
Abstract
The Japanese plum (Prunus salicina Lindl.) is one of the most important stone fruit crops in China. High-density linkage map is valuable resources which enhance functional genomics and genetic breeding studies. So far several Japanese plum linkage maps have been reported using different kinds of molecular markers; however, the marker numbers and chromosome coverage are limited. Recently, a newly developed strategy which genome sequencing towards specific-locus amplified fragments (SLAF) markers, has been proven to be powerful for rapid genotyping of genome-wide markers and for high-density genetic map construction. In this study, SLAF was used to genotype markers with 114 F1 seedlings from the ‘09–16’ × ‘Fortune’ cross. Suitable SLAF markers (160,344 out of 343,436,902 pair-end reads) were chosen to conduct genetic map construction, 16.31% of which were polymorphic. The overall integrated map contained 3,341 high quality SLAFs and 720 loci that were grouped in eight genetic linkage groups with a total length of 869.9 cM and an average distance of 1.21 cM, and only five gaps with a genetic distance > 5 cM between adjacent markers occurred in linkage group (LG) 3 and LG6. The number of markers with each LG ranged from 82.3 cM (LG3) to 138.3 cM (LG1). Aligning the map against the peach reference genome sequence (Prunus persica L.) indicated a strictly co-linear relationship between the LGs and peach genome, demonstrating the markers on ours LGs were well ordered. Overall, our studies identified large-scale of genetic markers and constructed high-density linkage maps for Japanese plum, which will obviously provide a solid foundation for marker-assisted selection and sequence assembly of the Japanese plum reference genome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Japanese plum (Prunus salicina Lindl.) has a diploid genome (2n = 2x = 16) and belongs to the subgenus Prunophora, genus Prunus, the Rosaceae family (Topp et al. 2012). The Japanese plum is a crop tree species native to China and it is widely cultivated from 23° N (the Guangxi and Yunnan provinces) to 45° N (Heihongjiang province). Due to its rich nutrients and unique flavour, the Japanese plum was popular in China, where the yield was the highest in the world (6,663,165 tonnes). Other major Japanese plum-producing countries include the USA (392,537 tonnes), followed by India (261,903 tonnes), Mexico (77,931 tonnes) and Japan (23,000 tonnes). In the Southern hemisphere plum production is dominated by Chile (294,873 tonnes) followed by Argentina (156,084 tonnes), South Africa (81,463 tonnes) and Australia (17,992 tonnes), also based on Japanese plum production (http://faostat.fao.org).
The highest plum production in China over the last century came from native cultivars, since they had good flavour and were able to adapt to complex environments, though they had poor appearance quality and shelf life. In this century, a number of breeding cultivars introduced from abroad (mainly the USA) quickly became the main planted cultivars in commercial production due to good appearance quality and suitable storage performance, though they bring a poor gene pool, flavour and adaptability (Liu et al. 2018). For over a hundred years, most Japanese plum breeding launched in the USA was based on a few cultivars, which were derived from a cross between P. salicina and several plum species of the same subgenus. Therefore, it is necessary to diversify the range of cultivars to accelerate breeding programs using Chinese native cultivars.
Marker-assisted selection (MAS) is an important biotechnological tool used in breeding programs to improve the efficiency of selected genotypes (Tavassolian et al. 2010). Genetic map construction is the basis for quantitative trait locus (QTL) mapping and MAS breeding, and it can also be used for genome-assisted assembly and evolutionary comparisons between closely related species. In the case of the Japanese plum, however, only a few studies have focused on genetic linkage map construction. A genetic linkage-mapping program was initiated by Vieira et al. (2005) and the genetic linkage maps of two parents contained only 56 markers and 84 markers using amplified fragment length polymorphism (AFLP) markers. Because of limited markers, these linkage maps were sparse with a total length of 905 cM (Chatard) and 1349.6 cM (Santa Rosa), respectively. Salazar et al. (2017) developed two genetic maps of ‘98–99’ (479 SNPs) and ‘Angeleno’ (502 SNPs) using SNP markers through genotyping by sequencing, (covering a genetic distance of 688.8 and 647.03 cM, respectively. The most saturated map is the ‘Angeleno’ x ‘Aurora’ map that has been reported by Carrasco et al. (2018). This consensus map was built using 732 SNPs, which spanned 617 cM with an average of 0.96 cM between adjacent markers; however, this map contained more large gaps (> 10 cM). All the parents described above are commercial varieties selected by crossbreeding. There may be more markers for the local varieties of Chinese plums that are heterozygous, so that more markers are mapped. In fact, the genetic linkage map for local varieties of Chinese plums has not been reported, possibly owing to a limited number of markers and suitable genetic populations.
High-density genetic linkage maps require not only appropriate genetic segregation populations but also high-throughput SNP detection technology, as they represent the whole genome information regarding molecular markers. In recent years, SNP markers using a high-throughput sequencing technique have gained popularity in genetic mapping because of their stability, heritability, low cost, high-throughput efficiency and abundant genetics variations (Guo et al. 2015). Specific-locus amplified fragment sequencing (SLAF-seq) markers is a high-throughput technique that has been widely adopted in different species for multiple purposes, including genetic diversity and population structure in sweet potato (Su et al. 2017) and genome-wide association studies in rapeseed (Zhou et al. 2017). In the last 5 years, SLAF-seq has been used successfully for construction of high-density genetic maps in many crops, including tea (Ma et al. 2015), tree peony (Cai et al. 2015), sorghum (Ji et al. 2017), sweet osmanthus (He et al. 2017) and poplars (Fang et al. 2018). Exploiting this approach to scan the whole Japanese plum genome has great importance for plum breeding through high density marker development and gene mining.
Over last two decades, we have performed Japanese plum hybridization breeding research, which has established a large number of segregation populations for the Japanese plum. In this study, we have drawn a high-density genetic linkage map in P. salicina derived from the native cultivar and a commercial cultivar using the SLAF-seq method. This genetic map makes it possible to help QTLs analysis and MAS strategies in breeding programs, facilitating future de novo chromosome assembly for the Japanese plum.
Materials and methods
Plant materials
An F1 population of 114 progenies was generated from the cross between ‘09–16’ and ‘Fortune’ cultivars in 2015. The diploid seed parent material “09–16” was the hybrid seedling of the ‘Wanshu Xiangjiaoli’ (a famous native cultivar) and ‘Akihime’ (a Japanese cultivar). The diploid pollen parent came from “Fortune”, which is an important breeding cultivar in commercial production. The seedlings of the F1 progeny were planted in the Chinese National Germplasm Repository for Plums and Apricots located at Liaoning Institute of Pomology (Liaoning, China) in 2015.
Young leaf samples were harvested from each individual F1 plant and the two parents. These samples were immediately frozen in liquid nitrogen and kept at − 80 °C. Genomic DNA was extracted using the CTAB method (Doyle and Doyle 1990). DNA samples were checked for quality and concentration and used for further experiments.
SLAF library construction and high-throughput sequencing
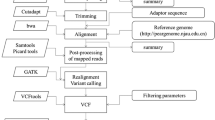
An improved SLAF-seq strategy was utilized in our experiment. Genomic DNA from each F1 individual and each parent was digested with the A1-2+A1-6 according to the Prunus persica genome (https://www.rosaceae.org/species/prunus_persica/genome_v2.0.a1) (Arús et al. 2012), the library was constructed utilizing the SLAF-seq strategy with some modifications, and an enzyme fragment length of 264–364 bp sequence was defined as the SLAF labels. The details of SLAF-seq strategy have been described by Zhang et al. (2015). The pair-end sequencing (125 bp on each end) was performed on an Illumina HiSeq 2500 system (Illumina, Inc.; San Diego, CA, USA) according to the manufacturer’s recommendations.
Sequence data mapping and genotyping
The high-quality Illumina sequences were used for SLAF marker identification and genotyping with procedures described by Sun et al. (2013). The quality of the raw 160-bp pair-end data was evaluated using FastQC with default parameters. Adaptor sequences, low quality reads, and bases with a Phred score ≤ 20 were then removed with the Cutadapt program (version 1.10). The sequences with above 90% similarity would be grouped and indentified as one SLAF marker (locus). The alleles of each SLAF were defined according to their parents, and individuals were genotyped by sequence similarity to their parents. The marker codes of the polymorphic SLAFs were analysed according to the population type cp, which consisted of segregation types ‘aa×bb’. Single nucleotide polymorphism (SNP) loci of each SLAF locus were then detected between parents, and SLAFs with more or equal 3 SNPs were filtered out firstly. For diploid species, one SLAF locus can contain at most 4 genotypes, so SLAF loci with more than 4 alleles were defined as repetitive SLAFs and subsequently discarded. Only SLAFs with 2 to 4 alleles were identified as polymorphic and considered potential markers.
Genotype scoring was then performed using a Bayesian approach to further ensure the genotyping quality. Firstly, a posteriori (conditional) probability was calculated using the coverage of each allele and the number of single nucleotide polymorphisms. Then, the genotyping quality score translated from the probability was used to select qualified markers for subsequent analysis as described by Sun et al. (2013). Low-quality markers for each marker and each individual were counted, and the inefficient marker or individual were deleted during the dynamic process. When the average genotype quality scores of all SLAF markers reached the cutoff value, the process was stopped.
Three strict criteria were used for filtering individuals and SLAF markers in order to obtain the high-quality genetic mapping: (1) average sequence depths should > 10 fold in each progeny and > 20 fold in the parents; (2) More than 11 missing genotype markers were filtered out and (3) markers with significant segregation ratios (1:2:1 or 1:1) were initially excluded from the map construction using a chi-square test (P < 0.05).
We first defined a bin to be a locus within which two or more marker cannot be ordered (Vision et al. 2000). This bin, consisting of multiple markers with no breakpoints among any individual of a given set of progenies, has limited the increase in map density through the number of markers.
Genetic linkage map analysis
Loci were separated into three types: (1) those showing segregation for the female parent (heterozygous), (2) those showing segregation for the male parent (heterozygous), and (3) those heterozygous in for both. Heterozygous genetic markers present in one parent but not the other, plus markers heterozygous for both parents, were used to construct separate genetic linkage maps for the female and male parents, using the two-way pseudo-testcross strategy. Two approaches were used for map construction. Markers that showed a segregation-ratio distortion, departing from the Mendelian ratio at the 0.05 level, were added to the maps after mapping the markers that did not depart from the Mendelian ratio. A second approach consisted of both types of markers in the analysis. Marker loci were partitioned primarily into LGs by the modified logarithm of odds (MLOD) scores > 5.
Construction of the high-density and high-quality map used the HighMap strategy for map calculation (Li et al. 2008). Map distances in centimorgans were estimated using the Kosambi mapping function. Skewed markers were then added into this map by applying a multipoint method of maximum likelihood. The SMOOTH error correction strategy of was then conducted according to the parental contribution of genotypes, and a k-nearest neighbour algorithm was applied to impute missing genotypes (Van et al. 2005). Double recombination standard is based on a particular flanking locus defined window; the size of the window is each 15 loci before and after the locus. The observed marker score at locus is calculated as the weighted value according to (Van et al., 2005). The threshold value is set as 0.9, and if the threshold value is exceeded, the existence of double recombination recombination is judged. Haplotype maps and heat maps were used to evaluate the quality of the map following the description by West et al. (2006).
Collinearity analysis
In order to assess the correspondence of each LG in P. salicina to the chromosome in P. persica, the tag sequences used for mapping were extracted and aligned with the peach genome_v2.0 scaffolds (Arús et al. 2012) using BLAST. The Pearson correlation between the linkage groups and corresponding chromosomes was calculated using the R script (Fang et al. 2018), and the consensus of their positions was represented using a dot-plot diagram.
Results
SLAF sequencing
We generated 65.36 Gb high-quality cleaned data containing 343,436,902 pair-end reads by the SLAF library from the 114 progeny and the two parents using Illumina HiSeq 2500 system. The average high-quality data in parents and individuals were 2.33 and 0.53 Gb, respectively. After eliminating the index sequences, both ends of each read were about 30 bp in length. Among them, the average high-quality bases (Q Score > 20) ratio was 97.96 %, and the average guanine-cytosine (GC) content was 38.52 % (Table S1). Of these high-quality data, 1.97 Gb were from the female parent ‘09–16’ with 9,859,875 reads and 2.70 Gb from male parent ‘Fortune’ with 2,698,346 reads. We obtained 327.20 million reads from the libraries for the 114 individuals, where the number of reads for the individuals ranged from 1.54 to 6.67 M with an average of 2.67 M reads (Fig. 1).

Distribution of reads number for F1 individuals. The x-axis indicates the interval of reads number; the y-axis indicates the number of individuals
SLAF markers discovery and genotype definition
Based on sequence similarity, all the reads were clustered into SLAFs. After eliminating the low-depth and repeat-suspicious SLAFs, 46,086,386 high-quality reads were assigned to 160,344 SLAF loci, of which 107,678 (71.92 %) and 115,319 (67.15 %) were detected in the female and male parents, respectively. The numbers of reads for SLAFs detected in the female and male parents were 6,053,916 and 8,136,595, respectively, indicating an average read depth of ~ 56.2- and ~ 70.56-fold for each SLAF. In all the 114 F1 population, the number of SLAFs per individual ranged from 71,714 to 105,567 with an average of 88,893, while the reads depth was from ~ 11- to ~ 31-fold with an average of ~ 17.3-fold at the population level. The average sequencing depths were 104.8-fold in the parents and 21.54-fold in the offspring on linked markers. The integrity and depth of the markers were enough to guarantee the accuracy of the genetic map construction.
Among the 160,344 SLAFs that were defined, 68,032 (42.43%) were polymorphic, while 91,781 (57.34%) were non-polymorphic, and only 531 SLAFs were labeled as repetitive sequences. Based on parental genotypes, the loci missing information were flitted out. The 30,168 polymorphic SLAFs were obtained and successfully classified into eight segregation patterns according to the criteria in the “Materials and methods”. As shown in Fig. 2, only 4015 SLAF markers (13.31%) were homozygous in the two parents with genotype aa or bb, which were not segregated in the offspring. Most markers (86.69%) conformed to the CP population segregation codes, including ab×cd, ef×eg, hk×hk, lm×ll, nn×np, ab×cc and cc×ab. Stringent filtering criteria were applied to identify and remove markers, with low sequence depths (less than 10-fold) or > = 2 SNPs SLAF tags, a high level of missing data or Mendelian errors. After the chi-square test, 5,134 (7.55%) SLAFs were used to construct the final genetic map. The marker codes lm×ll and nn×np represent markers with one parent heterozygous (providing 1:1 segregation ratios), where hk×hk or ef×eg represents markers with both parents heterozygous (providing 1:2:1 segregation ratios). Of these 5,134 markers, 1,118 (21.78 %) were present in the male configuration lm×ll (i.e. heterozygous only in ‘Fortune’), 3,410 (66.42 %) in the female configuration nn×np (i.e. heterozygous only in ‘09-16’), and the remaining 395 (7.69 %) in the bi-parent configuration hk×hk or ef×eg (i.e. heterozygous in both parents), agreeing with the higher heterozygosity observed in ‘09–16’ with respect to ‘Fortune’.

Distribution of SLAF markers in eight segregation patterns
Linkage map construction
Using HighMap to construct the genetic map, 3,341 SNP markers were assigned to eight linkage groups (LG1–LG8) for the integrated map of the Japanese plum based on the ‘09–16’ × ‘Fortune’ progeny with a grouping LOD value of 5 (Figure S1 and Table S2). The genetic map spanned 869.9 cM in whole genomic region, implying an average markers distance of 0.27 cM. Of the mapped SNP markers, the marker numbers of per loci varied from 1 to 45 with an average 4.64. After linkage analysis, 3,166 (94.76 %) mapped markers were grouped into 545 bins and 175 markers were singletons, indicating there were the total 720 loci on the map with an interval between two adjacent loci of 1.21 cM (Table 1 and Fig. 3). The longest chromosome was LG1 with 138.3 cM, whereas the shortest chromosome was the LG3 with 82.3 cM in the integrated map.
A high-quality integrated linkage map should have short distances between adjacent loci and evenly distributed markers. The average intervals between two adjacent mapped loci ranged from 1.09 (LG4) to 1.62 cM (LG6) for this integrated maps (Table 1). Our results show that most of the SLAF markers were evenly mapped in eight groups. Among the 417 intervals (58.57%) were smaller than 1 cM, and only 4 intervals (0.56%) were larger than 5 cM. The largest gap in no marker coverage on this map was located in LG3 with a length of 7.5 cM. These results indicate that this genetic map is of high quality and high resolution.

Integrated linkage map of the Japanese plum for “‘09–-16”’ × ×“‘Fortune”’ composed of 720 loci. The font of the locus name in red indicates that the locus has at least a segregation distortion marker. An asterisk in the locus name indicates that only segregation distortion markers are present at the locus.
Map evaluation
We developed an integrated genetic map of the Japanese plum with 3,341 SLAF markers, which shows a high depth of coverage in the parents (mean of ~ 106-fold) and the F1 progeny (mean of ~ 22-fold), as well as high genotype integrity in the mapping population with an average value of 99.93% (Table S3). The results of the χ2 test indicated that 245 (7.33%) of the 3,341 markers showed significant segregation distortion (P < 0.05) on the integrated linkage map (Figure S1 and Table S2), where most of the distorted markers were distributed unevenly on seven out of eight LGs, and not distributed on LG4. A total of 84, 56 and 45 distorted markers were clustered on LG2, LG6 and LG3, respectively. The majority (94.29%) of distorted markers mapped were similarly located on some particular LG.
From the haplotype map (Figure S2), the results show that the source of each individual was consistent in the large segment of each linkage group. This result shows that there is almost no double recombination in individual off-spring. As can be seen from the heat map, the markers in each linkage group were ordered (Figure S3), indicating the high quality of the genetic map.
Collinearity analysis
The Japanese plum (P. salicina) and peach (P. persica) both belong to the genus Prunus, and have a high level of genetic similarity. The publication of the peach genome v2.0a1 provides an excellent opportunity for genetic and genomic studies of the Japanese plum. In order to assess the quality of this genetic map, the tags used for mapping were compared with the peach reference genome. Eight hundred and eighty six of the SLAF markers in the integrated map were mapped to different physical chromosome of the peach genome V2.1a using BLAST (Table S4). A dot-plot diagram was used to evaluate the physical distances of the mapped SLAF tags on the peach genome sequence plotted against their genetic positions on the Japanese plum genetic map (Fig. 4). The Spearman correlation coefficient (r2 value) of LGs ranged from 0.714 (LG1) to 0.980 (LG5) showing all linkage groups have good linear agreement between the physical and genetic maps (Table 2). However, there was also nonconformity of some regions, such as LG1 (0.714) and LG2 (0.808). The reason might be genome sequence differences between the different species between Japanese plum and peach.

Collinear analysis of the consensus between integrated Japanese plum genetic maps and peach genome. The x-axis indicates the genetic position of each SLAF marker; the y-axis indicates the physical position of each SLAF marker
Discussion
Due to abundant genetic variation and heritability, SNP markers have become increasingly popular in genetic mapping, evolutionary relationship analysis, population structure analysis and association analysis. With the development of next-generation sequencing (NGS) technologies, large-scale SNPs have been successfully discovered in Japanese plum genomes (Salazar et al. 2017; Marti et al. 2018; Carrasco et al. 2018). The SLAF-seq approach is one of the most important SNP detection technologies. SLAF-seq provides an accurate, effective and low-cost method for developing molecular markers. In this study, SLAF sequencing was successfully employed for large-scale SNP discovery and genotyping in an F1 population of Japanese plum. We constructed a SLAF library of the Japanese plum, and obtained 68.6 Gb of data containing 343 M pair-end reads from this library, and 160,344 SLAF tags were detected, and 68,032 polymorphic markers were identified, such that the SLAF markers in this study were far more abundant than the SNP markers obtained through GBS technology (Salazar et al. 2017; Carrasco et al. 2018). This result indicates that the markers from SLAF library would be a useful tool for genetic linkage map, genetic diversity and association analysis.
Because of the uniformly distributed genome, SLAF-seq has been used successfully over the last 5 years for high-density genetic map construction in many perennial plants, such as tea (Ma et al. 2015), tree peony (Cai et al. 2015), sweet osmanthus (He et al. 2017) and poplars (Fang et al. 2018). In the absence of genomic information, Cai et al. (2015) constructed an integrated genetic map, which comprised 1189 SLAF markers on the five linkage groups and spanned 920.70 cM with an average inter-marker distance of 0.77 cM. Based on SLAF sequencing technology, Ma et al. (2015) developed a high-density genetic map of tea with a total map length of 3,965 cM and an average inter-locus distance of 1.0 cM, where the 99.2% distance between adjacent markers was smaller than 5 cM. These results demonstrate the value of SLAF-seq for the development of large numbers of mapping SNP markers and for the construction of the highly saturated linkage maps. In this study, we successfully constructed a saturated integrated genetic map of the Japanese plum using a ‘09–16’ × ‘Fortune’ F1 population. The total length of the integrated linkage group reached 869.9 cM with 720 SLAF loci and an average distance of 1.21 cM. On the integrated map, only five gaps with a genetic distance > 5 cM between adjacent markers occurred in LG3 and LG6. Compared with the maps of AFLP markers (Vieira et al. 2005) or SNPs markers (Salazar et al. 2017; Carrasco et al. 2018), this linkage map had the longest length, the highest density and was the finest map, making it a powerful tool for research involving marker-assisted selection of traits of the Japanese plum.
The improvement in sequencing depth could not only reduce the error rate of SNP detection or false positive, but also improve the sequencing coverage of tags and the number of markers (Sun et al. 2013). In previous reports, the average sequencing depth of the offspring population in tree peony (Cai et al. 2015), tea (Ma et al. 2015), sweet osmanthus (He et al. 2017) and sorghum (Ji et al. 2017) was 2.41-, 6.0-, 5.27- and 3.3-fold, respectively. Mapping of the SLAF markers in this study produced, an average sequencing depth in the offspring population of 21.5-fold, which was significantly higher than the depth of the studies described above. The sequencing depth of the SNP markers was 15.0-fold in the plum population, as determined through genotyping by sequencing (Salazar et al. 2017), and the mapping marker number of the genetic linkage map was 981 SNP markers. The number of SLAF markers in this article was much higher than the SNP markers in the previous study. As recombinational crossover sites or fragmentation sites were limited in a certain genetic population (Vision et al. 2000), it was difficult to detect new recombination hotspots by simply adding to the number of mapping markers. Therefore, the much greater number of markers in this study was clustered to a single bin, which cannot be ordered relative to on another without supplementary marker information. This result indicates that the SLAF markers adhered to the requirements of the high-density linkage map in this study.
The length of the linkage map in the present study is significantly different from previously published genetic maps for the in Japanese plum (Salazar et al. 2017; Carrasco et al. 2018). The results in this paper were similar to the conclusion by Salazar et al. (2017), where the longest linkage group was LG1. This supposes nearly 45 Mb for LG1 and below 20 Mb for LG5 by Arús et al. (2012), however, the length of LG5 in this paper was 95.2 cM. In the LG5, the number of the 312 mapped markers in this paper was much bigger than the number of 54 markers in ‘Angeleno’ (Salazar et al. 2017) and 121 markers in a consensus map of the ‘Angeleno’ and ‘Aurora’ cultivars (Carrasco et al. 2018). There are two possible reasons for this result. First, the differences may be caused by differences in recombination hotspots of the populations among different parents. The material used in Salazar et al. (2017) and Carrasco et al. (2018) were two cultivars from a hybrid of the Burbank series, but the materials used in this study were a local Chinese cultivar and a cultivar of the Burbank series, where the latter results in a higher recombination frequency and more marker polymorphism. The genetic distance refers to the recombination frequency between two linked genetic markers, not a physical distance. Second, this may also be related to the number of mapping markers. In tea tree, Ma et al. (2015) observed that the LG size was significantly correlated with the marker numbers (r2 = 0.88). The 3441 markers (720 loci) mapped on the integrated map in this paper, which was much higher than the 479 and 502 for flower and pollen parent maps in Salazar et al. (2017), respectively.The mapped markers in this study not only expanded the length of the LGs but also increased the density of the integrated map. The largest gap in marker coverage on the integrated map in this paper was located in LG3 with a length of 7.5 cM. Therefore, this map will be a powerful tool for research involving both fine-scales and QTL mapping of important economic traits of the Japanese plum.
Owing to the influence of gametophyte and zygote genotype selection, partial separation of the map loci was a common phenomenon, which not related to the heterozygosity or the size of the population (Li et al. 2011). The linkage maps in apricot were about 35% of the markers and showed partial separation based on SSR and SRAP markers (Zhang et al. 2011). About 25.3% of the mapped markers deviated from the Mendelian segregation patterns in tea plant, and the distorted markers were clustered in specific LG regions (Ma et al. 2015), while 37.85% of the mapped markers in tree peony showed a clustered distribution in 54 segregation distortion regions, distributed between LGs (Cai et al. 2015). In this study, only 7.33% markers showed partial separation, and these markers were distributed unevenly on eight groups, expect LG4.
The genus Prunus has shown conserved intraspecific and intragenic co-linearity in the Rosaceae; therefore, we attempted a collinearity analysis between the peach and Japanese plum in order to compare these mapped marker sequences to the peach genome_v2.0 scaffolds (Arús et al. 2012). The results of collinearity show the correlation of all eight groups was greater than 0.8. These results indicated a high level of collinearity between the Japanese plum and peach genomes, in agreement with the results from Carrasco et al. (2018). Most of the markers showed good collinear agreement with the peach genome on LG5, LG7, and LG8. Some markers on LG1 and LG2 were uniform with the peach genome because of genome structure differences between the different species. These collinear markers on this map will provide a wealth of genomic information, which may aid in de novo genome assembly and comparative linkage mapping in the plum plant.
In conclusion, we have constructed the refined high-density linkage map for Japanese plum using the SLAF-seq method. The map spans 869.9 cM and is divided into eight linkage groups corresponding to the number of P. salicina chromosomes, with an average distance of 1.21 cM among 720 SLAF loci. Our results further demonstrate that SLAF-seq is a very effective method for developing markers and constructing high-density linkage maps. The most marker loci on the pseudo-chromosomes showed good collinear agreement with the physical maps of the Prunus persica. Our study provides a valuable genetic resource for QTL analysis, MAS, map-based gene cloning and the sequence assembly of the Japanese plum reference genome.
References
Arús P, Verde I, Sosinski B, Zhebentyayeva T, Abbott AG (2012) The peach genome. Tree Genetics & Genomes 8(3):531–547
Cai CF, Cheng FY, Wu J, Zhong Y, Liu GX (2015) The first high-density genetic map construction in tree peony (Paeonia Sect. Moutan) using genotyping by specific-locus amplified fragment sequencing. PloS one 10(5):e0128584
Carrasco B, González M, Gebauer M, García-González R, Maldonado J, Silva H (2018) Construction of a highly saturated linkage map in Japanese plum (Prunus salicina L.) using GBS for SNP marker calling. PloS one 13(12):e0208032
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Fang LC, Liu HL, Wei SY, Keefover-Ring K, Yin TM (2018) High-density genetic map of Populus deltoides constructed by using specific length amplified fragment sequencing. Tree Genetics & Genomes 14(5):79
Guo F, Yu HW, Tang Z, Jiang XL, Wang L, Wang X, Xu Q, Deng XX (2015) Construction of a SNP-based high-density genetic map for pummelo using RAD sequencing. Tree Genetics & Genomes 11:2
He YX, Yuan WJ, Dong MF, Han YJ, Shang FD (2017) The first genetic map in sweet osmanthus (Osmanthus fragrans Lour.) using specific locus amplified fragment sequencing. Frontiers in Plant Science 8:1621
Ji GS, Zhang QJ, Du RH, Lv P, Ma X, Fan S, Li SY, Hou SL, Han YC, Liu GQ (2017) Construction of a high-density genetic map using specific-locus amplified fragments in sorghum. BMC genomics 18(1):51
Li R, Li Y, Kristiansen K, Wang J (2008) SOAP: short oligonucleotide alignment program. Bioinformatics 24(5):713–714
Li XH, Wang XJ, Wei YL, Brummer EC (2011) Prevalence of segregation distortion in diploid alfalfa and its implications for genetics and breeding applications. Theoretical and Applied Genetics 123(4):667–679
Liu S, Xu M, Zhang YP, Zhang YJ, Ma XX, Zhang QP, Liu N, Liu WS (2018) Retrospect, problematic -cal issues and the prospect of plum breeding in China. Journal of fruit science 35(02):231–245
Ma JQ, Huang L, Ma CL, Jin JQ, Li CF, Wang RK, Zheng HK, Yao MZ, Chen L (2015) Large-scale SNP discovery and genotyping for constructing a high-density genetic map of tea plant using specific-locus amplified fragment sequencing (SLAF-seq). PloS one 10(6):e0128798
Marti AFI, Saski CA, Manganaris GA, Gasic K, Crisosto CH (2018) Genomic sequencing of Japanese plum (Prunus salicina Lindl.) mutants provides a new model for Rosaceae fruit ripening studies. Frontiers in. Plant Science 9:21
Salazar JA, Pacheco I, Shinya P, Zapata P, Silva C, Aradhya M, Velasco D, Ruiz D, Martínez-Gómez P, Infante R (2017) Genotyping by sequencing for snp-based linkage analysis and identification of qtls linked to fruit quality traits in Japanese plum (Prunus salicina Lindl.). Frontiers in Plant Science 8:476
Su WJ, Wang LJ, Lei J, Chai SS, Liu Y, Yang YY, Yang XS, Jiao CH (2017) Genome-wide assessment of population structure and genetic diversity and development of a core germplasm set for sweet potato based on specific length amplified fragment (SLAF) sequencing. PloS one 12(2):e0172066
Sun XW, Liu DY, Zhang XF, Li WB, Liu H, Hong WG, Jiang CB, Guan N, Ma CX, Zeng HP, Xu CH, Song J, Huang L, Wang CM, Shi JJ, Wang R, Zheng XH, Lu CY, Wang XW, Zheng HK (2013) SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PloS one 8(3):e58700
Tavassolian I, Rabiei G, Gregory D, Mnejja M, Wirthensohn MG, Hunt PW, Gibson JP, Ford CM, Sedgley M, Wu SB (2010) Construction of an almond linkage map in an Australian population Nonpareil × Lauranne. BMC genomics 11(1):551
Topp BL, Russell DM, Neumuller M, Dalbo M, Liu WS (2012) Plum. Badenes ML, Byrne DH. Fruit Breeding. Media: Springer Science Business: 571-621.
Van OH, Stam P, Visser RG, van Eck HJ (2005) SMOOTH: a statistical method for successful removal of genotyping errors from high-density genetic linkage data. Theoretical and Applied Genetics 112(1):187–194
Vieira EA, Nodari RO, Dantas A, Ducroquet JPHJ, Dalbó M, Borges CV (2005) Genetic mapping of Japanese plum. Crop Breed Appl Biotech 5:29–37
Vision TJ, Brown DG, Shmoys DB, Durrett RT, Tanksley SD (2000) Selective mapping: a strategy for optimizing the construction of high-density linkage maps. Genetics 155(1):407–420
West MAL, Van L, Leeuwen HV, Kozik A, Daniel JK, Doerge RW, DAS C, Michelmore RW (2006) High-density haplotyping with microarray-based expression and single feature polymorphism markers in Arabidopsis. Genome Research 16(6):787–795
Zhang J, Zhang QX, Cheng TR, Yang WR, Pan HT, Zhong JJ, Huang L, Liu EZ (2015) High-density genetic map construction and identification of a locus controlling weeping trait in an ornamental woody plant (Prunus mume Sieb. et Zucc). DNA Research 22(3):183–191
Zhang QP, Liu WS, Liu N, Zhang YP, Liu S, Wei X, Liu YC (2011) Egregation type of heterozygous loci in F1 and construction of molecular markers linkage map in apricot. Acta Horticulturae Sinica 38(10):1983–1990
Zhou QH, Zhou C, Zheng W, Mason AS, Fan SY, Wu CJ, Fu DH, Huang YJ (2017) Genome-wide SNP markers based on SLAF-seq uncover breeding traces in rapeseed (Brassica napus L.). Frontiers in Plant Science 8:648
Funding
This study was supported by: National Natural Science Foundation of China (31401826), the Program of Conservation and Utilization of Crop Germplasm Resources (2018NWB003) and National Crop Germplasm Resources Platform of China (NICGR2018-056).
Author information
Authors and Affiliations
Contributions
ZQP and LWS conceived and designed the experiments. ZQP and WX performed the experiments. ZQP, WX and LWS analyzed the data and wrote the paper. LN, ZYP, XM and ZYJ collected samples and managed the mapping population. MXX revised the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Additional information
Communicated by D. Chagné
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(XLSX 144 kb)
Figure S1
Integrated linkage map of the Japanese plum for ‘09-16’ × ‘Fortune’ composed of 5742 markers. The marker name with the word "seg" indicates that the marker is a segregation distortion marker. (PDF 226 kb)
Figure S2
Integrated linkage map of the Japanese plum for ‘09-16’ × ‘Fortune’ composed of 1357 loci. The font of the locus name in red indicates that the locus has at least a segregation distortion marker. An asterisk in the locus name indicates that only segregation distortion markers are present at the locus. (PDF 22304 kb)
Figure S3
Haplotype map of the integrated maps. Different individuals in rows were spaced by blank line, markers on the right were listed coordinate to the order in the linkage map. The swift of green and blue colors indicates the recombination events. The grey color represents missing marker. (PDF 164 kb)
Figure S4
Heat map of the integrated maps. Each cell represents the recombination rate of pair-wise markers. Yellow color indicates strong linkage, and changes of colors from yellow to purple indicating weaker linkage. (PDF 8569 kb)
Data archiving statement
The raw data of SLAF-seq have been submitted to NCBI SRA database under the BioProject ID Number PRJNA43000.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, Qp., Wei, X., Liu, N. et al. Construction of an SNP-based high-density genetic map for Japanese plum in a Chinese population using specific length fragment sequencing. Tree Genetics & Genomes 16, 18 (2020). https://doi.org/10.1007/s11295-019-1385-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-019-1385-y