
Abstract
The martensitic transformation in a high carbon steel was studied by a new experimental approach focusing on the nucleation and growth as well as the variant pairing of the early-formed martensite. A mixed microstructure with tempered early-formed martensite and fresh later-formed martensite was achieved by a heat treatment with an isothermal hold below the martensite start temperature. In-situ high-energy X-ray diffraction showed no further transformation of austenite to ferrite/martensite during the isothermal hold. The tempered early-formed martensite was characterized with a combination of light optical microscopy and local tetragonality determination by electron backscatter diffraction. The characterization allowed qualitative as well as quantitative analysis of the tempered early-formed martensite with regard to the prior austenite grain boundaries (PAGB) and variant pairing. The early-formed martensite was shown to grow predominantly along the PAGBs and clustering was observed indicating an autocatalytic nucleation mechanism. The variant pairing of the early-formed martensite had a stronger plate character compared to the later-formed martensite.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The martensitic transformation is a contributor to the large variety of applications for steels due to its high hardness and strength. Extensive investigations of martensite transformation mechanisms and kinetics[1,2,3,4,5] have been necessary for continuous improvements of heat treatments for optimal performance due to the complex nature of the martensitic transformation. The early stages of the transformation are of special interest for an improved understanding of the nucleation and growth mechanism that impacts the overall kinetics of the transformation. The early-formed martensite primarily forms at prior austenite grain boundaries (PAGB) and then grows through the prior austenite grain (PAG) in low carbon steel lath martensite[6] and in Fe–Ni–C plate martensite.[7,8] This is called the geometrical partitioning process[9] since it partitions the PAGs into smaller volumes, which leads to a mechanical stabilization of the austenite.[6,10,11,12] Van Bohemen et al.[13] studied the early martensite formation in high carbon steel in the bulk and at a free surface by tempering the early-formed martensite. In the bulk, the martensite seemed to form and grow along PAGBs, but a definite conclusion was not possible due to experimental limitations in PAG determination. The experiments at a free surface showed the same behavior as observed in the low carbon steel and Fe–Ni–C alloy with geometrical partitioning of PAGs. Hence, the growth behavior in the bulk for high carbon steel has not been elucidated yet.
High carbon steels with a carbon content around 0.6 to 1.0 wt pct C are in the mixed martensite region between lath and plate martensite.[1,14] Stormvinter et al.[15] showed a gradual transition in the variant pairing between lath and plate martensite and the mixed martensite region has components typical for lath and plate martensite as well as for the mixed region. However, the variant pairing at different stages of the transformation, e.g., early-formed and later-formed martensite, has not yet been investigated. Therefore, it is not clear if there are changes in the variant pairing of the formed martensite during the transformation.
The development of prior austenite orientation reconstruction algorithms[16,17,18,19,20] allows the determination of PAGs and PAGBs via electron backscatter diffraction (EBSD) for a detailed analysis of the early-formed martensite. The combination with local tetragonality determination by pattern matching[21,22,23] enables new possibilities of qualitative and more quantitative analysis of tempered martensite due to its lower tetragonality compared to fresh martensite.[24,25]
In this study, the early martensitic transformation in a 0.74C–1.15Mn–1.08Cr high carbon steel is investigated and new approaches for quantitative analysis are used to get a better understanding of the initially formed martensite. A mixed microstructure of tempered and fresh martensite is achieved by an isothermal hold below the martensite start temperature (Ms) followed by quenching. The combination of light optical microscopy (LOM) and EBSD with local tetragonality determination allows the separate qualitative and quantitative analysis of the tempered early-formed martensite. Based on this, the nucleation and growth in regard to PAGBs as well as the variant pairing of the early-formed martensite are investigated.
2 Experimental Methods
2.1 Material
A high carbon steel with a composition of 0.74C–1.15Mn–1.08Cr–0.19Si–Fe in weight percent was studied. The samples were cast, hot rolled at 900 \(^{\circ }\)C, homogenized under vacuum in a quartz tube at 1200 \(^{\circ }\)C for 24 hours, and machined into rectangular, \(10 \times 5 \times 2\) mm, dilatometer samples.
2.2 Heat Treatment
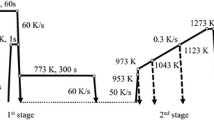
The early martensitic transformation was studied by carrying out two consecutive heat treatments in a dilatometer DIL 805 A/D from TA Instruments. The first heat treatment “Direct” consisted of austenitization at 960 \(^{\circ }\)C and subsequent cooling to room temperature, see Figure 1. The cooling sequence was divided into three steps with different cooling rates: 30 K/s to 550 \(^{\circ }\)C, 15 K/s to 220 \(^{\circ }\)C, and 5 K/s to room temperature. The Ms temperature was determined to be 194 \(^{\circ }\)C for the “Direct” sample by the offset method.[26]
In the second heat treatment “Hold,” the sample was austenitized and cooled as in the first heat treatment with an additional isothermal hold at 184 \(^{\circ }\)C slightly below Ms for 600 s followed by quenching to room temperature with 5 K/s. 184\(^{\circ }\) C was chosen as hold temperature to study the martensite formed around an additional maximum in the first derivative of expansion for “Direct,” see Section III–A. The isothermal hold was used to achieve a mixture of tempered and fresh martensite, where the martensite formed before the isothermal hold became tempered and the fresh martensite formed during cooling from the holding period was untempered. The tempering allowed the separate investigation of early-formed martensite in the final microstructure, see Section II–D.

Temperature profile over time for “Direct” and “Hold” heat treatments with isothermal hold at 184 \(^{\circ }\)C
2.3 In-Situ HEXRD
In-situ high-energy X-ray diffraction (HEXRD) experiments were carried out at the beamline P07b, DESY, Hamburg, operated by the Helmholtz–Zentrum Hereon[27] to study any possible further martensitic transformation during the isothermal hold. The used experimental setup is described in detail in Reference 28. The samples were heat treated as described in Section II–B in combination with the recording of transmission diffraction pattern with acquisition rates of 0.25 and 5 Hz. The use of a beam energy of 87.1 keV (0.14236 Å) in combination with a 2D PerkinElmer detector at a distance of 1392 mm behind the samples allowed for measurement of the first 4 martensite and 5 austenite peaks. The recorded 2D diffraction patterns were integrated over 360 \(^{\circ }\) to 1D patterns using the Fit2D[29] software. The phase fraction and tetragonality c/a of martensite during the isothermal hold was evaluated by Rietveld refinement using the software package Topas-academic.[30] For comparison, the martensite phase fraction was determined for the start of the martensitic transformation, 160 \(^{\circ }\)C to 195 \(^{\circ }\)C, for the “Direct” heat treatment.
As overall diffraction patterns (or peak shapes) are a convolution of background, sample, and instrument, the standard lanthanum hexaboride NIST660C was measured under the same experimental configurations as the samples for calibration and instrument profile function extraction purpose. Further, the Rietveld refinements were performed to determine the profile function. After a satisfactory fit was obtained from the lanthanum hexaboride data, the instrument profile function parameters were fixed together with zero error correction. Finally, batch Rietveld refinements were carried out to refine the structure of various phases to define best possible unit cell parameters at a given temperature and time.
The peak profiles were modeled with a modified Thompson–Cox–Hastings pseudo-Voigt (pV-TCHZ) profile function.[30] The background was fitted with a Chebyshev function with seven coefficients. The Rietveld fitting was performed on the full diffraction pattern range and one typical fit is shown in Figure 2. The crystal structure information of the various phases needed for Rietveld analysis was obtained from the Inorganic Crystal Structure Database (ICSD).[31]

Typical measured diffraction data (purple) with corresponding Rietveld fitting result (red) and their difference (gray) (Color figure online)
2.4 Characterization of Tempered Martensite
The tempered early-formed martensite in the final microstructure after the second heat treatment was characterized with LOM as well as EBSD and imaging using the forward scattering detectors (FSD).
The samples were prepared by mechanical grinding and polishing using 9, 3, and 1 \({\mu }\)m diamond suspension with subsequent electropolishing at 12 V and 10 \(^{\circ }\)C for 60 seconds with Struers A3 electrolyte for EBSD investigation. The electropolishing resulted in a contrast between tempered and fresh martensite units in FSD. For confirmation, the surface was carefully repolished and etched with Nital after the EBSD measurements to obtain a clear contrast between tempered and fresh martensite in LOM. The careful repolishing resulted in minimal material removal and it was possible to study almost the same microstructure as in EBSD. The ImageJ[32] plugins for extended depth of field[33] and image stitching[34] were used for the LOM images.
For the EBSD investigation, a FEI Nova 600 NanoLab dual-beam system with an Oxford Instruments Symmetry CMOS detector with an acceleration voltage of 20 kV, a current of 9.5 nA, and an acquisition time of 10 ms was used and the data were analyzed using the MTEX toolbox.[35,36] The raw EBSD patterns were recorded with a resolution of \(156 \times 128\) pixels (\(8 \times 8\) binning) for analysis by the projective Kikuchi pattern matching algorithm from Winkelmann et al.[21,22] to determine the local tetragonality c/a. Only data points with a normalized cross-correlation coefficient r defined in References 21, 22 above 0.5 were taken for the tetragonality c/a analysis in order to exclude inadequate matches of experimental and simulated patterns. A total of three measurements were recorded with a step size of 150 nm and each having a size of 1\(26 \times 111 \mu \)m.
In addition to LOM and FSD imaging, the local tetragonality c/a was used to determine the position of tempered martensite units via threshold values of low c/a in the EBSD data for quantitative analysis. This analysis is based on the lower tetragonality of tempered compared to fresh martensite.[24,25] Three different cut-off values for low tetragonality c/a were chosen 1.01, 1.0125, and 1.015. The tempered martensite units were determined for the conditions that the total average tetragonality c/a ratio of the unit was below the respective cut-off value and that the tetragonality determination was successful for the majority of pixels of the unit. The second condition was chosen to ensure that the average tetragonality c/a was representative for each unit. The resulting tempered martensite units were compared with the corresponding FSD image as a control and the best fit for the three threshold values was used for more in-depth characterization of the variant pairing and unit size.
Furthermore, the prior austenite orientation reconstruction algorithm of Nyyssönen[19] was used. The reconstructed orientation of austenite allows the calculation of PAGs in MTEX[36] and thereby the exact determination of PAGBs. Additionally, Nyyssönen’s algorithm[19] was used to calculate the variant pairing between martensite units after the definition of Morito.[37] The analysis was expanded for the selection of specific boundaries, e.g., between tempered martensite units and calculating their variant pairing from the complete data set. This allowed the comparison between tempered and all formed martensite.
The determination of PAGBs and variant pairing in combination with the local tetragonality c/a allows an in-depth quantitative analysis of the tempered early-formed martensite in addition to qualitative observations. The distance of each low tetragonality c/a pixel to the nearest PAGB in comparison to all data points was used to quantify the location of the tempered martensite. The variant pairing between the tempered martensite units was determined to study differences between martensite formed at an early or later stage, respectively. Finally, the unit size distribution of tempered martensite was compared with the complete data sets. The statistical analysis of variant pairing, distance to nearest PAGB and unit size was summed up over all three data sets. The results of the two additional data sets as well as tetragonality c/a cut-off values are shown in the electronic supplementary.
3 Results
3.1 Dilatometer
The dilatometer results during the heat treatments “Direct” and “Hold” (Figure 1) are shown in Figure 3. For both heat treatments, the results show the same behavior before the isothermal hold with a linear decrease in length change \(\Delta L\) above Ms and a sharp increase at the onset of the bulk martensitic transformation around 185 \(^{\circ }\)C. For “Hold,” a continuous length increase is observed during the isothermal hold, further discussed in Section III–B, followed by a nearly linear contraction before the restart of martensite transformation on subsequent cooling. This results in a deviation in the dilatation behavior between the two heat treatments after the isothermal hold.
The observed behavior of “Direct” can be seen more clearly in the corresponding dilatation rate \({\text{d}}\Delta L/{\text{d}}T\) with \({\text{d}}T\) = 2.5 \(^{\circ }\)C in Figure 3. The curve has two maxima, the first at approximately 185 \(^{\circ }\)C, which coincides with the sharp increase at the transformation onset. The second is the global maximum for the complete transformation at around 140 \(^{\circ }\)C. A local minimum in expansion rate around 175 \(^{\circ }\)C is observed between the two maxima. The hold temperature of the second heat treatment “Hold” was selected to be at a temperature slightly below the first maximum of \({\text{d}}\Delta L/{\text{d}}T\) to allow the study of the martensite formed at the maximum.

Length change \(\Delta L\) vs temperature for heat treatments “Direct and “Hold” and dilatation rate \({\text{d}}\Delta L/{\text{d}}T\) for \({\text{d}}T = 2.5 ^{\circ }\)C for “Direct” during cooling below 250 \(^{\circ }\)C
3.2 In-Situ HEXRD
The martensite phase fraction from Rietveld refinement, tetragonality c/a, and temperature evolution during the isothermal hold are presented in Figure 4. After a strong initial transformation no significant further martensite formation is observed during the isothermal hold, see Figure 4(a). This significant transformation at the start coincides with the end of temperature equilibration as can be seen in Figure 4(b). The tetragonality c/a decreases continuously until reaching a constant value around 1.01, Figure 4(a). The final tetragonality c/a after quenching to room temperature is around 1.029, which shows a clear difference compared to the tetragonality of the martensite formed before the isothermal hold. The change in scatter and amount of data points at the end of Figure 4(a) is due to the increase in acquisition rate from 0.25 to 5 Hz. In addition, the martensite phase fraction from Rietveld refinement and the dilatation \(\Delta L\) for the “Direct” heat treatment are shown in Figure 4(c). The phase fraction shows a slower transformation start compared to the dilatation \(\Delta L\) until around 180 \(^{\circ }\)C, where a strong increase to approximately 10 pct of martensite is observed. This strong increase can be seen more clearly in diffraction patterns for the {110} martensite peaks from 191.7 \(^{\circ }\)C to 171.3 \(^{\circ }\)C in Figure 4(d), where the early stages 191.7 \(^{\circ }\)C to 182.4 \(^{\circ }\)C show a very slow intensity increase followed by a stronger increase between 182.4 and 178.9 \(^{\circ }\)C. For lower temperatures, the behavior of \(\Delta L\) and phase fraction are nearly identical upon further cooling.

(a) Martensite phase fraction (○) and tetragonality c/a ( ) vs time during the isothermal hold, (b) Martensite phase fraction (○) and temperature (
) vs time during the isothermal hold, (b) Martensite phase fraction (○) and temperature ( ) vs time at the start of the isothermal hold, (c) Length change \(\Delta L\) and martensite phase fraction vs temperature for “Direct” heat treatment at the initial stage of the transformation, (d) 1D diffraction pattern of {110} martensite peaks for ’Direct’ from 191.7 to 171.3 \(^{\circ }\)C with approximately 1 \(^{\circ }\)C steps and a focus on 182.4 to 178.9 \(^{\circ }\)C (Color figure online)
) vs time at the start of the isothermal hold, (c) Length change \(\Delta L\) and martensite phase fraction vs temperature for “Direct” heat treatment at the initial stage of the transformation, (d) 1D diffraction pattern of {110} martensite peaks for ’Direct’ from 191.7 to 171.3 \(^{\circ }\)C with approximately 1 \(^{\circ }\)C steps and a focus on 182.4 to 178.9 \(^{\circ }\)C (Color figure online)
3.3 Characterization of Tempered Martensite
3.3.1 Formation with respect to PAG
The tempered martensite units appear darker in LOM than fresh martensite units and have a slight topographic contrast in FSD images as shown in Figures 5(a) and (b). The FSD images show typical spatial distortion[38,39,40,41] compared to LOM, but the tempered martensite units can be matched almost perfectly between the two methods. The small differences between both images can mainly be contributed to a different depth position due to the polishing step between FSD and LOM as well as the different contrast methods. For further analysis, the FSD images are mainly compared with EBSD since these two are from the same measurement.
The microstructure in Figure 5(c) shows multiple large martensite units, up to 10s of \(\mu \)m, in combination with small units. The majority of the tempered martensite units identified in Figures 5(a) and (b) can be clearly seen in the EBSD map in Figure 5(c) with the exception of a few smaller units.

(a) ×1000 LOM image of Nital etched surface, (b) forward scattered image from EBSD detector, (c) orientation map of martensite, (d) tetragonality c/a colormap together with PAGB (red) and numbering of units 1–3; (Color figure online)
The calculated local tetragonality c/a shown in Figure 5(d) is only successful for part of the microstructure, but the majority of the tempered martensite units in Figure 5(b) can be identified. These units generally have a c/a value around 1.01, which is significantly lower than the general c/a. Units with low tetragonality c/a are observed dominantly along or close to PAGBs marked in red, Figure 5(d). There are a few exceptions with units growing within the PAGs, but no units clearly dividing a PAG are observed. The tempered martensite units are generally observed in groups consisting of multiple units, where the groups in most cases are clearly separated from each other. The clustering of tempered martensite can be more clearly seen in the 200x LOM image in Figure 6.

×200 LOM image of Nital etched surface with circles marking 2 examples of clusters of tempered martensite units (red, bottom) and 1 area with no tempered martensite units (blue, top) (Color figure online)
The reconstructed tempered martensite units with an average tetragonality below 1.0125 as well as the local tetragonality for values below 1.0125 are shown together with PAGB in Figures 7(a) and (b). For Figure 7(b) only pixels that are part of a reconstructed unit are selected. The reconstructed units show an overall good agreement for larger units with the tempered martensite in the FSD and LOM image (Figure 5(b)) while the smaller units are not clearly observed in the FSD or LOM images. Additionally, there are larger units that cannot be identified as tempered in FSD and/or LOM image or vice versa, e.g., position 1 in Figures 5(a) and (b), which according to FSD is untempered but in LOM appears as tempered. These units are tempered martensite units according to Figure 7(a) due to their low average tetragonality c/a value that can be seen in Figures 5(d) and 7(b).
The tempered martensite units in Figure 7(a) are generally larger compared to Figure 7(b) since the tempered martensite units are calculated through an average as well as that only the majority of pixels of each unit needs to have a successful c/a value for the used definition in Section II–D. The threshold value of 1.0125 was chosen since it shows the best qualitative agreement with the FSD image in Figure 5(b). The results for tetragonality c/a values below 1.01 and 1.015 as well as the two additional data sets are shown in the electronic supplementary Figures S3–S6.
In Figure 7(c), the distribution of the distance to the closest PAGB is presented for the low tetragonality pixels in Figure 7(b) and for all martensite data points. It can be observed that the low tetragonality c/a pixels have a higher probability to be closer to PAGB compared to all data points.

(a) Plot of tempered martensite units with average tetragonality \(c/a < 1.0125\) together with PAGB (red), (b) local tetragonality c/a values below 1.0125 with PAGB (red), (c) Distribution of the distance of pixels with a tetragonality c/a below 1.0125 and all martensite pixels to their nearest PAGB (Color figure online)
3.3.2 Variant pairing
The variant pairing for all martensite units as well as only between the tempered martensite units from Figure 7(a) is shown in Figure 8. The tempered martensite units show a stronger preference of V1–V16 pairing compared to all units. The V1–16 variant pairing takes up around 6 pct of all martensitic units while it is around 32 pct between tempered martensite units. It is interesting to note that there is no significant change in the amount of twin boundaries V1–V2.

Variant pairing of (a) all martensite units and (b) tempered martensite units
3.3.3 Comparison with large units
The tempered martensite units make up a significant part of the large units of the complete microstructure, which can be clearly seen by comparing the unit size colormaps for all and only tempered martensite units in Figures 9(a) and (b). The large units (>1500 px) are dominantly tempered with the exception of units 2 and 3, see Figure 9(a). These units as well as the largest single observed unit, see electronic supplementary Figures S2d and S9a, show a mixture of low and high tetragonality c/a in Figure 5(d). The statistical comparison of units with a size over 500 px in Figure 9(c) shows a good agreement for the majority of large units between tempered and all martensite units. This agreement can also be seen in Figure 9(d) where the fraction of large martensite units that are tempered is shown for the three tetragonality c/a threshold values and the cut-off sizes 500, 750, and 1000 px. It can be seen that the fraction of tempered martensite increases continuously for increasing cut-off size and threshold value of c/a, e.g., \(c/a = 1.0125\) having a fraction around 50 pct for 750 and 65 pct for 1000 px.

Size map for (a) all martensite units together with numbering of units 2–3 and (b) tempered martensite units, (c) Martensite unit size distribution for large units over 500 px (inset over 1000 px) for all and tetragonality \(c/a < 1.0125\), (d) Fraction of large martensite units that are tempered
4 Discussion
The combination of dilatometer, in-situ HEXRD, and microstructure analysis gives new insights into the early stage of the martensitic transformation in a 0.74C–1.15Mn–1.08Cr high carbon steel. The early-formed martensite can be distinguished from the martensite formed after the isothermal hold since it becomes tempered during the isothermal hold and there is no significant further transformation during the hold as shown in Figures 4(a) and (b). Therefore, the continuous volume increase during the isothermal hold in Figure 3 cannot be connected to a phase transformation from fcc to bcc/bct and has to be due to another, not yet understood, effect. Other effects like tempering of martensite or reduction in martensite tetragonality can be excluded as well since they result in a volume decrease instead of an increase.[42] The initial phase fraction increase with time observed in Figure 4(a) can be best explained by an athermal martensitic transformation due to temperature equilibration, see Figure 4(b). The effect of temperature equilibration is further supported by the slower transformation onset in the HEXRD phase fraction for the “Direct” heat treatment compared to the dilatation \(\Delta L\) in Figures 4(c) and (d). The HEXRD results are limited to smaller sample volumes compared to dilatometer and are more susceptive to local temperature variations that are known to be present for fast quenching experiments.[43,44] Overall, no further transformation of austenite to martensite is observed during the isothermal hold allowing the differentiation of the martensite formed before and after.
The tempered martensite is dominantly observed in and along the PAGBs, Figures 5 and 7(c), which shows that the transformation preferably starts and progresses there. This observation is supported by the distances of tempered martensite to PAGB in comparison to all martensite in Figure 7(b). The preferred nucleation at grain boundaries is known,[45,46] but the growth along PAGBs is in contrast to the previously reported behavior in low carbon steels[6] and Fe–Ni–C alloys,[7,8] where geometrical partitioning[9] of the PAGs into smaller grains by the early-formed martensite was observed. This study does not show any significant evidence for this behavior. Differences in the martensite structure, lath/plate vs mixed,[14] due to the different carbon and nickel content could be an explanation. Another explanation could be that the start of the martensitic transformation is separated into multiple stages, starting with nucleation and growth along PAGBs observed in this study and followed by geometrical partitioning.[6,7,8,9]. For clarification of this effect further investigations of the bulk transformation at different hold temperatures would be necessary. Van Bohemen et al.[13] studied the early transformation behavior in a 0.8 wt pct C steel, where the bulk behavior was not clearly identified due to experimental limitations, but the free surface experiments showed results similar to References 6,7, through 8.
The formation of large units at the start of the transformation and smaller units at later transformation stages is expected due to space restrictions[9] and mechanical stabilization of austenite.[13,47,48] Based on this, a two-step transformation indicated by the presence of two maxima in dilatation rate \({\text{d}}\Delta L/{\text{d}}T\) in Figure 3 is proposed, with the large martensite units forming during the first maxima followed by the formation of smaller martensite units in the second step. This explanation is supported by the tempering of the majority of large martensite units in Figure 9 as well as the mixed tetragonality c/a of the large martensite units in Figure 5(d) that are not tempered according to Figure 5(b). This mixed tetragonality c/a is typical for martensite formed at higher temperatures due to auto-tempering/-partitioning effects[49] and these units should therefore be formed at the early stages of the transformation directly after the isothermal hold.
The proposed two-step process is supported by the variant pairing analysis in Figure 8 that shows a significantly higher content of V1–V16 pairing between tempered martensite units compared to the later-formed martensite. This V1–V16 pairing is typical for high carbon plate martensite[15] and therefore indicates that the early-formed large martensite units have a stronger plate martensite crystallographic character compared to the later-formed units. The plate character of martensite is expected from Ms calculation in Thermo-Calc[50,51,52,53] for the used composition, where Ms is 198.4 \(^{\circ }\)C for plate and 187.3 \(^{\circ }\)C for lath martensite. The later-formed martensite has the typical character shown in the mixed region between lath and plate martensite.[15]
The clustering of tempered martensite units in Figures 5 and 6 indicates an autocatalysis mechanism[9,54,55,56] with preferred nucleation close to the already formed martensite. It is important to note that no isothermal martensite formed during the isothermal hold in Figures 4(a) and (b). The presence of autocatalysis in Fe–Ni and Fe–Ni–C alloys is well documented,[54,55,56] but is experimentally not well understood for Fe–C alloys with carbon contents around 0.8 wt pct C. Van Bohemen et al.[57] found no indication of an autocatalysis mechanism for the athermal martensite transformation of a 0.80 wt pct C steel in continuous cooling experiments, but discussed the possibility of autocatalysis processes on very short time scales that cannot be experimentally resolved in dilatometer analysis. Guimaraes[56,58] has proposed models with an autocatalytic part for the athermal transformation in Fe–Ni–C and Fe–C alloys. Part of this preferred nucleation can also be explained by the elastic stress fields of the formed martensite and the resulting preferred nucleation in neighboring PAGs[59] since multiple clusters have PAGBs within them, see Figure 7(a).
The quantitative analysis of the tempered martensite is based on the correct determination of low c/a values. The comparison with the FSD and LOM images shows that this method works well for the majority of the martensite units in Figures 5 and 7(a) with some exceptions. Large units with mixed tetragonality c/a that are not observed as tempered in the FSD image can be seen. This discrepancy can be explained by the auto-tempering/-partitioning observed in reference[49] after the initial isothermal hold. Overall, the determination of tempered martensite via tetragonality c/a is a powerful complementary technique for quantitative analysis of tempered martensite in a mixed structure.
5 Conclusion
The early martensitic transformation in a 0.74C–1.15Mn–1.08Cr high carbon steel was studied with a combination of dilatometer, in-situ HEXRD, LOM as well as qualitative and new quantitative microstructure characterization methods based on the local tetragonality determination by EBSD.
The early martensitic transformation was shown to start at the PAGBs and to predominantly grow along them instead of growing through PAGs. The formation of martensite seems to induce the preferred further transformation leading to clusters of early-formed martensite. This clustering indicates autocatalytic nucleation of martensite at an early stage.
The transformation kinetics indicate a two-step transformation where the early-formed martensite exhibits a strong plate martensite character in their variant pairing compared to the later-formed martensite and consists mainly of the large units of the microstructure. The expansion rate decrease after the first step could be connected to the stop of formation of large martensite units.
The results show a new perspective on the early martensitic transformation in high carbon steel regarding the nucleation and growth along PAGBs as well as the mixed character between lath and plate martensite.
References
G. Krauss: Mater. Sci. Eng. A, 1999, vol. 273–275, pp. 40–57.
A. Roitburd, G. Kurdjumov: Mater. Sci. Eng., 1979, vol. 39(2), pp. 141–67.
G. Olson, M. Cohen: Metall. Trans. A, 1976, vol. 7(12), pp. 1905–14.
G. Olson, M. Cohen: Metall. Trans. A, 1976, vol. 7(12), pp. 1915–23.
W. Bleck, F. Brühl, Y. Ma, C. Sasse: BHM, 2019, vol. 164(11), pp. 466–74.
C. Celada-Casero, J. Sietsma, M. Santofimia: Mater. Design, 2019, vol. 167, p. 107625.
J. Guimarães, J. Gomes: Acta Metall., 1978, vol. 26(10), pp. 1591–96.
J. Guimaraes, P. Rios: Metall. Mater. Trans. A, 2010, vol. 41(8), pp. 1928–35.
J. Fisher, J. Hollomon, D. Turnbull: JOM, 1949, vol. 1(10), pp. 691–700.
S. Chatterjee, H.-S. Wang, J. Yang, H. Bhadeshia: Mater. Sci. Technol., 2006, vol. 22(6), pp. 641–44.
T. Furuhara et al.: ISIJ Int., 2008, vol. 48(8), pp. 1038–45.
S. Takaki, K. Fukunaga, J. Syarif, T. Tsuchiyama: Mater. Trans., 2004, vol. 45(7), pp. 2245–51.
S. Van Bohemen, J. Sietsma: Metall. Mater. Trans. A, 2009, vol. 40(5), pp. 1059–68.
A. Marder, G. Krauss: Trans. ASM, 1967, vol. 60, pp. 651–60.
A. Stormvinter, G. Miyamoto, T. Furuhara, P. Hedström, A. Borgenstam: Acta Mater., 2012, vol. 60(20), pp. 7265–74.
C. Cayron, B. Artaud, L. Briottet: Mater. Charact., 2006, vol. 57(4–5), pp. 386–401.
C. Cayron: J. Appl. Crystal., 2007, vol. 40(6), pp. 1183–88.
G. Miyamoto, N. Iwata, N. Takayama, T. Furuhara: Acta Mater., 2010, vol. 58(19), pp. 6393–6403.
T. Nyyssönen, M. Isakov, P. Peura, V.-T. Kuokkala: Metall. Mater. Trans. A, 2016, vol. 47(6), pp. 2587–90.
E. Gomes de Araujo, H. Pirgazi, M. Sanjari, M. Mohammadi, L. Kestens: J. Appl. Crystal., 2021, vol. 54(2).
A. Winkelmann, G. Nolze, G. Cios, T. Tokarski: Phys. Rev. Mater., 2018, vol. 2(12), pp. 1–7. https://doi.org/10.1103/PhysRevMaterials.2.123803
G. Nolze, A. Winkelmann, G. Cios, T. Tokarski: Mater. Charact., 2021. https://doi.org/10.1016/j.matchar.2021.111040
T. Tanaka, N. Maruyama, N. Nakamura, A. Wilkinson: Acta Mater., 2020, vol. 195, pp. 728–38.
D. Buffum, J. Hollomon, L. Jaffe: Tech. Rep, Watertown Arsenal Labs MA Watertown Arsenal, 1945
F. Werner, B. Averbach, M. Cohen: JOM, 1956, vol. 8(10), pp. 1484–84.
H.-S. Yang, H. Bhadeshia: Mater. Sci. Technol., 2007, vol. 23(5), pp. 556–60.
N. Schell et al.: Mater. Sci. Forum, 2014, vol. 772, pp. 57–61. https://doi.org/10.4028/WWW.SCIENTIFIC.NET/MSF.772.57
S. Aoued et al.: Metals, 2020, vol. 10(1), p. 137. https://doi.org/10.3390/met10010137
A. Hammersley: J. Appl. Crystal., 2016, vol. 49(2), pp. 646–52. https://doi.org/10.1107/S1600576716000455
A. Coelho: J. Appl. Crystal., 2018, vol. 51(1), pp. 210–18.
G. Bergerhoff, R. Hundt, R. Sievers, I. Brown: J. Chem. Inf. Comput. Sci., 1983, vol. 23(2), pp. 66–69.
J. Schindelin et al.: Nat. Methods, 2012, vol. 9(7), pp. 676–82.
B. Forster, D. Van De Ville, J. Berent, D. Sage, M. Unser: Microsc. Res. Tech., 2004, vol. 65(1–2), pp. 33–42.
S. Preibisch, S. Saalfeld, P. Tomancak: Bioinformatics, 2009, vol. 25(11), pp. 1463–65.
F. Niessen, T. Nyyssönen, A. Gazder, R. Hielscher: J. Appl. Crystal., 2022, vol. 55(1).
F. Bachmann, R. Hielscher, H. Schaeben: Ultramicroscopy, 2011, vol. 111(12), pp. 1720–33.
S. Morito et al.: Acta Mater., 2003, vol. 51(6), pp. 1789–99.
J. Kapur, D. Casasent: in Geometric Correction of Sem Images, vol. 4044, International Society for Optics and Photonics, 2000, pp. 165–76
G. Nolze: Mater. Sci. Technol., 2006, vol. 22(11), pp. 1343–51.
G. Nolze: Ultramicroscopy, 2007, vol. 107(2–3), pp. 172–83.
V. Tong, T. Britton: Ultramicroscopy, 2021, vol. 221, p. 113130.
E. Mittemeijer, L. Cheng, P. Van der Schaaf, C. Brakman, B. Korevaar: Metall. Trans. A, 1988, vol. 19(4), pp. 925–32.
T. Kop, Y. Van Leeuwen, J. Sietsma, S. Van Der Zwaag: ISIJ Int., 2000, vol. 40(7), pp. 713–18.
T. Kop, J. Sietsma, S. Van Der Zwaag: J. Mater. Sci., 2001, vol. 36(2), pp. 519–26.
S. Kajiwara: Metall. Mater. Trans. A, 1986, vol. 17(10), pp. 1693–1702.
T. Song, B. De Cooman: ISIJ Int., 2014, vol. 54(10), pp. 2394–2403.
G. Miyamoto, A. Shibata, T. Maki, T. Furuhara: Acta Mater., 2009, vol. 57(4), pp. 1120–31.
L. Morsdorf, C. Tasan, D. Ponge, D. Raabe: Acta Mater., 2015, vol. 95, pp. 366–77.
T. Kohne. et al.: unpublished work, 2022
A. Borgenstam, M. Hillert: Acta Mater., 1997, vol. 45(5), pp. 2079–91.
J.-O. Andersson, T. Helander, L. Höglund, P. Shi, B. Sundman: Calphad, 2002, vol. 26(2), pp. 273–312.
A. Stormvinter, A. Borgenstam, J. Ågren: Metall. Mater. Trans. A, 2012, vol. 43(10), pp. 3870–79.
F. Huyan, P. Hedström, L. Höglund, A. Borgenstam: Metall. Mater. Trans. A, 2016, vol. 47(9), pp. 4404–10.
E. Machlin, M. Cohen: JOM, 1951, vol. 3(9), pp. 746–54.
M. Lin, G. Olson, M. Cohen: Metall. Trans. A, 1992, vol. 23(11), pp. 2987–98.
J. Guimaraes: Mater. Sci. Eng., 1987, vol. 95, pp. 217–24.
S. Van Bohemen, J. Sietsma: Mater. Sci. Technol. (UK), 2014, vol. 30(9), pp. 1024–33. https://doi.org/10.1179/1743284714Y.0000000532
J. Guimaraes:Mater. Sci. Eng. A, 2008, vol. 476(1–2), pp. 106–111.
T. Tomida: Acta Mater., 2018, vol. 146, pp. 25–41.
Acknowledgments
We acknowledge the members of the Vinnova “Controlled quenching at case hardening for optimal performance -QuenchCool” project for their help with sample manufacturing and discussions. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at the beamline P07b and we would like to thank Dr. Norbert Schell for assistance.
Funding
Open access funding provided by Royal Institute of Technology. We acknowledge Vinnova for funding the project “Controlled quenching at case hardening for optimal performance- QuenchCool” within the programme “Strategic Vehicle Research and Innovation” (FFI) of which this research was part of. A.W. was supported by the Polish National Science Centre (NCN) Grant number 2020/37/B/ST5/03669.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted January 18, 2022; accepted May 16, 2022.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file 2 (avi 5032 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kohne, T., Maimaitiyili, T., Winkelmann, A. et al. Early Martensitic Transformation in a 0.74C–1.15Mn–1.08Cr High Carbon Steel. Metall Mater Trans A 53, 3034–3043 (2022). https://doi.org/10.1007/s11661-022-06724-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-022-06724-z