 2018;70:308–24.
Podcasy JL, Epperson CN. Considering sex and gender in Alzheimer disease and other dementias. Dialogues Clin Neurosci. 2016;18(4):437–46.
Villa A, et al. Estrogens, neuroinflammation, and neurodegeneration. Endocr Rev. 2016;37(4):372–402.
Yue X, et al. Brain estrogen deficiency accelerates Abeta plaque formation in an Alzheimer’s disease animal model. Proc Natl Acad Sci U S A. 2005;102(52):19198–203.
Carroll JC, et al. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J Neurosci. 2007;27(48):13357–65.
Wang C, et al. Estrogen receptor-alpha is localized to neurofibrillary tangles in Alzheimer’s disease. Sci Rep. 2016;6:20352.
Kim JY, et al. Mitigating effect of estrogen in Alzheimer’s disease-mimicking cerebral organoid. Front Neurosci. 2022;16: 816174.
Srivastava DP, Woolfrey KM, Penzes P. Insights into rapid modulation of neuroplasticity by brain estrogens. Pharmacol Rev. 2013;65(4):1318–50.
Simpkins JW, et al. The potential for estrogens in preventing Alzheimer’s disease and vascular dementia. Ther Adv Neurol Disord. 2009;2(1):31–49.
Goodman Y, et al. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66(5):1836–44.
Toran-Allerand CD, et al. Estrogen receptors colocalize with low-affinity nerve growth factor receptors in cholinergic neurons of the basal forebrain. Proc Natl Acad Sci U S A. 1992;89(10):4668–72.
Vegeto E, Benedusi V, Maggi A. Estrogen anti-inflammatory activity in brain: a therapeutic opportunity for menopause and neurodegenerative diseases. Front Neuroendocrinol. 2008;29(4):507–19.
Ospina JA, et al. Estrogen suppresses IL-1beta-mediated induction of COX-2 pathway in rat cerebral blood vessels. Am J Physiol Heart Circ Physiol. 2004;286(5):H2010–9.
Brown CM, et al. Production of proinflammatory cytokines and chemokines during neuroinflammation: novel roles for estrogen receptors alpha and beta. Endocrinology. 2010;151(10):4916–25.
Shivers KY, et al. Estrogen alters baseline and inflammatory-induced cytokine levels independent from hypothalamic-pituitary-adrenal axis activity. Cytokine. 2015;72(2):121–9.
Gatson JW, et al. Estrogen treatment following severe burn injury reduces brain inflammation and apoptotic signaling. J Neuroinflammation. 2009;6:30.
Baez-Jurado E, et al. Molecular mechanisms involved in the protective actions of Selective Estrogen Receptor Modulators in brain cells. Front Neuroendocrinol. 2019;52:44–64.
Baker AE, Brautigam VM, Watters JJ. Estrogen modulates microglial inflammatory mediator production via interactions with estrogen receptor beta. Endocrinology. 2004;145(11):5021–32.
Hoozemans JJ, et al. Cyclooxygenase expression in microglia and neurons in Alzheimer’s disease and control brain. Acta Neuropathol. 2001;101(1):2–8.
Ghazanfari N, et al. Is cyclooxygenase-1 involved in neuroinflammation? J Neurosci Res. 2021;99(11):2976–98.
Woodling NS, Andreasson KI. Untangling the web: toxic and protective effects of neuroinflammation and PGE2 signaling in Alzheimer’s disease. ACS Chem Neurosci. 2016;7(4):454–63.
Heery M, et al. Precautions for patients taking aromatase inhibitors. J Adv Pract Oncol. 2020;11(2):184–9.
Goncalves RM, et al. COX-2 promotes mammary adipose tissue inflammation, local estrogen biosynthesis, and carcinogenesis in high-sugar/fat diet treated mice. Cancer Lett. 2021;502:44–57.
Castardo-de-Paula JC, et al. Effects of inducible nitric oxide synthase inhibition on cardiovascular risk of adult endotoxemic female rats: role of estrogen. Front Physiol. 2018;9:1020.
•• Xiong J, et al. FSH blockade improves cognition in mice with Alzheimer’s disease. Nature 2022;603 Suppl 7901: 470–476. This article uncovers a novel connection between FSH (Follicle Stimulating Hormone) and AD, highlighting an unexpected intersection between reproductive hormones and cognitive health. This groundbreaking research not only offers a fresh perspective on AD pathogenesis but also opens new avenues for therapeutic strategies that target bone health and cognition simultaneously.
Kubota T, Matsumoto H, Kirino Y. Ameliorative effect of membrane-associated estrogen receptor G protein coupled receptor 30 activation on object recognition memory in mouse models of Alzheimer’s disease. J Pharmacol Sci. 2016;131(3):219–22.
Gera S, et al. FSH-blocking therapeutic for osteoporosis. Elife. 2022;11:e78022.
Gera S, et al. First-in-class humanized FSH blocking antibody targets bone and fat. Proc Natl Acad Sci U S A. 2020;117(46):28971–9.
Altmann A, et al. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann Neurol. 2014;75(4):563–73.
Ferretti MT, et al. Sex differences in Alzheimer disease - the gateway to precision medicine. Nat Rev Neurol. 2018;14(8):457–69.
Lewandowski CT, Maldonado Weng J, LaDu MJ. Alzheimer’s disease pathology in APOE transgenic mouse models: the who, what, when, where, why, and how. Neurobiol Dis. 2020;139:104811.
Ungar L, Altmann A, Greicius MD. Apolipoprotein E, gender, and Alzheimer’s disease: an overlooked, but potent and promising interaction. Brain Imaging Behav. 2014;8(2):262–73.
Giunta B, et al. Inflammaging as a prodrome to Alzheimer’s disease. J Neuroinflammation. 2008;5:51.
Burton DG. Cellular senescence, ageing and disease. Age (Dordr). 2009;31(1):1–9.
Franceschi C, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128(1):92–105.
Furman D, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25(12):1822–32.
DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139 Suppl 2:136–153.
Cisbani G, Rivest S. Targeting innate immunity to protect and cure Alzheimer’s disease: opportunities and pitfalls. Mol Psychiatry. 2021;26(10):5504–15.
Heneka MT, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405.
Thakur S, et al. Neuroinflammation in Alzheimer’s disease: current progress in molecular signaling and therapeutics. Inflammation. 2023;46(1):1–17.
Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17(3):157–72.
Morales I, et al. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front Cell Neurosci. 2014;8:112.
Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353(6301):777–83.
Cherry JD, et al. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun. 2016;4(1):112.
Bouvier DS, et al. High resolution dissection of reactive glial nets in Alzheimer’s disease. Sci Rep. 2016;6:24544.
Perez-Nievas BG, Serrano-Pozo A. Deciphering the astrocyte reaction in Alzheimer’s disease. Front Aging Neurosci. 2018;10:114.
Di Benedetto G, et al. Role of microglia and astrocytes in Alzheimer’s disease: from neuroinflammation to Ca(2+) homeostasis dysregulation. Cells 2022;11(17):2728.
Lee CY, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm (Vienna). 2010;117(8):949–60.
Daria A, et al. Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 2017;36(5):583–603.
Wolf Y, et al. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci. 2013;7:26.
Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011;3 Suppl 77:77sr1.
Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200(6):629–38.
Belaya I, et al. Astrocyte remodeling in the beneficial effects of long-term voluntary exercise in Alzheimer’s disease. J Neuroinflammation. 2020;17(1):271.
Liddelow SA, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–7.
Siracusa R, Fusco R, Cuzzocrea S. Astrocytes: role and functions in brain pathologies. Front Pharmacol. 2019;10:1114.
Jiwaji Z, Hardingham GE. Good, bad, and neglectful: astrocyte changes in neurodegenerative disease. Free Radic Biol Med. 2022;182:93–9.
Andreasson KI, et al. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J Neurochem. 2016;138(5):653–93.
Bloom GS. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014;71(4):505–8.
Eikelenboom P, et al. Neuroinflammation - an early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegener Dis. 2010;7(1–3):38–41.
Novoa C, et al. Inflammation context in Alzheimer’s disease, a relationship intricate to define. Biol Res. 2022;55(1):39.
Krstic D, Knuesel I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol. 2013;9(1):25–34.
Weller I, Schatzker J. Hip fractures and Alzheimer’s disease in elderly institutionalized Canadians. Ann Epidemiol. 2004;14(5):319–24.
Amarasekara DS, et al. Regulation of osteoclast differentiation by cytokine networks. Immune Netw. 2018;18(1): e8.
Roodman GD. Role of cytokines in the regulation of bone resorption. Calcif Tissue Int. 1993;53(Suppl 1):S94–8.
Madel MB, et al. Specific targeting of inflammatory osteoclastogenesis by the probiotic yeast S. boulardii CNCM I-745 reduces bone loss in osteoporosis. Elife. 2023;12:e82037.
Da W, Tao L, Zhu Y. The role of osteoclast energy metabolism in the occurrence and development of osteoporosis. Front Endocrinol (Lausanne). 2021;12: 675385.
Baum R, Gravallese EM. Impact of inflammation on the osteoblast in rheumatic diseases. Curr Osteoporos Rep. 2014;12(1):9–16.
Epsley S, et al. The effect of inflammation on bone. Front Physiol. 2020;11: 511799.
Cizza G, Primma S, Csako G. Depression as a risk factor for osteoporosis. Trends Endocrinol Metab. 2009;20(8):367–73.
Amasi-Hartoonian N, et al. Cause or consequence? Understanding the role of cortisol in the increased inflammation observed in depression. Curr Opin Endocr Metab Res. 2022;24: 100356.
Komoltsev IG, Gulyaeva NV. Brain trauma, glucocorticoids and neuroinflammation: dangerous liaisons for the hippocampus. Biomedicines. 2022;10(5):1139.
Holmes C, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73(10):768–74.
Yun AJ, Lee PY. Maldaptation of the link between inflammation and bone turnover may be a key determinant of osteoporosis. Med Hypotheses. 2004;63(3):532–7.
Bastian O, et al. Systemic inflammation and fracture healing. J Leukoc Biol. 2011;89(5):669–73.
Safavynia SA, Goldstein PA. The role of neuroinflammation in postoperative cognitive dysfunction: moving from hypothesis to treatment. Front Psychiatry. 2018;9:752.
Kline RP, et al. Surgery and brain atrophy in cognitively normal elderly subjects and subjects diagnosed with mild cognitive impairment. Anesthesiology. 2012;116(3):603–12.
Rundshagen I. Postoperative cognitive dysfunction. Dtsch Arztebl Int. 2014;111(8):119–25.
Brodier EA, Cibelli M. Postoperative cognitive dysfunction in clinical practice. BJA Educ. 2021;21(2):75–82.
Lu B, et al. Effects of different types of non-cardiac surgical trauma on hippocampus-dependent memory and neuroinflammation. Front Behav Neurosci. 2022;16: 950093.
Yang T, Velagapudi R, Terrando N. Neuroinflammation after surgery: from mechanisms to therapeutic targets. Nat Immunol. 2020;21(11):1319–26.
Alam A, et al. Surgery, neuroinflammation and cognitive impairment. EBioMedicine. 2018;37:547–56.
Subramaniyan S, Terrando N. Neuroinflammation and perioperative neurocognitive disorders. Anesth Analg. 2019;128(4):781–8.
Danielson M, et al. Neuroinflammatory markers associate with cognitive decline after major surgery: findings of an explorative study. Ann Neurol. 2020;87(3):370–82.
Baker NL, et al. Hip fracture risk and subsequent mortality among Alzheimer’s disease patients in the United Kingdom, 1988–2007. Age Ageing. 2011;40(1):49–54.
Allia J, et al. Early mortality and morbidity of odontoid fractures after 70 years of age. Orthop Traumatol Surg Res. 2020;106(7):1399–403.
Zhang DL, et al. Age-adjusted Charlson comorbidity index predicts postoperative mortality in elderly patients with hip fracture: a prospective cohort. Front Med (Lausanne). 2023;10:1066145.
Zhao Y, Shen L, Ji HF. Alzheimer’s disease and risk of hip fracture: a meta-analysis study. ScientificWorldJournal. 2012;2012: 872173.
Xiao T, et al. Association of bone mineral density and dementia: the rotterdam study. Neurology. 2023;100(20):e2125–33.
Dev K, et al. Prevalence of falls and fractures in Alzheimer’s patients compared to general population. Cureus. 2021;13(1): e12923.
Tolppanen AM, et al. Incident hip fractures among community dwelling persons with Alzheimer’s disease in a Finnish nationwide register-based cohort. PLoS ONE. 2013;8(3): e59124.
• Kim SY, et al. Increased risk of dementia after distal radius, hip, and spine fractures. Medicine (Baltimore). 2020;99 Suppl 10:e19048. This article provides compelling evidence linking fractures in specific sites - distal radius, hip, and spine - with an elevated risk of dementia. This article begins to explore if there is a connection between fractures and Alzheimer’s Disease progression.
Rigler SK, et al. Fracture risk in nursing home residents initiating antipsychotic medications. J Am Geriatr Soc. 2013;61(5):715–22.
Panel on Prevention of Falls in Older Persons, A.G.S. and S. British Geriatrics. Summary of the Updated American Geriatrics Society/British Geriatrics Society clinical practice guideline for prevention of falls in older persons. J Am Geriatr Soc. 2011;59 Suppl 1:148–57.
Fostinelli S, et al. Eating behavior in aging and dementia: the need for a comprehensive assessment. Front Nutr. 2020;7: 604488.
Sliwinska S, Jeziorek M. The role of nutrition in Alzheimer’s disease. Rocz Panstw Zakl Hig. 2021;72(1):29–39.
Meng Q, Lin MS, Tzeng IS. Relationship between exercise and Alzheimer’s disease: a narrative literature review. Front Neurosci. 2020;14:131.
Vlad SC, et al. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70(19):1672–7.
Zandi PP, Breitner JC. Do NSAIDs prevent Alzheimer’s disease? And, if so, why? The epidemiological evidence. Neurobiol Aging. 2001;22(6):811–7.
McGeer PL, McGeer EG. NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging. 2007;28(5):639–47.
Szekely CA, et al. No advantage of A beta 42-lowering NSAIDs for prevention of Alzheimer dementia in six pooled cohort studies. Neurology. 2008;70(24):2291–8.
Kotake S, et al. Effects of NSAIDs on differentiation and function of human and murine osteoclasts - crucial ‘human osteoclastology.’ Pharmaceuticals (Basel). 2010;3(5):1394–410.
Su B, O’Connor JP. NSAID therapy effects on healing of bone, tendon, and the enthesis. J Appl Physiol (1985). 2013;115 Suppl 6:892–9.
Jha NK, et al. Nuclear factor-kappa beta as a therapeutic target for Alzheimer’s disease. J Neurochem. 2019;150(2):113–37.
Abu-Amer Y. NF-kappaB signaling and bone resorption. Osteoporos Int. 2013;24(9):2377–86.
Takakura N, et al. A novel inhibitor of NF-kappaB-inducing kinase prevents bone loss by inhibiting osteoclastic bone resorption in ovariectomized mice. Bone. 2020;135: 115316.
Tian H, et al. Nur77 prevents osteoporosis by inhibiting the NF-kappaB signalling pathway and osteoclast differentiation. J Cell Mol Med. 2022;26(8):2163–76.
Shen G, et al. Plumbagin is a NF-kappaB-inducing kinase inhibitor with dual anabolic and antiresorptive effects that prevents menopausal-related osteoporosis in mice. J Biol Chem. 2022;298(4): 101767.
Kong F, et al. Forsythoside B attenuates memory impairment and neuroinflammation via inhibition on NF-kappaB signaling in Alzheimer’s disease. J Neuroinflammation. 2020;17(1):305.
Hanley DA, et al. Denosumab: mechanism of action and clinical outcomes. Int J Clin Pract. 2012;66(12):1139–46.
Dubois EA, Rissmann R, Cohen AF. Denosumab. Br J Clin Pharmacol. 2011;71(6):804–6.
Shimamura M, et al. OPG/RANKL/RANK axis is a critical inflammatory signaling system in ischemic brain in mice. Proc Natl Acad Sci U S A. 2014;111(22):8191–6.
Lewiecki EM. Role of sclerostin in bone and cartilage and its potential as a therapeutic target in bone diseases. Ther Adv Musculoskelet Dis. 2014;6(2):48–57.
Marini F, et al. Role of Wnt signaling and sclerostin in bone and as therapeutic targets in skeletal disorders. Osteoporos Int. 2023;34(2):213–38.
Kobayakawa T, et al. Denosumab versus romosozumab for postmenopausal osteoporosis treatment. Sci Rep. 2021;11(1):11801.
Liu CC, et al. Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer’s disease. Neuron. 2014;84(1):63–77.
Zheng H, et al. TREM2 promotes microglial survival by activating Wnt/beta-catenin pathway. J Neurosci. 2017;37(7):1772–84.
Li W, et al. Intracerebroventricular injection of sclerostin reduced social hierarchy and impaired neuronal dendritic complexity in mice. Neurosci Lett. 2022;773: 136514.
Chang R, et al. Blood-brain barrier penetrating biologic TNF-alpha inhibitor for Alzheimer’s disease. Mol Pharm. 2017;14(7):2340–9.
Zhou M, et al. Tumor Necrosis Factor (TNF) blocking agents are associated with lower risk for Alzheimer’s disease in patients with rheumatoid arthritis and psoriasis. PLoS ONE. 2020;15(3): e0229819.
Redlich K, et al. Repair of local bone erosions and reversal of systemic bone loss upon therapy with anti-tumor necrosis factor in combination with osteoprotegerin or parathyroid hormone in tumor necrosis factor-mediated arthritis. Am J Pathol. 2004;164(2):543–55.
Saito H, et al. A tumor necrosis factor receptor loop peptide mimic inhibits bone destruction to the same extent as anti-tumor necrosis factor monoclonal antibody in murine collagen-induced arthritis. Arthritis Rheum. 2007;56(4):1164–74.
Kawai VK, et al. Effects of anti-tumor necrosis factor alpha agents on bone. Curr Opin Rheumatol. 2012;24(5):576–85.
Shaftel SS, Griffin WS, O’Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation. 2008;5:7.
Pacifici R, et al. The role of interleukin-1 in postmenopausal bone loss. Exp Gerontol. 1990;25(3–4):309–16.
Kimble RB, et al. Interleukin-1 receptor antagonist decreases bone loss and bone resorption in ovariectomized rats. J Clin Invest. 1994;93(5):1959–67.
Lampson LA. Monoclonal antibodies in neuro-oncology: Getting past the blood-brain barrier. MAbs. 2011;3(2):153–60.
Barrio P, Hidalgo D, Udina M. Bipolar depressive recurrence following treatment with the human monoclonal antibody denosumab: a case report. Biol Psychiatry. 2013;74(12):e37–8.
Pardridge WM. Kinetics of blood-brain barrier transport of monoclonal antibodies targeting the insulin receptor and the transferrin receptor. Pharmaceuticals (Basel). 2021;15(1):3.
Zhao P, Zhang N, An Z. Engineering antibody and protein therapeutics to cross the blood-brain barrier. Antib Ther. 2022;5(4):311–31.
Song YJ, et al. The effect of estrogen replacement therapy on Alzheimer’s disease and Parkinson’s disease in postmenopausal women: a meta-analysis. Front Neurosci. 2020;14:157.
Compton J, van Amelsvoort T, Murphy D. HRT and its effect on normal ageing of the brain and dementia. Br J Clin Pharmacol. 2001;52(6):647–53.
Gambacciani M, Levancini M. Hormone replacement therapy and the prevention of postmenopausal osteoporosis. Prz Menopauzalny. 2014;13(4):213–20.
Levin VA, Jiang X, Kagan R. Estrogen therapy for osteoporosis in the modern era. Osteoporos Int. 2018;29(5):1049–55.
Sun L, et al. FSH directly regulates bone mass. Cell. 2006;125(2):247–60.
Patel A, et al. Genetic depletion of amylin/calcitonin receptors improves memory and learning in transgenic Alzheimer’s disease mouse models. Mol Neurobiol. 2021;58(10):5369–82.
Papiri G, et al. Vasoactive neuropeptides and Alzheimer’s disease: a systematic review focusing on calcitonin gene-related peptide. J Integr Neurosci. 2021;20(4):1059–65.
Munoz-Torres M, Alonso G, Raya MP. Calcitonin therapy in osteoporosis. Treat Endocrinol. 2004;3(2):117–32.
Wattiez AS, Sowers LP, Russo AF. Calcitonin gene-related peptide (CGRP): role in migraine pathophysiology and therapeutic targeting. Expert Opin Ther Targets. 2020;24(2):91–100.
Kamm K, Straube A, Ruscheweyh R. Calcitonin gene-related peptide levels in tear fluid are elevated in migraine patients compared to healthy controls. Cephalalgia. 2019;39(12):1535–43.
Cernuda-Morollon E, et al. Interictal increase of CGRP levels in peripheral blood as a biomarker for chronic migraine. Neurology. 2013;81(14):1191–6.
Lassen LH, et al. CGRP may play a causative role in migraine. Cephalalgia. 2002;22(1):54–61.
Deen M, et al. Blocking CGRP in migraine patients - a review of pros and cons. J Headache Pain. 2017;18(1):96.
Canto C, Auwerx J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol Metab. 2009;20(7):325–31.
Guarente L. Calorie restriction and sirtuins revisited. Genes Dev. 2013;27(19):2072–85.
Vachharajani VT, et al. Sirtuins link inflammation and metabolism. J Immunol Res. 2016;2016:8167273.
Grabowska W, Sikora E, Bielak-Zmijewska A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology. 2017;18(4):447–76.
Julien C, et al. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68(1):48–58.
Donmez G. The effects of SIRT1 on Alzheimer’s disease models. Int J Alzheimers Dis. 2012;2012: 509529.
Jiao F, Gong Z. The beneficial roles of SIRT1 in neuroinflammation-related diseases. Oxid Med Cell Longev. 2020;2020:6782872.
Mishra P, et al. SIRT1 promotes neuronal fortification in neurodegenerative diseases through attenuation of pathological hallmarks and enhancement of cellular lifespan. Curr Neuropharmacol. 2021;19(7):1019–37.
Yang Y, et al. Regulation of SIRT1 and its roles in inflammation. Front Immunol. 2022;13: 831168.
Zainabadi K, et al. SIRT1 is a positive regulator of in vivo bone mass and a therapeutic target for osteoporosis. PLoS ONE. 2017;12(9): e0185236.
Wang H, et al. Sirt1 promotes osteogenic differentiation and increases alveolar bone mass via Bmi1 activation in mice. J Bone Miner Res. 2019;34(6):1169–81.
Dadwal UC, et al. The effects of SRT1720 treatment on endothelial cells derived from the lung and bone marrow of young and aged, male and female mice. Int J Mol Sci. 2021;22(20):11097.
Cao K, et al. The neuroprotective effects of SIRT1 in mice carrying the APP/PS1 double-transgenic mutation and in SH-SY5Y cells over-expressing human APP670/671 may involve elevated levels of alpha7 nicotinic acetylcholine receptors. Aging (Albany NY). 2020;12(2):1792–807.
Zhu L, et al. SIRT1 is involved in the neuroprotection of pterostilbene against amyloid beta 25–35-induced cognitive deficits in mice. Front Pharmacol. 2022;13: 877098.
Xu J, et al. Brain SIRT1 mediates metabolic homeostasis and neuroprotection. Front Endocrinol (Lausanne). 2018;9:702.
Hadar A, et al. SIRT1, miR-132 and miR-212 link human longevity to Alzheimer’s disease. Sci Rep. 2018;8(1):8465.
Zhang M, Tang Z. Therapeutic potential of natural molecules against Alzheimer’s disease via SIRT1 modulation. Biomed Pharmacother. 2023;161: 114474.
Lee HR, et al. Cilostazol suppresses beta-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-beta. J Neurosci Res. 2014;92(11):1581–90.
Lutz MI, et al. Distinct patterns of sirtuin expression during progression of Alzheimer’s disease. Neuromolecular Med. 2014;16(2):405–14.
Iyer S, et al. Sirtuin1 (Sirt1) promotes cortical bone formation by preventing beta-catenin sequestration by FoxO transcription factors in osteoblast progenitors. J Biol Chem. 2014;289(35):24069–78.
Cohen-Kfir E, et al. Sirt1 is a regulator of bone mass and a repressor of Sost encoding for sclerostin, a bone formation inhibitor. Endocrinology. 2011;152(12):4514–24.
Kotekar N, Shenkar A, Nagaraj R. Postoperative cognitive dysfunction - current preventive strategies. Clin Interv Aging. 2018;13:2267–73.
Funding
Funding for these studies was provided in part by the NIH AG060621/AG060621-05S1/AG060621-05S2 (MAK), U54AG054345 (AO), AG078861/AG078861-S1 (LIP), AG064003 (AM), AG068595 (AM), and T35HL110854 (HW)). This work was also supported in part by Indiana University School of Medicine, the Indiana Clinical and Translational Sciences Institute (funded in part by NIH UM1TR004402), the Indiana Center for Musculoskeletal Health, the Stark Neuroscience Research Institute, and the Department of Orthopaedic Surgery. This material is also the result of work supported with resources and the use of facilities at the Richard L. Roudebush VA Medical Center, Indianapolis, IN: VA Merit I01BX006399 (MAK) and I01RX003552 (MAK) and I01BX005154 (LIP). The presented contents are solely the responsibility of the authors and do not necessarily represent the official views of any of the aforementioned agencies.
Author information
Authors and Affiliations
Contributions
This review article was conceived by MAK, LIP JCF, AM, and ALO. SJK performed the literature search and drafted/edited the manuscript. All authors revised the manuscript critically for important intellectual content, take responsibility for all aspects of the work, and approve of the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
Dr. Kacena is the Editor-in-Chief of Current Osteoporosis Reports. Drs. Fehrenbacher and Plotkin serve as section editors for Current Osteoporosis Reports.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Karnik, S.J., Margetts, T.J., Wang, H.S. et al. Mind the Gap: Unraveling the Intricate Dance Between Alzheimer’s Disease and Related Dementias and Bone Health. Curr Osteoporos Rep 22, 165–176 (2024). https://doi.org/10.1007/s11914-023-00847-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-023-00847-x