
Abstract
Hyperinflammation plays an important role in severe and critical COVID-19. Using inconsistent criteria, many researchers define hyperinflammation as a form of very severe inflammation with cytokine storm. Therefore, COVID-19 patients are treated with anti-inflammatory drugs. These drugs appear to be less efficacious than expected and are sometimes accompanied by serious adverse effects. SARS-CoV-2 promotes cellular ATP release. Increased levels of extracellular ATP activate the purinergic receptors of the immune cells initiating the physiologic pro-inflammatory immune response. Persisting viral infection drives the ATP release even further leading to the activation of the P2X7 purinergic receptors (P2X7Rs) and a severe yet physiologic inflammation. Disease progression promotes prolonged vigorous activation of the P2X7R causing cell death and uncontrolled ATP release leading to cytokine storm and desensitisation of all other purinergic receptors of the immune cells. This results in immune paralysis with co-infections or secondary infections. We refer to this pathologic condition as hyperinflammation. The readily available and affordable P2X7R antagonist lidocaine can abrogate hyperinflammation and restore the normal immune function. The issue is that the half-maximal effective concentration for P2X7R inhibition of lidocaine is much higher than the maximal tolerable plasma concentration where adverse effects start to develop. To overcome this, we selectively inhibit the P2X7Rs of the immune cells of the lymphatic system inducing clonal expansion of Tregs in local lymph nodes. Subsequently, these Tregs migrate throughout the body exerting anti-inflammatory activities suppressing systemic and (distant) local hyperinflammation. We illustrate this with six critically ill COVID-19 patients treated with lidocaine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hyperinflammation and acute respiratory distress syndrome (ARDS) caused by coronavirus disease 2019 (COVID-19) have become the world’s number 1 challenge. The exponential pattern in the number of severe cases in the second and third waves of the SARS-CoV-2 pandemic has shown to reach nations’ maximum ICU capacities in weeks rather than months after outbreak of the disease irrespective of rigorous population-based preventive measures. In a recently published systematic review, the case fatality rates in patients in the ICU across 7 countries vary between 14.9 and 66.7%, while the case fatality rates among those who required mechanical ventilation vary between 16.7 and 97.0% [1]. In addition, the case fatality rate in a cohort of 1035 critically ill COVID-19 patients requiring extracorporeal membrane oxygenation (ECMO, artificial lungs) is alarmingly high (37.4%) [2].
The clinical manifestations of severe COVID-19 consist of pneumonia with dyspnoea and hyperinflammation. Hyperinflammation is thought to be the basis of the development of severe and critical COVID-19 [3,4,5]. Currently, a clear-cut definition of hyperinflammation is lacking. Some authors describe the condition of hyperinflammation as a form of very severe inflammation with cytokine storm [6]. The criteria of hyperinflammation are not consistent and include clinical data and/or different combinations of the parameters of the activation of the pro-inflammatory response of the immune system (i.e. fever, rapid respiratory deterioration, cytokine, ferritin and/or CRP concentrations, changes in blood levels of several types of immune cells, etc., examples are presented in Table 1) [3, 4, 6,7,8, 9, 10,11,12,13,14]. In addition, the current definitions of hyperinflammatory syndrome do not provide an explanation for the frequently observed co-infections or secondary infections in COVID-19 [15, 16].
The results of non-randomised cohort studies with controls and of retrospective observational studies suggest that IL-1 receptor blockade (anakinra) [9, 17], monoclonal antibodies against IL-6 receptors [18,19,20,21] and the combination of both drugs [22, 23] may improve survival rate in at least a subgroup of patients with COVID-19. However, in prospective randomised controlled trials with the exception of one trial with tocilizumab and sarilumab in critically patients [24], anti-inflammatory therapy with anakinra [25] or tocilizumab [26,27,28,29,30,31,32] did not improve the outcome in moderate, severe and critically ill COVID-19. On December 10, 2020, an editorial commented that it is disappointing that nearly 10 months into the COVID-19 pandemic, a breakthrough treatment has not been identified [33]. Researchers of the US National Institute of Allergy and Infectious Diseases stated that although Remdesivir is effective to reduce time to recovery in hospitalised COVID-19 patients [34] and dexamethasone reduces mortality in critically ill COVID-19 patients [35], there is no treatment for early or mild infection [36]. Moreover, dexamethasone raises concerns because it increased the 28-day mortality in patients who did not receive respiratory support [35] and it dampens the “alarm phase” of the inflammation process including the capacity of detecting pathogens in mammals by the immune system [37]. In addition, administration of methylprednisolone (1 mg/kg/day intravenously) in COVID-19 reduced the blood levels of NK cells, CD4+ and CD8+T-cells and increases the duration of throat viral RNA detectability indicating immune cell dysfunction [38]. Furthermore, targeted anti-viral medication has failed to treat COVID-19 effectively [39]. According to the World Health Organisation, after a record-breaking development, vaccine deployment is slow and has many challenges to overcome [40]. Vaccine hesitancy is relatively high [41] even among health care workers [42, 43]. It could take more than a year to vaccinate enough people required to make an impact on SARS-CoV-2 spreading, while therapeutic measures that can immediately attenuate the course of SARS-CoV-2-related lung damage are promptly needed on a global scale. To make the matters worse, many scientists expect that SARS-CoV-2 may become endemic and is here to stay [44].
In this report, we developed a novel definition of hyperinflammation based on purinergic signalling. Subsequently, we describe our discovery of an old drug capable of attenuating hyperinflammation and illustrate this with six critically ill patients suffering from COVID-19. Finally, we present the future development of a new and more accessible administration route for this drug as shown in Fig. 1.

A graphical summary of the future development of the administration of lipophilic lidocaine base in the sublingual region or elsewhere in the oral cavity. We postulate that selective inhibition of the P2X7Rs of the immune cells of the lymphatic system by lidocaine suppresses hyperinflammation in two stages. Stage 1: The selective inhibition of the P2X7Rs of the immune cells residing in the lymph nodes (stage 1a) induces clonal expansion of Tregs with improved function in these lymph nodes (stage 1b); Stage 2: Subsequently, these Tregs migrate throughout the body exerting anti-inflammatory activities reducing systemic and (distant) local hyperinflammation. See text under the heading “Future development” for explanation
Purinergic signalling
In 1929 adenylic acid (identical to adenosine) was identified [45], and in the same year, the adenosine triphosphate (ATP) molecule was discovered and isolated [46]. Ten years later (1939), researchers contributed to the understanding of intracellular ATP as an intracellular energy transport molecule [47,48,49,50]. In 1948 and in 1959, it was reported that extracellular ATP has a different function than ATP within the cytoplasm [51, 52]. The authors showed that extracellular ATP molecules have an intercellular signalling function. The intercellular signalling by nucleotides (ATP, ADP, UTP and UDP) and nucleoside (adenosine) is referred to as purinergic signalling. The purinergic co-transmission in neurons was discovered by Geoffrey Burnstock in 1972 [53]. It took over 20 years for the importance of purinergic signalling to be accepted [54, 55]. Finally, researchers of the University of Ferrara first reported that the P2Z receptor (the former name of the P2X7R) plays an intriguing role in immunity, inflammation and cell death [56].
The intracellular levels of ATP are high at millimolar concentrations (2–8 mM) [57], and the ATP concentrations in synaptic vesicles are even higher in the range of 5 to 100 mM [58]. In contrast, under normal resting conditions, the extracellular levels of ATP are quite low at nanomolar concentrations (<3 nM) [57, 59]. Under specific conditions, ATP release can rise by more than 1000-fold [53, 57, 60, 61] and leads to a significant increase in the extracellular levels of ATP. The resulting significant increase in extracellular nucleotides and adenosine concentrations activates their purinergic receptors inducing certain cellular functions. Examples of such conditions are membrane depolarisation (i.e. sympathetic neuron endings) [53], mechanical stress (i.e. high mechanical power ventilation) [59,60,61,62,63], hypoxia [64], hyperosmosis, hypotonic and isotonic stress of endothelial cells [65,66,67,68], inflammation [69, 70], surfactant release by alveolar epithelial type II cells [59,60,61], mucine release by airway smooth muscle cells [71], insulin release by pancreatic islet beta-cells [72, 73], etc. There is an exception to this concept: Although a spontaneous ATP-induced inward Ca2+ current through the P2X7R could not be detected below extracellular ATP levels of 200 μmol/ml [74], low tonic basal activation of P2X7R at nanomolar extracellular ATP concentrations promotes serum independent cellular proliferation [75], promotes closure of the wound area in scratch wound assay [76], protects from apoptosis [77], initiates anaerobic glycolysis independent of the oxygen contents [78], etc. (Table 2, rows 80–85). However, low tonic basal activation of the P2X7Rs by extracellular ATP does not cause a pro-inflammatory response of the immune system. Therefore, this topic is beyond the scope of this paper and will not be discussed here.
Clearance of the ATP molecule in order to avoid accumulation in the extracellular space is performed by enzymes attached to the outside of the cell membranes (ecto-enzymes) and by soluble enzymes excreted to the extracellular space (Fig. 2) [57, 272,273,274,275]. A proportion of the enzymatic-breakdown product of ATP adenosine enters the cells via the equilibrative nucleoside transporters (ENT1 and ENT2) and concentrative nucleoside transporters (CNT1 and CNT2) (Fig. 2) [57, 60, 61]. The release and subsequently clearance of the extracellular nucleotides and adenosine cause fluctuation in the extracellular levels of ATP, other nucleotides and adenosine. These fluctuations in extracellular concentrations are indispensable for the receptor resensitisation after desensitisation following receptor activation as discussed below.

Clearance of extracellular ATP and adenosine by ectonucleotidases and soluble extracellular nucleotidases [272,273,274275]. This process is indispensable to enable receptors to recover from desensitisation following receptor activation (resensitisation, see text under the heading “Purinergic signalling in inflammation and hyperinflammation” for explanation). CD39,Ecto-nucleoside triphosphate diphosphohydrolase 1-3 (ENTPD 1-3); CD73, Ecto-5′-nucleotidase (5’-NT); NPP, nucleotide pyrophosphatase/phosphodiesterase; TNAP, tissue nonspecific alkaline phosphatase; ADA, adenosine deaminase; ADK, adenosine kinase; HGPRT, hypoxanthine-guanine phosphoribosyltransferase; ATP, adenosine triphosphate; ADP, adenosine diphosphate; AMP, adenosine monophosphate; ADO, adenosine; ENTs, equilibrative nucleoside transporters; CNTs, concentrative nucleoside transporters
Purinergic signalling in inflammation and hyperinflammation
The purinergic control of cellular processes including the pro-inflammatory and anti-inflammatory responses of the immune system is depending on the activation and the desensitisation phenomenon of the nucleotides and adenosine receptors of the immune cells [74, 276,277,278,279,280,281,282]. Except for the P2X7R, all other purinergic receptors, i.e. P2XRs, P2YRs and P1 receptors (adenosine receptors—AdoRs), are subject to desensitisation [279,280,281,282,283]. In addition, a certain extent of desensitisation occurs after every activation, and this desensitisation requires time to return to the state of complete resensitisation [279, 280]. The higher and the longer the stimulus of the activation, the higher the extent of desensitisation and the longer the recovery time to the state of complete resensitisation [278]. One of the P2 receptors, the P2X7 receptor, is not prone to desensitisation, and apart from the low tonic basal activation of this receptor at low nanomolar concentrations as mentioned above, the extracellular concentration of ATP required to activate this receptor is much higher. Activation of the P2X7R starts at 100 μM with an EC50 of >1 mM [74, 279, 284].
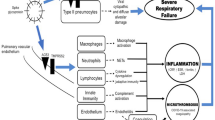
Summary of the effects of extracellular nucleotides and nucleoside on the innate and adaptive immune system through different purinergic receptors is presented in Table 2, rows 1–163. Coronaviruses can induce inflammation by the activation of the intracellular sensing molecules IRIG1/MDA5 [285, 286]. Reportedly, acute inflammation [69, 70] and infection with SARS-CoV-2 virus induce ATP release [287]. The vesicular exocytosis-mediated release of ATP, connexin-43 (Cx43)-mediated ATP release and pannexin-1 (Panx-1)-mediated ATP release can be triggered by the activation of Toll-like receptor 4 (TLR4) and TLR2 by pathogen-associated molecular patterns (PAMPs) and by the activation of P2X7Rs [180,181,182, 187]. In turn, activation of the P2X7Rs upregulates the protein expression of TLR 2, TLR3, TLR4 and TLR 5 [288]. Additionally, increased levels of TNF-α during inflammation induce ATP release via Panx-1 [289]. Pro-inflammatory immune response is initiated by the increase in the extracellular ATP, ADP and adenosine levels in the microenvironment of immune cells activating the P2XRs, P2YRs and AdoRs (Fig. 3) [57, 60, 169, 290]. In this case, ATP acts as a danger-associated molecular pattern (DAMP) [291, 292]. Increased ADP levels promote platelet activation and intravascular thrombosis (Table 2, rows 125 and 126). Reportedly, the pathological changes in the lung in patients with COVID-19 pneumonia showed marked microvascular thrombosis [293]. The EC50 for AdoRs is in the range of 26 nM to 1.4 μM [281] and for ATP, UTP or ADP receptors (P2XRs and P2YRs with the exception of the P2X7R) in the range of 0.01 nM to 10 μM [284, 294]. Obviously, the extent of the cellular ATP release is proportional to the severity of the infection. A severe infection with SARS-CoV-2 causes massive extracellular ATP release by the infected cells. This may be confined to the airway mucosa and the lung or may be extensive in multiple organs. Although increased extracellular ATP concentrations upregulate the expression of ecto-nucleotidases [295] these high ATP concentrations exceed the capacity of these ecto-enzymes (CD39, CD73, etc.) to clear the extracellular space from ATP molecules [60] ending in ATP concentrations of >1 mM. This is demonstrated in a report where the authors show that TLR-mediated CD39 internalisation (causing the deactivation of the ecto-enzyme CD39) in mice bone marrow-derived dendritic cells (BMDCs) leads to the accumulation of extracellular ATP to 1.4 mM [296]. The activation of P2X7Rs in ongoing inflammation is the hallmark of severe pro-inflammatory immune response (Table 2, rows 74, 86–119 and Fig. 3) [297] including COVID-19 [298]. If these levels of extracellular ATP are accompanied by the absence of the required fluctuations for other purinergic receptor to recover from desensitisation, all P1 and P2 (other than P2X7) purinergic receptors will become fully desensitised demarcating the initiation of hyperinflammation (Fig.3 and Table 2, rows 120–123) [279,280,281,282,283].

A schematic presentation of the activation of the purinergic receptors of the immune cells causing a pro-inflammatory response leading to hyperinflammation. Viral infection drives the controlled cellular release of ATP molecules. Increased extracellular nucleotides levels activate P2XRs and P2YRs. Upregulation of the extracellular ATP hydrolysing enzymes as depicted in Fig. 2 results in the increase of extracellular adenosine levels followed by the activation of the adenosine receptors (AdoRs). These processes initiate the physiologic pro-inflammatory response of the immune system. The green line at the bottom of the graph represents the extracellular ATP levels. The ascending part is caused by the ATP release, and the descending part results from the clearance of ATP by the extracellular or membrane-bound ATP hydrolysing enzymes. As the disease progresses and extracellular ATP levels increase above 1 mM, the P2X7R is additionally and effectively activated leading to a severe immune response. Except for P2X7Rs, all these receptors are known to be subject to desensitisation. Desensitisation of a receptor is defined as being unresponsive to activation by the ligand, resulting in (near) zero transmembrane signal transduction. A certain extent of desensitisation occurs after every activation, and this desensitisation requires time to return to the state of complete resensitisation. Increasing intensity and duration of the activation stimuli leads to increasing extent of desensitisation and duration of the recovery time to the state of complete resensitisation (represented by brown boxes with increasing size at the bottom of the graph). Severe viral infection can increase the controlled ATP release beyond the capacity of the extracellular enzymes to clear ATP and adenosine molecules. This causes a sustained high extracellular ATP and adenosine levels preventing the purinergic receptors from recovering from the state of desensitisation. The capacity to clear invading microorganisms diminishes leading to immune paralysis. In addition, prolonged high extracellular levels of ATP and activation of the P2X7R lead to macropore formation and cell death with uncontrolled release of ATP. In turn, this leads to vigorous activation of the P2X7R of the immune cells promoting massive production of cytokines ending in a cytokine storm and hyperinflammation
Hyperinflammation is characterised by the activation of P2X7Rs and desensitisation of other P2 receptors and AdoRs
As mentioned above, hyperinflammation starts when fluctuation of the extracellular nucleotides and adenosine no longer occurs and leads to prolonged activation of the P2X7Rs of the immune cells. Prolonged vigorous activation of the P2X7Rs leads to macropore formation and cytolysis with uncontrolled ATP release [222, 223, 227, 299] (Table 2, rows 120–123) causing hyperinflammation with massive pro-inflammatory immune response, massive pro-inflammatory and anti-inflammatory cytokine release: the cytokine storm (Fig. 3). In the early phase of COVID-19, hyperinflammation may be confined to the site of viral entry (i.e. airway mucosa and conjunctivae) but as viral replication and viral spreading progress, systemic hyperinflammation devel ops.
The upregulation of the expression of ectonucleotidases also leads to an increase in the concentrations of other nucleotides (i.e. ADP) and adenosine. These high extracellular concentrations of nucleotides and adenosine do not show concentration fluctuations required for the recovery (resensitisation) time from desensitisation causing a state of persistent desensitisation of all P2XRs, P2YRs [279, 280, 283, 300, 301] and AdoRs [282] with the exception of P2X7Rs. Consequently, the physiological function in the affected organs and inflamma tory response of the immune system are deactivated. This leads to the failure of organ function (i.e. ARDS in the lungs as we reported earlier [61]) and the immune system (immune paralysis) rendering the host susceptible to secondary co-infections(Fig.3). Sepsis-induced immunosuppression [302, 303] or compensatory anti-inflammatory response syndrome (CARS) in critically ill patients [304] was already raised by researchers in 1996 [305] and is a well-known phenomenon in critically ill patients [302]. Secondary bacterial infections occurred in 34.4% of 274 surviving elderly patients (age over 60 years) with COVID-19 and in 81.7% of 65 deceased patients [15]. In addition, it was found that 76 co-infections with other respiratory pathogens occurred in another cohort of 354 COVID-19 patients (16 of 115 mild cases (13.9%), 33 of 155 severe cases (21.3%) and 27 in 84 critical cases (32%)) [16]. In a meta-analysis involving 118 scientific reports on patients with COVID-19, co-infection with other pathogens at admission was observed in 19% and superinfection with other pathogens during admission in the hospital in 24% [306].
Control of hyperinflammation is annihilated by the downregulation of Tregs through the activation of P2X7R and the desensitisation of adenosine receptors
Tregs are key elements in the control of hyperinflammation [307]. Activation of AdoRA2As promotes the differentiation of naïve T-cells towards regulatory T-cells(Tregs) [112], increases the frequency of Tregs and the expression of CTLA-4 receptor and upregulates ecto-enzymes CD39 and CD73 expression accelerating adenosine generation from extracellular ATP [118] (Table 2, rows 25, 29, 30 and 33). This process is upset in case of desensitisation of AdoRs. In addition, activation of P2X7Rs inhibits the suppressive potential and stability of Tregs, inhibits the clonal expansion of Tregs, promotes Treg death, induces Treg depletion and reduces Treg IL-10 production (Table 2, rows 86–88, 106 and 107). In COVID-19 patients, significant lower Treg frequencies [308,309,310], lower expression of forkhead box protein P3 (FoxP3), lower expression of transforming growth factor-β(TGF-β) and lower cytokine TGF-β secretion [309] are observed compared to healthy control. Additionally, a reduced proportion of specific SARS-CoV-2-reactive Tregs was reported [311]. The desensitisation of AdoRs and the activation of P2X7Rs may well be the underlying mechanism of the low Tregs frequency in severe and critically ill COVID-19.
P2X7R antagonist restores the reduced Tregs population and Tregs function in hyperinflammation
As stated above, infected cells release ATP into the extracellular space. Obviously, the P2X7R antagonist blocks the activation of the P2X7Rs. Because a significant proportion of the ATP release to the extracellular space is mediated by the P2X7R (Table 2, rows 77–79), P2X7R a ntagonism combined with the upregulated ATP hydrolysing activity of the ecto-enzymes results in the decrease of the extracellular ATP concentrations. This can potentially abrogate hyperinflammation and the concomitant immune paralysis. Moreover, P2X7R inhibition promotes the cell-autonomous conversion of CD4+ T cells into Tregs after stimulation of their T-cell receptors (TCRs) [190]. In addition, P2X7R knock-out mice, mimicking the state of complete P2X7R inhibition, show an increase in tissue Tregs, prevent Tregs death and the Tregs produce more IL-10 and TGF-β [191]. Experimental inhibition of P2X7Rs restores the Tregs levels and function (Table 2, rows 86–88, 106 and 107) [190,191,192]. Inhibition of the P2X7R or P2X7R knock-out can attenuate severe inflammation in abdominal sepsis [312] and in acute lung injury [313, 314]. Apparently, amelioration of hyperinflammation by P2X7R inhibition is based on the increased activation and clonal expansion of the anti-inflammatory Tregs population (Table 2, rows 86–88, 106 and 107).
Some authors proposed that the P2X7R is an ideal candida te to target in COVID-19-associated severe pneumonia [298, 315], and others suggested that hyperactivation of the P2X7R plays a key role in the neuropathology of COVID-19 and that P2X7R antagonism may prevent or treat neurological manifestations of COVID-19 [316].
Lidocaine is a P2X7R antagonist
In 2015 it was discovered that lidocaine is a P2X7R antagonist [74], and therefore, lidocaine can potentially reduce the clinical symptoms of hyperinflammation significantly. In experimental sepsis, lidocaine improves organ failure [317,318,319] and survival [317]. In septic patients, lidocaine reduces neutrophil recruitment by the mitigation of chemokine-induced arrest and transepithelial neutrophil migration [320]. Neutrophil recruitment is an important facilitating process in the pathogenesis of multiple organ failure [320] and hyperinflammation in COVID-19 [321,322,323,324]. In patients with skin lesions from atopic dermatitis, lidocaine increases the proportion of Tregs and upregulates the FoxP3 expression [325]. In addition, lidocaine increases the IL-10 levels in mechanically ventilated mice [326] and decreases the TNF-α in BAL, plasma and lung samples in pigs undergoing surgery for lung resection [327].
The P2X7R antagonist dose-response relationship of lidocaine is presented in Fig.4. The IC50 for the inhibition of the P2X7R by lidocaine is about 66.07 μg/ml (0.28 mM) [74] where IC50 is defined as the required extracellular concentrations of the receptor antagonist to reach an inhibitory effect of halfway between maximal activation and maximal inhibition (half-maximal inhibitory concentration). The main issue is that the IC50 for P2X7R inhibition is much higher than the maximal tolerable plasma concentration for mammals. The maximal tolerable plasma concentration in humans is about 4.7 μg/ml (0.02 mM); this corresponds with an IC10 or lower (<10% inhibitory concentration, Fig.4). Above this lidocaine plasma concentration, adverse effects in increasing severity occur as presented in Table 3 [328, 329]. Thus, systemic lidocaine plasma concentrations of >4.7 μg/ml must be avoided [328, 329]. Caveat: The inhibitory concentrations of lidocaine for P2X7R as presented in Fig.4 are not corrected for the series resistance (in the range of 1–3 MΩ) of the used whole-cell voltage clamp method with two puller microelectrodes [74]. One should bear in mind that after correction for series resistance, the reported inhibitory concentration values including IC50 are expected to be higher [330].

Dose-response relationship of lidocaine suppressing the ATP-induced currents in oocytes expressing P2X7R. We reconstructed the fitted curve from the inhibitory concentrations data of lidocaine for P2X7R from the original article: 7% inhibition: 0.01 mM (2.34 μg/ml); 11% inhibition: 0.03 mM (7.03 μg/ml); 35% inhibition: 0.10 mM (23.43 μg/ml); 50% inhibition (IC50): 0.28 mM (66.07 μg/ml); 55% inhibition: 0.30 mM (70.29 μg/ml); 74% inhibition: 1.00 mM (234.30 μg/ml); 91% inhibition: 3.00 mM (702.90 μg/ml); and 98% inhibition: 10.00 mM (2343.00 μg/ml), respectively. The usual plasma concentrations in clinical settings are indicated by the green box, and the targeted concentrations in the lymph nodes are indicated by the magenta box. Note that the maximal tolerable plasma levels for human (about 4.7 μg/ml–0.02 mmol/L) are much lower than the required extracellular concentrations of lidocaine to effectively inhibit the P2X7R. Source: Okura D, et al. [74]
In addition to the P2X7R antagonist properties, lidocaine is also known to have several other inhibitory pharmacological targets: the voltage-gated sodium channels (VGSC: Nav1.2 [331], Nav1.3 [332], Nav1.4[333], Nav1.5 [334], Nav1.7 [335], 1.8 [336] and Nav 1.9 [337]), the Toll-like receptor 2 (TLR 2) [338], TLR4 [318] and the N-methyl-D-aspartate receptor (NMDAR) [339].
VGSCs conduct sodium ions inward and are essential for the transduction of sensory stimuli, the generation of the action potential and the release of neurotransmitters from sensory neuron terminals. Lidocaine inhibition of VGSCs can effectively reduce pain signalling [340]. In addition, VGSCs are present on dendritic cells (maintain chemokine-induced migration) [341], macrophages (regulate phagocytosis and endosomal pH during LPS-mediated endosomal acidification) [342], microglia (regulate phagocytosis cytokine release ad migration) [343], neutrophils (regulate attachment, transmigration and chemotaxis) [344] and T-cells (regulate positive selection of CD4+ T cells) [345]. However, until date no relevant data have been published suggesting that other VGSC antagonists (such as HYP-17 [346], A-803467 [347, 348], PF-05089771 [349], phenytoin [350] or tetrodotoxin [351, 352]) may substitute non-steroidal anti-inflammatory drugs let alone may suppress COVID-19-related hyperinflammation [353]. A plausible reason is that during hyperinflammation—including hyperinflammation in COVID-19—the cytokine levels (i.e. IL-1β [354], IL-6, IL-10 [355, 356] and IL-12 [357]) are high. Reportedly, IL-1β [358] and IL-6 [359] inhibit sodium currents of VGSCs, and IL-10 downregulates the expression of VCSCs [360]. Moreover, activation of the P2X7R reduced the density and currents of VGSCs [361]. Therefore, we do not consider the inhibitory properties of lidocaine on VGSCs to be relevant for the treatment of hyperinflammation in COVID-19.
At first glance, the downregulation of the expression of TLR 2 [338] and TLR 4 [318] is an important anti-inflammatory mechanism directly induced by lidocaine. But at a closer look, it appeared that activation of P2X7R by the agonist cathelicidin (LL-37) leads to the upregulation of the protein expression of TLR2, TLR3, TLR4 and TLR 5 [288]. This is in line with the MyD88 (myeloid differentiation primary-response protein 88)-dependent activation of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) following the activation of the P2X7R by BzATP [362, 363]. The MyD88-dependent activation of NF-κB is part of the TLR4/NF-κB pathway. Therefore, it is unsurprising that the inhibition of P2X7R by its antagonists (Brilliant Blue G, A-438079 and A-740003) neutralises the above-mentioned P2X7R-induced upregulation of TLRs [362]. Consequently, we argue that lidocaine inhibits inflammation directly by blocking P2X7Rs independent from the neutralisation of the P2X7R-induced upregulated TLR2 and TLR4.
The subpopulation of NMDA receptors present on the peripheral neurons are involved in nociception, and their number increases during inflammation contributing to the sensitisation of peripheral nerves to nociceptive stimuli. NMDA receptor antagonists have anaesthetic-like effects [364]. In addition, NMDA receptor antagonist can prevent hypoxic neuronal death, IL-1β and TNFα release [365], reduce the activation of inflammatory experimental colitis [366] and suppress glial pro-inflammatory cytokine expression [367]. Moreover, the NMDA receptor antagonist memantine can increase IL-10 production in BCR/CD40-activatedB-cells [368]. Lidocaine inhibits NMDA receptors [339, 369, 370], and thus the anti-inflammatory properties of lidocaine could be attributed to the inhibition of NMDA receptors. However, it has been reported that the anti-inflammatory effect in T-cell functions (inhibition of antigen-specific T-cell proliferation, T-cell cytotoxicity, T-cell migration towards chemokines and decrease in IL-2 and IFN-γ production by Th1 effector cells in favour of IL-10 and IL-13 production by Th2 cells) of the NMDA receptor antagonist ifenprodil is effective both in wild-type and in NMDA receptor (GluN1) knockout mice [371]. Moreover, it was found that KN-62, an inhibitor of Ca2+/calmodulin-dependent kinase type II and a potent P2X7R antagonist, provides neuroprotection against NMDA-induced cell death [372]. Therefore, we argue that the anti-inflammatory properties of NMDA receptor antagonists (including lidocaine) should be attributed to the inhibition of P2X7Rs rather than to the inhibition of NMDA receptors.
Selective inhibition of the P2X7Rs of the immune cells in the lymphatics avoids exceeding the maximal tolerable plasma concentration of lidocaine and inhibits hyperinflammation in two stages
As mentioned above, the main issue is that the IC50 for P2X7R inhibition is much higher than the maximal tolerable plasma concentration for mammals because P2X7Rs are indispensable for normal physiological functions (i.e. in the central nervous system [373], the peripheral nervous system [374] and in the lungs [60, 61]). Therefore, intravenous or oral administration aimed at achieving an effective concentration of lidocaine to inhibit P2X7Rs in serum and in target organs will hamper organ functions and is potentially dangerous.
The lymphatic system is populated exclusively by trafficking immune cells, i.e. naïve T cells, activated T cells, B cells [375], dendritic cells [376], monocytes [377], macrophages [378], neutrophils [379], mast cells [380], eosinophils [381] and basophils [382]. We postulate that selective inhibition of the P2X7Rs of the immune cells of the lymphatic system by lidocaine suppresses hyperinflammation in two stages: stage 1, the selective inhibition of the P2X7Rs of the immune cells residing in the lymph nodes induces clonal expansion of Tregs in these lymph nodes; stage 2, subsequently, these Tregs migrate throughout the body exerting anti-inflammatory activities reducing systemic and (distant) local hyperinflammation (Fig. 1).
The endothelium of the dermal capillaries of the skin belongs to the structural type “continuous endothelium” [383]. Although capillary walls can transport substances from blood to tissue, the absorption of substances from tissue to blood is, if any, extremely low [384]. Apparently, specialised initial lymphatics harbouring one-way valve leaflets capable of absorbing fluid and molecules from the interstitium are localised in the dermis. The absorbed lymph fluid is then propelled forward in the lymphatic network by collecting lymphatic vessels harbouring a rhythmic contracting muscle layer [385]. This system brings fluids and particles into the lymph nodes where numerous immune processes take place. The administration route to target the lymphatic system in a domestic swine model is illustrated by the subcutaneous or intradermal injection of compounds (isosulfan blue, fluorescein and radioactive technetium-99 isotope—Tc99) and by tracing the extent and the transit time of the distribution of these compounds using whole body scintigraphy in pigs [386]. The absorption of intradermal application of radioactive Tc99 into the lymph nodes is 10 times faster than after deep subcutaneous application and leads to higher concentrations in the lymph nodes related to these lymphatic vessels [386]. Radionuclide lymphoscintigraphy with molecules of different sizes after intradermal and subcutaneous injections showed that smaller particles (i.e. 99mTc-dextran and 99mTc-human serum albumin) migrate more rapidly towards the lymphatic vessels and lymphatic nodes than larger particles (i.e. radiocolloids of larger molecular size) [387]. The rate of clearance of 99mTc-pertechnetate and 99mTcDTPA after subcutaneous and intradermal administration in the back of the hand in humans is 1 %/min and 8 to 10 %/min, respectively [387].
The additional advantage is that the plasma concentrations of subcutaneously administered lidocaine are much lower than intravenously administered lidocaine. Intravenous administration of 2 mg/kg lidocaine in cats is almost immediately followed by a peak plasma concentration of 3.6 μg/mL [388]. In contrast, the achieved mean peak plasma concentrations after the subcutaneous administration of 30 mg/kg, 20 mg/kg and 10 mg/kg lidocaine are much lower: 1.69, 1.07 and 0.77 μg/mL, respectively [389]. Note that the applied subcutaneous dose [389] is 15, 10 and 5 times higher than the intravenous dose, respectively [388, 389]. Reportedly, the difference in the plasma concentrations after intravenous and subcutaneous administration of lidocaine is caused by the fact that, in contrast to the intravenous administration, a large proportion of the subcutaneously administered lidocaine is drained into the lymphatic system [390,391,392]. Obviously, this slows down the release of lidocaine to the venous blood. This is confirmed for bevacizumab in mice [390], for trastuzumab in rats [391] and for docetaxel in rats by [392].
As stated above, lymphatic absorption after intradermal administration is much higher than after deep subcutaneous administration [386, 387]. Practically, the intradermal infusion with lidocaine is not an accepted administration route for lidocaine. Therefore, we argue that a subdermal administration of lidocaine using a catheter inserted just beneath the dermis (subdermal infusion, Fig. 5) will result in higher concentrations of lidocaine in the draining local lymph nodes than a deep subcutaneous or intravenous infusion as depicted in the schematic presentation of the putative distribution of lidocaine in Fig. 6.

The cannula for subdermal infusion of lidocaine is superficially positioned just below the dermis to promote the uptake of lidocaine by the initial lymphatics of the dermis and to avoid accumulation of lidocaine in the subcutaneous fat tissue

Schematic presentation of the putative distribution of intravenous, oral, transmucosal (i.e. in the oral cavity) and subdermal administered lidocaine. Administration of hydrophilic lidocaine (lidocaine HCL) through a (central) venous catheter or by oral intake results in concentration gradients with the highest value in the venous blood and the lowest value in the lymph nodes. The reason is that by the time lidocaine reaches the lymph nodes, the drug is massively diluted and may never reach the effective concentration required to adequately inhibit the P2X7Rs of the immune system. In contrast, after subdermal injection of hydrophilic lidocaine, apart from a minimal absorption by the dermal capillaries, almost all the lidocaine is absorbed by the lymphatic system via the initial lymphatics. Because the fluid in the afferent collecting lymphatics originates from the interstitial fluid of the tissues, dilution of the concentration of lidocaine occurs. This fluid is then drained into the local lymph nodes. The extent of the dilution of lidocaine in the targeted lymph nodes is far less drastic compared to the (central) venous administration of the drug. We postulate that with continuous subdermal infusion, we can achieve concentrations of lidocaine in the lymph nodes sufficient to effectively inhibit the P2X7Rs of the immune cells. Theoretically, similar results may be expected from transmucosal and transdermal administration of lipophilic lidocaine base with a high concentration. Obviously, the subdermal, transmucosal and transdermal administration routes may also apply to other P2X7R antagonists
In summary, by means of the subdermal administration of lidocaine, we can ensure high concentrations of lidocaine in the local lymph nodes enabling an effective inhibition of the P2X7R of the immune cells while keeping the lidocaine plasma concentrations <4.7 μg/ml(stage 1a and 1b in Fig. 1). The induced Tregs clonal expansion in these local lymph nodes produces Tregs which migrate throughout the body controlling the ongoing hyperinflammation (stage 2 in Fig. 1). Obviously, the subdermal administration route may also apply to other P2X7R antagonists.
Three other P2X7R antagonists have been tested in human: CE-224,535 500 (Pfizer), AZD-9056(Astra-Zeneca) and JNJ-54175446 (Johnson and Johnson). A phase IIa study with CE-224,535 in patients with rheumatoid arthritis not responding adequately to methotrexate was recently reported [393]. Patients in the treatment arm received oral CE-224,535 500 mg twice/day for 12 weeks. Although the safety and tolerability for the compound were acceptable, CE-224,535 was not effective in this group of patients. The results of a phase II study with AZD-9056 in patients with active rheumatoid arthritis despite treatment with methotrexate or sulphasalazine was published. The treatment arm consists of oral AZD-9056 100 or 400 mg/day for 6 months [394]. The AZD-9056 used in this trial is non-lipophilic as indicated by the fact that this compound cannot penetrate the blood-brain barrier [395]. The authors conceded that “AZD-9056 does not have significant efficacy in the treatment of RA, and the P2X7 receptor does not appear to be a therapeutically useful target in RA” [394]. Recently, a randomised, placebo controlled, sequential-group, single-centre ascending dose phase I study was reported. The patients in the 5 treatment arms received 0.5, 2.5, 10, 50, 150 and 300 mg JNJ-54175446, respectively. The authors reported dose-dependent plasma levels, no serious adverse events, ex vivo attenuation of lipopolysaccharide-induced IL-1β release in peripheral blood and confirmation of passive brain penetration of JNJ-54175446 [396]. The approach of the P2X7R antagonist therapy of the above-mentioned authors is quite different from ours: While these authors directly targeted the diseased organs via the gut absorption of the drug, we target the immune cells in local lymph nodes inducing an anti-inflammatory immune response which in turn targets the diseased organs (Fig. 1). This is illustrated by the following study concerning a placebo-controlled, multicentre, double-blind phase IIa study in patients with moderately to severely active Crohn’s disease. The patients in the treatment arm received oral AZD-9056 200 mg/day for 28 days. The authors found a significant improvement in the Crohn’s Disease Activity Index (CDAI) at day 28 [397]. In contrast to the skin, the endothelium of the mucosal capillaries of the mouth and the gastrointestinal tract are fenestrated allowing molecules to pass from the submucosal tissue into the capillaries [383]. Unlike the failure of the treatment of rheumatoid arthritis described above, the successful treatment of gut inflammation here can be attributed to the absorption of non-lipophilic oral AZD-9056 by the mucosa-associated lymphoid tissue (MALT). This is the inductive site of the mucosal immune system consisting of mesenteric lymph nodes, Peyer’s patches and isolated lymph follicles [398, 399]. Although lymphatic transport to the lymph nodes of the non-lipophilic oral AZD-9056 is limited [400, 401], AZD-9056 inhibits P2X7Rs of the local T-cells via absorption by the inductive sites of MALT. This induces a local anti-inflammatory immune response executed by the effector sites of MALT consisting of lamina propria lymphocytes and intraepithelial lymphocytes [398, 399].
Real-world subdermal administration of lidocaine in critically ill COVID-19 patients
Six COVID-19-induced ARDS patients
From April 2020 until end of July 2020, two of the authors of this report (AS and TK) have successfully treated six critically ill patients with COVID-19 admitted to the ICU of the Showa University in Tokyo, Japan, with lidocaine. The lidocaine treatment was based on off-label use. The Medical Ethical Committee of the Showa University, School of Medicine, Tokyo, approved the collection, analysis and publication of patients on mechanical ventilation admitted to the ICU (protocol number 3313). The administration was initially intravenously in the two first patients, followed by subdermally (a superficially inserted subcutaneous catheter as illustrated in Fig. 5). In the other four patients, only the subdermal administration was further applied. The concentration of the intravenous lidocaine infusion solution is 20 mg/ml (2%), the route for continuous administration of lidocaine commonly used in daily practice. The dose for intravenous administration is 0.6 mg/kg/h as recommended earlier [402]. Due to the limited efficacy of intravenous lidocaine and based on the hypothesis of selectively targeting the inhibition of the P2X7Rs of the immune cells, the infusion in both patients was converted to subdermal infusion of 1.0 mg/kg/h (dosage as reported by Japanese researchers [403]) after 7 and 6 days, respectively. The time course of clinical parameters of these six patients is presented in Figures 7, 8, 9, 10. In about 20% of the inserted subdermal cannulae, local subdermal indurations were observed. Whenever this occurred, the infusion cannula was removed and replaced with a new cannula at a different location.

Patient 1, the first of the six cases with severe COVID-19 treated with subdermal lidocaine in the ICU of the Showa University, Tokyo, Japan. A 63-year-old male with COVID-19-induced ARDS, was admitted to the hospital. The CT scan showed bilateral ground glass opacities. Co-morbidities: COPD, smoking 60 cigarettes per day for more than 40 years. About 40 years before admission, the patient suffered from pneumothorax. After admission the clinical condition deteriorated requiring an ICU admission and mechanical ventilation on day 4. On day 11, continuous intravenous lidocaine of 0.6 mg/kg/h was initiated, but the patient’s condition kept worsening with high pulmonary artery pressures and reduced aeration of the lung. On day 19, the continuous intravenous lidocaine of 0.6 mg/kg/h was changed to continuous subdermal lidocaine of 1 mg/kg/h. This was followed by improvement of the clinical condition, and on day 20, the aeration of the lung was improved, but the pulmonary artery pressures remained high. Despite this the P/F ratio was gradually improving, and ECMO weaning was done on day 50. No new ECG changes were observed during treatment with lidocaine. Blood metHb were within the normal range (0.3–0.8%). On day 99, he was weaned from the mechanical ventilator and was discharged from the ICU on day 121. CT scan on day 146 showed reduced ground glass opacities in both lungs and some interstitial change in upper and middle fields of the lung and improvement of the pneumothorax. The patient was discharged from the hospital on day 187, he went home, and he could walk but needed extra oxygen supply of 2L/min. Nine months after admission, the patient is doing well and has returned to work. The patient visited the hospital 3 months after discharge: He only uses oxygen 1 L/min to go shopping and during physical training (out-patient rehabilitation). He talked to the treating intensivist without requiring oxygen and had no shortness of breath or tachypnoea. The red-coloured labels of the legends refer to graph plots using the (left) primary Y-axis, and the black-coloured labels of the legends refer to graph plots using the (right) secondary Y-axis

Patient 2. A 68-year-old male with COVID-19-induced ARDS admitted to the ICU and required mechanical ventilation. The CT scan showed bilateral ground glass opacities. Co-morbidity: Asthma. After admission the patient’s condition was deteriorating. On day 5, continuous intravenous lidocaine of 0.6 mg/kg/h was initiated, but the clinical condition and the P/F ratio kept worsening. On day 11, the intravenous lidocaine of 0.6 mg/kg/h was changed to continuous subdermal lidocaine of 1 mg/kg/h. A few days later, this was followed by improvement of the clinical condition and the P/F ratio. No new ECG changes were observed during treatment with lidocaine. Blood metHb were within the normal range (0.1–0.6%). The patient was discharged from the ICU on day 30 home on day 37. At 3 months after admission, the patient is doing well. The red coloured labels of the legends refer to graph plots using the (left) primary Y-axis, and the black-coloured labels of the legends refer to graph plots using the (right) secondary Y-axis

Left graph: Patient 3. A 59-year-old male with respiratory distress and bilateral ground glass opacities on the CT scan. Co-morbidity: Obesity, diabetes mellitus and gout. No new ECG changes were observed during treatment with lidocaine. Blood metHb were within the normal range (0.1–0.4%). The patient was discharged from the ICU on day 8 and was discharged home on day 20. After 3 months, he is doing well. Right graph: Patient 4. A 51-year-old male with fever, dyspnoea and cough due to COVID-19. The CT scan showed bilateral ground glass opacities. Co-morbidity: none. No new ECG changes were observed during treatment with lidocaine. Blood metHb were within the normal range (0.1–0.3%). The patient was discharged from the ICU on day 8 and was disc harged home on day 28. At 3 months, he is doing well and has returned to work. The red-coloured labels of the legends refer to graph plots using the (left) primary Y-axis, and the black-coloured labels of the legends refer to graph plots using the (right) secondary Y-axis

Left gr aph: P atient 5. A 5 8 -year-o ld male with fever, dyspnoea and cough due to COVID-19. The CT scan showed bilateral ground glass opacities. Co-morbidity: Fatty liver. No new ECG changes were observed during treatment with lidocaine. Blood metHb were within the normal range (0.1–0.3%). On day 14, the patient was discharged from the ICU. On day 20, the patient was discharged home and is doing well at 3 months after admission. Right graph: Patien ts 6. A 59-year-old male with fever, dyspnoea and cough due to COVID-19. CT scan showed bilateral ground glass opacities. Co-morbidity: Hypertension on medication. No new ECG changes were observed during treatment with lidocaine. MetHb were within the normal range (0.1–0.3%). On day 13, the patient was discharged from the ICU. He was discharged from the hospital on day 20, and a t 3 months after admission, he is doing well, played golf regularly and has returned to work. The red-coloured labels of the legends refer to graph plots using the (left) primary Y-axis, and the black-coloured labels of the legends refer to graph plots using the (right) secondary Y-axis
The first patient (Fig.7), a 63-year-old male (75 kg, 168 cm), developed fever and nausea on March 27, 2020, and 3 days later, he started to cough and became dyspnoeic. After 5 days, the PCR SARS-Cov-2 test was positive, and he was admitted to the hospital with SARS-Cov-2-induced ARDS. Co-morbidities include COPD and smoking 60 cigarettes per day for more than 40 years. About 40 years earlier, the patient suffered from pneumothorax. On day 3, the patient deteriorated and was intubated and mechanically ventilated due to poor blood gases. No haemodynamic instability was observed. The CT scan showed bilateral ground glass opacities compatible with ARDS. On day 5, the patient was transferred to the ICU of the university hospital because of further respiratory deterioration. The patient received favipiravir for 14 days after admission; the patient did not receive dexamethasone. Prone position mechanical ventilation was initiated due to the progression of the respiratory disease with an extremely low PaO2/FiO2 ratio of 63.3 mm Hg (severe ARDS according to the Berlin definition. The Berlin definition of ARDS includes severe PaO2/FiO2 ratio ≤100 mm Hg, moderate PaO2/FiO2 >100 to 200 mm Hg, mild PaO2/FiO2 >200 to 300 mm Hg, no ARDS PaO2/FiO2 >300 mm Hg [404]). The initial ventilator settings include APRV, Phigh27 cm H2O, Thigh 7.0 s, Plow 0 cm H2O and Tlow 0.32 s. The PaCO2 was normal. The echocardiographic estimated pulmonary arterial systolic pressure (PASP) was 80 mm Hg. The Krebs von Lungen 6 (KL-6, a marker for lung fibrosis [405]) plasma level was highly elevated (1299 U/mL; normal value <425 U/mL), CRP was also high (40.4 mg/L; normal value <10 mg/L), and albumin was 2.2 g/dl. The white blood cell count, platelet count and urine production were normal. On day 4, the chest X-ray was not improved. On day 6, the PaO2/FiO2 ratio was slightly increased but remained low at 103 mm Hg, and the chest X-ray showed progression of the ARDS. ECMO was initiated due to exhausted ventilatory strategy. On day 9, the PaO2/FiO2 ratio improved but remained low at around 153 mm Hg, but the CRP declined to around 21.8 mg/L. The patient was put on muscle relaxants. The patient’s ARDS status had improved from severe to moderate ARDS. From day 10 until day 30, the ferritin levels were well >1000 ng/ml (>100 μg/dl, normal values <300 ng/ml). From day 11 until day 62, D-Dimer was very high reaching 121.9 nM/L day 14. On day 11, no improvement of the blood gases was observed, and it was decided to treat the patient with continuous intravenous lidocaine 0.6 mg/kg/h. The CRP showed a progressive decline from 19 (on day 12) to 12.8 (on day 16) and 7.4 (on day 19), but the PaO2/FiO2 ratio remained poor at around 90 mm Hg (severe ARDS according to the Berlin criteria) and the chest X-ray image on day 15, 3 days after the initiation of the intravenous lidocaine infusion, deteriorated dramatically. The lidocaine plasma concentrations were 3.4 μg/ml on day 13 and 5.4 μg/ml on day 14. On day 19, the continuous intravenous lidocaine infusion was replaced by continuous subdermal lidocaine infusion of 1 mg/kg/h. Although the PaO2/FiO2 ratio remained unchanged on day 20 (1 day after the switch to the continuous subdermal lidocaine), the chest X-ray improved clearly. On day 21, the lidocaine plasma concentration was 2.6 μg/ml, and albumin was 2.5 g/dl. From day 22, the PaO2/FiO2ratio was gradually improving reaching 151 mm Hg on day 34 (moderate ARDS). The KL-6 on day 22 dropped to 458 U/L (this is only slightly above the normal value of <450 U/l). On day 31, the CRP was low at 1 mg/L, and the lidocaine plasma concentration was 1.2 μg/ml. The muscle relaxants were discontinued. Albumin was 2.3 g/dl. On day 33, the chest X-ray was further improved, and the CRP remained low at 5.5 mg/L. The patient was awake and could communicate with the nurses. On day 38, the lidocaine plasma level was 2.3. On day 43, the PaO2/FiO2 ratio was increased to 214 mm Hg. According to the Berlin definition of ARDS [404], the patient’s ARDS status had changed from moderate to mild. Albumin was 2.8 g/dl. On day 50, the patient was weaned from ECMO. On day 51, the patient underwent tracheotomy. Because the clinical condition of the patient was stabilised with a low CRP of 6.3 mg/L on day 55, the continuous subdermal lidocaine was discontinued on day 57. On day 69, he developed pneumothorax requiring pleural drainage. On day 99, he was weaned from the mechanical ventilator and was discharged from the ICU on day 121. No new ECG changes were observed during treatment with lidocaine. Blood methaemoglobin (metHb) were within the normal range (0.3–0.8%). CT scan on day 146 showed reduced ground glass opacities in both lungs, some interstitial change in upper and middle fields of the lung and improvement of the pneumothorax. The patient left the hospital on day 187, he went home, and he could walk but needed extra oxygen supply of 2L/min. Nine months after admission, the patient is doing well and has returned to work. The patient visited the hospital 3 months after discharge: He only uses oxygen 1 L/min to go shopping and during physical training (out-patient rehabilitation). He talked to the treating intensivist without requiring oxygen and had no shortness of breath or tachypnoea.
The second patient (Fig.8) is a 68-year-old male (75 kg, 164 cm) with SARS-Cov-2-induced ARDS and positive SARS-Cov-2 PCR test admitted to the university hospital. Co-morbidity is asthma. The CT scan showed bilateral ground glass opacities. Haemodynamically the patient was stable. The patient received tocilizumab on day 8 and favipiravir for 14 days; he did not receive dexamethasone. On day 2, the respiratory conditions deteriorated, and the PaO2/FiO2 ratio is 118 mm Hg (moderate ARDS according to the Berlin ARDS definition [404]). The patient was intubated and required mechanical ventilation. The initial ventilator settings include pressure control, peak inspiratory pressure 28 cm H2O, PEEP 13 cm H2O and respiratory rate 30/min. CRP was 10.6 mg/L, and KL-6 was 486 U/ml. White blood cell count, platelet count and urine production were normal. The ferritin levels remained >1000 ng/ml (100 μg/dl) during the entire ICU stay. Albumin was 2.9 g/dl. In the following 3 days, the PaO2/FiO2 ratio improved to around 150 mm Hg. The PaO2/FiO2 ratio dropped from 152 on day 5 to 84 mm Hg on day 6. CRP was increased to 22.9, and the KL-6 was increased to 762 U/ml. The patient was put in prone position and given muscle relaxants. Continuous intravenous lidocaine of 0.6 ml/kg/h was started. Albumin was 1.8 g/dl. On day 7, the PaO2/FiO2 ratio increased to 128 mm Hg, CRP dropped to 10.3 mg/mL and the lidocaine plasma concentration was 2.2 μg/ml. From day 3 until discharge from the ICU, D-dimer values were elevated reaching 75 nM/L on day 14. On day 8, although the PaO2/FiO2 ratio improved from 84 to 125 mm Hg, the mechanical ventilatory strategies were exhausted, and the patient was put on ECMO. The KL-6 was increased to 845 U/L, and lidocaine plasma level was 2.9 μg/ml. The PaO2/FiO2 ratio improved to 238 mm Hg on day 9, but on day 10, a sharp drop of the PaO2/FiO2 ratio to 60 mm Hg was observed, and CRP was 2.0 mg/ml. The patient’s ARDS status had changed from moderate to severe according to the Berlin ARDS criteria [404]. Lidocaine treatment was switched from continuous intravenous to continuous subdermal (dosage: 1 mg/kg/h). On day 14, the lidocaine plasma level was 2.7 μg/ml. KL-6 dropped to 549 U/l. On day 17, the clinical condition of the patient was improving, and the PaO2/FiO2 ratio reached 158 mm Hg. The patient was weaned from ECMO. The PaO2/FiO2 ratio improved further reaching 291 mm Hg on day 21, and the patient’s ARDS status has changed from moderate to mild ARDS [404]. On day 22, mechanical ventilation was discontinued, and the patient was extubated. The patient was orientated, and no signs of confusion were detected. CT scan on day 25 showed persistent ground glass opacities in both lungs, some pulmonary effusion (right >left), and no signs of vascular thrombosis. In addition, no signs of deep venous thrombosis were found in the lower extremities. Lidocaine treatment was continued until discharge from the ICU on day 30. No new ECG changes were observed during treatment with lidocaine. Blood metHb were within the normal range (0.1–0.6%). The patient was discharged home on day 37. At 3 months after admission, the patient is doing well.
The third patient (Fig. 9left), a 59-year-old male (109 kg, 170 cm), was admitted to the university hospital with respiratory distress and bilateral ground glass opacities on the CT scan with a positive SARS-CoV-2 test. Co-morbidities include obesity (BMI 37.7 kg/m2), diabetes mellitus and gout. The patient required immediate intubation and mechanical ventilation. The patient received tocilizumab on day 3 and favipiravir for 15 days and did not receive dexamethasone. The initial ventilator settings are pressure control, peak inspiratory pressure 30 cm H2O, PEEP 15 cm H2O and respiratory rate 25/min. The PaO2/FiO2 ratio on admission was 160 mm Hg (moderate ARDS according to the Berlin definition [404]), CRP was 39.3 mg/L and KL-6 was 294 U/ml. White blood cell count was increased (13.10−9/L) and platelet count and urine production were normal. Albumin was 2.1 g/dl. Haemodynamic parameters were stable. On the admission day, continuous subdermal lidocaine was started at 1 mg/kg/h. On day 2, the PaO2/FiO2 ratio improved to 283 mm Hg, and the patient’s ARDS status had changed from moderate to mild ARDS. CRP was 41 mg/L, KL-6 was 268 U/L and the lidocaine plasma level was 3.7 μg/ml. Albumin was 1.7 g/dl. On day 4, the PaO2/FiO2 ratio was 302 mm Hg, and the patient’s ARDS status had changed from mild ARDS to no ARDS according to the Berlin ARDS criteria. On day 5, the PaO2/FiO2 ratio was improved further to 328 mm Hg, and CRP dropped to 16.4, and the patient was extubated. The patient was orientated, no signs of confusion were detected. The patient was discharged from the ICU on day 8; CRP was 2.3 mg/ml. Albumin was 2.5 g/dl. No new ECG changes were observed during treatment with lidocaine. Blood metHb were within the normal range (0.1–0.4%). The patient was discharged home on day 20. After 3 months, he is doing well.
The fourth patient (Fig. 9right) is a 51-year-old male (68 kg, 175 cm). Ten days before admission, he developed fever and 2 days before admission dyspnoea and coughing. On the day of admission, the PCR SARS-CoV-2 test was positive. The CT scan showed bilateral ground glass opacities. Co-morbidity is none. The patient was intubated and put on mechanical ventilation on admission. The patient received favipiravir for 14 days; he did not receive dexamethasone. On day 3, he was transferred to the university hospital because of deterioration of pulmonary condition. The initial ventilator settings include pressure control, peak inspiratory pressure 24 cm H2O, PEEP 12 cm H2O and respiratory rate 15/min. The haemodynamic conditions were stable. White blood cell count and platelet count were normal. Albumin was 2.6 g/dl. Continuous subdermal lidocaine was started immediately. On day 3, the PaO2/FiO2 ratio was 214 (moderate ARDS according to the Berlin definition [404]). KL-6 was 177 U/L, and CRP was 17.4 mg/L. On day 5, the PaO2/FiO2 ratio was increased to 382 (the patient’s ARDS status had changed from mild ARDS to no ARDS), and lidocaine plasma concentration was 5.2 μg/ml. CRP was 27.3mg/L. Lidocaine plasma levels on day 3 and 4 were 3.4 and 4.2 μg/ml, respectively. KL-6 was 163 U/L. The patient was extubated. The patient was orientated, and no signs of confusion were detected. The patient was discharged from the ICU on day 8, and the CRP was 9.3 mg/L. No new ECG changes were observed during treatment with lidocaine. Blood metHb were within the normal range (0.1–0.3%). He was discharged home on day 28. At 3 months, he is doing well and has returned to work.
The fifth patient (Fig. 10left) is a 58-year-old male (80 kg, 175 cm). Nine days before admission, he developed a sore throat. A day later, he developed fever. Two days before admission, he started coughing and was dyspnoeic. On the day of admission, the PCR SARS-Cov-2 test was positive. Co-morbidity includes fatty liver. The CT scan showed bilateral ground glass opacities. The patient was initially admitted to the hospital ward. The patient received tocilizumab on day 7 and favipiravir for 10 days; dexamethasone was not prescribed. On day 3, the patient deteriorated and had to be intubated and put on mechanical ventilation. On day 4, the patient was transferred to the university hospital due to deterioration of the pulmonary condition. The initial ventilator settings include pressure control, peak inspiratory pressure 27 cm H2O, PEEP 12 cm H2O and respiratory rate 25/min. PaO2/FiO2 ratio was 188 (moderate ARDS according to the Berlin definition). Haemodynamic parameters were stable, and CRP was 12.9 mg/ml. White blood cell count was increased (14.4.109/L), but platelet count was normal. KL-6 was 330 U/L. Continuous subdermal lidocaine was started at 1 mg/kg/h at arrival at the ICU of the university hospital. Albumin was 2.8 g/dl. On day 5, the PaO2/FiO2 ratio was unchanged, CRP was 10.4 mg/L and the lidocaine plasma level was 4 μg/ml. On day 6, the lidocaine plasma level was 3.2 μg/ml. KL-6 remained stable at 400 U/L. Albumin was 2.3 g/dl. On day 10, the respiratory insufficiency had cleared; although the PaO2/FiO2 ratio remained 184, the CRP dropped to 2.4 mg/L, and KL-6 was 322 U/L. The patient was extubated, and he was orientated; no signs of confusion were detected. On day 14, the patient was discharged from the ICU. No new ECG changes were observed during treatment with lidocaine. Blood metHb were within the normal range (0.1–0.3%). On day 20, the patient was discharged home and is doing well at 3 months after admission.
The sixth patient (Fig. 10right) is a 59-year-old male (65 kg, 175 cm) with fever, dyspnoea and cough due to COVID-19. CT scan showed bilateral ground glass opacities. Co-morbidity includes hypertension on medication. The patient was admitted to the general ward. KL-6 233 U/L, white blood cell count and platelet count were normal. Albumin was 3.6 g/dl. On day 3, there is a deterioration of the respiratory function necessitating a transfer to the ICU and mechanical ventilation. Tocilizumab was given on day 4. The patient received favipiravir for 11 days, and the patient did not receive dexamethasone. The initial ventilator settings include pressure control, peak inspiratory pressure 22 cm H2O, PEEP 10 cm H2O and respiratory rate 20/min. Continuous subdermal lidocaine of 1 mg/kg/h was initiated after admission to the ICU. Haemodynamic parameters were stable. CRP was 6.3 mg/L, and KL-6 was 263 U/L. On day 4, a progressive respiratory failure occurred requiring intubation and mechanical ventilation. PaO2/FiO2 ratio was 218 mm Hg; the haemodynamic parameters remained stable. CRP was 6.3 mg/L, and the white blood count and platelet count were normal. Lidocaine plasma level was 4.6 μg/ml. On day 5, the PaO2/FiO2 ratio dropped further to 164 mm Hg. Lidocaine plasma level was 3.4 μg/ml. Albumin was 3.2 g/dl. On day 9, the clinical condition of the patient improved. The ventilator settings could be decreased, the PaO2/FiO2 ratio remained 207 mm Hg during the weaning period, and CRP was 0.7 mg/L. On day 10, the patient was extubated, he was orientated, and no signs of confusion were detected. On day 13, the patient was discharged from the ICU. No new ECG changes were observed during treatment with lidocaine. Blood metHb were within the normal range (0.1–0.3%). He was discharged from the hospital on day 20, and at 3 months after admission, he is doing well, played golf regularly and has returned to work.
Additional 14 patients with COVID-19-induced ARDS
From July 2020 until beginning of December 2020, 14 additional critically ill patients with COVID-19-induced ARDS requiring mechanical ventilation were treated in the ICU of the Showa University with continuous subdermal lidocaine infusion (1 mg/kg/h) plus intravenous or oral dexamethasone (6 mg/day) as reported earlier [35]. Of these 20 patients, 19 survived, but an 87-year-old female patient died of invasive aspergillosis. No other patient developed secondary co-infections (unpublished data, personal communication by AS and TK).
Discussion of the clinical cases
After completing the novel definition of hyperinflammation, we developed a new approach to target the lymphatic system with continuous subdermal administration of lidocaine. This is meant to increase the anti-hyperinflammatory effect of lidocaine while avoiding toxic plasma levels. We described the treatment of six critically ill patients with COVID-19 with lidocaine. Two patients required mechanical ventilation and ECMO, and four patients were treated with mechanical ventilation. As mentioned under the heading “Introduction”, the case fatality rates of patients requiring mechanical ventilation and/or ECMO are alarmingly high [1, 2]. Patient 1 and patient 2 were older than 60 years. Additionally, patient 1 had COPD and had smoked 60 cigarettes per day for more than 40 years. Patient 3 suffered from obesity and diabetes mellitus. These are serious prognostic factors for bad outcome COVID-19 [406, 407]. Patient 1 and patient 2 were initially treated with continuous intravenous lidocaine through a central venous line. In both patients, the pulmonary conditions deteriorated after the initiation of intravenous lidocaine: Patient 1 who was already on ECMO showed progressive pulmonary deterioration on the chest X-rays, and patient 2 deteriorated further necessitating the initiation of ECMO therapy. Remarkably, the pulmonary conditions of both patients improved within 48 h after the switch from intravenous to subdermal continuous lidocaine. The lidocaine plasma levels remained around 5 μg/ml. To our knowledge, these six cases represent the first observations of the promising treatment of critically ill COVID-19 patients with lidocaine targeting P2X7Rs of the immune cells in the lymphatics. All patients recovered completely from their illness. None of the patients showed the feared side effect of cardiac arrhythmia and methaemoglobinaemia during lidocaine therapy. Our findings suggest that continuous subdermal lidocaine infusion at the rate of 1 mg/kg/h has the potential to mitigate hyperinflammation and ARDS in critically ill COVID-19-patients. Obviously, although all six patients appeared to respond positively to the treatment and no severe adverse effects were observed, no final conclusions can be made on the efficacy of lidocaine in critically ill COVID-19 patients.
Researchers from Lima, Peru, reported the treatment of 28 (three mild, 21 moderate and four severe) COVID-19 patients with 0.5% lidocaine HCL solution with an intravenous dose of 1 mg/kg once a day for 2 days and 2% lidocaine HCL solution with a subcutaneous dose of 1 mg/kg once a day for 2 days [408]. The authors aimed at the improvement of pain, cough, respiratory rate and oxygen saturation. They found improvement in most patients. In severe cases, this treatment did not improve the oxygen saturation. As expected, treatment with a low daily dose of lidocaine once per day for a total treatment duration of 2 days could not adequately inhibit the P2X7R-induced hyperinflammation in COVID-19.
Recently, a group of researchers from Strasbourg, France, announced a study entitled: “Impact of intravenous lidocaine on clinical outcomes of patients with ARDS during COVID-19 pandemia (LidoCovid): A structured summary of a study protocol for a randomised controlled trial” (ClinicalTrials.gov Identifier: NCT04609865) [409].
Lately, an extraordinary treatment of COVID-19 ARDS was reported [410]. The authors performed lung transplantations in three critically ill COVID-19 ARDS patients: a 28-year-old female, a 62-year-old male and a 43-year-old male. The first patient underwent lung transplantation after weeks on veno-venous ECMO support with elevated pulmonary arterial pressures and severe secondary Serratia marcescens pneumonia. The second patient underwent lung transplantation after 100 days on veno-venous ECMO support complicated by Pseudomonas aeruginosa pneumonia, haemothorax and empyema, while the third patient after 90 days on the mechanical ventilator. This patient suffered from many complications: asystolic cardiac arrest, heparin-induced thrombocytopenia, a left frontal lobe infarct of the cerebral cortex, Serratia marcescens-mediated pneumonia with bacteraemia, acute kidney injury, a left haemothorax requiring thoracotomy and lung decortication, a right pneumothorax requiring tube thoracostomy, hypernatremia associated with seizures and malnutrition. Before lung transplantation, the patient developed increasing clinical signs of pulmonary fibrosis and severe pulmonary hypertension. The first two patients are reported to have achieved independence in daily life activities several months after lung transplantation. Three months after lung transplantation, the third patient made improvements in the neurocognitive status and muscular strength at an inpatient rehabilitation centre.
Far less drastic is our proposed treatment of hyperinflammation in COVID-19-induced ARDS with lidocaine, an old drug that is readily available to hospitals all over the world at a low cost. In November 1948, Xylocaine was approved by the Food and Drug Administration (FDA) in the USA [411]. Lidocaine is used as a local anaesthetic [411], treatment of chronic neuropathic pain [412] but also for the prophylaxis or treatment of ventricular arrhythmia [328, 329]. Recently, intravenous lidocaine has been administered as general anaesthetic replacing opioids in the perioperative settings [413]. Potentially, lidocaine, as a P2X7R antagonist, can abrogate hyperinflammation, can restore the capacity of the immune system to combat secondary co-infections and can improve the clinical condition in critically ill COVID-19 patients. Despite several in vitro [326, 327, 414, 415], animal studies [319, 416,417,418,419,420] and patient cohorts [408, 421] on the anti-inflammatory properties of lidocaine, completed clinical trials which deliver a proof of concept (i.e. a randomised controlled trial) have not yet been performed. We postulate that because the maximal tolerable plasma concentration of lidocaine is much lower than the required extracellular concentration to effectively inhibit P2X7Rs, intravenous systemic administration of lidocaine simply cannot not be used to effectively treat hyperinflammation. This is a plausible reason why 5 years after the discovery of lidocaine as a P2X7R inhibitor (published in 2015) [74] the drug is still not used as an anti-hyperinflammatory treatment in clinical practice.
Concluding remarks
As stated in the introduction, therapeutic measures that can immediately attenuate the course of SARS-CoV-2-related lung damage are promptly needed on a global scale. In contrast to the investigational P2X7R antagonists described above, continuous subdermal infusion of 2% lidocaine solution to primarily deposit lidocaine into the lymphatics is readily available and can be used in the daily practice immediately and, in principle, even outside the ICU and is very well affordable. Therefore, this therapy deserves to be investigated in larger placebo controlled randomised clinical studies with COVID-19 patients.
Future development
However, our experience with subdermal administration of lidocaine in the ICU made clear that this method may not be routinely suitable outside hospital settings. Needless to say that high complexity and high-cost treatments (requiring highly skilled nurses and infusion pump equipment) are inaccessible to low-income COVID-19 patients in developing countries. Also, as the severity and case fatality rate of COVID-19 increase with age [406], the case fatality rate in elderly patients in nursing homes is strikingly high, and many residents have poor access to medical care [422]. This encouraged us to explore alternative uncomplicated methods of lidocaine administration accessible to everyone, particularly elderly COVID-19 patients and COVID-19 patients in developing countries.
Recently, researchers stated in their article on targeting the P2X7R in COVID-19 that the P2X7R antagonists for human use are available only in oral form and that this might be an inefficient route of drug delivery [298]. We found a solution to this problem. Permeability of the skin and mucous membrane to water, drugs, etc. is said to be dependent on the site of the administration [423, 424]. For example, the permeability constant of the floor of the mouth (sublingual mucosa), lateral border of the tongue and buccal mucosa for tritium-labelled water is 22, 17 and 13 times as high as human skin, respectively [423]. We argue that this also applies to lidocaine. As mentioned above, the endothelium of the mucosal capillaries of the mouth and the gastrointestinal tract belong to the structural type “fenestrated endothelium” allowing molecules to pass from the submucosal tissue into the capillaries [383]. Lidocaine hydrochloride is highly soluble in water (solubility of 680 mg/ml in water) [425] and therefore will mainly be absorbed by the submucosal capillary [426] and the inductive sites of MALT [398, 399]. In contrast, the highly lipophilic lidocaine base (solubility of 4 mg/ml in water, 760 mg/ml in 95% ethanol and 790 mg/ml in chloroform) [425] is preferably absorbed by the local initial lymphatics in the submucosal tissue [426, 427]. In addition, the lymphatic drainage of the floor of the mouth is extensive, involving many lymph nodes [428,429,430,431].
We estimate that with a sublingual administration of lipophilic lidocaine base (Fig. 1), we may reach the IC50 of the P2X7Rs in the draining lymph nodes to control systemic hyperinflammation and avoid toxic lidocaine plasma levels (Figs. 4 and 6). Obviously, such solution may also apply to other P2X7R antagonists. We stress that sublingual and buccal administration of lipophilic lidocaine is different from oral administration of lidocaine. Oral administration of lidocaine is aimed at the resorption of the drug in the gastrointestinal tract (Fig.6).
There are other methods of targeting the immune cells in the lymphatics, i.e. transdermal administration of lipophilic P2X7R antagonist with skin penetration enhancers (i.e. alpha-terpineol [432], ethanol [433] and lipid based nanoformulations [434]), intravenous administration of a P2X7R antagonist using nano-sized drug delivery systems [435], liposomes or polymer micelles [436] and oral administration of a P2X7R antagonist using delivery systems for intestinal lymphatic drug transport such as chylomicrons [437].
Data Availability
The datasets generated during and/or analysed during the current study are not publicly available due to privacy reasons but are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Quah P, Li A, Phua J (2020) Mortality rates of patients with COVID-19 in the intensive care unit: a systematic review of the emerging literature. Crit Care 24(1):285. https://doi.org/10.1186/s13054-020-03006-1
Barbaro RP, MacLaren G, Boonstra PS, Iwashyna TJ, Slutsky AS, Fan E, Bartlett RH, Tonna JE, Hyslop R, Fanning JJ, Rycus PT, Hyer SJ, Anders MM, Agerstrand CL, Hryniewicz K, Diaz R, Lorusso R, Combes A, Brodie D (2020) Extracorporeal membrane oxygenation support in COVID-19: an international cohort study of the Extracorporeal Life Support Organization registry. Lancet 396(10257):1071–1078. https://doi.org/10.1016/s0140-6736(20)32008-0
Manson JJ, Crooks C, Naja M, Ledlie A, Goulden B, Liddle T, Khan E, Mehta P, Martin-Gutierrez L, Waddington KE, Robinson GA, Ribeiro Santos L, McLoughlin E, Snell A, Adeney C, Schim van der Loeff I, Baker KF, Duncan CJA, Hanrath AT et al (2020)COVID-19-associated hyperinflammation and escalation of patient care: a retrospective longitudinal cohort study. Lancet Rheumatol 2(10):e594–e602. https://doi.org/10.1016/s2665-9913(20)30275-7
Gustine JN, Jones D (2021) Immunopathology of hyperinflammation in COVID-19. Am J Pathol 191(1):4–17. https://doi.org/10.1016/j.ajpath.2020.08.009
Afrin LB, Weinstock LB, Molderings GJ (2020)Covid-19 hyperinflammation and post-Covid-19 illness may be rooted in mast cell activation syndrome. Int J Infect Dis 100:327–332. https://doi.org/10.1016/j.ijid.2020.09.016
Fajgenbaum DC, June CH (2020) Cytokine Storm. N Engl J Med 383(23):2255–2273. https://doi.org/10.1056/NEJMra2026131
Webb BJ, Peltan ID, Jensen P, Hoda D, Hunter B, Silver A, Starr N, Buckel W, Grisel N, Hummel E, Snow G, Morris D, Stenehjem E, Srivastava R, Brown SM (2020) Clinical criteria for COVID-19-associated hyperinflammatory syndrome: a cohort study. Lancet Rheumatol 2(12):e754–e763. https://doi.org/10.1016/s2665-9913(20)30343-x
Cardone M, Yano M, Rosenberg AS, Puig M (2020) Lessons learned to date on COVID-19 hyperinflammatory syndrome: considerations for interventions to mitigate SARS-CoV-2 viral infection and detrimental hyperinflammation. Front Immunol 11:1131. https://doi.org/10.3389/fimmu.2020.01131
Bozzi G, Mangioni D, Minoia F, Aliberti S, Grasselli G, Barbetta L, Castelli V, Palomba E, Alagna L, Lombardi A, Ungaro R, Agostoni C, Baldini M, Blasi F, Cesari M, Costantino G, Fracanzani AL, Montano N, Monzani V et al (2021) Anakinra combined with methylprednisolone in patients with severe COVID-19 pneumonia and hyperinflammation: an observational cohort study. J Allergy Clin Immunol 147(2):561–566.e564. https://doi.org/10.1016/j.jaci.2020.11.006
Landewé RBM, Ramiro S, Mostard RLM (2021)COVID-19-induced hyperinflammation, immunosuppression, recovery and survival: how causal inference may help draw robust conclusions. RMD Open 7(1). https://doi.org/10.1136/rmdopen-2021-001638
Anka AU, Tahir MI, Abubakar SD, Alsabbagh M, Zian Z, Hamedifar H, Sabzevari A, Azizi G (2021) Coronavirus disease 2019 (COVID-19): an overview of the immunopathology, serological diagnosis and management. Scand J Immunol 93(4):e12998. https://doi.org/10.1111/sji.12998
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ (2020) COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. https://doi.org/10.1016/s0140-6736(20)30628-0
Freeman TL, Swartz TH (2020) Targeting the NLRP3 Inflammasome in severe COVID-19. Front Immunol 11:1518. https://doi.org/10.3389/fimmu.2020.01518
De Luca G, Cavalli G, Campochiaro C, Della-Torre E, Angelillo P, Tomelleri A, Boffini N, Tentori S, Mette F, Farina N, Rovere-Querini P, Ruggeri A, D'Aliberti T, Scarpellini P, Landoni G, De Cobelli F, Paolini JF, Zangrillo A, Tresoldi M et al (2020)GM-CSF blockade with mavrilimumab in severe COVID-19 pneumonia and systemic hyperinflammation: a single-centre, prospective cohort study. Lancet Rheumatol 2(8):e465–e473. https://doi.org/10.1016/s2665-9913(20)30170-3
Wang L, He W, Yu X, Hu D, Bao M, Liu H, Zhou J, Jiang H (2020) Coronavirus disease 2019 in elderly patients: Characteristics and prognostic factors based on 4-week follow-up. J Inf Secur. https://doi.org/10.1016/j.jinf.2020.03.019
Lv Z, Cheng S, Le J, Huang J, Feng L, Zhang B, Li Y (2020) Clinical characteristics and co-infections of 354 hospitalized patients with COVID-19 in Wuhan, China: a retrospective cohort study. Microbes Infect 22(4-5):195–199. https://doi.org/10.1016/j.micinf.2020.05.007
Kooistra EJ, Waalders NJB, Grondman I, Janssen NAF, de Nooijer AH, Netea MG, van de Veerdonk FL, Ewalds E, van der Hoeven JG, Kox M, Pickkers P (2020) Anakinra treatment in critically ill COVID-19 patients: a prospective cohort study. Crit Care 24(1):688. https://doi.org/10.1186/s13054-020-03364-w
Xu X, Han M, Li T, Sun W, Wang D, Fu B, Zhou Y, Zheng X, Yang Y, Li X, Zhang X, Pan A, Wei H (2020) Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci U S A 117(20):10970–10975. https://doi.org/10.1073/pnas.2005615117
Perrone F, Piccirillo MC, Ascierto PA, Salvarani C, Parrella R, Marata AM, Popoli P, Ferraris L, Marrocco-Trischitta MM, Ripamonti D, Binda F, Bonfanti P, Squillace N, Castelli F, Muiesan ML, Lichtner M, Calzetti C, Salerno ND, Atripaldi L et al (2020) Tocilizumab for patients with COVID-19 pneumonia. The single-arm TOCIVID-19 prospective trial. J Transl Med 18(1):405. https://doi.org/10.1186/s12967-020-02573-9
Luis BM, Miguel MB, Pedro DL, David IP, Itziar A, Ana GH, Enrique IJ, María LV, Noelia TF, Julio César BB, Marta UI, Rodrigo SL, María CB, Andrés LM, Javier MI, Juan Pablo GM, Gerardo HF, Carolina NF, Jorge BL et al (2021) Benefits of early aggressive immunomodulatory therapy (tocilizumab and methylprednisolone) in COVID-19: single center cohort study of 685 patients. J Transl Autoimmun 4:100086. https://doi.org/10.1016/j.jtauto.2021.100086
Gokhale Y, Mehta R, Kulkarni U, Karnik N, Gokhale S, Sundar U, Chavan S, Kor A, Thakur S, Trivedi T, Kumar N, Baveja S, Wadal A, Kolte S, Deolankar A, Pednekar S, Kalekar L, Padiyar R, Londhe C et al (2021) Tocilizumab improves survival in severe COVID-19 pneumonia with persistent hypoxia: a retrospective cohort study with follow-up from Mumbai, India. BMC Infect Dis 21(1):241. https://doi.org/10.1186/s12879-021-05912-3
Aomar-Millán IF, Salvatierra J, Torres-Parejo Ú, Faro-Miguez N, Callejas-Rubio JL, Ceballos-Torres Á, Cruces-Moreno MT, Gómez-Jiménez FJ, Hernández-Quero J, Anguita-Santos F (2021) Anakinra after treatment with corticosteroids alone or with tocilizumab in patients with severe COVID-19 pneumonia and moderate hyperinflammation. A retrospective cohort study. Intern Emerg Med 16(4):843–852. https://doi.org/10.1007/s11739-020-02600-z
Cavalli G, Larcher A, Tomelleri A, Campochiaro C, Della-Torre E, De Luca G, Farina N, Boffini N, Ruggeri A, Poli A, Scarpellini P, Rovere-Querini P, Tresoldi M, Salonia A, Montorsi F, Landoni G, Castagna A, Ciceri F, Zangrillo A, Dagna L (2021)Interleukin-1 and interleukin-6 inhibition compared with standard management in patients with COVID-19 and hyperinflammation: a cohort study. Lancet Rheumatol 3(4):e253–e261. https://doi.org/10.1016/s2665-9913(21)00012-6
Gordon AC, Mouncey PR, Al-Beidh F, Rowan KM, Nichol AD, Arabi YM, Annane D, Beane A, van Bentum-Puijk W, Berry LR, Bhimani Z, Bonten MJM, Bradbury CA, Brunkhorst FM, Buzgau A, Cheng AC, Detry MA, Duffy EJ, Estcourt LJ et al (2021)Interleukin-6 receptor antagonists in critically ill patients with Covid-19. N Engl J Med 384(16):1491–1502. https://doi.org/10.1056/NEJMoa2100433
The CORIMUNO-19 Collaborative group (2021) Effect of anakinra versus usual care in adults in hospital with COVID-19 and mild-to-moderate pneumonia (CORIMUNO-ANA-1): a randomised controlled trial. Lancet Respir Med 9(3):295–304. https://doi.org/10.1016/s2213-2600(20)30556-7
Hermine O, Mariette X, Tharaux PL, Resche-Rigon M, Porcher R, Ravaud P (2021) Effect of tocilizumab vs usual care in adults hospitalized with COVID-19 and moderate or severe pneumonia: a randomized clinical trial. JAMA Intern Med 181(1):32–40. https://doi.org/10.1001/jamainternmed.2020.6820
Ramiro S, Mostard RLM, Magro-Checa C, van Dongen CMP, Dormans T, Buijs J, Gronenschild M, de Kruif MD, van Haren EHJ, van Kraaij T, Leers MPG, Peeters R, Wong DR, Landewé RBM (2020) Historically controlled comparison of glucocorticoids with or without tocilizumab versus supportive care only in patients with COVID-19-associated cytokine storm syndrome: results of the CHIC study. Ann Rheum Dis 79(9):1143–1151. https://doi.org/10.1136/annrheumdis-2020-218479
Rosas IO, Bräu N, Waters M, Go RC, Hunter BD, Bhagani S, Skiest D, Aziz MS, Cooper N, Douglas IS, Savic S, Youngstein T, Del Sorbo L, Cubillo Gracian A, De La Zerda DJ, Ustianowski A, Bao M, Dimonaco S, Graham E et al (2021) Tocilizumab in hospitalized patients with severe Covid-19 pneumonia. N Engl J Med 384(16):1503–1516. https://doi.org/10.1056/NEJMoa2028700
Salama C, Han J, Yau L, Reiss WG, Kramer B, Neidhart JD, Criner GJ, Kaplan-Lewis E, Baden R, Pandit L, Cameron ML, Garcia-Diaz J, Chávez V, Mekebeb-Reuter M, Lima de Menezes F, Shah R, González-Lara MF, Assman B, Freedman J, Mohan SV (2021) Tocilizumab in patients hospitalized with Covid-19 pneumonia. N Engl J Med 384(1):20–30. https://doi.org/10.1056/NEJMoa2030340
Salvarani C, Dolci G, Massari M, Merlo DF, Cavuto S, Savoldi L, Bruzzi P, Boni F, Braglia L, Turrà C, Ballerini PF, Sciascia R, Zammarchi L, Para O, Scotton PG, Inojosa WO, Ravagnani V, Salerno ND, Sainaghi PP et al (2021) Effect of tocilizumab vs standard care on clinical worsening in patients hospitalized with COVID-19 pneumonia: a randomized clinical trial. JAMA Intern Med 181(1):24–31. https://doi.org/10.1001/jamainternmed.2020.6615
Soin AS, Kumar K, Choudhary NS, Sharma P, Mehta Y, Kataria S, Govil D, Deswal V, Chaudhry D, Singh PK, Gupta A, Agarwal V, Kumar S, Sangle SA, Chawla R, Narreddy S, Pandit R, Mishra V, Goel M, Ramanan AV (2021) Tocilizumab plus standard care versus standard care in patients in India with moderate to severe COVID-19-associated cytokine release syndrome (COVINTOC): an open-label, multicentre, randomised, controlled, phase 3 trial. Lancet Respir Med. https://doi.org/10.1016/s2213-2600(21)00081-3
Stone JH, Frigault MJ, Serling-Boyd NJ, Fernandes AD, Harvey L, Foulkes AS, Horick NK, Healy BC, Shah R, Bensaci AM, Woolley AE, Nikiforow S, Lin N, Sagar M, Schrager H, Huckins DS, Axelrod M, Pincus MD, Fleisher J et al (2020) Efficacy of tocilizumab in patients hospitalized with Covid-19. N Engl J Med. https://doi.org/10.1056/NEJMoa2028836
Huang E, Jordan SC (2020) Tocilizumab for Covid-19 - the ongoing search for effective therapies. N Engl J Med 383(24):2387–2388. https://doi.org/10.1056/NEJMe2032071
Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, Hohmann E, Chu HY, Luetkemeyer A, Kline S, Lopez de Castilla D, Finberg RW, Dierberg K, Tapson V, Hsieh L, Patterson TF, Paredes R, Sweeney DA, Short WR et al (2020) Remdesivir for the treatment of Covid-19 - final report. N Engl J Med 383(19):1813–1826. https://doi.org/10.1056/NEJMoa2007764
Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, Staplin N, Brightling C, Ustianowski A, Elmahi E, Prudon B, Green C, Felton T, Chadwick D, Rege K, Fegan C, Chappell LC, Faust SN, Jaki T et al (2021) Dexamethasone in hospitalized patients with Covid-19. N Engl J Med 384(8):693–704. https://doi.org/10.1056/NEJMoa2021436
Kim PS, Read SW, Fauci AS (2020) Therapy for early COVID-19: a critical need. JAMA 324(21):2149–2150. https://doi.org/10.1001/jama.2020.22813
Cain DW, Cidlowski JA (2017) Immune regulation by glucocorticoids. Nat Rev Immunol 17(4):233–247. https://doi.org/10.1038/nri.2017.1
Tang X, Feng YM, Ni JX, Zhang JY, Liu LM, Hu K, Wu XZ, Zhang JX, Chen JW, Zhang JC, Su J, Li YL, Zhao Y, Xie J, Ding Z, He XL, Wang W, Jin RH, Shi HZ, Sun B (2021) Early use of corticosteroid may prolong SARS-CoV-2 shedding in non-intensive care unit patients with COVID-19 pneumonia: a multicenter, single-blind, randomized control trial. Respiration 100(2):116–126. https://doi.org/10.1159/000512063
Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB (2020) Pharmacologic treatments for coronavirus disease 2019 (COVID-19): a review. JAMA 323(18):1824–1836. https://doi.org/10.1001/jama.2020.6019
World Health Organisation (2021)COVID-19 vaccines: resolving deployment challenges. Bull World Health Organ 99(3):174–175. https://doi.org/10.2471/blt.21.020321
Troiano G, Nardi A (2021) Vaccine hesitancy in the era of COVID-19. Public Health 194:245–251. https://doi.org/10.1016/j.puhe.2021.02.025
Paris C, Bénézit F, Geslin M, Polard E, Baldeyrou M, Turmel V, Tadié É, Garlantezec R, Tattevin P (2021)COVID-19 vaccine hesitancy among healthcare workers. Infect Dis Now. https://doi.org/10.1016/j.idnow.2021.04.001
Dzieciolowska S, Hamel D, Gadio S, Dionne M, Gagnon D, Robitaille L, Cook E, Caron I, Talib A, Parkes L, Dubé È, Longtin Y (2021)Covid-19 vaccine acceptance, hesitancy, and refusal among Canadian healthcare workers: a multicenter survey. Am J Infect Control. https://doi.org/10.1016/j.ajic.2021.04.079
Phillips N (2021) The coronavirus is here to stay - here's what that means. Nature 590(7846):382–384. https://doi.org/10.1038/d41586-021-00396-2
Drury AN, Szent-Gyorgyi A (1929) The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J Physiol 68(3):213–237. https://doi.org/10.1113/jphysiol.1929.sp002608
Lohmann K (1929) Über die Pyrophosphatfraktion im Muskel. Naturwissenschaften 17(31):624–625. https://doi.org/10.1007/BF01506215
Engelhardt WA, Ljubimowa MN (1939) Myosine and adenosinetriphosphatase. Nature 144(3650):668–669. https://doi.org/10.1038/144668b0
Lipmann F (1940) A phosphorylated oxygenation product of pyruvic acid. J Biol Chem 134:463–464
Lipmann F (1944) Enzymatic synthesis of acetyl phosphate. J Biol Chem 155:55–70
Lipmann F, Jones ME, Black S, Flynn RM (1953) The mechanism of the ATP-CoA-acetate reaction. J Cell Physiol Suppl 41(Suppl 1):109–112
Feldberg W, Hebb C (1948) The stimulating action of phosphate compounds on the perfused superior cervical ganglion of the cat. J Physiol 107(2):210–221
Holton P (1959) The liberation of adenosine triphosphate on antidromic stimulation of sensory nerves. J Physiol 145(3):494–504
Burnstock G (1972) Purinergic nerves. Pharmacol Rev 24(3):509–581
Burnstock G (2012) Purinergic signalling: its unpopular beginning, its acceptance and its exciting future. Bioessays 34(3):218–225. https://doi.org/10.1002/bies.201100130
Burnstock G (2014) Purinergic signalling: from discovery to current developments. ExpPhysiol 99(1):16–34
Di Virgilio F (1995) The P2Z purinoceptor: an intriguing role in immunity, inflammation and cell death. Immunol Today 16(11):524–528. https://doi.org/10.1016/0167-5699(95)80045-x
Eltzschig HK, Sitkovsky MV, Robson SC (2012) Purinergic signaling during inflammation. N Engl J Med 367(24):2322–2333
Burnstock G (2007) Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev 87(2):659–797. https://doi.org/10.1152/physrev.00043.2006
Patel AS, Reigada D, Mitchell CH, Bates SR, Margulies SS, Koval M (2005) Paracrine stimulation of surfactant secretion by extracellular ATP in response to mechanical deformation. Am J Physiol Lung Cell MolPhysiol 289(3):L489–L496
Hasan D, Blankman P, Nieman GF (2017) Purinergic signalling links mechanical breath profile and alveolar mechanics with the pro-inflammatory innate immune response causing ventilation-induced lung injury. Purinergic Signal 13(3):363–386. https://doi.org/10.1007/s11302-017-9564-5
Hasan D, Satalin J, van der Zee P, Kollisch-Singule M, Blankman P, Shono A, Somhorst P, den Uil C, Meeder H, Kotani T, Nieman GF (2018) Excessive extracellular ATP desensitizes P2Y2 and P2X4 ATP receptors provoking surfactant impairment ending in ventilation-induced lung injury. Int J Mol Sci 19(4). https://doi.org/10.3390/ijms19041185
Takahara N, Ito S, Furuya K, Naruse K, Aso H, Kondo M, Sokabe M, Hasegawa Y (2014)Real-time imaging of ATP release induced by mechanical stretch in human airway smooth muscle cells. Am J Respir Cell Mol Biol 51(6):772–782. https://doi.org/10.1165/rcmb.2014-0008OC
Furuya K, Tan JJ, Boudreault F, Sokabe M, Berthiaume Y, Grygorczyk R (2016)Real-time imaging of inflation-induced ATP release in the ex-vivo rat lung. Am J Phys Lung Cell Mol Phys. https://doi.org/10.1152/ajplung.00425.2015
Bodin P, Burnstock G (1995) Synergistic effect of acute hypoxia on flow-induced release of ATP from cultured endothelial cells. Experientia 51(3):256–259. https://doi.org/10.1007/bf01931108
Seminario-Vidal L, Kreda S, Jones L, O'Neal W, Trejo J, Boucher RC, Lazarowski ER (2009) Thrombin promotes release of ATP from lung epithelial cells through coordinated activation of rho- and Ca2+-dependent signaling pathways. J Biol Chem 284(31):20638–20648. https://doi.org/10.1074/jbc.M109.004762
Guzman-Aranguez A, Perez de Lara MJ, Pintor J (2017) Hyperosmotic stress induces ATP release and changes in P2X7 receptor levels in human corneal and conjunctival epithelial cells. Purinergic Signal 13(2):249–258. https://doi.org/10.1007/s11302-017-9556-5
Nandigama R, Padmasekar M, Wartenberg M, Sauer H (2006) Feed forward cycle of hypotonic stress-induced ATP release, purinergic receptor activation, and growth stimulation of prostate cancer cells. J Biol Chem 281(9):5686–5693. https://doi.org/10.1074/jbc.M510452200
Cheung-Flynn J, Alvis BD, Hocking KM, Guth CM, Luo W, McCallister R, Chadalavada K, Polcz M, Komalavilas P, Brophy CM (2019) Normal saline solutions cause endothelial dysfunction through loss of membrane integrity, ATP release, and inflammatory responses mediated by P2X7R/p38 MAPK/MK2 signaling pathways. PLoS One 14(8):e0220893. https://doi.org/10.1371/journal.pone.0220893
Bodin P, Burnstock G (1998) Increased release of ATP from endothelial cells during acute inflammation. Inflamm Res 47(8):351–354. https://doi.org/10.1007/s000110050341
Okada SF, Ribeiro CM, Sesma JI, Seminario-Vidal L, Abdullah LH, van Heusden C, Lazarowski ER, Boucher RC (2013) Inflammation promotes airway epithelial ATP release via calcium-dependent vesicular pathways. Am J Respir Cell Mol Biol 49(5):814–820. https://doi.org/10.1165/rcmb.2012-0493OC
Kim KC, Zheng QX, Van-Seuningen I (1993) Involvement of a signal transduction mechanism in ATP-induced mucin release from cultured airway goblet cells. Am J Respir Cell Mol Biol 8(2):121–125. https://doi.org/10.1165/ajrcmb/8.2.121
Tozzi M, Larsen AT, Lange SC, Giannuzzo A, Andersen MN, Novak I (2018) The P2X7 receptor and pannexin-1 are involved in glucose-induced autocrine regulation in beta-cells. Sci Rep 8(1):8926. https://doi.org/10.1038/s41598-018-27281-9
Jacques-Silva MC, Correa-Medina M, Cabrera O, Rodriguez-Diaz R, Makeeva N, Fachado A, Diez J, Berman DM, Kenyon NS, Ricordi C, Pileggi A, Molano RD, Berggren PO, Caicedo A (2010)ATP-gated P2X3 receptors constitute a positive autocrine signal for insulin release in the human pancreatic beta cell. Proc Natl Acad Sci U S A 107(14):6465–6470. https://doi.org/10.1073/pnas.0908935107
Okura D, Horishita T, Ueno S, Yanagihara N, Sudo Y, Uezono Y, Minami T, Kawasaki T, Sata T (2015) Lidocaine preferentially inhibits the function of purinergic P2X7 receptors expressed in Xenopus oocytes. Anesth Analg 120(3):597–605. https://doi.org/10.1213/ane.0000000000000585
Adinolfi E, Callegari MG, Ferrari D, Bolognesi C, Minelli M, Wieckowski MR, Pinton P, Rizzuto R, Di Virgilio F (2005) Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol Biol Cell 16(7):3260–3272. https://doi.org/10.1091/mbc.e04-11-1025
Ghazi K, Deng-Pichon U, Warnet JM, Rat P (2012) Hyaluronan fragments improve wound healing on in vitro cutaneous model through P2X7 purinoreceptor basal activation: role of molecular weight. PLoS One 7(11):e48351. https://doi.org/10.1371/journal.pone.0048351
Adinolfi E, Callegari MG, Cirillo M, Pinton P, Giorgi C, Cavagna D, Rizzuto R, Di Virgilio F (2009) Expression of the P2X7 receptor increases the Ca2+ content of the endoplasmic reticulum, activates NFATc1, and protects from apoptosis. J Biol Chem 284(15):10120–10128. https://doi.org/10.1074/jbc.M805805200
Amoroso F, Falzoni S, Adinolfi E, Ferrari D, Di Virgilio F (2012) The P2X7 receptor is a key modulator of aerobic glycolysis. Cell Death Dis 3:e370. https://doi.org/10.1038/cddis.2012.105
Cronstein BN, Daguma L, Nichols D, Hutchison AJ, Williams M (1990) The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J Clin Invest 85(4):1150–1157. https://doi.org/10.1172/jci114547
Rose FR, Hirschhorn R, Weissmann G, Cronstein BN (1988) Adenosine promotes neutrophil chemotaxis. J Exp Med 167(3):1186–1194
Felsch A, Stocker K, Borchard U (1995) Phorbol ester-stimulated adherence of neutrophils to endothelial cells is reduced by adenosine A2 receptor agonists. J Immunol 155(1):333–338
Thiel M, Chouker A (1995) Acting via A2 receptors, adenosine inhibits the production of tumor necrosis factor-alpha of endotoxin-stimulated human polymorphonuclear leukocytes. J Lab Clin Med 126(3):275–282
Salmon JE, Cronstein BN (1990) Fc gamma receptor-mediated functions in neutrophils are modulated by adenosine receptor occupancy. A1 receptors are stimulatory and A2 receptors are inhibitory. J Immunol 145(7):2235–2240
Zalavary S, Stendahl O, Bengtsson T (1994) The role of cyclic AMP, calcium and filamentous actin in adenosine modulation of Fc receptor-mediated phagocytosis in human neutrophils. Biochim Biophys Acta 1222(2):249–256
Zalavary S, Bengtsson T (1998) Adenosine inhibits actin dynamics in human neutrophils: evidence for the involvement of cAMP. Eur J Cell Biol 75(2):128–139. https://doi.org/10.1016/s0171-9335(98)80055-1
Xu X, Zheng S, Xiong Y, Wang X, Qin W, Zhang H, Sun B (2017) Adenosine effectively restores endotoxin-induced inhibition of human neutrophil chemotaxis via A1 receptor-p38 pathway. Inflamm Res 66(4):353–364. https://doi.org/10.1007/s00011-016-1021-3
Chen L, Fredholm BB, Jondal M (2008) Adenosine, through the A1 receptor, inhibits vesicular MHC class I cross-presentation by resting DC. Mol Immunol 45(8):2247–2254. https://doi.org/10.1016/j.molimm.2007.11.016
Schnurr M, Toy T, Shin A, Hartmann G, Rothenfusser S, Soellner J, Davis ID, Cebon J, Maraskovsky E (2004) Role of adenosine receptors in regulating chemotaxis and cytokine production of plasmacytoid dendritic cells. Blood 103(4):1391–1397. https://doi.org/10.1182/blood-2003-06-1959
Figueiro F, Muller L, Funk S, Jackson EK, Battastini AM, Whiteside TL (2016) Phenotypic and functional characteristics of CD39high human regulatory B cells (Breg). Oncoimmunology 5(2):e1082703. https://doi.org/10.1080/2162402x.2015.1082703
Ohtsuka T, Changelian PS, Bouis D, Noon K, Harada H, Lama VN, Pinsky DJ (2010)Ecto-5'-nucleotidase(CD73) attenuates allograft airway rejection through adenosine 2A receptor stimulation. J Immunol 185(2):1321–1329. https://doi.org/10.4049/jimmunol.0901847
Link AA, Kino T, Worth JA, McGuire JL, Crane ML, Chrousos GP, Wilder RL, Elenkov IJ (2000)Ligand-activation of the adenosine A2a receptors inhibits IL-12 production by human monocytes. J Immunol 164(1):436–442
Zhang JG, Hepburn L, Cruz G, Borman RA, Clark KL (2005) The role of adenosine A2A and A2B receptors in the regulation of TNF-alpha production by human monocytes. Biochem Pharmacol 69(6):883–889. https://doi.org/10.1016/j.bcp.2004.12.008
Thiel M, Chambers JD, Chouker A, Fischer S, Zourelidis C, Bardenheuer HJ, Arfors KE, Peter K (1996) Effect of adenosine on the expression of beta(2) integrins and L-selectin of human polymorphonuclear leukocytes in vitro. J Leukoc Biol 59(5):671–682
Wollner A, Wollner S, Smith JB (1993) Acting via A2 receptors, adenosine inhibits the upregulation of Mac-1 (Cd11b/CD18) expression on FMLP-stimulated neutrophils. Am J Respir Cell Mol Biol 9(2):179–185. https://doi.org/10.1165/ajrcmb/9.2.179
Cadieux JS, Leclerc P, St-Onge M, Dussault AA, Laflamme C, Picard S, Ledent C, Borgeat P, Pouliot M (2005) Potentiation of neutrophil cyclooxygenase-2 by adenosine: an early anti-inflammatory signal. J Cell Sci 118(Pt 7):1437–1447. https://doi.org/10.1242/jcs.01737
Sullivan GW, Linden J, Buster BL, Scheld WM (1999) Neutrophil A2A adenosine receptor inhibits inflammation in a rat model of meningitis: synergy with the type IV phosphodiesterase inhibitor, rolipram. J Infect Dis 180(5):1550–1560. https://doi.org/10.1086/315084
McColl SR, St-Onge M, Dussault AA, Laflamme C, Bouchard L, Boulanger J, Pouliot M (2006) Immunomodulatory impact of the A2A adenosine receptor on the profile of chemokines produced by neutrophils. FASEB J 20(1):187–189. https://doi.org/10.1096/fj.05-4804fje
Krump E, Lemay G, Borgeat P (1996) Adenosine A2 receptor-induced inhibition of leukotriene B4 synthesis in whole blood ex vivo. Br J Pharmacol 117(8):1639–1644
Krump E, Picard S, Mancini J, Borgeat P (1997) Suppression of leukotriene B4 biosynthesis by endogenous adenosine in ligand-activated human neutrophils. J Exp Med 186(8):1401–1406
Surette ME, Krump E, Picard S, Borgeat P (1999) Activation of leukotriene synthesis in human neutrophils by exogenous arachidonic acid: inhibition by adenosine A(2a) receptor agonists and crucial role of autocrine activation by leukotriene B(4). Mol Pharmacol 56(5):1055–1062
Flamand N, Boudreault S, Picard S, Austin M, Surette ME, Plante H, Krump E, Vallee MJ, Gilbert C, Naccache P, Laviolette M, Borgeat P (2000) Adenosine, a potent natural suppressor of arachidonic acid release and leukotriene biosynthesis in human neutrophils. Am J Respir Crit Care Med 161(2 Pt 2):S88–S94. https://doi.org/10.1164/ajrccm.161.supplement_1.ltta-18
Flamand N, Surette ME, Picard S, Bourgoin S, Borgeat P (2002) Cyclic AMP-mediated inhibition of 5-lipoxygenase translocation and leukotriene biosynthesis in human neutrophils. Mol Pharmacol 62(2):250–256
Sullivan GW, Rieger JM, Scheld WM, Macdonald TL, Linden J (2001) Cyclic AMP-dependent inhibition of human neutrophil oxidative activity by substituted 2-propynylcyclohexyl adenosine A(2A) receptor agonists. Br J Pharmacol 132(5):1017–1026. https://doi.org/10.1038/sj.bjp.0703893
Anderson R, Visser SS, Ramafi G, Theron AJ (2000) Accelerated resequestration of cytosolic calcium and suppression of the pro-inflammatory activities of human neutrophils by CGS 21680 in vitro. Br J Pharmacol 130(4):717–724. https://doi.org/10.1038/sj.bjp.0703344
Richter J (1992) Effect of adenosine analogues and cAMP-raising agents on TNF-, GM-CSF-, and chemotactic peptide-induced degranulation in single adherent neutrophils. J Leukoc Biol 51(3):270–275
Visser SS, Theron AJ, Ramafi G, Ker JA, Anderson R (2000) Apparent involvement of the A(2A) subtype adenosine receptor in the anti-inflammatory interactions of CGS 21680, cyclopentyladenosine, and IB-MECA with human neutrophils. Biochem Pharmacol 60(7):993–999
Walker BA, Rocchini C, Boone RH, Ip S, Jacobson MA (1997) Adenosine A2a receptor activation delays apoptosis in human neutrophils. J Immunol 158(6):2926–2931
Liu YW, Yang T, Zhao L, Ni Z, Yang N, He F, Dai SS (2016) Activation of adenosine 2A receptor inhibits neutrophil apoptosis in an autophagy-dependent manner in mice with systemic inflammatory response syndrome. Sci Rep 6:33614. https://doi.org/10.1038/srep33614
Ryzhov S, Goldstein AE, Matafonov A, Zeng D, Biaggioni I, Feoktistov I (2004)Adenosine-activated mast cells induce IgE synthesis by B lymphocytes: an A2B-mediated process involving Th2 cytokines IL-4 and IL-13 with implications for asthma. J Immunol 172(12):7726–7733
Kreckler LM, Gizewski E, Wan TC, Auchampach JA (2009) Adenosine suppresses lipopolysaccharide-induced tumor necrosis factor-alpha production by murine macrophages through a protein kinase A- and exchange protein activated by cAMP-independent signaling pathway. J Pharmacol Exp Ther 331(3):1051–1061. https://doi.org/10.1124/jpet.109.157651
Hassanian SM, Dinarvand P, Rezaie AR (2014) Adenosine regulates the proinflammatory signaling function of thrombin in endothelial cells. J Cell Physiol 229(9):1292–1300. https://doi.org/10.1002/jcp.24568
Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, Drake CG, Powell JD (2008) A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 111 (1):251-259. https://doi.org/10.1182/blood-2007-03-081646
Csoka B, Himer L, Selmeczy Z, Vizi ES, Pacher P, Ledent C, Deitch EA, Spolarics Z, Nemeth ZH, Hasko G (2008) Adenosine A2A receptor activation inhibits T helper 1 and T helper 2 cell development and effector function. FASEB J 22(10):3491–3499. https://doi.org/10.1096/fj.08-107458
Alam MS, Kurtz CC, Wilson JM, Burnette BR, Wiznerowicz EB, Ross WG, Rieger JM, Figler RA, Linden J, Crowe SE, Ernst PB (2009) A2A adenosine receptor (AR) activation inhibits pro-inflammatory cytokine production by human CD4+ helper T cells and regulates Helicobacter-induced gastritis and bacterial persistence. Mucosal Immunol 2(3):232–242. https://doi.org/10.1038/mi.2009.4
Hasko G, Kuhel DG, Chen JF, Schwarzschild MA, Deitch EA, Mabley JG, Marton A, Szabo C (2000) Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. FASEB J 14(13):2065–2074. https://doi.org/10.1096/fj.99-0508com
Erdmann AA, Gao ZG, Jung U, Foley J, Borenstein T, Jacobson KA, Fowler DH (2005) Activation of Th1 and Tc1 cell adenosine A2A receptors directly inhibits IL-2 secretion in vitro and IL-2-driven expansion in vivo. Blood 105(12):4707–4714. https://doi.org/10.1182/blood-2004-04-1407
Lappas CM, Rieger JM, Linden J (2005) A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J Immunol 174(2):1073–1080
Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M (2012) The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol 3:190. https://doi.org/10.3389/fimmu.2012.00190
Huang X, He Y, Chen Y, Wu P, Gui D, Cai H, Chen A, Chen M, Dai C, Yao D, Wang L (2016) Baicalin attenuates bleomycin-induced pulmonary fibrosis via adenosine A2a receptor related TGF-beta1-inducedERK1/2 signaling pathway. BMC Pulm Med 16(1):132. https://doi.org/10.1186/s12890-016-0294-1
Guillén-Gómez E, Silva I, Serra N, Caballero F, Leal J, Breda A, San Martín R, Pastor-Anglada M, Ballarín JA, Guirado L, Díaz-Encarnación MM (2020) From inflammation to the onset of fibrosis through A(2A) receptors in kidneys from deceased donors. Int J Mol Sci 21(22). https://doi.org/10.3390/ijms21228826
Shi L, Feng M, Du S, Wei X, Song H, Yixin X, Song J, Wenxian G (2019) Adenosine generated by regulatory T cells induces CD8(+) T cell exhaustion in gastric cancer through A2aR pathway. Biomed Res Int 2019:4093214. https://doi.org/10.1155/2019/4093214
Barbera-Cremades M, Baroja-Mazo A, Pelegrin P (2016) Purinergic signaling during macrophage differentiation results in M2 alternative activated macrophages. J Leukoc Biol 99(2):289–299. https://doi.org/10.1189/jlb.1A0514-267RR
Csoka B, Selmeczy Z, Koscso B, Nemeth ZH, Pacher P, Murray PJ, Kepka-Lenhart D, Morris SM Jr, Gause WC, Leibovich SJ, Hasko G (2012) Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J 26(1):376–386. https://doi.org/10.1096/fj.11-190934
Ferrante CJ, Pinhal-Enfield G, Elson G, Cronstein BN, Hasko G, Outram S, Leibovich SJ (2013) The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Ralpha) signaling. Inflammation 36(4):921–931. https://doi.org/10.1007/s10753-013-9621-3
Koscso B, Csoka B, Kokai E, Nemeth ZH, Pacher P, Virag L, Leibovich SJ, Hasko G (2013) Adenosine augments IL-10-induced STAT3 signaling in M2c macrophages. J Leukoc Biol 94(6):1309–1315. https://doi.org/10.1189/jlb.0113043
Hasko G, Pacher P (2012) Regulation of macrophage function by adenosine. Arterioscler Thromb Vasc Biol 32(4):865–869. https://doi.org/10.1161/atvbaha.111.226852
Koroskenyi K, Kiss B, Szondy Z (2016) Adenosine A2A receptor signaling attenuates LPS-induced pro-inflammatory cytokine formation of mouse macrophages by inducing the expression of DUSP1. Biochim Biophys Acta 1863(7 Pt A):1461–1471. https://doi.org/10.1016/j.bbamcr.2016.04.003
Majumdar S, Aggarwal BB (2003) Adenosine suppresses activation of nuclear factor-kappaB selectively induced by tumor necrosis factor in different cell types. Oncogene 22(8):1206–1218. https://doi.org/10.1038/sj.onc.1206184
Mirakaj V, Thix CA, Laucher S, Mielke C, Morote-Garcia JC, Schmit MA, Henes J, Unertl KE, Kohler D, Rosenberger P (2010)Netrin-1 dampens pulmonary inflammation during acute lung injury. Am J Respir Crit Care Med 181(8):815–824. https://doi.org/10.1164/rccm.200905-0717OC
Aherne CM, Collins CB, Masterson JC, Tizzano M, Boyle TA, Westrich JA, Parnes JA, Furuta GT, Rivera-Nieves J, Eltzschig HK (2012) Neuronal guidance molecule netrin-1 attenuates inflammatory cell trafficking during acute experimental colitis. Gut 61(5):695–705. https://doi.org/10.1136/gutjnl-2011-300012
van der Hoeven D, Wan TC, Gizewski ET, Kreckler LM, Maas JE, Van Orman J, Ravid K, Auchampach JA (2011) A role for the low-affinity A2B adenosine receptor in regulating superoxide generation by murine neutrophils. J Pharmacol Exp Ther 338(3):1004–1012. https://doi.org/10.1124/jpet.111.181792
Nemeth ZH, Lutz CS, Csoka B, Deitch EA, Leibovich SJ, Gause WC, Tone M, Pacher P, Vizi ES, Hasko G (2005) Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J Immunol 175(12):8260–8270
Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, Blackburn MR, Biaggioni I, Carbone DP, Feoktistov I, Dikov MM (2008) Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 112(5):1822–1831. https://doi.org/10.1182/blood-2008-02-136325
Wilson JM, Kurtz CC, Black SG, Ross WG, Alam MS, Linden J, Ernst PB (2011) The A2B adenosine receptor promotes Th17 differentiation via stimulation of dendritic cell IL-6. J Immunol 186(12):6746–6752. https://doi.org/10.4049/jimmunol.1100117
Liang D, Zuo A, Shao H, Chen M, Kaplan HJ, Sun D (2015) A2B adenosine receptor activation switches differentiation of bone marrow cells to a CD11c(+)Gr-1(+) dendritic cell subset that promotes the Th17 response. Immun Inflamm Dis 3(4):360–373. https://doi.org/10.1002/iid3.74
Karmouty-Quintana H, Philip K, Acero LF, Chen NY, Weng T, Molina JG, Luo F, Davies J, Le NB, Bunge I, Volcik KA, Le TT, Johnston RA, Xia Y, Eltzschig HK, Blackburn MR (2015) Deletion of ADORA2B from myeloid cells dampens lung fibrosis and pulmonary hypertension. FASEB J 29(1):50–60. https://doi.org/10.1096/fj.14-260182
Eckle T, Grenz A, Laucher S, Eltzschig HK (2008) A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest 118(10):3301–3315
Grant MB, Tarnuzzer RW, Caballero S, Ozeck MJ, Davis MI, Spoerri PE, Feoktistov I, Biaggioni I, Shryock JC, Belardinelli L (1999) Adenosine receptor activation induces vascular endothelial growth factor in human retinal endothelial cells. Circ Res 85(8):699–706
Zhong H, Wu Y, Belardinelli L, Zeng D (2006) A2B adenosine receptors induce IL-19 from bronchial epithelial cells, resulting in TNF-alpha increase. Am J Respir Cell Mol Biol 35(5):587–592. https://doi.org/10.1165/rcmb.2005-0476OC
Wilkinson PF, Farrell FX, Morel D, Law W, Murphy S (2016) Adenosine signaling increases proinflammatory and profibrotic mediators through activation of a functional adenosine 2B receptor in renal fibroblasts. Ann Clin Lab Sci 46(4):339–345
Zhou Y, Murthy JN, Zeng D, Belardinelli L, Blackburn MR (2010) Alterations in adenosine metabolism and signaling in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. PLoS One 5(2):e9224. https://doi.org/10.1371/journal.pone.0009224
Huang L, Fan J, Chen YX, Wang JH (2020) Inhibition of A(2B) Adenosine receptor attenuates intestinal injury in a rat model of necrotizing enterocolitis. Mediat Inflamm 2020:1562973. https://doi.org/10.1155/2020/1562973
Reyes AWB, Vu SH, Huy TXN, Min W, Lee HJ, Chang HH, Lee JH, Kim S (2020) Adenosine receptor Adora2b antagonism attenuates Brucella abortus 544 infection in professional phagocyte RAW 264.7 cells and BALB/c mice. Vet Microbiol 242:108586. https://doi.org/10.1016/j.vetmic.2020.108586
Feoktistov I, Ryzhov S, Goldstein AE, Biaggioni I (2003) Mast cell-mediated stimulation of angiogenesis: cooperative interaction between A2B and A3 adenosine receptors. Circ Res 92(5):485–492. https://doi.org/10.1161/01.res.0000061572.10929.2d
Joos G, Jakim J, Kiss B, Szamosi R, Papp T, Felszeghy S, Saghy T, Nagy G, Szondy Z (2017) Involvement of adenosine A3 receptors in the chemotactic navigation of macrophages towards apoptotic cells. Immunol Lett 183:62–72. https://doi.org/10.1016/j.imlet.2017.02.002
Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, Junger WG (2006) ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 314(5806):1792–1795
Inoue Y, Chen Y, Hirsh MI, Yip L, Junger WG (2008) A3 and P2Y2 receptors control the recruitment of neutrophils to the lungs in a mouse model of sepsis. Shock 30(2):173–177. https://doi.org/10.1097/shk.0b013e318160dad4
Tweedy L, Knecht DA, Mackay GM, Insall RH (2016)Self-generated chemoattractant gradients: attractant depletion extends the range and robustness of chemotaxis. PLoS Biol 14(3):e1002404. https://doi.org/10.1371/journal.pbio.1002404
Tweedy L, Susanto O, Insall RH (2016)Self-generated chemotactic gradients-cells steering themselves. Curr Opin Cell Biol 42:46–51. https://doi.org/10.1016/j.ceb.2016.04.003
Dona E, Barry JD, Valentin G, Quirin C, Khmelinskii A, Kunze A, Durdu S, Newton LR, Fernandez-Minan A, Huber W, Knop M, Gilmour D (2013) Directional tissue migration through a self-generated chemokine gradient. Nature 503(7475):285–289. https://doi.org/10.1038/nature12635
So H (2016) Where to go: breaking the symmetry in cell motility. PLoS Biol 14(5):e1002463. https://doi.org/10.1371/journal.pbio.1002463
Moissoglu K, Majumdar R, Parent CA (2014) Cell migration: sinking in a gradient. Curr Biol 24(1):R23–R25. https://doi.org/10.1016/j.cub.2013.10.075
Lee JY, Jhun BS, Oh YT, Lee JH, Choe W, Baik HH, Ha J, Yoon KS, Kim SS, Kang I (2006) Activation of adenosine A3 receptor suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of PI 3-kinase/Akt and NF-kappaB activation in murine BV2 microglial cells. Neurosci Lett 396(1):1–6. https://doi.org/10.1016/j.neulet.2005.11.004
Ren T, Qiu Y, Wu W, Feng X, Ye S, Wang Z, Tian T, He Y, Yu C, Zhou Y (2014) Activation of adenosine A3 receptor alleviates TNF-alpha-induced inflammation through inhibition of the NF-kappaB signaling pathway in human colonic epithelial cells. Mediat Inflamm 2014:818251. https://doi.org/10.1155/2014/818251
Hoskin DW, Butler JJ, Drapeau D, Haeryfar SM, Blay J (2002) Adenosine acts through an A3 receptor to prevent the induction of murine anti-CD3-activated killer T cells. Int J Cancer 99(3):386–395. https://doi.org/10.1002/ijc.10325
Ferreira-Silva J, Aires ID, Boia R, Ambrósio AF, Santiago AR (2020) Activation of adenosine A(3) receptor inhibits microglia reactivity elicited by elevated pressure. Int J Mol Sci 21(19). https://doi.org/10.3390/ijms21197218
Morschl E, Molina JG, Volmer JB, Mohsenin A, Pero RS, Hong JS, Kheradmand F, Lee JJ, Blackburn MR (2008) A3 adenosine receptor signaling influences pulmonary inflammation and fibrosis. Am J Respir Cell Mol Biol 39(6):697–705. https://doi.org/10.1165/rcmb.2007-0419OC
Ren TH, Lv MM, An XM, Leung WK, Seto WK (2020) Activation of adenosine A3 receptor inhibits inflammatory cytokine production in colonic mucosa of patients with ulcerative colitis by down-regulating the nuclear factor-kappa B signaling. J Dig Dis 21(1):38–45. https://doi.org/10.1111/1751-2980.12831
Oury C, Lecut C, Hego A, Wera O, Delierneux C (2015) Purinergic control of inflammation and thrombosis: role of P2X1 receptors. Comput Struct Biotechnol J 13:106–110. https://doi.org/10.1016/j.csbj.2014.11.008
Alarcón P, Manosalva C, Quiroga J, Belmar I, Álvarez K, Díaz G, Taubert A, Hermosilla C, Carretta MD, Burgos RA, Hidalgo MA (2020) Oleic and linoleic acids induce the release of neutrophil extracellular traps via pannexin 1-dependent ATP release and P2X1 receptor activation. Front Vet Sci 7:260. https://doi.org/10.3389/fvets.2020.00260
Woehrle T, Yip L, Elkhal A, Sumi Y, Chen Y, Yao Y, Insel PA, Junger WG (2010)Pannexin-1 hemichannel-mediated ATP release together with P2X1 and P2X4 receptors regulate T-cell activation at the immune synapse. Blood 116(18):3475–3484. https://doi.org/10.1182/blood-2010-04-277707
Bulanova E, Budagian V, Orinska Z, Koch-Nolte F, Haag F, Bulfone-Paus S (2009) ATP induces P2X7 receptor-independent cytokine and chemokine expression through P2X1 and P2X3 receptors in murine mast cells (Article retracted in 2011 due to figure irregularities). J Leukoc Biol 85(4):692–702. https://doi.org/10.1189/jlb.0808470
Manohar M, Hirsh MI, Chen Y, Woehrle T, Karande AA, Junger WG (2012) ATP release and autocrine signaling through P2X4 receptors regulate gammadelta T cell activation. J Leukoc Biol 92(4):787–794. https://doi.org/10.1189/jlb.0312121
Vazquez-Villoldo N, Domercq M, Martin A, Llop J, Gomez-Vallejo V, Matute C (2014) P2X4 receptors control the fate and survival of activated microglia. Glia 62(2):171–184. https://doi.org/10.1002/glia.22596
Ledderose C, Liu K, Kondo Y, Slubowski CJ, Dertnig T, Denicoló S, Arbab M, Hubner J, Konrad K, Fakhari M, Lederer JA, Robson SC, Visner GA, Junger WG (2018) Purinergic P2X4 receptors and mitochondrial ATP production regulate T cell migration. J Clin Invest 128(8):3583–3594. https://doi.org/10.1172/jci120972
Nguyen HM, di Lucente J, Chen YJ, Cui Y, Ibrahim RH, Pennington MW, Jin LW, Maezawa I, Wulff H (2020) Biophysical basis for Kv1.3 regulation of membrane potential changes induced by P2X4-mediated calcium entry in microglia. Glia 68(11):2377–2394. https://doi.org/10.1002/glia.23847
Ledderose C, Bromberger S, Slubowski CJ, Sueyoshi K, Junger WG (2020) Frontline Science: P2Y11 receptors support T cell activation by directing mitochondrial trafficking to the immune synapse. J Leukoc Biol. https://doi.org/10.1002/jlb.2hi0520-191r
Ledderose C, Bromberger S, Slubowski CJ, Sueyoshi K, Aytan D, Shen Y, Junger WG (2020) The purinergic receptor P2Y11 choreographs the polarization, mitochondrial metabolism, and migration of T lymphocytes. Sci Signal 13(651). https://doi.org/10.1126/scisignal.aba3300
Cekic C, Linden J (2016) Purinergic regulation of the immune system. Nat Rev Immunol 16(3):177–192
Lee BH, Hwang DM, Palaniyar N, Grinstein S, Philpott DJ, Hu J (2012) Activation of P2X(7) receptor by ATP plays an important role in regulating inflammatory responses during acute viral infection. PLoS One 7(4):e35812. https://doi.org/10.1371/journal.pone.0035812
Jo EK, Kim JK, Shin DM, Sasakawa C (2016) Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol 13(2):148–159
Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13(6):397–411. https://doi.org/10.1038/nri3452
Sakaki H, Fujiwaki T, Tsukimoto M, Kawano A, Harada H, Kojima S (2013) P2X4 receptor regulates P2X7 receptor-dependent IL-1beta and IL-18 release in mouse bone marrow-derived dendritic cells. Biochem Biophys Res Commun 432(3):406–411. https://doi.org/10.1016/j.bbrc.2013.01.135
Luna-Gomes T, Santana PT, Coutinho-Silva R (2015)Silica-induced inflammasome activation in macrophages: role of ATP and P2X7 receptor. Immunobiology 220(9):1101–1106. https://doi.org/10.1016/j.imbio.2015.05.004
Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E (2016) Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1beta secretion in response to ATP. Nat Commun 7:10555. https://doi.org/10.1038/ncomms10555
Eleftheriadis T, Pissas G, Karioti A, Antoniadi G, Golfinopoulos S, Liakopoulos V, Mamara A, Speletas M, Koukoulis G, Stefanidis I (2013) Uric acid induces caspase-1 activation, IL-1beta secretion and P2X7 receptor dependent proliferation in primary human lymphocytes. Hippokratia 17(2):141–145
Jeong YH, Walsh MC, Yu J, Shen H, Wherry EJ, Choi Y (2020) Mice lacking the purinergic receptor P2X5 exhibit defective inflammasome activation and early susceptibility to listeria monocytogenes. J Immunol 205(3):760–766. https://doi.org/10.4049/jimmunol.1901423
Wiley JS, Gu BJ (2012) A new role for the P2X7 receptor: a scavenger receptor for bacteria and apoptotic cells in the absence of serum and extracellular ATP. Purinergic Signal 8(3):579–586. https://doi.org/10.1007/s11302-012-9308-5
Gu BJ, Saunders BM, Petrou S, Wiley JS (2011) P2X(7) is a scavenger receptor for apoptotic cells in the absence of its ligand, extracellular ATP. J Immunol 187(5):2365–2375. https://doi.org/10.4049/jimmunol.1101178
Brandao-Burch A, Key ML, Patel JJ, Arnett TR, Orriss IR (2012) The P2X7 receptor is an important regulator of extracellular ATP levels. Front Endocrinol (Lausanne) 3:41. https://doi.org/10.3389/fendo.2012.00041
Gutierrez-Martin Y, Bustillo D, Gomez-Villafuertes R, Sanchez-Nogueiro J, Torregrosa-Hetland C, Binz T, Gutierrez LM, Miras-Portugal MT, Artalejo AR (2011) P2X7 receptors trigger ATP exocytosis and modify secretory vesicle dynamics in neuroblastoma cells. J Biol Chem 286(13):11370–11381. https://doi.org/10.1074/jbc.M110.139410
Suadicani SO, Brosnan CF, Scemes E (2006) P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J Neurosci 26(5):1378–1385. https://doi.org/10.1523/jneurosci.3902-05.2006
Henriquez M, Herrera-Molina R, Valdivia A, Alvarez A, Kong M, Munoz N, Eisner V, Jaimovich E, Schneider P, Quest AF, Leyton L (2011) ATP release due to Thy-1-integrin binding induces P2X7-mediated calcium entry required for focal adhesion formation. J Cell Sci 124(Pt 9):1581–1588. https://doi.org/10.1242/jcs.073171
Mishra A, Chintagari NR, Guo Y, Weng T, Su L, Liu L (2011) Purinergic P2X7 receptor regulates lung surfactant secretion in a paracrine manner. J Cell Sci 124(Pt 4):657–668
Ohshima Y, Tsukimoto M, Takenouchi T, Harada H, Suzuki A, Sato M, Kitani H, Kojima S (2010)gamma-Irradiation induces P2X(7) receptor-dependent ATP release from B16 melanoma cells. Biochim Biophys Acta 1800(1):40–46. https://doi.org/10.1016/j.bbagen.2009.10.008
Johnsen B, Kaschubowski KE, Nader S, Schneider E, Nicola JA, Fliegert R, Wolf IMA, Guse AH, Nikolaev VO, Koch-Nolte F, Haag F (2019)P2X7-mediated ATP secretion is accompanied by depletion of cytosolic ATP. Purinergic Signal 15(2):155–166. https://doi.org/10.1007/s11302-019-09654-5
Sáez PJ, Vargas P, Shoji KF, Harcha PA, Lennon-Duménil AM, Sáez JC (2017) ATP promotes the fast migration of dendritic cells through the activity of pannexin 1 channels and P2X(7) receptors. Sci Signal 10(506). https://doi.org/10.1126/scisignal.aah7107
Gilbert SM, Oliphant CJ, Hassan S, Peille AL, Bronsert P, Falzoni S, Di Virgilio F, McNulty S, Lara R (2019) ATP in the tumour microenvironment drives expression of nfP2X7, a key mediator of cancer cell survival. Oncogene 38(2):194–208. https://doi.org/10.1038/s41388-018-0426-6
Pellegatti P, Falzoni S, Donvito G, Lemaire I, Di Virgilio F (2011) P2X7 receptor drives osteoclast fusion by increasing the extracellular adenosine concentration. FASEB J 25(4):1264–1274. https://doi.org/10.1096/fj.10-169854
Schenk U, Frascoli M, Proietti M, Geffers R, Traggiai E, Buer J, Ricordi C, Westendorf AM, Grassi F (2011) ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci Signal 4(162):ra12. https://doi.org/10.1126/scisignal.2001270
Figliuolo VR, Savio LEB, Safya H, Nanini H, Bernardazzi C, Abalo A, de Souza HSP, Kanellopoulos J, Bobe P, Coutinho C, Coutinho-Silva R (2017) P2X7 receptor promotes intestinal inflammation in chemically induced colitis and triggers death of mucosal regulatory T cells. Biochim Biophys Acta Mol basis Dis 1863(6):1183–1194. https://doi.org/10.1016/j.bbadis.2017.03.004
Koo TY, Lee JG, Yan JJ, Jang JY, Ju KD, Han M, Oh KH, Ahn C, Yang J (2017) The P2X7 receptor antagonist, oxidized adenosine triphosphate, ameliorates renal ischemia-reperfusion injury by expansion of regulatory T cells. Kidney Int 92(2):415–431. https://doi.org/10.1016/j.kint.2017.01.031
Frascoli M, Marcandalli J, Schenk U, Grassi F (2012) Purinergic P2X7 receptor drives T cell lineage choice and shapes peripheral γδ cells. J Immunol 189(1):174–180. https://doi.org/10.4049/jimmunol.1101582
Romagnani A, Rottoli E, Mazza EMC, Rezzonico-Jost T, De Ponte CB, Proietti M, Perotti M, Civanelli E, Perruzza L, Catapano AL, Baragetti A, Tenedini E, Tagliafico E, Falzoni S, Di Virgilio F, Norata GD, Bicciato S, Grassi F (2020) P2X7 Receptor activity limits accumulation of T cells within tumors. Cancer Res 80(18):3906–3919. https://doi.org/10.1158/0008-5472.can-19-3807
Verhoef PA, Estacion M, Schilling W, Dubyak GR (2003) P2X7 receptor-dependent blebbing and the activation of Rho-effector kinases, caspases, and IL-1 beta release. J Immunol 170(11):5728–5738. https://doi.org/10.4049/jimmunol.170.11.5728
Pfeiffer ZA, Aga M, Prabhu U, Watters JJ, Hall DJ, Bertics PJ (2004) The nucleotide receptor P2X7 mediates actin reorganization and membrane blebbing in RAW 264.7 macrophages via p38 MAP kinase and Rho. J Leukoc Biol 75(6):1173–1182. https://doi.org/10.1189/jlb.1203648
Gu BJ, Wiley JS (2006) Rapid ATP-induced release of matrix metalloproteinase 9 is mediated by the P2X7 receptor. Blood 107(12):4946–4953. https://doi.org/10.1182/blood-2005-07-2994
de Torre-Minguela C, Barbera-Cremades M, Gomez AI, Martin-Sanchez F, Pelegrin P (2016) Macrophage activation and polarization modify P2X7 receptor secretome influencing the inflammatory process. Sci Rep 6:22586. https://doi.org/10.1038/srep22586
Csoka B, Nemeth ZH, Toro G, Idzko M, Zech A, Koscso B, Spolarics Z, Antonioli L, Cseri K, Erdelyi K, Pacher P, Hasko G (2015) Extracellular ATP protects against sepsis through macrophage P2X7 purinergic receptors by enhancing intracellular bacterial killing. FASEB J 29(9):3626–3637. https://doi.org/10.1096/fj.15-272450
Wareham KJ, Seward EP (2016) P2X7 receptors induce degranulation in human mast cells. Purinergic Signal 12(2):235–246. https://doi.org/10.1007/s11302-016-9497-4
Kawamura H, Aswad F, Minagawa M, Govindarajan S, Dennert G (2006) P2X7 receptors regulate NKT cells in autoimmune hepatitis. J Immunol 176(4):2152–2160
Pupovac A, Foster CM, Sluyter R (2013) Human P2X7 receptor activation induces the rapid shedding of CXCL16. Biochem Biophys Res Commun 432(4):626–631. https://doi.org/10.1016/j.bbrc.2013.01.134
Pupovac A, Geraghty NJ, Watson D, Sluyter R (2015) Activation of the P2X7 receptor induces the rapid shedding of CD23 from human and murine B cells. Immunol Cell Biol 93(1):77–85. https://doi.org/10.1038/icb.2014.69
Huang SW, Walker C, Pennock J, Else K, Muller W, Daniels MJ, Pellegrini C, Brough D, Lopez-Castejon G, Cruickshank SM (2017) P2X7 receptor-dependent tuning of gut epithelial responses to infection. Immunol Cell Biol 95(2):178–188. https://doi.org/10.1038/icb.2016.75
Yip L, Woehrle T, Corriden R, Hirsh M, Chen Y, Inoue Y, Ferrari V, Insel PA, Junger WG (2009) Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. FASEB J 23(6):1685–1693. https://doi.org/10.1096/fj.08-126458
Proietti M, Cornacchione V, Rezzonico Jost T, Romagnani A, Faliti CE, Perruzza L, Rigoni R, Radaelli E, Caprioli F, Preziuso S, Brannetti B, Thelen M, McCoy KD, Slack E, Traggiai E, Grassi F (2014)ATP-gated ionotropic P2X7 receptor controls follicular T helper cell numbers in Peyer's patches to promote host-microbiota mutualism. Immunity 41(5):789–801. https://doi.org/10.1016/j.immuni.2014.10.010
Hubert S, Rissiek B, Klages K, Huehn J, Sparwasser T, Haag F, Koch-Nolte F, Boyer O, Seman M, Adriouch S (2010) Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2-P2X7 pathway. J Exp Med 207(12):2561–2568. https://doi.org/10.1084/jem.20091154
Wilhelm K, Ganesan J, Muller T, Durr C, Grimm M, Beilhack A, Krempl CD, Sorichter S, Gerlach UV, Juttner E, Zerweck A, Gartner F, Pellegatti P, Di Virgilio F, Ferrari D, Kambham N, Fisch P, Finke J, Idzko M, Zeiser R (2010)Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med 16(12):1434–1438. https://doi.org/10.1038/nm.2242
Mishra A, Guo Y, Zhang L, More S, Weng T, Chintagari NR, Huang C, Liang Y, Pushparaj S, Gou D, Breshears M, Liu L (2016) A critical role for P2X7 receptor-induced VCAM-1 shedding and neutrophil infiltration during acute lung injury. JImmunol
Semenova S, Shatrova A, Vassilieva I, Shamatova M, Pugovkina N, Negulyaev Y (2020)Adenosine-5'-triphosphate suppresses proliferation and migration capacity of human endometrial stem cells. J Cell Mol Med 24(8):4580–4588. https://doi.org/10.1111/jcmm.15115
Wang J, Liu S, Nie Y, Wu B, Wu Q, Song M, Tang M, Xiao L, Xu P, Tan X, Zhang L, Li G, Liang S, Zhang C (2015) Activation of P2X7 receptors decreases the proliferation of murine luteal cells. Reprod Fertil Dev 27(8):1262–1271. https://doi.org/10.1071/rd14381
Wang CM, Chang YY, Sun SH (2003) Activation of P2X7 purinoceptor-stimulated TGF-beta 1 mRNA expression involves PKC/MAPK signalling pathway in a rat brain-derived type-2 astrocyte cell line, RBA-2. Cell Signal 15(12):1129–1137. https://doi.org/10.1016/s0898-6568(03)00112-8
Qu LP, Xue H, Yuan P, Zhou L, Yao T, Huang Y, Lu LM (2009) Adenosine 5'-triphosphate stimulates the increase of TGF-beta1 in rat mesangial cells under high-glucose conditions via reactive oxygen species and ERK1/2. Acta Pharmacol Sin 30(12):1601–1606. https://doi.org/10.1038/aps.2009.155
Fan X, Ma W, Zhang Y, Zhang L (2020) P2X7 receptor (P2X7R) of microglia mediates neuroinflammation by regulating (NOD)-like receptor protein 3 (NLRP3)inflammasome-dependent inflammation after spinal cord injury. Med Sci Monit 26:e925491. https://doi.org/10.12659/msm.925491
Wu H, Jiang M, Liu Q, Wen F, Nie Y (2020) lncRNA uc.48+ regulates immune and inflammatory reactions mediated by the P2X(7) receptor in type 2 diabetic mice. Exp Ther Med 20(6):230. https://doi.org/10.3892/etm.2020.9360
Wang W, Hu D, Feng Y, Wu C, Song Y, Liu W, Li A, Wang Y, Chen K, Tian M, Xiao F, Zhang Q, Chen W, Pan P, Wan P, Liu Y, Lan H, Wu K, Wu J (2020) Paxillin mediates ATP-induced activation of P2X7 receptor and NLRP3 inflammasome. BMC Biol 18(1):182. https://doi.org/10.1186/s12915-020-00918-w
Zhang C, Qin J, Zhang S, Zhang N, Tan B, Siwko S, Zhang Y, Wang Q, Chen J, Qian M, Liu M, Du B (2020) ADP/P2Y(1) aggravates inflammatory bowel disease through ERK5-mediated NLRP3 inflammasome activation. Mucosal Immunol 13(6):931–945. https://doi.org/10.1038/s41385-020-0307-5
Alarcón-Vila C, Baroja-Mazo A, de Torre-Minguela C, Martínez CM, Martínez-García JJ, Martínez-Banaclocha H, García-Palenciano C, Pelegrin P (2020) CD14 release induced by P2X7 receptor restricts inflammation and increases survival during sepsis. Elife 9. https://doi.org/10.7554/eLife.60849
Jiang M, Cui BW, Wu YL, Zhang Y, Shang Y, Liu J, Yang HX, Qiao CY, Zhan ZY, Ye H, Jin Q, Nan JX, Lian LH (2020) P2X7R orchestrates the progression of murine hepatic fibrosis by making a feedback loop from macrophage to hepatic stellate cells. Toxicol Lett 333:22–32. https://doi.org/10.1016/j.toxlet.2020.07.023
Xu SL, Lin Y, Liu W, Zhu XZ, Liu D, Tong ML, Liu LL, Lin LR (2020) The P2X7 receptor mediates NLRP3-dependent IL-1β secretion and promotes phagocytosis in the macrophage response to Treponema pallidum. Int Immunopharmacol 82:106344. https://doi.org/10.1016/j.intimp.2020.106344
Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ (2013) Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol 190(3):1217–1226. https://doi.org/10.4049/jimmunol.1202388
Pelegrin P, Surprenant A (2006)Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J 25(21):5071–5082. https://doi.org/10.1038/sj.emboj.7601378
Suh BC, Kim JS, Namgung U, Ha H, Kim KT (2001) P2X7 nucleotide receptor mediation of membrane pore formation and superoxide generation in human promyelocytes and neutrophils. J Immunol 166(11):6754–6763. https://doi.org/10.4049/jimmunol.166.11.6754
Tsukimoto M, Maehata M, Harada H, Ikari A, Takagi K, Degawa M (2006) P2X7 receptor-dependent cell death is modulated during murine T cell maturation and mediated by dual signaling pathways. J Immunol 177(5):2842–2850
Aswad F, Dennert G (2006) P2X7 receptor expression levels determine lethal effects of a purine based danger signal in T lymphocytes. Cell Immunol 243(1):58–65. https://doi.org/10.1016/j.cellimm.2006.12.003
Scheuplein F, Schwarz N, Adriouch S, Krebs C, Bannas P, Rissiek B, Seman M, Haag F, Koch-Nolte F (2009) NAD+ and ATP released from injured cells induce P2X7-dependent shedding of CD62L and externalization of phosphatidylserine by murine T cells. J Immunol 182(5):2898–2908. https://doi.org/10.4049/jimmunol.0801711
Surprenant A, Rassendren F, Kawashima E, North RA, Buell G (1996) The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272(5262):735–738
Shen D, Shen X, Schwarz W, Grygorczyk R, Wang L (2020) P2Y(13) and P2X(7) receptors modulate mechanically induced adenosine triphosphate release from mast cells. Exp Dermatol 29(5):499–508. https://doi.org/10.1111/exd.14093
Nylander S, Mattsson C, Ramstrom S, Lindahl TL (2004) Synergistic action between inhibition of P2Y12/P2Y1 and P2Y12/thrombin in ADP- and thrombin-induced human platelet activation. Br J Pharmacol 142(8):1325–1331. https://doi.org/10.1038/sj.bjp.0705885
Anderson R, Theron AJ, Steel HC, Nel JG, Tintinger GR (2020)ADP-mediated upregulation of expression of CD62P on human platelets is critically dependent on co-activation of P2Y1 and P2Y12 receptors. Pharmaceuticals (Basel) 13(12). https://doi.org/10.3390/ph13120420
Muller T, Fay S, Vieira RP, Karmouty-Quintana H, Cicko S, Ayata K, Zissel G, Goldmann T, Lungarella G, Ferrari D, Di Virgilio F, Robaye B, Boeynaems JM, Blackburn MR, Idzko M (2017) The purinergic receptor subtype P2Y2 mediates chemotaxis of neutrophils and fibroblasts in fibrotic lung disease. Oncotarget. https://doi.org/10.18632/oncotarget.16414
Muller T, Robaye B, Vieira RP, Ferrari D, Grimm M, Jakob T, Martin SF, Di Virgilio F, Boeynaems JM, Virchow JC, Idzko M (2010) The purinergic receptor P2Y2 receptor mediates chemotaxis of dendritic cells and eosinophils in allergic lung inflammation. Allergy 65(12):1545–1553. https://doi.org/10.1111/j.1398-9995.2010.02426.x
Vanderstocken G, Bondue B, Horckmans M, Di Pietrantonio L, Robaye B, Boeynaems JM, Communi D (2010) P2Y2 receptor regulates VCAM-1 membrane and soluble forms and eosinophil accumulation during lung inflammation. J Immunol 185(6):3702–3707. https://doi.org/10.4049/jimmunol.0903908
de la Rosa G, Gómez AI, Baños MC, Pelegrín P (2020) Signaling through purinergic receptor P2Y(2) enhances macrophage IL-1β production. Int J Mol Sci 21(13). https://doi.org/10.3390/ijms21134686
Thorstenberg ML, Martins MDA, Figliuolo V, Silva CLM, Savio LEB, Coutinho-Silva R (2020) P2Y(2) Receptor induces L. amazonensis infection control in a mechanism dependent on caspase-1 activation and IL-1β secretion. Mediat Inflamm 2020:2545682. https://doi.org/10.1155/2020/2545682
Ualiyeva S, Hallen N, Kanaoka Y, Ledderose C, Matsumoto I, Junger WG, Barrett NA, Bankova LG (2020) Airway brush cells generate cysteinyl leukotrienes through the ATP sensor P2Y2. Sci Immunol 5(43). https://doi.org/10.1126/sciimmunol.aax7224
Ohsawa K, Irino Y, Nakamura Y, Akazawa C, Inoue K, Kohsaka S (2007) Involvement of P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia 55(6):604–616. https://doi.org/10.1002/glia.20489
Irino Y, Nakamura Y, Inoue K, Kohsaka S, Ohsawa K (2008) Akt activation is involved in P2Y12 receptor-mediated chemotaxis of microglia. J Neurosci Res 86(7):1511–1519. https://doi.org/10.1002/jnr.21610
Sil P, Hayes CP, Reaves BJ, Breen P, Quinn S, Sokolove J, Rada B (2017) P2Y6 receptor antagonist MRS2578 inhibits neutrophil activation and aggregated neutrophil extracellular trap formation induced by gout-associated monosodium urate crystals. J Immunol 198(1):428–442. https://doi.org/10.4049/jimmunol.1600766
Uratsuji H, Tada Y, Kawashima T, Kamata M, Hau CS, Asano Y, Sugaya M, Kadono T, Asahina A, Sato S, Tamaki K (2012) P2Y6 receptor signaling pathway mediates inflammatory responses induced by monosodium urate crystals. J Immunol 188(1):436–444. https://doi.org/10.4049/jimmunol.1003746
Kimura T, Kobayashi S, Hanihara-Tatsuzawa F, Sayama A, MaruYama T, Muta T (2014) Responses of macrophages to the danger signals released from necrotic cells. Int Immunol 26(12):697–704. https://doi.org/10.1093/intimm/dxu080
Inoue K (2007) UDP facilitates microglial phagocytosis through P2Y6 receptors. Cell Adhes Migr 1(3):131–132
Morioka N, Tokuhara M, Harano S, Nakamura Y, Hisaoka-Nakashima K, Nakata Y (2013) The activation of P2Y6 receptor in cultured spinal microglia induces the production of CCL2 through the MAP kinases-NF-kappaB pathway. Neuropharmacology 75:116–125. https://doi.org/10.1016/j.neuropharm.2013.07.017
Xu Y, Hu W, Liu Y, Xu P, Li Z, Wu R, Shi X, Tang Y (2016) P2Y6 receptor-mediated microglial phagocytosis in radiation-induced brain injury. Mol Neurobiol 53(6):3552–3564. https://doi.org/10.1007/s12035-015-9282-3
Zhu J, Wang Z, Zhang N, Ma J, Xu SL, Wang Y, Shen Y, Li YH (2016) Protein interacting C-kinase 1 modulates surface expression of P2Y6 purinoreceptor, actin polymerization and phagocytosis in microglia. Neurochem Res 41(4):795–803. https://doi.org/10.1007/s11064-015-1754-3
Nakano M, Ito K, Yuno T, Soma N, Aburakawa S, Kasai K, Nakamura T, Takami H (2017)UDP/P2Y6 receptor signaling regulates IgE-dependent degranulation in human basophils. Allergol Int. https://doi.org/10.1016/j.alit.2017.02.014
Shin A, Toy T, Rothenfusser S, Robson N, Vorac J, Dauer M, Stuplich M, Endres S, Cebon J, Maraskovsky E, Schnurr M (2008) P2Y receptor signaling regulates phenotype and IFN-alpha secretion of human plasmacytoid dendritic cells. Blood 111(6):3062–3069. https://doi.org/10.1182/blood-2007-02-071910
Khine AA, Del Sorbo L, Vaschetto R, Voglis S, Tullis E, Slutsky AS, Downey GP, Zhang H (2006) Human neutrophil peptides induce interleukin-8 production through the P2Y6 signaling pathway. Blood 107(7):2936–2942. https://doi.org/10.1182/blood-2005-06-2314
Grbic DM, Degagne E, Larrivee JF, Bilodeau MS, Vinette V, Arguin G, Stankova J, Gendron FP (2012) P2Y6 receptor contributes to neutrophil recruitment to inflamed intestinal mucosa by increasing CXC chemokine ligand 8 expression in an AP-1-dependent manner in epithelial cells. Inflamm Bowel Dis 18(8):1456–1469. https://doi.org/10.1002/ibd.21931
Li R, Tan B, Yan Y, Ma X, Zhang N, Zhang Z, Liu M, Qian M, Du B (2014) Extracellular UDP and P2Y6 function as a danger signal to protect mice from vesicular stomatitis virus infection through an increase in IFN-beta production. J Immunol 193(9):4515–4526. https://doi.org/10.4049/jimmunol.1301930
Li Z, He C, Zhang J, Zhang H, Wei H, Wu S, Jiang W (2020) P2Y(6) Deficiency enhances dendritic cell-mediatedTh1/Th17 differentiation and aggravates experimental autoimmune encephalomyelitis. J Immunol 205(2):387–397. https://doi.org/10.4049/jimmunol.1900916
Wen RX, Shen H, Huang SX, Wang LP, Li ZW, Peng P, Mamtilahun M, Tang YH, Shen FX, Tian HL, Yang GY, Zhang ZJ (2020) P2Y6 receptor inhibition aggravates ischemic brain injury by reducing microglial phagocytosis. CNS Neurosci Ther 26(4):416–429. https://doi.org/10.1111/cns.13296
Vaughan KR, Stokes L, Prince LR, Marriott HM, Meis S, Kassack MU, Bingle CD, Sabroe I, Surprenant A, Whyte MK (2007) Inhibition of neutrophil apoptosis by ATP is mediated by the P2Y11 receptor. J Immunol 179(12):8544–8553
Alkayed F, Kashimata M, Koyama N, Hayashi T, Tamura Y, Azuma Y (2012) P2Y11 purinoceptor mediates the ATP-enhanced chemotactic response of rat neutrophils. J Pharmacol Sci 120(4):288–295
van der Weyden L, Conigrave AD, Morris MB (2000) Signal transduction and white cell maturation via extracellular ATP and the P2Y11 receptor. Immunol Cell Biol 78(4):369–374. https://doi.org/10.1046/j.1440-1711.2000.00918.x
Wilkin F, Duhant X, Bruyns C, Suarez-Huerta N, Boeynaems JM, Robaye B (2001) The P2Y11 receptor mediates the ATP-induced maturation of human monocyte-derived dendritic cells. J Immunol 166(12):7172–7177
Schnurr M, Toy T, Stoitzner P, Cameron P, Shin A, Beecroft T, Davis ID, Cebon J, Maraskovsky E (2003) ATP gradients inhibit the migratory capacity of specific human dendritic cell types: implications for P2Y11 receptor signaling. Blood 102(2):613–620. https://doi.org/10.1182/blood-2002-12-3745
Meis S, Hamacher A, Hongwiset D, Marzian C, Wiese M, Eckstein N, Royer HD, Communi D, Boeynaems JM, Hausmann R, Schmalzing G, Kassack MU (2010) NF546 [4,4'-(carbonylbis(imino-3,1-phenylene-carbonylimino-3,1-(4-methyl-phenylene)-car bonylimino))-bis(1,3-xylene-alpha,alpha'-diphosphonic acid) tetrasodium salt] is a non-nucleotide P2Y11 agonist and stimulates release of interleukin-8 from human monocyte-derived dendritic cells. J Pharmacol Exp Ther 332(1):238–247. https://doi.org/10.1124/jpet.109.157750
Sakaki H, Tsukimoto M, Harada H, Moriyama Y, Kojima S (2013) Autocrine regulation of macrophage activation via exocytosis of ATP and activation of P2Y11 receptor. PLoS One 8(4):e59778. https://doi.org/10.1371/journal.pone.0059778
Satonaka H, Nagata D, Takahashi M, Kiyosue A, Myojo M, Fujita D, Ishimitsu T, Nagano T, Nagai R, Hirata Y (2015) Involvement of P2Y12 receptor in vascular smooth muscle inflammatory changes via MCP-1 upregulation and monocyte adhesion. Am J Physiol Heart Circ Physiol 308(8):H853–H861. https://doi.org/10.1152/ajpheart.00862.2013
Ben Addi A, Cammarata D, Conley PB, Boeynaems JM, Robaye B (2010) Role of the P2Y12 receptor in the modulation of murine dendritic cell function by ADP. J Immunol 185(10):5900–5906. https://doi.org/10.4049/jimmunol.0901799
Lou N, Takano T, Pei Y, Xavier AL, Goldman SA, Nedergaard M (2016) Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc Natl Acad Sci U S A 113(4):1074–1079. https://doi.org/10.1073/pnas.1520398113
Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D (2006) The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci 9(12):1512–1519. https://doi.org/10.1038/nn1805
Moore CS, Ase AR, Kinsara A, Rao VT, Michell-Robinson M, Leong SY, Butovsky O, Ludwin SK, Seguela P, Bar-Or A, Antel JP (2015) P2Y12 expression and function in alternatively activated human microglia. Neurol Neuroimmunol Neuroinflamm 2(2):e80. https://doi.org/10.1212/nxi.0000000000000080
Tozaki-Saitoh H, Miyata H, Yamashita T, Matsushita K, Tsuda M, Inoue K (2017) P2Y12 receptors in primary microglia activate nuclear factor of activated T-cell signaling to induce C-C chemokine 3 expression. J Neurochem 141(1):100–110. https://doi.org/10.1111/jnc.13968
Albayati S, Vemulapalli H, Tsygankov AY, Liverani E (2020) P2Y(12) antagonism results in altered interactions between platelets and regulatory T cells during sepsis. J Leukoc Biol. https://doi.org/10.1002/jlb.3a0220-097r
Jing F, Zhang Y, Long T, He W, Qin G, Zhang D, Chen L, Zhou J (2019) P2Y12 receptor mediates microglial activation via RhoA/ROCK pathway in the trigeminal nucleus caudalis in a mouse model of chronic migraine. J Neuroinflammation 16(1):217. https://doi.org/10.1186/s12974-019-1603-4
Wang L, Olivecrona G, Gotberg M, Olsson ML, Winzell MS, Erlinge D (2005) ADP acting on P2Y13 receptors is a negative feedback pathway for ATP release from human red blood cells. Circ Res 96(2):189–196. https://doi.org/10.1161/01.res.0000153670.07559.e4
Arase T, Uchida H, Kajitani T, Ono M, Tamaki K, Oda H, Nishikawa S, Kagami M, Nagashima T, Masuda H, Asada H, Yoshimura Y, Maruyama T (2009) The UDP-glucose receptor P2RY14 triggers innate mucosal immunity in the female reproductive tract by inducing IL-8. J Immunol 182(11):7074–7084. https://doi.org/10.4049/jimmunol.0900001
Barrett MO, Sesma JI, Ball CB, Jayasekara PS, Jacobson KA, Lazarowski ER, Harden TK (2013) A selective high-affinity antagonist of the P2Y14 receptor inhibits UDP-glucose-stimulated chemotaxis of human neutrophils. Mol Pharmacol 84(1):41–49. https://doi.org/10.1124/mol.113.085654
Li H, Jiang W, Ye S, Zhou M, Liu C, Yang X, Hao K, Hu Q (2020) P2Y(14) receptor has a critical role in acute gouty arthritis by regulating pyroptosis of macrophages. Cell Death Dis 11(5):394. https://doi.org/10.1038/s41419-020-2609-7
Kaunitz JD, Yamaguchi DT (2008) TNAP, TrAP, ecto-purinergic signaling, and bone remodeling. J Cell Biochem 105(3):655–662. https://doi.org/10.1002/jcb.21885
Burnstock G, Arnett TR, Orriss IR (2013) Purinergic signalling in the musculoskeletal system. Purinergic Signal 9(4):541–572. https://doi.org/10.1007/s11302-013-9381-4
Sebastián-Serrano Á, de Diego-García L, Martínez-Frailes C, Ávila J, Zimmermann H, Millán JL, Miras-Portugal MT, Díaz-Hernández M (2015)Tissue-nonspecific alkaline phosphatase regulates purinergic transmission in the central nervous system during development and disease. Comput Struct Biotechnol J 13:95–100. https://doi.org/10.1016/j.csbj.2014.12.004
Zimmermann H (2021) History of ectonucleotidases and their role in purinergic signaling. Biochem Pharmacol 187:114322. https://doi.org/10.1016/j.bcp.2020.114322
Jacobson KA, Muller CE (2016) Medicinal chemistry of adenosine, P2Y and P2X receptors. Neuropharmacology 104:31–49. https://doi.org/10.1016/j.neuropharm.2015.12.001
North RA (2016) P2X receptors. Philos Trans R Soc Lond Ser B Biol Sci 371(1700). https://doi.org/10.1098/rstb.2015.0427
Coddou C, Stojilkovic SS, Huidobro-Toro JP (2011) Allosteric modulation of ATP-gated P2X receptor channels. Rev Neurosci 22(3):335–354. https://doi.org/10.1515/rns.2011.014
Coddou C, Yan Z, Obsil T, Huidobro-Toro JP, Stojilkovic SS (2011) Activation and regulation of purinergic P2X receptor channels. Pharmacol Rev 63(3):641–683. https://doi.org/10.1124/pr.110.003129
Sanabria P, Ross E, Ramirez E, Colon K, Hernandez M, Maldonado HM, Silva WI, Jimenez-Rivera CA, Gonzalez FA (2008) P2Y2 receptor desensitization on single endothelial cells. Endothelium 15(1):43–51. https://doi.org/10.1080/10623320802092294
Schulte G, Fredholm BB (2000) Human adenosine A(1), A(2A), A(2B), and A(3) receptors expressed in Chinese hamster ovary cells all mediate the phosphorylation of extracellular-regulated kinase 1/2. Mol Pharmacol 58(3):477–482
Klaasse EC, Ijzerman AP, de Grip WJ, Beukers MW (2008) Internalization and desensitization of adenosine receptors. Purinergic Signal 4(1):21–37. https://doi.org/10.1007/s11302-007-9086-7
North RA (2002) Molecular physiology of P2X receptors. Physiol Rev 82(4):1013–1067. https://doi.org/10.1152/physrev.00015.2002
Illes P, Müller CE, Jacobson KA, Grutter T, Nicke A, Fountain SJ, Kennedy C, Schmalzing G, Jarvis MF, Stojilkovic SS, King BF, Di Virgilio F (2021) Update of P2X receptor properties and their pharmacology: IUPHAR review 30. Br J Pharmacol 178(3):489–514. https://doi.org/10.1111/bph.15299
Fischer W, Urban N, Immig K, Franke H, Schaefer M (2014) Natural compounds with P2X7 receptor-modulating properties. Purinergic Signal 10(2):313–326. https://doi.org/10.1007/s11302-013-9392-1
Roth-Cross JK, Bender SJ, Weiss SR (2008) Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J Virol 82(20):9829–9838. https://doi.org/10.1128/jvi.01199-08
Swayne LA, Johnstone SR, Ng CS, Sanchez-Arias JC, Good ME, Penuela S, Lohman AW, Wolpe AG, Laubach VE, Koval M, Isakson BE (2020) Consideration of pannexin 1 channels in COVID-19 pathology and treatment. Am J Phys Lung Cell Mol Phys 319(1):L121–l125. https://doi.org/10.1152/ajplung.00146.2020
Agier J, Brzezińska-Błaszczyk E, Żelechowska P, Wiktorska M, Pietrzak J, Różalska S (2018) Cathelicidin LL-37 affects surface and intracellular toll-like receptor expression in tissue mast cells. J Immunol Res 2018:7357162. https://doi.org/10.1155/2018/7357162
Lohman AW, Leskov IL, Butcher JT, Johnstone SR, Stokes TA, Begandt D, DeLalio LJ, Best AK, Penuela S, Leitinger N, Ravichandran KS, Stokes KY, Isakson BE (2015) Pannexin 1 channels regulate leukocyte emigration through the venous endothelium during acute inflammation. Nat Commun 6:7965. https://doi.org/10.1038/ncomms8965
Idzko M, Ferrari D, Eltzschig HK (2014) Nucleotide signalling during inflammation. Nature 509(7500):310–317
Di Virgilio F, Sarti AC, Coutinho-Silva R (2020) Purinergic signaling, DAMPs, and inflammation. Am J Phys Cell Phys 318(5):C832–C835. https://doi.org/10.1152/ajpcell.00053.2020
Venereau E, Ceriotti C, Bianchi ME (2015) DAMPs from cell death to new life. Front Immunol 6:422. https://doi.org/10.3389/fimmu.2015.00422
McGonagle D, O'Donnell JS, Sharif K, Emery P, Bridgewood C (2020) Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol 2(7):e437–e445. https://doi.org/10.1016/s2665-9913(20)30121-1
Jacobson KA, Delicado EG, Gachet C, Kennedy C, von Kügelgen I, Li B, Miras-Portugal MT, Novak I, Schöneberg T, Perez-Sen R, Thor D, Wu B, Yang Z, Müller CE (2020) Update of P2Y receptor pharmacology: IUPHAR Review 27. Br J Pharmacol 177(11):2413–2433. https://doi.org/10.1111/bph.15005
Eckle T, Fullbier L, Wehrmann M, Khoury J, Mittelbronn M, Ibla J, Rosenberger P, Eltzschig HK (2007) Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol 178(12):8127–8137
Zhao R, Qiao J, Zhang X, Zhao Y, Meng X, Sun D, Peng X (2019)Toll-like receptor-mediated activation of CD39 internalization in BMDCs leads to extracellular ATP accumulation and facilitates P2X7 receptor activation. Front Immunol 10:2524. https://doi.org/10.3389/fimmu.2019.02524
Burnstock G (2016) P2X ion channel receptors and inflammation. Purinergic Signal 12(1):59–67. https://doi.org/10.1007/s11302-015-9493-0
Di Virgilio F, Tang Y, Sarti AC, Rossato M (2020) A rationale for targeting the P2X7 receptor in coronavirus disease 19 (Covid-19). Br J Pharmacol. https://doi.org/10.1111/bph.15138
Savio LEB, de Andrade MP, da Silva CG, Coutinho-Silva R (2018) The P2X7 receptor in inflammatory diseases: angel or demon? Front Pharmacol 9:52. https://doi.org/10.3389/fphar.2018.00052
Flores RV, Hernandez-Perez MG, Aquino E, Garrad RC, Weisman GA, Gonzalez FA (2005)Agonist-induced phosphorylation and desensitization of the P2Y2 nucleotide receptor. Mol Cell Biochem 280(1-2):35–45. https://doi.org/10.1007/s11010-005-8050-5
Sromek SM, Harden TK (1998)Agonist-induced internalization of the P2Y2 receptor. Mol Pharmacol 54(3):485–494
Hotchkiss RS, Monneret G, Payen D (2013)Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13(12):862–874. https://doi.org/10.1038/nri3552
Hotchkiss RS, Monneret G, Payen D (2013) Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 13(3):260–268. https://doi.org/10.1016/s1473-3099(13)70001-x
Ward NS, Casserly B, Ayala A (2008) The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med 29(4):617–625, viii. https://doi.org/10.1016/j.ccm.2008.06.010
Bone RC (1996) Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med 24(7):1125–1128. https://doi.org/10.1097/00003246-199607000-00010
Musuuza JS, Watson L, Parmasad V, Putman-Buehler N, Christensen L, Safdar N (2021) Prevalence and outcomes of co-infection and superinfection with SARS-CoV-2 and other pathogens: a systematic review and meta-analysis. PLoS One 16(5):e0251170. https://doi.org/10.1371/journal.pone.0251170
Hori S, Carvalho TL, Demengeot J (2002) CD25+CD4+ regulatory T cells suppress CD4+ T cell-mediated pulmonary hyperinflammation driven by Pneumocystis carinii in immunodeficient mice. Eur J Immunol 32(5):1282–1291.
Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, Wang T, Zhang X, Chen H, Yu H, Zhang X, Zhang M, Wu S, Song J, Chen T, Han M, Li S, Luo X, Zhao J, Ning Q (2020) Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest 130(5):2620–2629. https://doi.org/10.1172/jci137244
Sadeghi A, Tahmasebi S, Mahmood A, Kuznetsova M, Valizadeh H, Taghizadieh A, Nazemiyeh M, Aghebati-Maleki L, Jadidi-Niaragh F, Abbaspour-Aghdam S, Roshangar L, Mikaeili H, Ahmadi M (2020) Th17 and Treg cells function in SARS-CoV2 patients compared with healthy controls. J Cell Physiol. https://doi.org/10.1002/jcp.30047
Wang F, Hou H, Luo Y, Tang G, Wu S, Huang M, Liu W, Zhu Y, Lin Q, Mao L, Fang M, Zhang H, Sun Z (2020) The laboratory tests and host immunity of COVID-19 patients with different severity of illness. JCI Insight 5(10). https://doi.org/10.1172/jci.insight.137799
Meckiff BJ, Ramírez-Suástegui C, Fajardo V, Chee SJ, Kusnadi A, Simon H, Eschweiler S, Grifoni A, Pelosi E, Weiskopf D, Sette A, Ay F, Seumois G, Ottensmeier CH, Vijayanand P (2020) Imbalance of regulatory and cytotoxic SARS-CoV-2-reactive CD4(+) T cells in COVID-19. Cell. https://doi.org/10.1016/j.cell.2020.10.001
Wu X, Ren J, Chen G, Wu L, Song X, Li G, Deng Y, Wang G, Gu G, Li J (2017) Systemic blockade of P2X7 receptor protects against sepsis-induced intestinal barrier disruption. Sci Rep 7(1):4364. https://doi.org/10.1038/s41598-017-04231-5
Wang S, Zhao J, Wang H, Liang Y, Yang N, Huang Y (2015) Blockage of P2X7 attenuates acute lung injury in mice by inhibiting NLRP3 inflammasome. Int Immunopharmacol 27(1):38–45. https://doi.org/10.1016/j.intimp.2015.04.035
Galam L, Rajan A, Failla A, Soundararajan R, Lockey RF, Kolliputi N (2016) Deletion of P2X7 attenuates hyperoxia-induced acute lung injury via inflammasome suppression. Am J Phys Lung Cell Mol Phys 310(6):L572–L581. https://doi.org/10.1152/ajplung.00417.2015
Pacheco PAF, Faria RX (2021) The potential involvement of P2X7 receptor in COVID-19 pathogenesis: a new therapeutic target? Scand J Immunol 93(2):e12960. https://doi.org/10.1111/sji.12960
Ribeiro DE, Oliveira-Giacomelli Á, Glaser T, Arnaud-Sampaio VF, Andrejew R, Dieckmann L, Baranova J, Lameu C, Ratajczak MZ, Ulrich H (2021) Hyperactivation of P2X7 receptors as a culprit of COVID-19 neuropathology. Mol Psychiatry 26(4):1044–1059. https://doi.org/10.1038/s41380-020-00965-3
Wang HL, Xing YQ, Xu YX, Rong F, Lei WF, Zhang WH (2013) The protective effect of lidocaine on septic rats via the inhibition of high mobility group box 1 expression and NF-κB activation. Mediat Inflamm 2013:570370. https://doi.org/10.1155/2013/570370
Liu J, Zhang H, Qi Z, Zheng X (2014) Lidocaine protects against renal and hepatic dysfunction in septic rats via downregulation of Toll-like receptor 4. Mol Med Rep 9(1):118–124. https://doi.org/10.3892/mmr.2013.1799
Gallos G, Jones DR, Nasr SH, Emala CW, Lee HT (2004) Local anesthetics reduce mortality and protect against renal and hepatic dysfunction in murine septic peritonitis. Anesthesiology 101(4):902–911. https://doi.org/10.1097/00000542-200410000-00015
Berger C, Rossaint J, Van Aken H, Westphal M, Hahnenkamp K, Zarbock A (2014) Lidocaine reduces neutrophil recruitment by abolishing chemokine-induced arrest and transendothelial migration in septic patients. J Immunol 192(1):367–376. https://doi.org/10.4049/jimmunol.1301363
Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, Daßler-Plenker J, Guerci P, Huynh C, Knight JS, Loda M, Looney MR, McAllister F, Rayes R, Renaud S, Rousseau S, Salvatore S, Schwartz RE, Spicer JD et al (2020) Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med 217(6). https://doi.org/10.1084/jem.20200652
Narasaraju T, Tang BM, Herrmann M, Muller S, Chow VTK, Radic M (2020) Neutrophilia and NETopathy as key pathologic drivers of progressive lung impairment in patients with COVID-19. Front Pharmacol 11:870. https://doi.org/10.3389/fphar.2020.00870
Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, Mostyka M, Baxter-Stoltzfus A, Borczuk AC, Loda M, Cody MJ, Manne BK, Portier I, Harris ES, Petrey AC, Beswick EJ, Caulin AF, Iovino A, Abegglen LM et al (2020) Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 136(10):1169–1179. https://doi.org/10.1182/blood.2020007008
Tomar B, Anders HJ, Desai J, Mulay SR (2020) Neutrophils and neutrophil extracellular traps drive necroinflammation in COVID-19. Cells 9(6). https://doi.org/10.3390/cells9061383
Li H, Li C, Zhang H, Zhang L, Cheng R, Li M, Guo Y, Zhang Z, Lu Z, Zhuang Y, Yan M, Gu Y, Feng X, Liang J, Yu X, Wang H, Yao Z (2016) Effects of lidocaine on regulatory T cells in atopic dermatitis. J Allergy Clin Immunol 137(2):613–617.e615. https://doi.org/10.1016/j.jaci.2015.07.039
Van Der Wal S, Vaneker M, Steegers M, Van Berkum B, Kox M, Van Der Laak J, Van Der Hoeven J, Vissers K, Scheffer GJ (2015) Lidocaine increases the anti-inflammatory cytokine IL-10 following mechanical ventilation in healthy mice. Acta Anaesthesiol Scand 59(1):47–55. https://doi.org/10.1111/aas.12417
Garutti I, Rancan L, Simón C, Cusati G, Sanchez-Pedrosa G, Moraga F, Olmedilla L, Lopez-Gil MT, Vara E (2014) Intravenous lidocaine decreases tumor necrosis factor alpha expression both locally and systemically in pigs undergoing lung resection surgery. Anesth Analg 119(4):815–828. https://doi.org/10.1213/ane.0000000000000360
Hermanns H, Hollmann MW, Stevens MF, Lirk P, Brandenburger T, Piegeler T, Werdehausen R (2019) Molecular mechanisms of action of systemic lidocaine in acute and chronic pain: a narrative review. Br J Anaesth 123(3):335–349. https://doi.org/10.1016/j.bja.2019.06.014
Weinberg L, Peake B, Tan C, Nikfarjam M (2015) Pharmacokinetics and pharmacodynamics of lignocaine: a review. World J Anesthesiol 4(2):17–29. https://doi.org/10.5313/wja.v4.i2.17
Montnach J, Lorenzini M, Lesage A, Simon I, Nicolas S, Moreau E, Marionneau C, Baró I, De Waard M, Loussouarn G (2021) Computer modeling of whole-cell voltage-clamp analyses to delineate guidelines for good practice of manual and automated patch-clamp. Sci Rep 11(1):3282. https://doi.org/10.1038/s41598-021-82077-8
Wang Y, Jones PJ, Batts TW, Landry V, Patel MK, Brown ML (2009)Ligand-based design and synthesis of novel sodium channel blockers from a combined phenytoin-lidocaine pharmacophore. Bioorg Med Chem 17(19):7064–7072. https://doi.org/10.1016/j.bmc.2008.10.031
Cheng KI, Lai CS, Wang FY, Wang HC, Chang LL, Ho ST, Tsai HP, Kwan AL (2011) Intrathecal lidocaine pretreatment attenuates immediate neuropathic pain by modulating Nav1.3 expression and decreasing spinal microglial activation. BMC Neurol 11:71. https://doi.org/10.1186/1471-2377-11-71
Gingrich KJ, Wagner LE 2nd (2016)Fast-onset lidocaine block of rat NaV1.4 channels suggests involvement of a second high-affinity open state. Biochim Biophys Acta 1858(6):1175–1188. https://doi.org/10.1016/j.bbamem.2016.02.033
Elajnaf T, Baptista-Hon DT, Hales TG (2018) Potent inactivation-dependent inhibition of adult and neonatal NaV1.5 channels by lidocaine and levobupivacaine. Anesth Analg 127(3):650–660. https://doi.org/10.1213/ane.0000000000003597
Sheets PL, Jarecki BW, Cummins TR (2011) Lidocaine reduces the transition to slow inactivation in Na(v)1.7 voltage-gated sodium channels. Br J Pharmacol 164(2b):719–730. https://doi.org/10.1111/j.1476-5381.2011.01209.x
Mert T, Gunes Y (2012) Antinociceptive activities of lidocaine and the nav1.8 blocker a803467 in diabetic rats. J Am Assoc Lab Anim Sci 51(5):579–585
Dusmez D, Cengiz B, Yumrutas O, Demir T, Oztuzcu S, Demiryurek S, Tutar E, Bayraktar R, Bulut A, Simsek H, Daglı SN, Kılıc T, Bagcı C (2014) Effect of verapamil and lidocaine on TRPM and NaV1.9 gene expressions in renal ischemia-reperfusion. Transplant Proc 46(1):33–39. https://doi.org/10.1016/j.transproceed.2013.10.036
Wang L, Wang M, Li S, Wu H, Shen Q, Zhang S, Fang L, Liu R (2018) Nebulized lidocaine ameliorates allergic airway inflammation via downregulation of TLR2. Mol Immunol 97:94–100. https://doi.org/10.1016/j.molimm.2018.03.010
Nishizawa N, Shirasaki T, Nakao S, Matsuda H, Shingu K (2002) The inhibition of the N-methyl-D-aspartate receptor channel by local anesthetics in mouse CA1 pyramidal neurons. Anesth Analg 94(2):325–330, table of contents. https://doi.org/10.1097/00000539-200202000-00017
Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD (2019) The role of voltage-gated sodium channels in pain signaling. Physiol Rev 99(2):1079–1151. https://doi.org/10.1152/physrev.00052.2017
Kis-Toth K, Hajdu P, Bacskai I, Szilagyi O, Papp F, Szanto A, Posta E, Gogolak P, Panyi G, Rajnavolgyi E (2011)Voltage-gated sodium channel Nav1.7 maintains the membrane potential and regulates the activation and chemokine-induced migration of a monocyte-derived dendritic cell subset. J Immunol 187(3):1273–1280. https://doi.org/10.4049/jimmunol.1003345
Carrithers LM, Hulseberg P, Sandor M, Carrithers MD (2011) The human macrophage sodium channel NaV1.5 regulates mycobacteria processing through organelle polarization and localized calcium oscillations. FEMS Immunol Med Microbiol 63(3):319–327. https://doi.org/10.1111/j.1574-695X.2011.00853.x
Black JA, Waxman SG (2012) Sodium channels and microglial function. Exp Neurol 234(2):302–315. https://doi.org/10.1016/j.expneurol.2011.09.030
Poffers M, Bühne N, Herzog C, Thorenz A, Chen R, Güler F, Hage A, Leffler A, Echtermeyer F (2018) Sodium channel Nav1.3 is expressed by polymorphonuclear neutrophils during mouse heart and kidney ischemia in vivo and regulates adhesion, transmigration, and chemotaxis of human and mouse neutrophils in vitro. Anesthesiology 128(6):1151–1166. https://doi.org/10.1097/aln.0000000000002135
Lo WL, Donermeyer DL, Allen PM (2012) A voltage-gated sodium channel is essential for the positive selection of CD4(+) T cells. Nat Immunol 13(9):880–887. https://doi.org/10.1038/ni.2379
Lee JY, Kam YL, Oh J, Kim DH, Choi JS, Choi HY, Han S, Youn I, Choo HP, Yune TY (2017) HYP-17, a novel voltage-gated sodium channel blocker, relieves inflammatory and neuropathic pain in rats. Pharmacol Biochem Behav 153:116–129. https://doi.org/10.1016/j.pbb.2016.12.013
Jarvis MF, Honore P, Shieh CC, Chapman M, Joshi S, Zhang XF, Kort M, Carroll W, Marron B, Atkinson R, Thomas J, Liu D, Krambis M, Liu Y, McGaraughty S, Chu K, Roeloffs R, Zhong C, Mikusa JP et al (2007) A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci U S A 104(20):8520–8525. https://doi.org/10.1073/pnas.0611364104
Joshi SK, Honore P, Hernandez G, Schmidt R, Gomtsyan A, Scanio M, Kort M, Jarvis MF (2009) Additive antinociceptive effects of the selective Nav1.8 blocker A-803467 and selective TRPV1 antagonists in rat inflammatory and neuropathic pain models. J Pain 10(3):306–315. https://doi.org/10.1016/j.jpain.2008.09.007
Bankar G, Goodchild SJ, Howard S, Nelkenbrecher K, Waldbrook M, Dourado M, Shuart NG, Lin S, Young C, Xie Z, Khakh K, Chang E, Sojo LE, Lindgren A, Chowdhury S, Decker S, Grimwood M, Andrez JC, Dehnhardt CM et al (2018) Selective Na(V)1.7 Antagonists with long residence time show improved efficacy against inflammatory and neuropathic pain. Cell Rep 24(12):3133–3145. https://doi.org/10.1016/j.celrep.2018.08.063
Sun H, Jiang J, Gong L, Li X, Yang Y, Luo Y, Guo Z, Lu R, Li H, Li J, Zhao J, Yang N, Li Y (2019)Voltage-gated sodium channel inhibitor reduces atherosclerosis by modulating monocyte/macrophage subsets and suppressing macrophage proliferation. Biomed Pharmacother 120:109352. https://doi.org/10.1016/j.biopha.2019.109352
Beloeil H, Ababneh Z, Chung R, Zurakowski D, Mulkern RV, Berde CB (2006) Effects of bupivacaine and tetrodotoxin on carrageenan-induced hind paw inflammation in rats (Part 1): hyperalgesia, edema, and systemic cytokines. Anesthesiology 105(1):128–138. https://doi.org/10.1097/00000542-200607000-00022
Beloeil H, Ji RR, Berde CB (2006) Effects of bupivacaine and tetrodotoxin on carrageenan-induced hind paw inflammation in rats (Part 2): cytokines and p38 mitogen-activated protein kinases in dorsal root ganglia and spinal cord. Anesthesiology 105(1):139–145. https://doi.org/10.1097/00000542-200607000-00023
Alcántara Montero A, Sánchez Carnerero CI (2021)Voltage-gated sodium channel blockers: new perspectives in the treatment of neuropathic pain. Neurologia 36(2):169–171. https://doi.org/10.1016/j.nrl.2020.02.004
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395(10223):497–506. https://doi.org/10.1016/S0140-6736(20)30183-5
Dhar SK, K V, Damodar S, Gujar S, Das M (2021)IL-6 and IL-10 as predictors of disease severity in COVID-19 patients: results from meta-analysis and regression. Heliyon 7(2):e06155. https://doi.org/10.1016/j.heliyon.2021.e06155
Udomsinprasert W, Jittikoon J, Sangroongruangsri S, Chaikledkaew U (2021) Circulating levels of interleukin-6 and Interleukin-10, but not tumor necrosis factor-alpha, as potential biomarkers of severity and mortality for COVID-19: systematic review with meta-analysis. J Clin Immunol 41(1):11–22. https://doi.org/10.1007/s10875-020-00899-z
Sun D, Li H, Lu XX, Xiao H, Ren J, Zhang FR, Liu ZS (2020) Clinical features of severe pediatric patients with coronavirus disease 2019 in Wuhan: a single center's observational study. World J Pediatr. https://doi.org/10.1007/s12519-020-00354-4
Zhou C, Qi C, Zhao J, Wang F, Zhang W, Li C, Jing J, Kang X, Chai Z (2011)Interleukin-1β inhibits voltage-gated sodium currents in a time- and dose-dependent manner in cortical neurons. Neurochem Res 36(6):1116–1123. https://doi.org/10.1007/s11064-011-0456-8
Li X, Chen W, Sheng J, Cao D, Wang W (2014)Interleukin-6 inhibits voltage-gated sodium channel activity of cultured rat spinal cord neurons. Acta Neuropsychiatr 26(3):170–177. https://doi.org/10.1017/neu.2013.49
Shen KF, Zhu HQ, Wei XH, Wang J, Li YY, Pang RP, Liu XG (2013)Interleukin-10 down-regulates voltage gated sodium channels in rat dorsal root ganglion neurons. Exp Neurol 247:466–475. https://doi.org/10.1016/j.expneurol.2013.01.018
Del Puerto A, Fronzaroli-Molinieres L, Perez-Alvarez MJ, Giraud P, Carlier E, Wandosell F, Debanne D, Garrido JJ (2015)ATP-P2X7 receptor modulates axon initial segment composition and function in physiological conditions and brain injury. Cereb Cortex 25(8):2282–2294. https://doi.org/10.1093/cercor/bhu035
Huang C, Chi XS, Li R, Hu X, Xu HX, Li JM, Zhou D (2017) Inhibition of P2X7 receptor ameliorates nuclear factor-kappa B mediated neuroinflammation induced by status epilepticus in rat hippocampus. J Mol Neurosci 63(2):173–184. https://doi.org/10.1007/s12031-017-0968-z
Liu Y, Xiao Y, Li Z (2011) P2X7 receptor positively regulates MyD88-dependent NF-κB activation. Cytokine 55(2):229–236. https://doi.org/10.1016/j.cyto.2011.05.003
Petrenko AB, Yamakura T, Baba H, Shimoji K (2003) The role of N-methyl-D-aspartate(NMDA) receptors in pain: a review. Anesth Analg 97(4):1108–1116. https://doi.org/10.1213/01.ane.0000081061.12235.55
Di Loreto S, Balestrino M, Pellegrini P, Berghella AM, Del Beato T, Di Marco F, Adorno D (1997) Blockade of N-methyl-D-aspartate receptor prevents hypoxic neuronal death and cytokine release. Neuroimmunomodulation 4(4):195–199. https://doi.org/10.1159/000097338
Érces D, Varga G, Fazekas B, Kovács T, Tőkés T, Tiszlavicz L, Fülöp F, Vécsei L, Boros M, Kaszaki J (2012)N-methyl-D-aspartate receptor antagonist therapy suppresses colon motility and inflammatory activation six days after the onset of experimental colitis in rats. Eur J Pharmacol 691(1-3):225–234. https://doi.org/10.1016/j.ejphar.2012.06.044
Liu CH, Cherng CH, Lin SL, Yeh CC, Wu CT, Tai YH, Wong CS (2011)N-methyl-D-aspartate receptor antagonist MK-801 suppresses glial pro-inflammatory cytokine expression in morphine-tolerant rats. Pharmacol Biochem Behav 99(3):371–380. https://doi.org/10.1016/j.pbb.2011.05.016
Simma N, Bose T, Kahlfuss S, Mankiewicz J, Lowinus T, Lühder F, Schüler T, Schraven B, Heine M, Bommhardt U (2014)NMDA-receptor antagonists block B-cell function but foster IL-10 production in BCR/CD40-activated B cells. Cell Commun Signal 12:75. https://doi.org/10.1186/s12964-014-0075-5
Sugimoto M, Uchida I, Mashimo T (2003) Local anaesthetics have different mechanisms and sites of action at the recombinant N-methyl-D-aspartate(NMDA) receptors. Br J Pharmacol 138(5):876–882. https://doi.org/10.1038/sj.bjp.0705107
Hahnenkamp K, Durieux ME, Hahnenkamp A, Schauerte SK, Hoenemann CW, Vegh V, Theilmeier G, Hollmann MW (2006) Local anaesthetics inhibit signalling of human NMDA receptors recombinantly expressed in Xenopus laevis oocytes: role of protein kinase C. Br J Anaesth 96(1):77–87. https://doi.org/10.1093/bja/aei271
Kahlfuß S, Simma N, Mankiewicz J, Bose T, Lowinus T, Klein-Hessling S, Sprengel R, Schraven B, Heine M, Bommhardt U (2014) Immunosuppression by N-methyl-D-aspartate receptor antagonists is mediated through inhibition of Kv1.3 and KCa3.1 channels in T cells. Mol Cell Biol 34(5):820–831. https://doi.org/10.1128/mcb.01273-13
Hajimohammadreza I, Probert AW, Coughenour LL, Borosky SA, Marcoux FW, Boxer PA, Wang KK (1995) A specific inhibitor of calcium/calmodulin-dependent protein kinase-II provides neuroprotection against NMDA- and hypoxia/hypoglycemia-induced cell death. J Neurosci 15(5 Pt 2):4093–4101. https://doi.org/10.1523/jneurosci.15-05-04093.1995
Cotrina ML, Nedergaard M (2009) Physiological and pathological functions of P2X7 receptor in the spinal cord. Purinergic Signal 5(2):223–232. https://doi.org/10.1007/s11302-009-9138-2
Alves LA, Bezerra RJ, Faria RX, Ferreira LG, da Silva FV (2013) Physiological roles and potential therapeutic applications of the P2X7 receptor in inflammation and pain. Molecules 18(9):10953–10972. https://doi.org/10.3390/molecules180910953
Hunter MC, Teijeira A, Halin C (2016) T cell trafficking through lymphatic vessels. Front Immunol 7:613. https://doi.org/10.3389/fimmu.2016.00613
Teijeira A, Russo E, Halin C (2014) Taking the lymphatic route: dendritic cell migration to draining lymph nodes. Semin Immunopathol 36(2):261–274. https://doi.org/10.1007/s00281-013-0410-8
Lund H, Boysen P, Åkesson CP, Lewandowska-Sabat AM, Storset AK (2016) Transient migration of large numbers of CD14(++) CD16(+) monocytes to the draining lymph node after onset of inflammation. Front Immunol 7:322. https://doi.org/10.3389/fimmu.2016.00322
Louie DAP, Liao S (2019) Lymph node subcapsular sinus macrophages as the frontline of lymphatic immune defense. Front Immunol 10:347. https://doi.org/10.3389/fimmu.2019.00347
Hampton HR, Chtanova T (2016) The lymph node neutrophil. Semin Immunol 28(2):129–136. https://doi.org/10.1016/j.smim.2016.03.008
Wang HW, Tedla N, Lloyd AR, Wakefield D, McNeil PH (1998) Mast cell activation and migration to lymph nodes during induction of an immune response in mice. J Clin Invest 102(8):1617–1626. https://doi.org/10.1172/jci3704
Shi HZ, Humbles A, Gerard C, Jin Z, Weller PF (2000) Lymph node trafficking and antigen presentation by endobronchial eosinophils. J Clin Invest 105(7):945–953. https://doi.org/10.1172/jci8945
Kim S, Prout M, Ramshaw H, Lopez AF, LeGros G, Min B (2010) Cutting edge: basophils are transiently recruited into the draining lymph nodes during helminth infection via IL-3, but infection-induced Th2 immunity can develop without basophil lymph node recruitment or IL-3. J Immunol 184(3):1143–1147. https://doi.org/10.4049/jimmunol.0902447
Tse D, Stan RV (2010) Morphological heterogeneity of endothelium. Semin Thromb Hemost 36(3):236–245. https://doi.org/10.1055/s-0030-1253447
Kretsos K, Kasting GB (2005) Dermal capillary clearance: physiology and modeling. Skin Pharmacol Physiol 18(2):55–74. https://doi.org/10.1159/000083706
Breslin JW, Yang Y, Scallan JP, Sweat RS, Adderley SP, Murfee WL (2018) Lymphatic vessel network structure and physiology. Compr Physiol 9(1):207–299. https://doi.org/10.1002/cphy.c180015
Kersey TW, Van Eyk J, Lannin DR, Chua AN, Tafra L (2001) Comparison of intradermal and subcutaneous injections in lymphatic mapping. J Surg Res 96(2):255–259. https://doi.org/10.1006/jsre.2000.6075
Karimeddini MK (1989) Intradermal pathways. In: Spencer RP (ed) New procedures in nuclear medicine. CRC Press, Inc., Boca Raton, pp 208–211
Thomasy SM, Pypendop BH, Ilkiw JE, Stanley SD (2005) Pharmacokinetics of lidocaine and its active metabolite, monoethylglycinexylidide, after intravenous administration of lidocaine to awake and isoflurane-anesthetized cats. Am J Vet Res 66(7):1162–1166
Hatef DA, Brown SA, Lipschitz AH, Kenkel JM (2009) Efficacy of lidocaine for pain control in subcutaneous infiltration during liposuction. Aesthet Surg J 29(2):122–128. https://doi.org/10.1016/j.asj.2009.01.014
Wu F, Tamhane M, Morris ME (2012) Pharmacokinetics, lymph node uptake, and mechanistic PK model of near-infrared dye-labeled bevacizumab after IV and SC administration in mice. AAPS J 14(2):252–261. https://doi.org/10.1208/s12248-012-9342-9
Dahlberg AM, Kaminskas LM, Smith A, Nicolazzo JA, Porter CJ, Bulitta JB, McIntosh MP (2014) The lymphatic system plays a major role in the intravenous and subcutaneous pharmacokinetics of trastuzumab in rats. Mol Pharm 11(2):496–504. https://doi.org/10.1021/mp400464s
Worley DR, Hansen RJ, Wittenburg LA, Chubb LS, Gustafson DL (2016) Docetaxel accumulates in lymphatic circulation following subcutaneous delivery compared to intravenous delivery in rats. Anticancer Res 36(10):5071–5078. https://doi.org/10.21873/anticanres.11076
Stock TC, Bloom BJ, Wei N, Ishaq S, Park W, Wang X, Gupta P, Mebus CA (2012) Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J Rheumatol 39(4):720–727. https://doi.org/10.3899/jrheum.110874
Keystone EC, Wang MM, Layton M, Hollis S, McInnes IB (2012) Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann Rheum Dis 71(10):1630–1635. https://doi.org/10.1136/annrheumdis-2011-143578
Bhattacharya A (2018) Recent advances in CNS P2X7 physiology and pharmacology: focus on neuropsychiatric disorders. Front Pharmacol 9:30. https://doi.org/10.3389/fphar.2018.00030
Timmers M, Ravenstijn P, Xi L, Triana-Baltzer G, Furey M, Van Hemelryck S, Biewenga J, Ceusters M, Bhattacharya A, van den Boer M, van Nueten L, de Boer P (2018) Clinical pharmacokinetics, pharmacodynamics, safety, and tolerability of JNJ-54175446, a brain permeable P2X7 antagonist, in a randomised single-ascending dose study in healthy participants. J Psychopharmacol 32(12):1341–1350. https://doi.org/10.1177/0269881118800067
Eser A, Colombel JF, Rutgeerts P, Vermeire S, Vogelsang H, Braddock M, Persson T, Reinisch W (2015) Safety and efficacy of an oral inhibitor of the purinergic receptor P2X7 in adult patients with moderately to severely active Crohn's disease: a randomized placebo-controlled, double-blind, phase iia study. Inflamm Bowel Dis 21(10):2247–2253. https://doi.org/10.1097/mib.0000000000000514
Shakweh M, Ponchel G, Fattal E (2004) Particle uptake by Peyer's patches: a pathway for drug and vaccine delivery. Expert Opin Drug Deliv 1(1):141–163. https://doi.org/10.1517/17425247.1.1.141
Zgair A, Wong JCM, Gershkovich P (2016) Targeting immunomodulatory agents to the gut-associated lymphoid tissue. Neuro-Immuno-Gastroenterology:237–261. https://doi.org/10.1007/978-3-319-28609-9_14
Trevaskis NL, Charman WN, Porter CJ (2008)Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Deliv Rev 60(6):702–716. https://doi.org/10.1016/j.addr.2007.09.007
Trevaskis NL, Charman WN, Porter CJ (2010) Targeted drug delivery to lymphocytes: a route to site-specific immunomodulation? Mol Pharm 7(6):2297–2309. https://doi.org/10.1021/mp100259a
Hsu YW, Somma J, Newman MF, Mathew JP (2011) Population pharmacokinetics of lidocaine administered during and after cardiac surgery. J Cardiothorac Vasc Anesth 25(6):931–936. https://doi.org/10.1053/j.jvca.2011.03.008
Mori K, Ito H, Toda Y, Hashimoto T, Miyazaki M, Saijo T, Kuroda Y (2004) Successful management of intractable epilepsy with lidocaine tapes and continuous subcutaneous lidocaine infusion. Epilepsia 45(10):1287–1290. https://doi.org/10.1111/j.0013-9580.2004.17304.x
Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS (2012) Acute respiratory distress syndrome: the Berlin Definition. Jama 307(23):2526–2533. https://doi.org/10.1001/jama.2012.5669
Wang J, Zheng P, Huang Z, Huang H, Xue M, Liao C, Sun B, Zhong N (2020) Serum SP-A and KL-6 levels can predict the improvement and deterioration of patients with interstitial pneumonia with autoimmune features. BMC Pulm Med 20(1):315. https://doi.org/10.1186/s12890-020-01336-y
Mallapaty S (2020) The coronavirus is most deadly if you are older and male - new data reveal the risks. Nature 585(7823):16–17. https://doi.org/10.1038/d41586-020-02483-2
Izcovich A, Ragusa MA, Tortosa F, Lavena Marzio MA, Agnoletti C, Bengolea A, Ceirano A, Espinosa F, Saavedra E, Sanguine V, Tassara A, Cid C, Catalano HN, Agarwal A, Foroutan F, Rada G (2020) Prognostic factors for severity and mortality in patients infected with COVID-19: A systematic review. PLoS One 15(11):e0241955. https://doi.org/10.1371/journal.pone.0241955
Diaz-Vera M, Terrones Santa Cruz J, Forttini Headrington A, Cerna Paz JA, Quintanilla Rios L, Medina Melendez MP, Del Águila Torres J (2020) Lidocaine to reduce the severity of covid-19 cases. Therapia Neural. Sabadell. Barcelona, Spain. http://www.terapianeural.com/articulos/28-studies-estudios/495-lidocaine-to-reduce-the-severity-of-covid-19-cases. Accessed 19-11 2020
Muller M, Lefebvre F, Harlay ML, Glady L, Becker G, Muller C, Aberkane O, Tawk M, Julians M, Romoli A, Hecketsweiler S, Schneider F, Pottecher J, Chamaraux-Tran TN (2021) Impact of intravenous lidocaine on clinical outcomes of patients with ARDS during COVID-19 pandemia (LidoCovid): a structured summary of a study protocol for a randomised controlled trial. Trials 22(1):131. https://doi.org/10.1186/s13063-021-05095-x
Bharat A, Querrey M, Markov NS, Kim S, Kurihara C, Garza-Castillon R, Manerikar A, Shilatifard A, Tomic R, Politanska Y, Abdala-Valencia H, Yeldandi AV, Lomasney JW, Misharin AV, Budinger GRS (2020) Lung transplantation for patients with severe COVID-19. Sci Transl Med. https://doi.org/10.1126/scitranslmed.abe4282
Gordh T (2010) Lidocaine: the origin of a modern local anesthetic. 1949. Anesthesiology 113(6):1433–1437. https://doi.org/10.1097/ALN.0b013e3181fcef48
Derry S, Wiffen PJ, Moore RA, Quinlan J (2014) Topical lidocaine for neuropathic pain in adults. Cochrane Database Syst Rev 2014(7):CD010958. https://doi.org/10.1002/14651858.CD010958.pub2
Beaussier M, Delbos A, Maurice-Szamburski A, Ecoffey C, Mercadal L (2018) Perioperative use of intravenous lidocaine. Drugs 78(12):1229–1246. https://doi.org/10.1007/s40265-018-0955-x
Su D, Gu Y, Wang Z, Wang X (2010) Lidocaine attenuates proinflammatory cytokine production induced by extracellular adenosine triphosphate in cultured rat microglia. Anesth Analg 111(3):768–774. https://doi.org/10.1213/ANE.0b013e3181e9e897
Yuan T, Li Z, Li X, Yu G, Wang N, Yang X (2014) Lidocaine attenuates lipopolysaccharide-induced inflammatory responses in microglia. J Surg Res 192(1):150–162. https://doi.org/10.1016/j.jss.2014.05.023
Flondor M, Listle H, Kemming GI, Zwissler B, Hofstetter C (2010) Effect of inhaled and intravenous lidocaine on inflammatory reaction in endotoxaemic rats. Eur J Anaesthesiol 27(1):53–60. https://doi.org/10.1097/EJA.0b013e32832b8a70
Chen LJ, Ding YB, Ma PL, Jiang SH, Li KZ, Li AZ, Li MC, Shi CX, Du J, Zhou HD (2018) The protective effect of lidocaine on lipopolysaccharide-induced acute lung injury in rats through NF-κB and p38 MAPK signaling pathway and excessive inflammatory responses. Eur Rev Med Pharmacol Sci 22(7):2099–2108. https://doi.org/10.26355/eurrev_201804_14743
Feng G, Liu S, Wang G-L, Liu G-J(2008) Lidocaine attenuates lipopolysaccharide-induced acute lung injury through inhibiting NF-kappaB activation. Pharmacology 81(1):32–40. https://doi.org/10.1159/000107792
Huang TK, Uyehara CF, Balaraman V, Miyasato CY, Person D, Egan E, Easa D (2004) Surfactant lavage with lidocaine improves pulmonary function in piglets after HCl-induced acute lung injury. Lung 182(1):15–25. https://doi.org/10.1007/s00408-003-1041-y
Kiyonari Y, Nishina K, Mikawa K, Maekawa N, Obara H (2000) Lidocaine attenuates acute lung injury induced by a combination of phospholipase A2 and trypsin. Crit Care Med 28(2):484–489. https://doi.org/10.1097/00003246-200002000-00033
Pedersen JL, Callesen T, Møiniche S, Kehlet H (1996) Analgesic and anti-inflammatory effects of lignocaine-prilocaine(EMLA) cream in human burn injury. Br J Anaesth 76(6):806–810. https://doi.org/10.1093/bja/76.6.806
Werner RM, Hoffman AK, Coe NB (2020)Long-term care policy after Covid-19 - solving the nursing home crisis. N Engl J Med 383(10):903–905. https://doi.org/10.1056/NEJMp2014811
Lesch CA, Squier CA, Cruchley A, Williams DM, Speight P (1989) The permeability of human oral mucosa and skin to water. J Dent Res 68(9):1345–1349. https://doi.org/10.1177/00220345890680091101
Goyal AK, Singh R, Chauhan G, Rath G (2018)Non-invasive systemic drug delivery through mucosal routes. Artif Cells Nanomed Biotechnol 46(sup2):539–551. https://doi.org/10.1080/21691401.2018.1463230
Gröningsson K, Lindgren JE, Lundberg E, Sandberg R, Wahlén A (1985) Lidocaine base and hydrochloride. In: Florey K (ed) Analytical profiles of drug substances, vol 14. Academic Press, pp 207-243. https://doi.org/10.1016/S0099-5428(08)60582-1
Charman WNA, Stella VJ (1986) Estimating the maximal potential for intestinal lymphatic transport of lipophilic drug molecules. Int J Pharm 34(1):175–178. https://doi.org/10.1016/0378-5173(86)90027-X
Porter CJ, Trevaskis NL, Charman WN (2007) Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov 6(3):231–248. https://doi.org/10.1038/nrd2197
Gvetadze SR, Ilkaev KD (2020) Lingual lymph nodes: anatomy, clinical considerations, and oncological significance. World J Clin Oncol 11(6):337–347. https://doi.org/10.5306/wjco.v11.i6.337
Ananian SG, Gvetadze SR, Ilkaev KD, Mochalnikova VV, Zayratiants GO, Mkhitarov VA, Yang X, Ciciashvili AM (2015)Anatomic-histologic study of the floor of the mouth: the lingual lymph nodes. Jpn J Clin Oncol 45(6):547–554. https://doi.org/10.1093/jjco/hyv029
Fujimura A, Sato Y, Shoji M, Onodera M, Nozaka Y (2007) Lymphatic architecture of the oral region - beneath the buccal mucosa. Microvasc Rev Commun 1(1):9–11. https://doi.org/10.14532/mvrc.1.9
Ossoff RH, Sisson GA (1981) Lymphatics of the floor of the mouth and neck: anatomical studies related to contralateral drainage pathways. Laryngoscope 91(11):1847–1850. https://doi.org/10.1288/00005537-198111000-00008
Mohammadi-Samani S, Jamshidzadeh A, Montaseri H, Rangbar-Zahedani M, Kianrad R (2010) The effects of some permeability enhancers on the percutaneous absorption of lidocaine. Pak J Pharm Sci 23(1):83–88
Sakdiset P, Kitao Y, Todo H, Sugibayashi K (2017)High-throughput screening of potential skin penetration-enhancers using stratum corneum lipid liposomes: preliminary evaluation for different concentrations of ethanol. J Pharm (Cairo) 2017:7409420. https://doi.org/10.1155/2017/7409420
Ali Khan A, Mudassir J, Mohtar N, Darwis Y (2013) Advanced drug delivery to the lymphatic system: lipid-based nanoformulations. Int J Nanomedicine 8:2733–2744. https://doi.org/10.2147/ijn.s41521
Cho HY, Lee YB (2014)Nano-sized drug delivery systems for lymphatic delivery. J Nanosci Nanotechnol 14(1):868–880. https://doi.org/10.1166/jnn.2014.9122
Zhang XY, Lu WY (2014) Recent advances in lymphatic targeted drug delivery system for tumor metastasis. Cancer Biol Med 11(4):247–254. https://doi.org/10.7497/j.issn.2095-3941.2014.04.003
Ahn H, Park JH (2016) Liposomal delivery systems for intestinal lymphatic drug transport. Biomater Res 20:36. https://doi.org/10.1186/s40824-016-0083-1
Acknowledgements
We dedicate this report to the memory of Professor Geoffrey Burnstock, the discoverer of purinergic signalling. This report makes clear that purinergic signalling is indispensable to see through the complicated pathophysiology of hyperinflammation in COVID-19.
We would like to thank Coosje van der Pol, PhD, for her valuable and constructive editing suggestions during the writing of this research work.
Author information
Authors and Affiliations
Contributions
Conception, design and data handling: DH, AS, CvK, EPK and TK
Substantial contributions to the conception and design of the manuscript: DH, AS, CvK and EPK
Acquisition, analysis or interpretation of data for the manuscript: DH, AS, EPK and TK
Drafting and revising the manuscript: DH, AS, CvK, PJvdS, EPK and TK
Drafting the manuscript: DH, AS, CvK, PJvdS, EPK and TK
Revising the manuscript critically for intellectual content: DH, AS, CvK, PJvdS, EPK and TK
Final approval of the version to be published: DH, AS, CvK, PJvdS, EPK and TK
Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: DH, AS, CvK, PJvdS, EPK and TK
Corresponding author
Ethics declarations
Ethics approval, consent to participate and consent for publication
The Medical Ethical Committee of the Showa University, School of Medicine, Tokyo, approved the collection, analysis and publication of patients on mechanical ventilation admitted to the ICU (protocol number 3313).
Conflicts of interest
DH: The author declares to have filed patent applications that are based on the content of the paper.
AS: none.
CvK: none.
PJvdS: none.
EPK: none.
TK: none.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hasan, D., Shono, A., van Kalken, C.K. et al. A novel definition and treatment of hyperinflammation in COVID-19 based on purinergic signalling. Purinergic Signalling 18, 13–59 (2022). https://doi.org/10.1007/s11302-021-09814-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-021-09814-6