
Abstract
The novel coronavirus disease (COVID-19) pandemic has caused an unprecedented worldwide socio-economic and health impact. There is increasing evidence that a combination of inflammation and hypercoagulable state are the main mechanisms of respiratory failure in these patients. This narrative review aims to summarize currently available evidence on the complex interplay of immune dysregulation, hypercoagulability, and thrombosis in the pathogenesis of respiratory failure in COVID-19 disease. In addition, we will describe the experience of anticoagulation and anti-inflammatory strategies that have been tested. Profound suppression of the adaptive and hyperactivity of innate immune systems with macrophage activation appears to be a prominent feature in this infection. Immune dysregulation together with endotheliitis and severe hypercoagulability results in thromboinflammation and microvascular thrombosis in the pulmonary vasculature leading to severe respiratory distress. Currently, some guidelines recommend the use of prophylactic low molecular weight heparin in all hospitalized patients, with intermediate dose prophylaxis in those needing intensive care, and the use of therapeutic anticoagulation in patients with proven or suspected thrombosis. Strong recommendations cannot be made until this approach is validated by trial results. To target the inflammatory cascade, low-dose dexamethasone appears to be helpful in moderate to severe cases and trials with anti-interleukin agents (e.g., tocilizumab, anakinra, siltuximab) and non-steroidal anti-inflammatory drugs are showing early promising results. Potential newer agents (e.g., Janus kinase inhibitor such as ruxolitinib, baricitinib, fedratinib) are likely to be investigated in clinical trials. Unfortunately, current trials are mostly examining these agents in isolation and there may be a significant delay before evidence-based practice can be implemented. It is plausible that a combination of anti-viral drugs together with anti-inflammatory and anti-coagulation medicines will be the most successful strategy in managing severely affected patients with COVID-19.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Immune dysregulation in COVID-19 infection includes suppression of adaptive but hyperactivation of innate immune system. |
Hypercoagulability combined with endotheliitis and hyperinflammation results in significant incidence of pulmonary microvascular thrombosis. |
Aggressive prophylactic anticoagulation strategy is likely to be effective in sick patients in preventing thrombosis and improving outcome. |
Clinical and laboratory biomarker of disease severity will help to identify who will benefit from combination of antiviral, anti inflammatory and anticoagulation drugs. |
Urgent international collaboration is necessary to run large trials to find which drugs and in what combination will be most effective in treating COVID-19 patients. |
Introduction
A novel coronavirus infection caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), now called Coronavirus disease-19 (COVID-19), has led to a global pandemic in 2020. It has affected 10,302,867 people worldwide with 505,518 deaths directly attributed to the virus at the time of writing this paper (June 30, 2020), and the rate of new infections continues to increase at a very fast pace [1]. While most of those infected have mild cases of the disease, up to 14% can have a severe clinical picture (i.e., dyspnea, tachypnea, hypoxemia and/or lung infiltrates on radiological imaging), and approximately 5% of the infected patients will require admission to the intensive care unit (ICU) for respiratory failure, septic shock, and/or multiple organ failure [2].
The severe respiratory failure caused by COVID-19 infection results in acute hypoxemia, which is associated with severe ventilation/perfusion (V/Q) mismatch and overt intrapulmonary shunting [3, 4]. However, the exact mechanism of respiratory failure remains unclear and there is evidence that pulmonary microvascular thrombosis due to thrombo-inflammation may be a major factor contributing to respiratory decompensation in these patients [5].
This narrative review aims to summarize the current available evidence on the interplay between hypercoagulability, thrombo-inflammation, and pulmonary microvascular thrombosis in COVID-19 infection resulting in respiratory failure and how this information can be used to design clinical trials to optimize patient outcomes. We begin with a concise review of the unique properties of SARS-CoV-2 that impacts the coagulation and inflammation pathways before proceeding with an in-depth discussion on the current available medical literature. We will also discuss anticoagulation and other pharmacological strategies described for these patients that pertain to the role of the coagulation pathway in the prevention and management of COVID-19-induced respiratory failure. For the purpose of this review, we performed a literature search on PubMed using the following keywords: COVID-19, coronavirus, SARS-CoV-2, thrombosis, and respiratory failure. This review is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
The Virulence of SARS-CoV-2
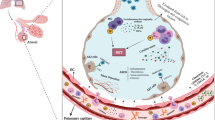
SARS-CoV-2 is a novel betacoronavirus [large ribonucleic acid (RNA) virus] that is similar to the earlier SARS-CoV virus that caused the severe acute respiratory distress (SARS) outbreak in 2003 [6]. Multiple spike glycoproteins (S) protrude from the viral surface and give it a halo-like appearance (hence corona). The spike S protein enables the virus to engage with its target cell receptor, angiotensin-converting enzyme 2 (ACE2) [7]. The host’s transmembrane protease serine 2 (TMPRSS2) primes the spike S protein of the virus to facilitate its internalization into the cell [8] (Fig. 1). After binding to the ACE2 receptor, the virus enters the cell through endocytosis, releases ribonucleic acid (RNA) into the cytosol, replicates by using the cell machinery, and finally, by the process of exocytosis, is excreted from the cell [9]. ACE2 is mostly expressed in the airway and type II pneumocytes explaining the tropism of the virus to the respiratory system. Not surprisingly, one of the major features of COVID-19 infection is involvement of the respiratory system. Importantly, ACE2 receptors are also expressed by vascular endothelial cells [10]. While most patients will have symptoms of respiratory tract infection and start to improve within a week, sudden clinical deterioration 7–8 days after initial symptom onset and development of severe respiratory failure often combined with multi-organ failure is commonly recognized [11]. The group at highest risk of deterioration includes the elderly population, people with pre-existing chronic health conditions (e.g., cancer, chronic kidney disease, chronic obstructive pulmonary disease, immunocompromised state, obesity, cardiovascular comorbidities, sickle cell disease and type 2 diabetes mellitus) [12]. In addition, patients from certain ethnicity groups (e.g., Afro-Americans, South Asians, and minority ethnic groups) and blood groups (e.g., blood group A with 3p21.31 gene cluster) have been shown to be at higher risk of worse outcomes [13,14,15] (Fig. 2).

Indirect and direct injury to the lungs by the severe acute respiratory syndrome coronavirus 2 involving the interplay between coagulation and inflammation pathways. ACE2 angiotensin-converting enzyme 2, CRP C-reactive protein, ESR erythrocyte sedimentation rate, LDH lactate dehydrogenase, NETS neutrophil extracellular traps, SARS-COV-2 severe acute respiratory syndrome coronavirus 2, TMPRSS2 transmembrane protease serine 2

Currently known clinical and laboratory biomarkers of severity to predict disease progression, combining with timely antiviral, anti-inflammatory, and anticoagulation intervention to optimize outcome. CHD chronic heart disease, CLD chronic lung disease, CKD chronic kidney disease, DOACS direct oral anticoagulants, FDPs fibrinogen degradation products, HTN hypertension, IFN interferon, JAK Janus kinase, LDH lactate dehydrogenase, LMWH low molecular weight heparin, NSAIDS non-steroidal anti-inflammatory drugs, PT prothrombin time, TNF tumor necrosis factor, VW Ag Von Willebrand antigen
Current literature suggests that a unique pattern of immune dysfunction is responsible for this disease progression in high-risk patients. Suppression of lymphocytes by the virus leads to a complicated immune mechanism with activated macrophages which releases cytokines (Fig. 1). This creates a cytokine storm [mostly interleukin IL-1β and IL-6], promoting expression of adhesion molecules for endothelial activation, inflammatory cell infiltration, and vascular inflammation. Endothelial cells release pro-inflammatory cytokines that contribute to propagation of microcirculatory lesions. The dysfunctional endothelium then becomes pro-adhesive and pro-coagulant [16]. Presence of viral elements within endothelial cells causes endotheliitis and accumulation of inflammatory cells with increasing cell death, leading to impairment of microcirculatory function in different vascular beds and their consequent clinical effects [17]. A study that examined the serum of 50 COVID-19 patients found elevated markers of neutrophil extracellular traps (NETs) [18]. NETs are microbicidal proteins, extracellular webs of chromatin, and oxidant enzymes that are released by neutrophils to contain infections. NETs have potential to cause inflammation and microvascular thrombosis in the lungs, causing respiratory failure. Highly specific markers of NETs such as myeloperoxidase (MPO)-deoxyribonucleic acid (DNA), and citrullinated histone H3 (Cit-H3) were found to be elevated in this study. These collective findings suggest a unique ability of the virus to cause intense inflammatory reaction that makes the virus lethal.
Severe hypercoagulability with respiratory failure is a recognized feature of COVID-19 patients admitted in intensive care units (ICU) [19]. A combination of profound inflammation and microvascular thrombosis appears to be responsible for the clinical picture that leads to progressive multi-organ failure in a small percentage of patients, ultimately causing fatalities.
Complex Immune Dysregulation in COVID-19 Infection
Initial response to the viral infection is by the activation of the innate immune system with neutrophils, macrophages, and cytokines. This results in elevation of acute-phase reactants like C reactive protein (CRP), erythrocyte sedimentation rate (ESR), and ferritin [20, 21]. This can be considered as the first cytokine wave or hypercytokinemia, which is essential in controlling viral infection. Dysregulated type 1 interferon response has been demonstrated in mice models of SARS-CoV infection [22]. It is postulated that similar suppressed type 1 interferon response is present in COVID-19 infection, which when combined with suppression of the adaptive immune system [T,B and natural killer (NK) lymphocytes] creates an immunodeficient state that results in poor and delayed viral clearance [23]. Initial hypercytokinemia, suppressed type 1 interferon response, and the inability of the adaptive immune system to clear the virus sets the stage for hyper activation of the innate immune system and cytokine storm in some patients. Cytokine dysregulation is a recognized feature of this infection. Studies in hospitalized patients have shown that plasma interleukin1 beta(IL1β), IL-1 receptor antagonist (IL1RA), IL7, IL8, IL9, IL10, granulocyte colony-stimulating factor (GCSF), granulocyte monocyte colony-stimulating factor (GMCSF), interferon gamma (IFNγ), interferon gamma-induced protein 10 (IP10), monocyte chemoattractant protein 1 (MCP1), macrophage inflammatory protein 1 alpha (MIP1A), macrophage inflammatory protein 1 beta (MIP1B), platelet-derived growth factor (PDGF), tumor necrosis factor alpha (TNFα), and vascular endothelial growth factor (VEGF) concentrations on admission were higher in patients with COVID-19 infection compared to healthy adults [21]. In the same study, plasma concentrations of IL2, IL7, IL10, GCSF, IP10, MCP1, MIP1A, and TNFα were found to be higher in patients needing ICU admission compared to those who did not. Disease severity has been found to positively correlate with serum levels of TNFα, IL6, and IL10, and there was also a strong negative correlation of levels of TNFα and IL6 with the number of CD4+, CD8+ T lymphocytes [24]. In a study by Diao et al., higher levels of the exhausted marker PD-1 were seen in COVID-19 patients, suggesting not only that lymphocyte numbers are lower, but existing T cells are functionally exhausted. In a larger study of 452 patients, of whom 286 were classified as having severe disease, dysregulation of the immune system was seen with a high leucocyte count and high neutrophil-to-lymphocyte ratio [25]. Regulatory T cells that help control inflammation were also found to be low in this study, especially in patients with severe disease. These mechanisms partly explain the unabated progression of the inflammatory process, the ongoing cytokine storm, and the progressive tissue damage. The similarity of severe COVID-19 disease to thrombotic microangiopathic anemia with elevated lactate dehydrogenase (LDH), D-dimer and bilirubin, decreased platelets, anemia, and renal and cardiac injury is suggestive of excessive complement activation. Hence, complement inhibition as a therapeutic option has also been suggested [26].
Another multi-center study conducted in Europe investigated immune dysregulation in 54 patients (28 with severe respiratory failure). Clinical features suggestive of macrophage activation syndrome (MAS) associated with low CD4+ T-cells, CD19+ B-cells, and NK cells were found in all the patients. Sustained elevation of TNFα and IL-6 was also found, representing a hyperactive monocyte macrophage system [11]. In another study, lymphocyte subset analysis in COVID-19 patients showed a statistically significant lower number of CD4+ helper T cells, CD8+ cytotoxic T cells, NK cells, and B cells when compared to healthy controls [27]. In this study, patients with severe disease (32% cases) had significantly lower CD4+ and CD8+ T cells and B cells, although no significant differences were seen in the CD4/CD8 ratio or NK cell numbers when compared to those with mild symptoms.
These studies suggest that in this disease, a suppression of the adaptive immune system along with hyper activation of the innate immune system causes a disease phenomenon that is similar to MAS/hemophagocytic lymphohistiocytosis (HLH). Comparing the laboratory features, hyperferritinemia and high lactate dehydrogenase (LDH) levels are common in both COVID-19 patients and patients with primary HLH. On the other hand, low fibrinogen and cytopenias due to bone marrow hemophagocytosis is not commonly found in COVID-19 patients [28]. It is likely that the pathophysiology of COVID-19 overlaps with low-grade HLH, and further studies addressing this aspect will be important to evaluate the role of immunosuppression and HLH type therapy in these patients.
COVID-19-Associated Coagulopathy: More Thrombosis, Less Bleeding
The acronym COVID-19-associated coagulopathy (CAC) is being used to describe the coagulation changes in infected patients [29]. Abnormal coagulation parameters including increased D-dimer, fibrin degradation products, and prolonged pro-thrombin time have been found to be associated with poor prognosis [30]. In a study of 183 patients with COVID-19 disease, 15 out of 21 (71.4%) non-survivors had disseminated intravascular coagulation (DIC) as per the criteria set by the International Society of Thrombosis and Haemostasis (ISTH). This was in contrast to only one (0.6%) survivor fulfilling the criteria for DIC during the period of hospitalization, indicating that non-survivors have a higher incidence of DIC. Described incidence of thrombocytopenia has been variable, reported as 12% in an initial case series of 99 patients [31] and 36.2% in another series of 1099 patients [32]. A meta-analysis analyzing pooled data from nine studies of 1779 patients including 399 with severe disease has demonstrated that thrombocytopenia is prognostic of the severity of the disease. Although there was notable heterogeneity between studies, severe thrombocytopenia was common in non-survivors [33] and increased mortality has been associated with lower platelet counts [34]. Activated partial thromboplastin time (aPTT)-based clot waveform analysis (CWA) is an easily available form of global hemostatic assay and remarkably high CWA were noted despite prophylactic anticoagulation in patients admitted to the ICU although there was no thrombosis or death in a study from Singapore [35]. A study on 24 critically ill patients analyzed whole-blood thromboelastography (TEG) which showed decreased R (reaction time) and K (kinetics time) values but increased K angle and MA (maximum amplitude) values, which is consistent with hypercoagulability. Factor VIII, Von Willebrand factor, fibrinogen, D-dimer, platelets, and protein C were increased in most patients, and antithrombin marginally decreased, while prothrombin time (PT) and APTT were mostly normal [36]. The authors concluded that the above findings do not support features of acute DIC but are consistent with severe hypercoagulability secondary to inflammation. Another similar study in 44 patients admitted to the ICU with COVID-19 showed fibrinolysis shutdown as evidenced by markedly raised D-dimer and complete absence of clot lysis in TEG at 30 min; this was predictive of high rate of venous thrombosis) and need for hemodialysis [37]. This may suggest that functional coagulation assessment like TEG may be more useful in assessing hypercoagulability rather than conventional coagulation tests in patients with COVID-19.
A high rate of venous thrombosis has been observed even in anti-coagulated patients with this disease. In a French study of 26 patients on mechanical ventilation, prophylactic anticoagulation was used in eight (31%) cases while therapeutic dose anticoagulation with low molecular weight heparin (LMWH) was started on admission in 18 (69%) patients [38]. All of these patients were screened by Doppler ultrasound of the lower limbs. Despite anticoagulation, high rates of deep vein thrombosis were noted in all patients in the prophylactic dose group and 56% in the therapeutic dose group that was associated with high rates of pulmonary embolism [eight (23%) in the whole cohort]. A larger study involving 184 patients admitted in three hospitals in the Netherlands and treated with prophylactic enoxaparin reported an incidence of 31% and 25% of deep vein or arterial thrombosis and pulmonary embolism, respectively [39]. A follow-up of the same cohort of patients was published, which confirmed the high incidence of pulmonary embolism (65/75, 87%) in those patients diagnosed with deep vein thrombosis; and pulmonary embolism was associated with higher risk of all-cause mortality [40].
A high rate (45%) of lupus anticoagulant activity was identified in a study of 56 patients when screened by a sensitive APTT test and direct Russel viper venom test (DRVVT). This may be a contributing factor in the development of thrombosis in some patients [41]. Peripheral arterial disease in the form of arteriosclerosis obliterans of lower extremity in combination with deep vein thrombosis has also been reported in severe infection, which confirms the presence of a severe hypercoagulable status in these patients [42].
Pulmonary Thrombo-Inflammation as One of the Major Pathogenic Mechanisms of Respiratory Failure
The pathophysiological mechanism of respiratory failure in COVID-19 infection is not clear at present but there is an increasing body of evidence suggesting microthrombosis of the pulmonary vasculature is a key player (Fig. 1). In COVID-19 pneumonitis, histological changes showed pauci-inflammatory septal capillary injury with significant septal capillary fibrin deposition and neutrophil infiltration of alveolar septa [43]. Viral cytopathic and classical acute respiratory distress syndrome (ARDS) changes such as diffuse alveolar damage with hyaline membranes or hyperplasia of type II pneumocyte were not prominent. Deposits of terminal complement components C5b-9, C4d, and mannose binding lectin-associated serine protease (MASP) in the microvasculature was noted, suggesting activation of the alternative and lectin-based complement pathways. Another study with lung autopsy findings from ten patients showed predominantly proliferative diffuse alveolar damage, epithelial viral cytopathic effects of small airway epithelium, but minimal lymphocytic infiltration [44]. The lung injury observed in this study was secondary to formation of fibrinous thrombi in small pulmonary arteriole, endothelial tumefaction, and megakaryocyte aggregation in pulmonary capillaries, suggesting activation of coagulation cascade with microthrombus formation. The term “microvascular COVID-19 lung vessels obstructive thrombo-inflammatory syndrome (microCLOTS)” has been proposed to describe the thrombo-inflammatory lung pathology [5]. The working hypothesis is that in individuals at risk of severe disease, an inflammatory reaction and pulmonary microvascular thrombosis leading to alveolar damage is initiated. Because of the systemic endotheliitis, this thrombo-inflammatory syndrome may progress to involve a microvascular bed of other major organs contributing to multi-organ failure. It is increasingly recognized that in this infection, pulmonary thrombus develops due to a local inflammatory reaction. Platelets interacting with the vascular wall, leukocytes, factor XIIa, Von Willebrand factor and complement leads to an intense thrombo-inflammation, which is a direct consequence of vascular damage associated with viral infection [45].
Antithrombotic Therapy in COVID-19
Because of the unusually high incidence of thrombotic complications despite prophylactic anticoagulation, some centers recommend a higher dose for prophylactic anticoagulation (e.g., enoxaparin 40 mg twice a day rather than the conventional dosing of 40 mg once a day) [39]. Although there appears to be a consensus about treating all hospitalized patients with some form of anticoagulation, dosing strategies are not yet clear. The ISTH interim guidelines and some other expert opinions have proposed that routine therapeutic anticoagulation is not advised (even in patients who required mechanical ventilation); instead, prophylactic doses of LMWH should be considered in the absence of contraindications [29, 46, 47. The dose recommended remains controversial, with some experts advocating intermediate doses (0.5 mg/kg/dose twice a day). However, this approach of either standard or intermediate dose prophylaxis has been questioned and systemic anticoagulation with unfractionated heparin infusion is routinely used for critically ill patients in some centers [48]. This approach is recommended because CAC is considered to be an overwhelmingly thrombotic DIC with pulmonary embolism being difficult to diagnose in patients who require mechanical ventilation. Also, bleeding is noted to be rare even in sick patients, and hence the risk of therapeutic anticoagulation is not as great as other patients with sepsis-induced coagulopathy. Hyperfibrinogenemia with fibrin deposition in the air spaces and lung parenchyma due to a hypofibrinolytic state is well recognized in these patients [49]. Fibrin deposition can occur even before appearance of symptoms of infection as identified in lung biopsy specimens of two patients with lung cancer [50]. There is limited experience in the use of fibrinolytic therapy with tissue plasminogen activator (TPA). It has been used in off-label fashion in three patients with a noted improvement in oxygenation [increase in partial pressure of oxygen (PaO2)/fraction of inspired oxygen (FiO2) (P/F) ratio] [51]. Nebulized TPA may be effective with less risk of bleeding and this is currently being examined in a clinical trial (NCT04356833). A recent review has recommended standard prophylactic dose of anticoagulation in hospitalized but well patients, intermediate dose prophylaxis to those admitted in ICU and ARDS, and therapeutic anticoagulation to those with presumed or confirmed deep vein thrombosis [29]. Currently, there are 19 trials registered with clinicaltrials.gov that are investigating anticoagulation in these patients [52] (Table 1). These are mainly adult trials with only one pediatric study. One registered study aims to look at antiplatelet agents to prevent cardiac complications. Outcomes from these trials (Table 1) will be important for our understanding of the optimum anticoagulation strategy in this infection to prevent thrombosis-associated complications.
Anti-Inflammatory Therapy Experience in COVID-19
Glucocorticoids have been investigated in the treatment of COVID-19 disease. Unfortunately, the first randomized controlled trial of methylprednisolone against standard of care in Beijing, China (NCT04244591) had to be stopped because of lack of patients, with the pandemic ending in China. At the time of writing this review, reports but not the full peer-reviewed manuscript for the Randomised Evaluation of COVID-19 Therapy (RECOVERY) conducted in the United Kingdom provided preliminary data that low-dose dexamethasone reduced deaths by one-third in ventilated patients and by one-fifth in other patients receiving oxygen, but made no difference to those with milder disease [53]. There are several other ongoing trials of systemic steroids (NCT04348305, NCT04360876, NCT0435524, NCT04323592) and some others looking at efficacy of inhaled steroids (NCT04331054, NCT04355637, NCT04331470).
Similar to patients with SARS and Middle Eastern Respiratory Syndrome (MERS), the cytokine responses seen in patients with COVID-19 have features of a cytokine storm. One study involving 43 adult patients found significant correlation of increased baseline IL6 levels with disease severity [54] and this is supported by similar findings in another study [20]. Hence, some investigators have proposed using IL6 as a biomarker for assessing disease severity and blocking IL6 as a therapeutic strategy has been suggested [55]. Tocilizumab, a monoclonal antibody against the IL6 receptor, has been used safely and effectively in patients with rheumatoid arthritis [56]. A pilot trial of adding tocilizumab therapy in patients with severe COVID-19 disease in combination with standard therapy of lopinavir, methylprednisolone, and oxygen therapy showed remarkable short-term clinical responses in 20 out of 21 patients within 2 weeks of tocilizumab therapy [57]. No adverse drug reactions attributable to tocilizumab were noted. Following the success of this preliminary trial, a larger multicenter clinical trial is currently being undertaken (ChiCTR2000029765). Another trial in Belgium has been designed to observe the efficacy of the IL6 blockers tocilizumab and siltuximab individually and in combination with the IL1 blocker anakinra (NCT04330638). Indeed, IL1, IL18 blockade, IFN-γ inhibition, and TNFα inhibition are also amongst consideration for randomized control trials in patients with severe disease. High-dose intravenous immunoglobulin (IVIG) has been used as an immunomodulator in patients with severe disease with good outcomes [58]. There are several trials currently exploring the utility of IVIG (NCT04411667, NCT04400058, NCT04261426, NCT04403269, and NCT04350580). The role of non-steroidal anti-inflammatory drugs (NSAIDS) as therapy is extremely controversial. Ibuprofen is a commonly used NSAID, which is used for fever control, usually with acetaminophen. However, ibuprofen increases the synthesis of ACE2 enzyme and may potentially increase the risk of severity and fatality of COVID 19 infection [59]. However, currently there is no definite evidence for or against the use of ibuprofen. The European Medicine Agency (EMA) guidelines published on March 31, 2020 suggest using acetaminophen for fever but also had advised not to stop ibuprofen if the patient is on it for other reasons [60]. Currently, there are ongoing trials to assess the efficacy of inhaled ibuprofen (NCT04382768) and also oral lipid formulation of ibuprofen (LIBERATE trial, Lipid ibuprofen versus standard of care for acute hypoxemic respiratory failure due to COVID19, NCT04334629). The efficacy and safety of other NSAIDS are currently being evaluated, the details of which are beyond the scope of this article.
Combining anti-inflammatory agents with anti-viral agents is another attractive option. Artificial intelligence (AI)-powered algorithm predictions have identified baricitinib, ruxolitinib, and fedratinib as potential candidates [61]. These drugs belong to the Janus kinase (JAK) inhibitor class of medications that have been approved and used safely in conditions like myelofibrosis and rheumatoid arthritis. These drugs are powerful anti-inflammatory agents that target the JAK-STAT pathways and are likely to be effective against the deleterious consequences of elevated cytokines. Of particular interest is baricitinib, which is predicted to interfere with the ability of the virus to enter and infect lung cells by receptor-mediated endocytosis [62]. Due to the favorable pharmacological and safety profile of baricitinib, it can be considered in combination with direct-acting anti-viral agents like lopinavir, remdesivir, and ritonavir to decrease the viral replication, infectivity, as well as to calm the cytokine responses. Statins, another group of drugs that have anti-inflammatory and antithrombotic properties, have also been studied in COVID-19 patients; a retrospective study showed a favorable recovery profile and lower all-cause mortality with the use of statins in 1219 hospitalized patients with COVID-19 [63]. Convalescent plasma has historically been used to improve outcomes in a variety of viral epidemics [64]. In theory, recovered COVID-19 patients’ plasma containing anti COVID-19 neutralizing antibodies should provide passive immunity to patients with ongoing disease. However, a recent systematic review failed to demonstrate any benefit of this approach in patients hospitalized with COVID-19, in addition to uncertainty of the safety profile of convalescent plasma therapy [65].
Conclusions
It has been a steep learning curve for the medical fraternity in learning how to manage critically ill patients with COVID-19. The existing knowledge of abnormalities in inflammatory and coagulation pathways offers opportunities for identifying biomarkers for severe disease, which combined with clinical risk factors will help in prompt identification of patients at risk of severe disease who require early and targeted intervention. Better understanding of the pathophysiology suggests suppression of the adaptive immune system on one hand and a hyperactive innate immune system on the other as the two principle severity-driving mechanisms in COVID-19 infection. This leads to a MAS-like picture associated with a hypercoagulable state that causes both micro and macrovascular thrombosis in the pulmonary as well the systemic circulation. This understanding will help us develop treatment protocols targeting various inflammatory pathways as well as to optimize anticoagulation therapies. Current anticoagulation practice suggests that all hospitalized patients should receive prophylactic anticoagulation, those with severe disease needing intensive care with respiratory compromise should have intermediate dose prophylaxis and therapeutic anticoagulation reserved for those with proven or suspected thrombosis. Results of prospective trials will only answer if this strategy is optimum in preventing thrombotic complications and improving pulmonary and overall outcome. A multi-pronged approach combining anti-inflammatory drugs and anticoagulation with specific antiviral medications is likely the approach that will work in critically ill patients (Fig. 1). Many trials are currently trying to address efficacy of different antiviral, anti-inflammatory, and anticoagulation agents in isolation. Unfortunately, these trials may take a significant amount of time for completion and analysis before the evidence-based recommendations can be produced. Because of the enormous number of new patients over a short period, we have a unique opportunity to develop an international collaborative network and conduct large trials with many combination arms to answer the all-important question of which combination of antiviral, anti-inflammatory drugs with anticoagulation therapies are going to be the most effective in treating patients with severe disease. Early intervention strategy is likely to be helpful in preventing disease progression and thereby reducing the morbidity and mortality from this pandemic.
References
https://coronavirus.jhu.edu/map.html [Internet].
Wu Z, McGoogan JM. COVID-19 outbreak in China: summary of a report of 72,314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020. https://doi.org/10.1001/jama.2020.2648.
Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020. https://doi.org/10.1016/S2213-2600(20)30079-5.
Zhang T, Sun LX, Feng RE. Comparison of clinical and pathological features between severe acute respiratory syndrome and coronavirus disease 2019. Zhonghua Jie He He Hu Xi Za Zhi. 2020;43:E040.
Ciceri F, Beretta L, Scandroglio AM, Colombo S, Landoni G, Ruggeri A, et al. Microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome (MicroCLOTS): an atypical acute respiratory distress syndrome working hypothesis. Crit Care Resusc. 2020;22(2):95–7.
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–33.
Liu PP, Blet A, Smyth D, Li H. The science underlying COVID-19: implications for the cardiovascular system. Circulation. 2020;142:68–78.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–80.e8.
Shereen MA, Khan S, Kazmi A, Bashir N, Siddique R. COVID-19 infection: origin, transmission, and characteristics of human coronaviruses. J Adv Res. 2020;24:91–8.
Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111(20):2605–10.
Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020. https://doi.org/10.1016/j.chom.2020.04.009.
Clark A, Jit M, Warren-Gash C, Guthrie B, Wang HH, Mercer SW, Sanderson C, et al. Global, regional, and national estimates of the population at increased risk of severe COVID-19 due to underlying health conditions in 2020: a modelling study. Lancet Glob Health. 2020;8(8):E1003–17. https://doi.org/10.1016/S2214-109X(20)30264-3.
Pan D, Sze S, Minhas JS, Bangash MN, Pareek N, Divall P, et al. The impact of ethnicity on clinical outcomes in COVID-19: a systematic review. EClinicalMedicine. 2020;23:100404.
Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, Invernizzi P, et al. Genomewide association study of severe Covid-19 with respiratory failure. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2020283.
Latz CA, DeCarlo C, Boitano L, Png CYM, Patell R, Conrad MF, et al. Blood type and outcomes in patients with COVID-19. Ann Hematol. 2020. https://doi.org/10.1007/s00277-020-04169-1.
Boisramé-Helms J, Kremer H, Schini-Kerth V, Meziani F. Endothelial dysfunction in sepsis. Curr Vasc Pharmacol. 2013;11(2):150–60.
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020. https://doi.org/10.1016/S0140-6736(20)30937-5.
Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, Blair C, Weber A, Barnes BJ, Egeblad M, Woods RJ. Neutrophil extracellular traps (NETs) as markers of disease severity in COVID-19. medRxiv. 2020. https://doi.org/10.1101/2020.04.09.20059626.
Spiezia L, Boscolo A, Poletto F, Cerruti L, Tiberio I, Campello E, et al. COVID-19-related severe hypercoagulability in patients admitted to intensive care unit for acute respiratory failure. Thromb Haemost. 2020. https://doi.org/10.1055/s-0040-1710018.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–62.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506.
Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19(2):181–93.
Jamilloux Y, Henry T, Belot A, Viel S, Fauter M, El Jammal T, et al. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun Rev. 2020;19:102567.
Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol. 2020;11:827.
Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis. 2020. https://doi.org/10.2139/ssrn.3541136.
Campbell CM, Kahwash R. Will complement inhibition be the new target in treating COVID-19–related systemic thrombosis? Circulation. 2020;141(22):1739–41.
Wang F, Nie J, Wang H, Zhao Q, Xiong Y, Deng L, et al. Characteristics of peripheral lymphocyte subset alteration in COVID-19 pneumonia. J Infect Dis. 2020;221(11):1762–9.
Iba T, Levy JH, Levi M, Connors JM, Thachil J. Coagulopathy of coronavirus disease 2019. Crit Care Med. 2020. https://doi.org/10.1097/CCM.0000000000004458.
Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020. https://doi.org/10.1182/blood.2020006000.
Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(4):844–7.
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507–13.
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708–20.
Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta. 2020;506:145–8.
Yang X, Yang Q, Wang Y, Wu Y, Xu J, Yu Y, et al. Thrombocytopenia and its association with mortality in patients with COVID-19. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.14848.
Tan CW, Low JGH, Wong WH, Chua YY, Goh SL, Ng HJ. Critically ill COVID-19 infected patients exhibit increased clot waveform analysis parameters consistent with hypercoagulability. Am J Hematol. 2020;95(7):E156–E158158.
Panigada M, Bottino N, Tagliabue P, Grasselli G, Novembrino C, Chantarangkul V, et al. Hypercoagulability of COVID-19 patients in intensive care unit. A report of thromboelastography findings and other parameters of hemostasis. J Thromb Haemost. 2020;18(7):1738–42.
Wright FL, Vogler TO, Moore EE, Moore HB, Wohlauer MV, Urban S, et al. Fibrinolysis shutdown correlation with thromboembolic events in severe COVID-19 infection. J Am Coll Surg. 2020;231:193–203.
Llitjos JF, Leclerc M, Chochois C, Monsallier JM, Ramakers M, Auvray M, et al. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J Thromb Haemost. 2020;18:1743–6.
Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMP, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020. https://doi.org/10.1016/j.thromres.2020.04.041.
Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb Res. 2020;191:148–50.
Harzallah I, Debliquis A, Drénou B. Lupus anticoagulant is frequent in patients with Covid-19. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.14867.
Zhou B, She J, Wang Y, Ma X. Venous thrombosis and arteriosclerosis obliterans of lower extremities in a very severe patient with 2019 novel coronavirus disease: a case report. J Thromb Thrombolysis. 2020. https://doi.org/10.1007/s11239-020-02084-w.
Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement-associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020. https://doi.org/10.1016/j.trsl.2020.04.007.
Dolhnikoff M, Duarte-Neto AN, de Almeida Monteiro RA, Ferraz da Silva LF, Pierre de Oliveira E, Nascimento Saldiva PH, et al. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J Thromb Haemost. 2020;18:1517–9. https://doi.org/10.1111/jth.14844.
Danzi GB, Loffi M, Galeazzi G, Gherbesi E. Acute pulmonary embolism and COVID-19 pneumonia: a random association? Eur Heart J. 2020. https://doi.org/10.1093/eurheartj/ehaa254.
Thachil J, Tang N, Gando S, Falanga A, Cattaneo M, Levi M, et al. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J Thromb Haemost. 2020;18(5):1023–6.
Bikdeli B, Madhavan MV, Jimenez D, Chuich T, Dreyfus I, Driggin E, et al. COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-Up: JACC state-of-the-art review. J Am Coll Cardiol. 2020;75(23):2950–73.
Barrett CD, Moore HB, Yaffe MB, Moore EE. ISTH interim guidance on recognition and management of coagulopathy in COVID-19: a comment. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.14860.
Whyte CS, Morrow GB, Mitchell JL, Chowdary P, Mutch NJ. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.14872.
Tian S, Hu W, Niu L, Liu H, Xu H, Xiao SY. Pulmonary pathology of early-phase 2019 novel coronavirus (COVID-19) pneumonia in two patients with lung cancer. J Thorac Oncol. 2020;15(5):700–4.
Wang J, Hajizadeh N, Moore EE, McIntyre RC, Moore PK, Veress LA, et al. Tissue plasminogen activator (tPA) treatment for COVID-19-associated acute respiratory distress syndrome (ARDS): a case series. J Thromb Haemostasis. 2020. https://doi.org/10.1111/jth.14828.
Wilkinson E. RECOVERY trial: the UK covid-19 study resetting expectations for clinical trials. BMJ. 2020;369:m1626.
Gao Y, Li T, Han M, Li X, Wu D, Xu Y, et al. Diagnostic utility of clinical laboratory data determinations for patients with the severe COVID-19. J Med Virol. 2020. https://doi.org/10.1002/jmv.25770.
Liu B, Li M, Zhou Z, Guan X, Xiang Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease (COVID-19)-induced cytokine release syndrome (CRS)? J Autoimmun. 2019;2020:102452.
Scott LJ. Tocilizumab: a review in rheumatoid arthritis. Drugs. 2017;77(17):1865–79.
Fu B, Xu X, Wei H. Why tocilizumab could be an effective treatment for severe COVID-19? J Transl Med. 2020;18(1):164.
Cao W, Liu X, Bai T, Fan H, Hong K, Song H, et al. High-dose intravenous immunoglobulin as a therapeutic option for deteriorating patients with coronavirus disease 2019. Open Forum Infect Dis. 2020;7(3):102.
Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med. 2020;8(4):e21.
EMA advice on the use of NSAIDs for Covid-19. Drug Ther Bull. 2020;58(5):69.
Stebbing J, Phelan A, Griffin I, Tucker C, Oechsle O, Smith D, et al. COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect Dis. 2020;20(4):400–2.
Richardson P, Griffin I, Tucker C, Smith D, Oechsle O, Phelan A, et al. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet. 2020;395(10223):e30–e3131.
Zhang XJ, Qin JJ, Cheng X, Shen L, Zhao YC, Yuan Y, et al. In-hospital use of statins is associated with a reduced risk of mortality among individuals with COVID-19. Cell Metab. 2020. https://doi.org/10.1016/j.cmet.2020.06.015.
Marano G, Vaglio S, Pupella S, Facco G, Catalano L, Liumbruno GM, et al. Convalescent plasma: new evidence for an old therapeutic tool? Blood Transfus. 2016;14(2):152–7.
Piechotta V, Chai KL, Valk SJ, Doree C, Monsef I, Wood EM, et al. Convalescent plasma or hyperimmune immunoglobulin for people with COVID-19: a living systematic review. Cochrane Database Syst Rev. 2020;7:13600.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Rajat Bhattacharyya, Prasad Iyer, Ghee Chee Phua, and Lee Jan Hau have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital Features To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12739562.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bhattacharyya, R., Iyer, P., Phua, G.C. et al. The Interplay Between Coagulation and Inflammation Pathways in COVID-19-Associated Respiratory Failure: A Narrative Review. Pulm Ther 6, 215–231 (2020). https://doi.org/10.1007/s41030-020-00126-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41030-020-00126-5