
Abstract
The status of genomics and genetics research in the Myrtaceae, a large family of dicotyledonous woody plants, is reviewed with Eucalyptus as the focal genus. The family contains over 5,650 species in 130 to 150 genera, predominantly of neo-tropical and Southern Hemisphere distribution. Several genera are well known for their economic importance worldwide. Myrtaceae are typically diploids with small to intermediate genome size. Microsatellites have been developed for several genera while higher throughput marker systems such as diversity arrays technology and single nucleotide polymorphism are available for Eucalyptus. Molecular data have been fundamental to current perspectives on the phylogeny, phylogeography and taxonomy of the Myrtaceae, while numerous studies of genetic diversity have been carried out particularly as it relates to endangered, rare, fragmented, overharvested or economically important species. Large expressed sequence tag collections for species of Eucalyptus have recently become public to support the annotation of the Eucalyptus grandis genome. Transcriptomics in Eucalyptus has advanced by microarrays and next-generation sequencing focusing on wood development. Linkage maps for Eucalyptus display high synteny across species and have been extensively used to map quantitative trait loci for a number of traits including growth, wood quality, disease and insect resistance. Candidate gene-based association genetics have successfully found marker–trait associations for wood and fiber traits. Genomic selection experiments have demonstrated clear potential to improve the efficiency of breeding programs while freeze-tolerant transgenic Eucalyptus trials have recently been initiated. The recently released E. grandis genome, sequenced to an average coverage of 8×, will open up exceptional opportunities to advance Myrtaceae genetics and genomics research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Myrtaceae (Myrtle, eucalypts, clove, or guava family) is a large family of dicotyledonous woody plants placed within the order Myrtales containing over 5,650 species organized in 130 to 150 genera (Govaerts et al. 2008). Recognized as the eighth largest flowering plant family, it comprises several genera of outstanding ecological and economic relevance worldwide. The family occurs mainly in the Southern Hemisphere. It has centers of diversity in the wet tropics, particularly South America, Australia, and tropical Asia with occurrences in Africa and Europe (Fig. 1). The family is commonly found in many of the world's biodiversity hotspots such as south western Australia, and the Cerrado and Atlantic Rainforest in Brazil where up to 90 species of Myrtaceae per hectare, many of which are not described, can be found (Govaerts et al. 2008).

World distribution of the family Myrtaceae; adapted from (Heywood 1996)
In this review, we present the current standing of genomics and other related fields in the Myrtaceae. Among over 130 genera in the Myrtaceae family, Eucalyptus stands out as the pivotal genus for which genomic resources have been developed and currently represent the bulk of the genomics literature for the family. A number of reviews in the last few years have examined the advances of Eucalyptus genome research including applications to breeding and conservation (Byrne 2008; Grattapaglia and Kirst 2008; Myburg et al. 2007; Poke et al. 2005; Shepherd and Jones 2004). However, such reviews have not extended to other genera within Myrtaceae that have yet to experience close attention from the tree genetics and genomics community. Such genera will likely benefit from the rapid genomic and technological developments made in Eucalyptus soon to culminate with the forthcoming announcement of a complete genome sequence for Eucalyptus grandis.
This compendium article is structured according to genomic-oriented topics rather than taxonomy. In each theme, the status of knowledge in species of Myrtaceae for which genomic information has been reported is presented with Eucalyptus as the focal genus. The reader is initially introduced to the main features of the Myrtaceae, including estimates of genome size, chromosome numbers, and available molecular marker resources. A sequence of themes then covers studies on molecular phylogenetics and population genetics, linkage and association mapping, quantitative trait loci (QTL) analysis, transcriptomics, proteomics, and metabolomics, and, finally, molecular breeding. In closing, a snapshot of the current status of the Eucalyptus genome sequencing project is presented, highlighting its anticipated role as a key driver of future genomic undertakings in species of Myrtaceae.
Taxonomy and relevance of the main taxa targeted by genomic research
Myrtaceae is generally distinguished by a combination of features that include the presence of oil glands on leathery evergreen leaves; flower parts in multiples of four or five, generally numerous stamens; phloem located on both sides of the xylem, not just outside as in most other plants; and vestured pits on the xylem vessels (Wilson et al. 2001). The family was considered to be naturally divisible in two subfamilies: the Myrtoideae, with fleshy fruits and opposite leaves and Leptospermoideae, with capsular fruits and alternate leaves (Niedenzu 1893). This classification was first challenged by Johnson and Briggs (1984) based on a cladistic analysis using morphological characters, later followed by molecular phylogenetic studies that proposed a new infra-familial classification that recognized only two subfamilies (Myrtoideae and Psiloxyloideae) and 17 tribes, with Myrtoideae comprising the vast majority of genera (Wilson et al. 2005) (Fig. 2).

Approximately half of the species of Myrtaceae are in the tribes Syzygieae and Myrteae and comprise fleshy-fruited species associated with wet forests across the tropics, particularly in South-East Asia, and Central and South Americas. Some of the prominent genera include Syzygium, Eugenia, and Psidium (Biffin et al. 2010). A second large group comprises the tribes Leptospermae, Eucalyptae, and Chamelauciae, which are woody-fruited species that have radiated in Australia and include Leptospermum, Eucalyptus, Melaleuca, and Chamelacium (wax flower) (Wilson et al. 2001) (Fig. 2).
Overall, the Myrtaceae is characterized by having a number of species-rich genera. For example, Syzygium contains between 1,200 and 1,500 species (Craven and Biffin 2010), Eugenia approximately 1,050 species, and Eucalyptus about 700 species (Brooker 2000). Species richness in the tribes Syzygieae and Myrteae may have arisen through biotic dispersal mediated by a diversity of animal vectors possibly promoting allopatric speciation or reducing risk of extinction (Biffin et al. 2010). In Eucalyptus, the ability of epicormic buds to re-sprout so effectively after fire may have allowed eucalypts to dominate multiple niches and speciate widely (Crisp et al. 2011). Whatever the explanation, the consequence of this species-richness is that the taxonomy of the family is difficult, and the distinctiveness of many widely recognized genera is being questioned (e.g., Biffin et al. 2010; Edwards et al. 2010). However, this may also mean that synteny within the family (at least in members that are diploid) (da Costa and Forni-Martins 2006) is high and significant insight into other groups may be possible from the genome of Eucalyptus.
Several species of Myrtaceae stand out for their key ecological role in some specific ecosystems. For example, in some wet forests of eastern Brazil, Myrtaceae is the dominant family in terms of the number of species, individuals, and total basal area (Mori et al. 1983). In Australia, eucalypts are the dominant or co-dominant species of virtually all vegetation types except rainforest, the central arid zone, and higher mountain regions (Wiltshire 2004) and are considered keystone species for ecological studies in their natural ranges (Williams and Woinarski 1997).
Several genera of Myrtaceae are well known for their economic importance and are cultivated worldwide for their fleshy fruit such as guava (Psidium), jaboticaba (Myrciaria), rose apple (Syzygium), and pitanga (Eugenia) trees; spices such as clove (Syzygium) and allspice (Pimenta); antiseptic oils from eucalypt and tea tree (Melaleuca); or as important sources of timber or fiber for multiple industrial purposes including pulp, paper, and energy production (Eucalyptus and Corymbia) (Fig. 3). The acknowledged economic importance of these genera has been driving most of the genomic research in the Myrtaceae. Some general aspects of these genera where the vast majority of genomic efforts have been undertaken are presented below.

Some representative species of the main Myrtaceae genera for which genomic studies have been developed. a E. regnans is the tallest flowering plant in the world with the tallest tree (‘Centurian’) occurring on the island of Tasmania. It is 99.6 m tall and has a stem volume of 268 m3 (photo courtesy of Forestry Tasmania); b E. grandis (rose gum or flooded gum), BRASUZ1 (Brasil Suzano 1), the 17-year old S1 tree developed in Brazil whose genome has been sequenced (photo courtesy of Suzano S.A.); c Flower of E. globulus (Tasmanian blue gum) (photo courtesy of R. Wiltshire); d Myrciaria cauliflora (Jaboticaba) (photo courtesy of Instituto de Biociências, Letras e Ciências Exatas UNESP); e Leptospermum scoparium (New Zealand tea tree) (photo courtesy of R. Wiltshire); f Psidium gujava (guava) (photo courtesy of Baixaki.com); g Corymbia citriodora (bloodwood) (photo courtesy of M. Shepherd); h Melaleuca squamosa (tea tree) (photo courtesy of R. Wiltshire); i Syzygium jambos (jambo) (photo courtsey of E. Lucas, Royal Botanic Gardens, Kew); j Acca sellowiana (feijoa, pineapple guava) (photo courtsey of E. Lucas, Royal Botanic Gardens, Kew); k Eugenia uniflora (Pitanga) (photo courtsey of E. Nascimento, Saude pelas plantas)
Eucalyptus
The major impetus for genomic studies in the Myrtaceae has been to understand the genetic control of economically important traits, and, in that regard, the focus has been on Eucalyptus. The tribe Eucalypteae contains Eucalyptus and other groups including Angophora and Corymbia along with monotypic genera including Arillastrum from New Caledonia, and three genera from mesic habitats Allosyncarpia (Australia), Stockwellia (north-eastern Australia), and Eucalyptopsis (two species from New Guinea) (Ladiges et al. 2003). Within Eucalyptus, ten subgenera have been described with the most important being Symphyomyrtus (commonly referred to as the "symphyomyrts," comprising about 470 species) and Eucalyptus (formerly Monocalyptus and commonly referred to as the "monocalypts") comprising about 108 species. Eucalyptus is highly diverse and displays significant adaptability and phenotypic plasticity with individuals of some species able to grow from sea level to the tree line (e.g., Eucalyptus pauciflora) and on substrates ranging from rich volcanic soils to deep sand (e.g., Eucalyptus microcorys). This genus includes the tallest flowering plant species in the world, Eucalyptus regnans (99.6 m, Fig. 3a). While predominantly an Australian genus, Eucalyptus trees are grown throughout the world and are the major hardwoods used in the world’s industrial pulp wood plantations (Doughty 2000). Although the many species that grow as forest trees are best known, other species are shrubs (or “mallees”) at maturity and may only grow in very poor, restricted sites. Williams and Brooker (1997) provide a broad introduction to the diversity of the genus. Four species of Eucalyptus (e.g., Eucalyptus deglupta, Eucalyptus orophila, Eucalyptus urophylla, and Eucalyptus wetarensis) have their natural range completely outside Australia in Indonesia, the southern Philippines, and New Guinea. Natural hybridization in eucalypts is widely agreed to have played a major role in the evolution of the present diversity of species (Barbour et al. 2008; McKinnon et al. 2004a; Potts and Reid 1990; Pryor and Johnson 1981). Genomic studies would help us to understand which genes are associated with important adaptive traits, the degree to which they are under selection, and how these genes contribute to the persistence of hybrids. There is a single site in Australia at Currency Creek near Adelaide where an attempt has been made to cultivate the whole genus in a common garden (http://www.dn.com.au/, accessed on 1/4/2011), and although some of the tropical and cold-tolerant species have not thrived at this site, it still represents an enormous resource for those interested in the diversity of the eucalypts. The worldwide importance of species of Eucalyptus together with its relatively modest genome size, varying between 600 and 700 Mbp (Grattapaglia and Bradshaw 1994) has propelled whole-genome sequencing efforts, and to date, the genomes of three species of Eucalyptus have been sequenced at variable coverages (see Section “The status of the E. grandis genome”).
Corymbia
is a group of about 115 species that have a largely tropical and arid zone distribution in Australia, although the most important species industrially, the spotted gum (Corymbia maculata), grows on poor sandy soils on the east coast of Australia. The most recent formal classification of eucalypts treated Corymbia and Angophora as subgenera of Eucalyptus (Brooker 2000), although they are treated as separate genera in the more recent comprehensive electronic flora treatment (Slee et al. 2006) (see Section “Molecular phylogenetics”). The morphological distinctions between Eucalyptus and Corymbia are based on a combination of characters including ovule arrangement, leaf venation, operculum structure, and the presence of unicellular hairs associated with oil glands. Molecular evidence has also been reported for the separation of Corymbia and Eucalyptus (see Section “Molecular phylogenetics”).
Melaleuca
With about 260 species, Melaleuca is one of the major radiations of the Myrtaceae and is the second most speciose genus in Australia after Eucalyptus. Most species of Melaleuca are diploid, but polyploidy has been reported. Species of Melaleuca can be trees or small shrubs, which tend to occupy the wetland niche and are commonly dominant in the understory of eucalypt forest. They occur widely in Australia, Southeast Asia, and New Caledonia. The genus is characterized by long racemes of flowers that resemble a bottlebrush, and there are proposals for the remaining recognized genera of the Melaleuceae to be synonymized with Melaleuca resulting in a genus of 330–350 species (Brown et al. 2001; Edwards et al. 2010).
Psidium
Of the fleshy-fruited members of the Myrtaceae, guava (Psidium guajava L.) has received most attention because of its economic importance as the source of the guava fruit, which is a versatile product that has a significant market worldwide. Although P. guajava is diploid, other members of this genus have been reported to be polyploid. Like any plant family, the Myrtaceae is host to many pathogens and diseases, but, one known as guava rust (Puccinia psidii), is particularly noteworthy because of its very wide host range within the family (Zauza et al. 2010). Guava rust is endemic to parts of South America, but it has the potential to devastate susceptible plant communities where it is a new invader (Coutinho et al. 1998). Recently a member of the guava rust complex (Uredo rangelii) has become established in Australia. It has already had a severe impact on several non-eucalypts (e.g. Backhousia citriodora, Rhodomytus sp) and turned into a fairly widespread occurrence along the east coast of Australia (http://www.dpi.nsw.gov.au/biosecurity/plant/myrtle-rust). Genomic studies to identify resistance factors will be essential to maintain Australian industries based on native Myrtaceae.
Cytogenetics and genome size
Knowledge of the chromosome number, ploidy level, genome size, and structure of a species has important implications for genomic research. This information is generally available for the main genera of Myrtaceae, although to a much larger extent for species of Eucalyptus and closely related genera which have received greater attention due to their keystone ecological role and global economic importance.
Cytogenetics
Overall, the Myrtaceae are typically regular diploids with largely constant chromosome number and small to intermediate genome size. "The Myrtaceae are, on the basis of the species thus far studied, an unlikely subject for intensive use of cytotaxonomic methods." This introductory discussion statement of the first report on chromosome number variation in a set of Myrtaceae species Atchison (1947), provided an early indication of the limited variation in chromosome number in the family, later confirmed by several studies. Furthermore, Atchison (1947) also showed that chromosomes in Myrtaceae are generally small, ranging from 1.2 to 2.5 μm, making detailed cytogenetic analysis difficult, though improved methods have recently been successfully applied to Eucalyptus (Gamage and Schmidt 2009). In that original study by Atchison (1947), species of Psidium showed chromosome counts ranging from 2n = 22 to 88, strongly suggesting the occurrence of polyploidy. Twenty-three Eucalyptus species were found to have 2n = 22, but with two apparent aneuploid exceptions (2n = 24), later dismissed after observing that chromosome breaks during metaphase are commonly seen in Eucalyptus (Oudjehih and Bentouati 2006).
Several studies of chromosome number in Myrtaceae species followed since that early study. It is now well established that the basic haploid chromosome number in Myrtaceae is n = 11, and the vast majority of species are diploid with 2n = 22. Eucalypts are ideal representatives of diploids speciation with a homogeneous haploid chromosome number of n = 11 (2n = 22) based on the analysis of 135 distinct species to date. In spite of its wide ecological and morphological diversity, the genus Eucalyptus can therefore be considered as a vast karyological continuum that probably adopts a process of evolution based fundamentally on chromosome alterations (Oudjehih and Bentouati 2006). While polyploidy in Eucalyptus has not yet been observed in nature, it has been artificially induced (Janaki-Ammal and Khosla 1969), and a renewed interest in this strategy to potentially develop fast growing trees has recently emerged (Lin et al. 2010).
Outside the Eucalyptus, however, some level of karyotypic variation has been observed. Chromosome counts for 150 Western Australian species of the Myrtaceae, confirmed the base chromosome number of n = 11 but also revealed rare diploid variations as low as n = 5 (Rye 1979). Such variations were later confirmed by reports of 2n = 14, 16, 18, and 20 for species of Homoranthus (Copeland et al. 2008). Occasional polyploids (2n = 44) were reported in genus Leptospermum in New Zealand (Dawson 1987), although polyploidy is now generally considered rare in capsular-fruited taxa. On the other hand, karyotypes have been more variable in fleshy-fruited neotropical taxa (Myrteae), with relatively common reports of polyploidy. Although a predominance of 2n = 22 is also seen, occasional triploids (2n = 3× = 33), tetraploids (2n = 4× = 44), and hexaploids (2n = 6× = 66) were reported in Eugenia and Psidium, suggesting that polyploidy is of importance in the evolution of fleshy-fruited Myrteae (da Costa and Forni-Martins 2006, 2007).
Nuclear genome size
Relatively few genera and species of Myrtaceae have had their genome size estimated. Studies have been limited to species of some economic or ecological interest (Table 1). The first estimates of nuclear DNA content in the family were reported for P. guajava (0.33 pg/1 C) (Bennett and Smith 1976). Some level of discrepancy exists in the literature among some published estimates of nuclear DNA content. Ohri (2002) estimated the DNA content of Melaleuca leucadendra to be 1.13 pg/1 C, but this was previously reported to be 0.6 pg/1 C by Bennett and Leitch (1997). P. guajava had an estimated content of 0.62 pg/1 C by (Ohri 2002), an estimate disparate from the early estimate of 0.33 pg/1 C by Bennett and Smith (1976) (Table 1). These discrepancies are likely due to the use of Feulgen microdensitometry, a method that has been shown to be affected by plant polyphenols and therefore prone to be less reliable than the currently preferred and most widely employed flow cytometry (FCM) technique (Greilhuber 2008). The first estimates of nuclear DNA content and respective genome sizes for Eucalyptus were reported for Eucalyptus globulus at 1.13 pg/2 C (Marie and Brown 1993) and for a number of additional species by Grattapaglia and Bradshaw (1994). Estimates based on flow cytometry varied from a low of 1.09/2 C for E. globulus (530 Mbp) to 1.33/2 C for E. grandis (640 Mbp) and 1.47/2 C for E. saligna (710 Mbp). Corymbia citriodora, at the time still classified as belonging to the genus Eucalyptus, displayed a surprisingly smaller genome with only 0.77/2 C (370 Mbp), an unnoticed hint, at the time, to its clearly separate phylogenetic position within the Myrtaceae. Later studies in Eucalyptus largely matched those initial estimates, although with some variation, possibly due to methodological reasons (Pinto et al. 2004). A recent study compared FCM with image cytometry using three different internal standards and re-estimated the nuclear DNA content of three Eucalyptus species. After a detailed analysis of the various methodological issues involved, the most accurate 2 C values for E. grandis, E. urophylla, and E. globulus were 1.25 ± 0.025, 1.28 ± 0.030, and 1.10 ± 0.026, respectively, very close to the original estimates, corroborating that the temperate E. globulus (498 Mbp) has in fact a slightly smaller genome size than the tropical and sub-tropical E. grandis (611 Mbp) and E. urophylla (625 Mbp).
These nuclear DNA content estimates for species of Eucalyptus are within the reported range for hardwood capsular-fruited species of Myrtaceae reported to vary from a low of 0.4 pg/1 C to a high of 0.8/1 C (Ohri et al. 2004). Recently, however, the average nuclear DNA content of 32 species from ten genera of fleshy-fruited Myrtaceae was reported to be significantly smaller when compared with capsular-fruited Eucalypteae and Melaleuceae. Nuclear DNA content for species of genera Eugenia, Myrciaria, Calyptranthes, Acca, Campomanesia, and Psidium, all regular diploids (2n = 22), averaged around 0.5/2 C, while two tetraploid species of Psidium presented the expected doubled amount of DNA, around 1.0 pg/2 C (da Costa et al. 2008). Based on a wider sampling of genera provided by all these studies, the range of nuclear DNA content (0.25/1 C to 0.8/1 C) of Myrtaceae is significantly below the average for diploid Rosids (2.17/1 C) (Leitch and Bennett 2004) and small to intermediate by the classification of Soltis et al. (2003). The average genomes of the most "genomically relevant" genera of Myrtaceae were either smaller or equivalent in size to other model tree species such as Populus (0.6 pg/1 C; 570 Mbp) (Tuskan et al. 2006) and Prunus (0.305 pg/1 C; 300 Mbp) (Arumuganathan and Earle 1991), ranging from a low of 247 Mbp for Psidium gujava and 370 Mbp for C. citriodora to 498 Mbp for E. globulus. As regular diploid, with a relatively small number of chromosomes and small genome sizes, the main genera of Myrtaceae are strong candidates for genomic undertakings that demand physical manipulation of DNA such as positional cloning. Moreover, the upcoming availability of a reference genome sequence for E. grandis, together with increasingly powerful high-throughput sequencing technologies will provide exceptional opportunities for whole-genome comparative and evolutionary studies across genera in the Myrtaceae.
Organellar genomes
Both the chloroplast and mitochondrial genomes of Myrtaceae appear to be inherited exclusively in a maternal fashion as verified in Eucalyptus (Byrne et al. 1993; Vaillancourt et al. 2004) and Chamelaucium (Ma et al. 2004). A large-scale study in E. globulus and its hybrids with Eucalyptus nitens using a polymerase chain reaction (PCR) technique confirmed this observation after analyzing 425 offspring individuals in 40 families derived from controlled crosses with no evidence of pollen-mediated chloroplast-DNA (cpDNA) transmission (McKinnon et al. 2001b). Although virtually no data exist in other genera, it is reasonable to speculate that a similar pattern will be found elsewhere in the family.
The complete chloroplast genome sequence of E. globulus (GenBank accession no. AY780259) with an estimated size of 160,286 bp was reported by Steane (2005). It ranks among the larger land plant chloroplast genomes with a typical structure of most plastids: an inverted repeat (IR) (26,393 bp) separated by a large single copy (LSC) region of 89,012 bp and a small single copy (SSC) region of 18,488 bp. The E. globulus chloroplast genome has a GC content of 36.9%, comparable with that of other vascular plant plastids and essentially collinear with that of Nicotiana tabacum and Populus trichocarpa with a few exceptions. A total of 128 genes was annotated, 112 individual genes and 16 genes duplicated in the IR, coding for 30 transfer RNAs, four ribosomal RNAs, and 78 proteins. The chloroplast genome of E. grandis was also completely sequenced (Paiva et al. 2011; GenBank accession ID NC_014570), displaying a size of 160,137 bp, close to the one seen in E. globulus with an IR of 26,390 bp, an SSC of 18,478 bp, and the LSC with 88,879 bp in size. The nucleotide sequence similarity to the E. globulus chloroplast genome was remarkably high (99.57%) with a complete congruence in gene organization.
Although very little data exist on the genome structure and sequence of chloroplast genomes of other genera in Myrtaceae, it is likely that a significant conservation of sequence and gene order exists across genera. Evidences come from the various studies that used the cpDNA as a rich source of markers for phylogenetic, phylogeographic, and population genetics research in comparative studies going across the main genera of Myrtaceae (see Sections “Molecular phylogenetics” and “Molecular population genetics”).
Molecular marker resources
Molecular markers are the main working tools for genomics surveys. They have been used extensively in the Myrtaceae to characterize genetic variation in natural and breeding populations of a number of species. When connected to phenotypes by linkage or association mapping, molecular markers have assisted tree breeding, native forest management, and conservation purposes. This section provides a detailed profile of DNA marker resources in the Myrtaceae, recent trends in marker development, and future resources and applications. Specific applications of molecular markers in the Myrtaceae are detailed in subsequent sections.
Microsatellite or Simple Sequence Repeats (SSR) markers are the most broadly used molecular markers in the Myrtaceae, and a catalogue of this resource is provided (Table 2 and Electronic supplementary material). Mining of the recently released eucalypt genome will provide an abundant additional source of microsatellite markers that can be targeted to exact genomic regions of interest. Studies of both eucalypts and non-eucalypt Myrtaceae will benefit from a eucalypt genome sequence because transfer of microsatellite markers among Myrtaceous genera is likely to be sufficiently high to facilitate population and evolutionary genetic studies widely across the family. Resequencing the genome of a number of individuals and species will also provide an abundance of single-nucleotide polymorphism (SNP) markers, greatly increasing the prospects of finding markers associated with functional variation and thus of economic or adaptive importance.
Myrtaceous DNA markers profiled by genera and marker type
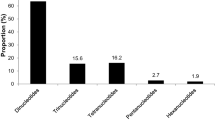
Using journal papers as a guide to research effort and directions, a sample of 152 papers published between 1993 and 2010 were identified that showed DNA markers developed for 11 Myrtaceae genera (Eucalyptus, Corymbia, Eugenia, Metrosideros, Melaleuca, Myrtus, Psidium, Calothamnus, Chamelaucium, Syzygium, and Acca) (Table 2 and Electronic supplementary material; NB. papers solely on isozymes or DNA sequencing were excluded from the sample).
Not surprisingly, the vast majority of effort on the development or application of markers has focused on Eucalyptus (109 papers), whereas there were eight papers each on Eugenia and Metrosideros, seven on Corymbia, six on Myrtus and Melaleuca, and three or less on Psidium, Calothamnus, Chamelaucium, Syzygium, and Acca. Across the Myrtaceae, most papers were associated with the development or use of microsatellite markers (65) followed by random amplified polymorphic DNA (RAPD, 27), amplified fragment length polymorphism (AFLP, 20), cpDNA (19), and then restriction fragment length polymorphism (RFLP, 9). For eucalypts, there were also six papers on SNP, three on inter-simple sequence repeats (ISSR), and one each on diversity arrays technology (DArT) and mitochondrial-DNA (mtDNA) markers.
Microsatellite markers are available for species in four of the 17 Myrtaceous tribes. A catalog of the current resource of microsatellite markers by genera is given in the Electronic supplementary material. A resource of 366 microsatellite markers has been published for Eucalyptus (Brondani et al. 1998, 2002, 2006; Byrne et al. 1996; Ottewell et al. 2005; Steane et al. 2001; Thamarus et al. 2002) and a set of 28 for the related genus Corymbia (Jones et al. 2001; Shepherd et al. 2006). In the non-eucalypt Myrtaceae, 113 microsatellite markers have been developed from Melaleuca (Miwa et al. 2000; Rossetto et al. 1999a), 24 for Metrosideros (Crawford et al. 2008; Kaneko et al. 2007), nine for Eugenia (Ferreira-Ramos et al. 2008), 14 for Myrtus (Albaladejo et al. 2010), ten for Calothamnus (Elliott and Byrne 2005), eight for Syzygium (Hillyer et al. 2007), and 13 for Acca (Santos et al. 2008).
DArT microarray genotyping
A marker technology based on DNA-hybridization in microarray format, the DArT, has recently been developed for eucalypts (Sansaloni et al. 2010). Operational arrays with 7,680 selected polymorphic DArT probes have been designed providing on average 1,000–2,000 polymorphic markers in bi-parental mapping populations and over 4,000 in genetically wider natural or breeding populations. The DArT microarray was shown to work widely across Eucalyptus species with optimum performance for species from the subgenera that include most of the commercial species, Symphyomyrtus and Eucalyptus (Sansaloni et al. 2010). The large number of markers supplied by this first version of a DArT genotyping array has permitted the development of high-density genetic maps for many species at relatively low cost (see Section “Genetic mapping, QTL, and eQTL identification”). Such maps are expected to be of most value for comparative mapping across species, high-resolution mapping, and co-location of QTL with candidate genes derived from the eucalypt reference genome. DArT markers are bi-allelic and display dominant inheritance and thus have less information content than co-dominant microsatellites or SNPs. For a number of applications, however, this limitation should be largely offset by the very large number of loci available and high throughput.
SNP-based markers
SNPs are the most abundant form of DNA polymorphism, with recent studies finding rates ranging from one per 16 to 33 bp across four species of eucalypts, some of the highest levels found for woody plants and plants in general (Külheim et al. 2009). Like DArT, they are largely bi-allelic, but, similarly, their high abundance should ameliorate this limitation. Published accounts of the SNP resource are still rare for eucalypts, but sequencing of the transcriptome and resequencing genome fractions or genes using conventional or next-generation sequencing has confirmed that large numbers of SNPs will be available (Külheim et al. 2009; Novaes et al. 2008; Poke et al. 2003). Assay methods have been used that accommodate the complexity of SNP genotyping in the highly heterozygous eucalypt genome (Sexton et al. 2010b), although at a relatively low throughput of less than 100 multiplexed SNPs.
Recently, a dataset of >1.2 million mixed expressed sequence tags (EST) dataset including Sanger and 454 sequences from multiple Eucalyptus species was used to develop the first collection of 768 genome-wide SNPs for Eucalyptus using the Illumina GGGT (Golden Gate genotyping technology) (Grattapaglia et al. 2011a). Assay success rates according to the GGGT recommended parameters were close to 95% for all five species genotyped (E. grandis, E. globulus, E. camaldulensis, E. urophylla, E. nitens). The assay conversion rate, i.e., the rate of polymorphic SNPs among the reliable ones, was 71% for all species together, and around 40% to 50% for each species separately, except for E. globulus (22.2%), a phylogenetically more distant species from E. grandis, the foremost source of the original EST collection. Results also indicated that a large proportion of SNPs will be reliable between the two main species, E. grandis and E. globulus (89%) and even across the additional four species (81%). However, the rate of polymorphic SNPs declines significantly across species. Only ~20% of the most reliable SNPs were polymorphic (minimum allele frequency ≥ 0.05) both in E. grandis and E. globulus and only ~10% when assessed across the remaining species.
Although the very high nucleotide diversity in Eucalyptus provides a rich source of SNPs in silico, it may complicate actual in vitro SNP assay because of uncharacterized sequence variation in the vicinity of the target SNP. Using the Sequenom mass spectrometry platform (Sexton et al. 2010b) overcame this problem by generating fewer but much longer template amplicons instead of several short ones. When developing SNPs for the GGGT, stringent requirements of conservation in flanking sequences to the SNP both within and across species significantly enhanced SNP reliability and polymorphism of the assay, although such requirements reduced the number of assayable SNPs (Grattapaglia et al. 2011a). This approach, although successful, showed that in silico assessment of polymorphism and interspecific transferability of the SNP requires high sequence coverage and representation to successfully provide reliable selective power for actual SNP assay development. In this respect, the upcoming reference genome for E. grandis will be vital. Recently, next-generation resequencing of reduced genomic representations at high coverage from multiple individuals of E. grandis and E. globulus allowed the discovery of over 42,000 high quality SNPs simultaneously polymorphic in both species (Grattapaglia et al. 2011b). This same scenario for SNP development will likely be found for other genera of Myrtaceae.
Transfer of microsatellites within eucalyptus and to other genera
Microsatellite transfer among species of eucalypts (genera Eucalyptus and Corymbia) shows an expected trend, where transfer declines with the degree of taxonomic divergence (Table 3). Transfer among the sister genera Corymbia and Eucalyptus ranges between 25 and 50%, with higher rates possibly achieved where methodological strategies designed to maximize transfer rates were adopted (Kirst et al. 1997; Shepherd et al. 2006). Transfer among the major Eucalyptus subgenera has been variable and ranged between 40 and 96%, but transfer within a subgenus (Symphyomyrtus) has been significantly higher (80–100%).
Wide transfer among the Myrtaceae has also been possible. Within the Melaleuceae tribe, transfer of 35 Melaleuca sp. derived microsatellite markers to Callistemon sp. was 74% and inter-tribal transfer between Melaleuca and Eucalyptus was 45% (Rossetto et al. 2000). Inter-tribal transfer, however, will likely be highly variable and often much lower as a large study using 346 Eucalyptus microsatellites found only ten loci transferred to Eugenia dysenterica (Zucchi et al. 2002). Nonetheless, for some Myrtaceous species and some applications, it may be more efficient to screen large sets of markers identified in eucalypts for transfer rather than develop markers de novo, although, with the declining cost and longer reads of next-generation sequencing, it has become easy to develop microsatellite markers from genomic sequences, without enrichment.
SNP markers, adaptive variation, and balancing selection
SNPs offer the advantages of abundance, a simpler and better understood mutational mechanism than microsatellites, and relatively low rates of genotyping error, and because they are more often gene-linked, they will be particularly useful for studies of adaptive variation and selection processes (Ryyanen et al. 2007). The confluence of massively parallel sequencing technologies and whole-genome or transcriptome sequences has given rise to the field of population genomics, where genome-wide scans of populations are undertaken to find genes underlying adaptive differentiation (Gonzalez-Martinez et al. 2006; Luikart et al. 2003). A recent empirical demonstration of the approach scanned the entire genomes of populations of Arabidopsis sp. for genes underlying ecotypic adaptation to serpentine soils (Turner et al. 2010). In eucalypts, comparative analysis of trans-specific SNP has revealed ancient polymorphism persisting across subgenera due to balancing selection (Sexton et al. 2010a) and signatures of positive selection (Külheim et al. 2009). These studies are providing insight into the processes that maintain diversity or purge variation in Eucalyptus and thus how the genome of this extraordinarily diverse group has evolved.
Molecular phylogenetics
Molecular data have been fundamental to current perspectives on the phylogeny, phylogeography, and taxonomy of the Myrtaceae (Stevens 2008). As with most angiosperm families (Calonje et al. 2009), higher-level (e.g., family, tribes, and sections) molecular phylogenetics has focused on markers derived from commonly used short sequences in non-coding DNA regions (intergenic spacers or introns) amplified using PCR primers derived from highly conserved flanking regions in plastid DNA (e.g., gene regions [matK, ndhF, psbA, rbcL, trnK, trnL, trnH], introns [rpl16] and various inter- and intra-genic spacers [trnK-matK, psbA-trnH, psbA-trnK, atpβ–rbcL, trnL-trnF]) and nuclear ribosomal DNA (e.g., internal [ITS] or external [ETS] transcribed spacers) (Table 4). Plastid DNA phylogenies from matK and an adjacent spacer sequence define the modern Myrtaceae tribes (Wilson et al. 2001, 2005), and relationships have been further resolved by combining matK with other gene sequences (e.g., plastid rbcL and ndhF (Sytsma et al. 2004); nuclear ITS (Biffin et al. 2010; Fig. 2; Table 4). The recent phylogenetic analysis of inter-tribe relationships based on matK and ITS provided good support for the modern tribal classification of the Myrtaceae (Biffin et al. 2010; Fig. 2). It showed that fleshy fruits have evolved independently from dry fruits in the Syzygieae and Myrteae tribes.
Tribal phylogenies
Molecular phylogenic studies are rapidly accumulating at all taxonomic levels in the Myrtaceae (Table 4). Many challenge the traditional morphology-based taxonomy and phylogenies to some degree, and the taxonomic classification in many tribes is still in flux. For example, in a recent study of the tribe Melaleuceae, the genus Melaleuca was shown not to be monophyletic in its matK phylogeny, and its species are distributed across most of the other genera (Edwards et al. 2010). This result was also consistent with a previous ITS study (Brown et al. 2001), and it is argued that all species of the tribe should be included within Melaleuca (Edwards et al. 2010). Similarly, molecular and morphological data have been used to argue that five genera in the tribe Syzygieae would be better placed within the large genus Syzygium (Craven 2006; Craven and Biffin 2005; Parnell et al. 2007). A subsequent nuclear and plastid DNA sequence phylogeny has recently resulted in the recognition of six subgenera and seven sections within Syzygium (Craven and Biffin 2010). In the tribe Chamelaucieae, analysis of sequence data from the matK gene and the atpβ-rbcL intergenic spacer showed no support for previously recognized sub-alliances based on fruit type (i.e., indehiscent fruit appear to have arisen in multiple lineages); some genera were monophyletic while others were not, such as Babingtonia (Wilson et al. 2007), and several clades required recognition as new genera (Lam et al. 2002).
Delineating the genus Eucalyptus
One of the first challenges for molecular studies of Myrtaceae phylogeny was in the eucalypt group (tribe Eucalypteae; Table 4, Fig. 2). The genus Corymbia (commonly referred to as the bloodwoods) had been recently split from Eucalyptus (Hill and Johnson 1995), but this treatment was not subsequently adopted in the formal taxonomic classification (Brooker 2000; Ladiges and Udovicic 2000). Most of these bloodwoods have operculate flowers, and resolution of this issue partly lay in the affinities of Corymbia to the genus Angophora that has flowers with free petals and sepals (Ladiges 1997). The marked divergence of the Corymbia lineage from Eucalyptus s. s. and its closer affinities to Angophora were demonstrated in numerous molecular studies (Sale et al. 1993; Steane et al. 1999, 2002; Udovicic et al. 1995). However, in the large molecular phylogenetic analysis of the eucalypt lineage involving ITS sequences from 90 species, Angophora was nested within the Corymbia clade (Steane et al. 2002). The vagaries of phylogenies built using a single gene were clearly apparent because, in subsequent studies using microsatellites, nuclear and/or plastid DNA sequence, Corymbia and Angophora were monophyletic sister groups (Ochieng et al. 2007a, b; Parra-O et al. 2006, 2009). This work led to the discovery of multiple nuclear ribosomal pseudogenes within the eucalypt group with differing phylogenetic signals revealed by their ITS sequence potentially complicating phylogenetic reconstruction through comparison of paralogous sequences (Bayly and Ladiges 2007; Bayly et al. 2008; Ochieng et al. 2007a). Monophyly of Corymbia was evident with a non-functional paralogue of ITS (Bayly et al. 2008) but not with the functional form sequenced by Steane et al. (2002). Nuclear ribosomal pseudogenes have now been detected in multiple genera within the eucalypt group and some pseudogenes represent “deep” duplications that appear to predate the divergence of Eucalyptus s. s., Corymbia and Angophora (Bayly and Ladiges 2007; Bayly et al. 2008). While using single-copy nuclear genes avoids complications associated with paralogy in multiple-copy regions of DNA, they may also present problems for phylogenetic reconstruction. This was seen clearly in the study of the cinnamoyl CoA reductase gene (CCR), involved in lignin synthesis, in section Maidenaria where one of the two clades detected appeared to contain evidence of a historical recombination event involving gene sequences from another taxonomic section (McKinnon et al. 2005; Poke et al. 2006).
Delineating the eucalyptus subgenera and sections
Within Eucalyptus s. s., the large ITS study of Steane et al. (2002) showed that, while the major subgenera defined by Brooker (2000) (subgenus Eucalyptus, subgenus Symphyomyrtus, and subgenus Eudesmia) were well-differentiated (Fig. 4), only Eudesmia appeared to be monophyletic (see also Gibbs et al. 2009). The previously recognized small subgenus Minutifructus (subgenus Telocalyptus of Pryor and Johnson (1971) comprising four tropical boxes species (including the extra-Australian E. deglupta) was a grouping of diverse lineages nested within Symphyomyrtus, and its over-ranking was confirmed with a subsequent cpDNA phylogeny (Whittock et al. 2003); see also Ladiges and Udovicic (2005). Symphyomyrtus is the largest subgenus of Eucalyptus s. s. The majority of the world’s eucalypt plantations involve species from this subgenus (Eldridge et al. 1993) and the three species for which genome sequencing is in progress belong to it (E. grandis from section Latoangulatae; E. camaldulensis from section Exsertaria, and E. globulus from section Maidenaria; Fig. 4). The relationships of species within this subgenus are, thus, important to understand and are the subject of ongoing research (Steane et al. 2011; Rebecca Jones personal communication). The intersectional affinities emerging from molecular phylogenetics of subgenus Symphyomyrtus differ markedly from historical perspectives (Brooker 2000; Ladiges 1997; Pryor and Johnson 1981). In Steane et al. (2002; 2011) only section Maidenaria appeared to be monophyletic. Sections Exsertaria and Latoangulatae were poorly differentiated, suggesting that they may need to be combined into a single section. Together, these three sections were well differentiated from the other major sections in the subgenus. The largest section, section Bisectae, was polyphyletic and divided into two distinct lineages, one of which had clear affinities to the clade that included section Adnataria and section Dumaria.

Summary of Splitstree4 analysis from genome-wide genotyping of 94 species in Eucalyptus s.s. with 8,354 DArT markers (provided by D. Steane using data presented in Steane et al. 2011). The subgeneric and sectional names follow Brooker (2000) and clades within subgenus Symphyomyrtus follow those identified in Steane et al. (1999, 2002, 2007). The alignment of the subgenera names with previous classifications is detailed in Table 1 of Byrne (2008). The DArT phylogeny provided results that were largely congruent with traditional taxonomy and ITS-based phylogenies, but provided more resolution within major clades than had been obtained previously. Most of the industrial plantations of the world are based on just a few eucalypt species and their hybrids, and the nine main species are indicated (Harwood 2011)
Phylogenetic relationships within the Eucalyptus sections
Below the sectional level in Eucalyptus, molecular phylogenies using ITS (Table 4) are poorly resolved (e.g., subgenus Symphyomyrtus section Maidenaria; Steane et al. 1999, 2002). Enhanced resolution was achieved by integrating ITS, ETS, and chloroplast sequence with morphological data in a recent phylogeny of subgenus Eudesmia (Gibbs et al. 2009). At these phylogenetic levels, more variable and robust marker systems are required to avoid problems with duplication (paralogy), recombination, single gene bias, and lack of sequence variability. High-throughput genome-wide SNP arrays or next-generation genotyping-by-sequencing approaches have the potential to offer such systems with very large numbers of markers of known position and function. Genome-wide genotyping with high-density marker systems have already been tested in Eucalyptus. Genotyping using AFLPs and the newly developed and mapped DArT markers (Sansaloni et al. 2010) show great promise for robust phylogenetic reconstruction at multiple scales (McKinnon et al. 2008; Steane et al. 2011) (Fig. 4). McKinnon et al. (2008) used 930 AFLPs to examine relationships among Tasmanian taxa of section Maidenaria. Analyses resolved species into clusters largely concordant with series defined in the most recent taxonomic revision of Eucalyptus (Brooker 2000). Some departures from current taxonomy were noted, indicating possible cases of morphological convergence and character reversion. Although the resolution obtained using AFLP was greatly superior to that of single sequence markers, the data demonstrated high homoplasy and incomplete resolution of closely related species. However, this is the level at which molecular phylogenetics and population genetics (see Section “Molecular population genetics”) intersect, with continuous variation often occurring between closely related taxa.
Population genetic approaches are increasingly being used to resolve the phylogenetic affinities and differentiation of species, particularly at the intra-series levels (e.g., Byrne 2008; Cook et al. 2008; Drummond et al. 2000; McKinnon et al. 2005; McKinnon et al. 2008; Ochieng et al. 2007a; Percy et al. 2008). In eucalypts, the frequent occurrence of geographic replacement series involving closely related taxa argues for recent speciation through allopatric processes (Butcher et al. 2009; Byrne 2008; Ladiges 1997). Geographically isolated species or subspecies can usually be differentiated on neutral molecular markers (Butcher et al. 2009; Byrne 2008; Byrne and Macdonald 2000; Jones et al. 2002; Le et al. 2009; McDonald et al. 2009). However, when co-occurring, such closely related species are often poorly differentiated, e.g. Corymbia (Ochieng et al. 2010; Shepherd et al. 2008a), Eucalyptus (Holman et al. 2003; Jones et al. 2002), although there are exceptions which argue for barriers to gene flow, e.g. Eucalyptus (McGowen et al. 2001) and Melaleuca (Broadhurst et al. 2004). Many recognized taxa form species complexes in which morphological and molecular variation is continuous, and there is often incongruence between molecular signals of phylogenetic relatedness and morphological and ecological divergence, e.g. Myrceugenia fernandeziana (Jensen et al. 2002); Metrosideros polymorpha (James et al. 2004, Harbaugh et al. 2009); Eucalyptus angustissima complex (Elliott and Byrne 2004); E. globulus (Jones 2009; Jones et al. 2002). Such discrepancies are no better exemplified than in a recent study of five species in the Hawaiian Metrosideros complex (Harbaugh et al. 2009), which suggests that major taxonomic revisions would be required to reflect the genetic structure revealed by microsatellite markers.
Evolution at lower taxonomic levels in Eucalyptus—species complexes
A number of nuclear microsatellite studies are also revealing an absence of consistent differentiation between closely related, co-occurring species in Corymbia (Ochieng et al. 2008, 2010; Shepherd et al. 2008a) and in Eucalyptus (Holman et al. 2003; Hudson 2007; Le et al. 2009). The spotted gums (genus Corymbia, section Politaria) show a species replacement series along the eastern seaboard of Australia, with distributions marked by regions of disjunction and sympatry. These population-based microsatellite studies showed that the southern C. maculata was resolved as a taxon. Three geographically concordant clusters were evident within the more northern taxa, but the alignment with taxonomic groupings was poor. The large-fruited spotted gum eucalypt Corymbia henryi occurs sympatrically with small-fruited spotted gum C. citriodora subsp. variegata over a large portion of its range on the east coast of Australia. However, these taxa could not be differentiated with microsatellites. In fact, differentiation between populations of the same taxon was greater than between co-occurring taxa (Ochieng et al. 2008). A similar situation is evident in Eucalyptus among the endemic peppermints (series Piperitae subgenus Eucalyptus), which are poorly differentiated in chloroplast (McKinnon et al. 1999) and nuclear (Sale et al. 1996; Turner et al. 2000) DNA markers. In a more extreme case, two morphologically very distinctive species, Eucalyptus amygdalina and Eucalyptus risdonii, growing on a single hill exhibited as much molecular differentiation between populations within species as there was between species (Sale et al. 1996). Morphological differentiation in the absence of neutral marker differentiation argues for recent divergence and a genic view of speciation, with observed phenotypic differences attributable to rare differences in the genome or resulting from epistatic interactions (Ochieng et al. 2010). At these lower taxonomic levels, recent adaptive radiation may be superimposed on historical and/or contemporary gene flow, with molecular phylogenetic reconstruction complicated by (1) shared ancestral polymorphisms (e.g., lineage sorting), (2) an inherent low rate of divergence in the sequences studied, (3) recent rapid radiation of species, (4) small population processes (drift and inbreeding), and (5) hybridization (Byrne 2008; Cook et al. 2008; Gibbs et al. 2009; McKinnon et al. 2001a; Percy et al. 2008; Steane et al. 1999).
Hybridization and reticulate evolution
Reticulate evolution appears to be occurring in many of the Myrtaceae lineages and may well explain some of the discrepancies between plastid and nuclear marker phylogenies. Natural and artificial hybridization are well documented for several genera such as Corymbia (Barbour et al. 2008), Kunzea [including inter-genus hybrids] (De Lange et al. 2005; Tierney and Wardle 2008), and particularly Eucalyptus (Potts et al. 2003).
Species of Corymbia and Angophora are unable to hybridize with Eucalyptus (Barbour et al. 2008; Ellis et al. 1991; Griffin et al. 1988). Within Eucalyptus, hybridization does not occur between the major subgenera. Within these subgenera, intra-sectional hybridization is more common than inter-sectional hybridization in nature, and endogenous post-zygotic barriers to hybridization are generally weaker within than between sections (Griffin et al. 1988; Potts and Dungey 2004; Myburg et al. 2004). There are numerous examples of hybrid swarms and zones of intergradation between species from the same section in the wild (Butcher et al. 2009; Pryor and Johnson 1971; Sale et al. 1996) as well as increasing molecular evidence of reticulate evolution in Eucalyptus (Byrne 2008; Byrne and Macdonald 2000; Jackson et al. 1999; McKinnon et al. 2001a, 2004a) and other Myrtaceae genera, e.g., Metrosideros (Gardner et al. 2004) and Melaleuca (Cook et al. 2008). Gene flow between divergent lineages may impact phylogenetic reconstructions, particularly when based on plastid sequences, which are maternally inherited. Chloroplast haplotype distributions more reflect geography than species boundaries in many eucalypt groups (McKinnon et al. 2001a; Nevill et al. 2008; Steane et al. 1998), although this pattern may be due to either lineage sorting or chloroplast capture through hybridization (Byrne 2008). However, in the most studied eucalypt examples, there was strong evidence that the widespread E. globulus has captured the chloroplast of the rare endemic E. cordata on the island of Tasmania (McKinnon et al. 2004b). Only a trace of introgression was detected in the nuclear genome using AFLP markers, and selection appeared to determine which DNA fragments persisted (McKinnon et al. 2010). Nevertheless, despite chloroplast capture and some AFLP marker sharing due to introgression, there was no overlap in the genome-wide nuclear genotypes of these two species (McKinnon et al. 2008, 2010).
Dating phylogenetic divergence
With the great diversification and widespread distribution of the Myrtaceae across three Gondwanan continents, a major challenge is linking molecular phylogenies with fossil, geological, and biogeographic evidence to date the evolutionary radiation of the Myrtaceae. A recent study using “relaxed clock methodology” dates the crown of the Myrtales lineage at between 89 and 99 Mya (million years ago) (Bell et al. 2010). Several studies attempt to date the deep lineage divergence within the order Myrtales. Sytsma et al. (2004) identified two major chloroplast rbcL lineages within the Myrtales, one of which included the Myrtaceae lineage, and argued that the ancestor of the order evolved in the mid-Cretaceous (ca. 100 Mya) in Southeast Africa (west Gondwana), rather than in Australasia. The Myrtaceae lineage (as defined with the chloroplast sequences from matK, ndhF, and rbcL) was dated at 70–80 Mya, and they argue it diversified in Australasia with more recent shifts to the Americas, Africa, the Mediterranean (e.g., 30 Mya), and possible subsequent dispersals back to Australasia. The majority of the genera they sampled were thought to be present by the mid-Oligocene at 30 Mya. A recent study by Biffin et al. (2010) produced age estimates for the divergence of the various tribes comparable in many cases with those obtained by Sytsma et al. (2004), but there were several discrepancies.
Dating the eucalypt lineage
The eucalypt lineage appears to have diverged early within the subfamily Lepidospermoideae [late Cretaceous; Sytsma et al. (2004)]. ITS-derived chronograms suggest that the two major lineages—Angophora/Corymbia and Eucalyptus—diverged about 60 Mya and the major subgenera 46–41 Mya (Eocene) (Crisp et al. 2004). The occurrence of probable fossil fruit tentatively assigned to subgenus Symphyomyrtus in South America of Early Eocene age (52 Mya) (Wilf et al. 2003) falls between these ages. Molecular dating suggests that diversification of the eucalypt lineage proceeded steadily for at least 30 million years prior to Australia becoming isolated from Antarctica (Crisp et al. 2004). Sections within subgenus Symphyomyrtus appear to have diverged 30–13 Mya. Diversification within sections coincided with the onset of a drier more seasonal climate on the Australian continent between 25 and 15 Mya, and more recent divergences coincide with the onset of severe aridity about 3 Mya. More recent dates are given for sectional-level divergence and that of specific taxa (e.g., E. deglupta) within Symphyomyrtus by Ladiges et al. (2003). There is also evidence of more recent speciation and diversification of eucalypts through the climatically unstable Quaternary (0 to 2.6 Mya) (Byrne 2007, 2008; McKinnon et al. 2004a). However, there are clearly many uncertainties associated with dating molecular phylogenies (Biffin et al. 2010; Crisp et al. 2005; Ladiges and Udovicic 2005), which will no doubt be better resolved in the near future. Phylogenetic analyses based on whole-genome re-sequencing of a number of species in the eucalypt lineage should prove extremely informative to this end.
Molecular population genetics
Genetic diversity
Numerous studies of genetic diversity have been carried-out in Myrtaceae, particularly as it relates to endangered, rare, fragmented, overharvested, or economically important species (see Table 5). Genetic diversity was low while genetic differentiation was high in the endangered Metrosideros boninensis of West Pacific Islands (Kaneko et al. 2008), thus indicating that, to conserve its genetic diversity, many populations need to be conserved. The level of genetic variation in Italian populations of Myrtus communis was highly correlated with the size of the population (r = 0.92), which appears to be caused by overharvesting of native populations for liquor production (Agrimonti et al. 2007). However, in many species, there is only a weak or no correlation between genetic diversity and population size, e.g., in Myrciaria floribunda, a common tree species in the Amazonian Atlantic Forest (Franceschinelli et al. 2007) or in Luma apiculata a tree species in north-western Patagonia (Caldiz and Premoli 2005), probably because the cause of the decrease in population size is relatively recent. It is important to characterize germplasm collections with molecular methods in order to manage genetic diversity and correct pedigree errors, e.g. Ugni molinae (Seguel et al. 2000); Feijoa sellowiana (Nodari et al. 1997) and/or decide on a sampling strategy for a new breeding program (e.g., E. dysenterica (Zucchi et al. 2003) (see Section “Molecular breeding”)).
Mating system and gene flow
The Myrtaceae flower is hermaphrodite, which increases the possibility of selfing. While most studied species appear to have a mixed-mating system, there is tremendous variation in outcrossing rates between species and plants within species, e.g., Eugenia uniflora (Franzon et al. 2010); C. citriodora (Bacles et al. 2009); Metrosideros excelsa (Schmidt-Adam et al. 2000); E. globulus (Mimura et al. 2009); M. communis (Gonzalez-Varo et al. 2009), Calothamnus quadrifidus (Byrne et al. 2007), possibly due to variation in levels of self-incompatibility, population fragmentation, and other factors. While some species of Syzygium use apomixis, this may be rare in the family (Lughadha and Proenca 1996). Most Myrtaceae are pollinated by animals; this includes insects, birds, bats, and even mammal (Southerton et al. 2004) and lizards, in rare cases (Godinez-Alvarez 2004). Thus, the mating pattern and gene flow of any one species and/or population can be highly idiosyncratic. For example, even though fragmentation is generally found to increase inbreeding, it may result in enhanced pollen flow (Mimura et al. 2009) or increased levels of seed-mediated gene flow (Albaladejo et al. 2009) in some cases, although these “positive” effects were probably insufficient to counteract the genetic erosion caused by habitat destruction. In eucalypt, pollen dispersal is believed to be much more important for gene flow than seed dispersal (Byrne 2008). This may be because eucalypt seed does not have special adaptation for dispersal, and thus, in genera that are fleshy fruited and/or those that have winged seeds (Metrosideros), this could be quite different.
Genetic structure
The study of genetic structure within species is important in order to help elaborate conservation measures, design germplasm collection, structure breeding populations, and/or association genetic studies. In many Myrtaceae species, such as in the fleshy-fruited, Myrceugenia fernandeziana, genetic distance between populations is correlated with geographic distance (Jensen et al. 2002), and significant population structure, as measured by F ST or G ST, has been found in many of these, but the large number of studies in the eucalypts (reviewed by Byrne 2008; Moran 1992; Potts and Wiltshire 1997) allow some generalizations to be made. Species with large population size and lack of disjunction tend to have low differentiation (Le et al. 2009; Shepherd et al. 2008a; Shepherd and Raymond 2010) while those with large disjunction (Rathbone et al. 2007) or small population size (Jones et al. 2005) or both (Byrne and Hopper 2008) tend to have higher differentiation. For example, a high level of differentiation, G ST = 0.61 (Bruna et al. 2007), is found between the highly disjunct populations of M. communis around the Mediterranean. These populations can be grouped into geographically consistent clusters (Bruna et al. 2007), and within clusters, further subdivision between bioclimatic zones is possible (Messaoud et al. 2007). This is similar to that found in E. globulus, where a race classification based on quantitative traits has been nearly validated using simple sequence repeats (SSR) markers (Steane et al. 2006b) even though F ST in this species was much smaller (F ST = 0.09). That study showed that analysis of population structure using, presumably neutral, molecular markers can be complemented by analysis of quantitative traits in field trials (F ST vs Q ST), which allows detecting the effect of natural selection on population differentiation (Steane et al. 2006b).
Genetic structure in plant species may also be found to coincide with ecotone boundaries. In the case of the dwarf ecotype of E. globulus, which is found on three exposed granite headlands in south-eastern Australia (Foster et al. 2007), analysis of molecular markers and flowering time found that the dwarf populations had evolved in parallel from the local tall ecotypes. This study showed that small marginal populations of eucalypts were capable of developing reproductive isolation from nearby larger populations, making parapatric speciation possible. In the case of Metrosideros polymorpha, a Hawaiian endemic known for its high levels of morphological diversity and localized adaptation, a molecular study was undertaken across ecotones from bogs to forests on multiple islands, sampling individuals exhibiting morphological extremes within a few meters of each other. Partitioning of the genetic diversity indicated that the between-islands variation was smaller than the variation resulting from microhabitat types within islands (Wright and Ranker 2010).
Genetic structure can also be found within populations. For example, there is a strong correlation between genetic and geographic distance between trees of the fleshy-fruited E. dysenterica, and this is best explained by restricted gene flow (Zucchi et al. 2004), thus even though its seeds are dispersed by animals, limited dispersal causes the genetic variation to be structured. In eucalypts, molecular markers combined with spatial autocorrelation analysis often reveals clustering of related individuals (family group), which is usually believed to be due to poor seed dispersal (Byrne 2008). A study of different-aged cohorts of individuals (mature trees vs. small suppressed seedlings) within E. globulus forest using microsatellite markers allowed detection of a shift in the spatial distribution of the family structure of approximately 10 m between the two cohorts. As this shift coincided with the prevailing winds direction, it was argued to be due to limited effective seed dispersal (Jones et al. 2007).
Phylogeography
There is a need for more southern hemisphere phylogeography studies (Beheregaray 2008), and the family Myrtaceae with its strong southern distribution offers many good candidates. Chloroplast DNA has been the main source of phylogeographic information. In Eucalyptus, the junction of the large single copy with repeat A (JLA region) of the chloroplast has been useful to study species in subgenus Symphyomyrtus (Freeman et al. 2001; Vaillancourt and Jackson 2000), while chloroplast microsatellites have proven useful in subgenus Eucalyptus (Steane et al. 2005; Nevill et al. 2010). The mitochondrial genome has not been used in phylogeographic studies in Myrtaceae, to our knowledge.
The first phylogeographic study in Myrtaceae using chloroplast RFLP markers was in E. nitens (Byrne and Moran 1994). Much of the variation within the species was due to population differentiation. In a follow-up study, Steane et al. (1998) found that the biogeographic distribution of chloroplast haplotypes in E. nitens was best explained by invoking a combination of processes including interspecific hybridization and convergent evolution in addition to drift in isolated populations. Biogeographic as well as phylogenetic conclusions must be drawn carefully in genera such as Eucalyptus where the propensity of closely related species to hybridize must be taken into consideration. However, this has not prevented single-species studies from being informative. For example, migration of E. globulus onto the island of Tasmania is believed to have occurred through a land bridge connecting the west coast of Tasmania to the mainland which formed during glacial periods (Freeman et al. 2001). A study of E. regnans, which occupies some of the same regions as E. globulus, provided further evidence for migration by seed through the western side of the land bridge connecting Tasmania to the mainland, during glacials (Nevill et al. 2010). In both studies, regions of low chloroplast (haplotypes) diversity were interpreted as indicating recolonized areas since the last glacial maximum and regions of high diversity to indicate putative glacial refugia. In Australia, the climate during the last glacial maximum was not only colder but also drier, which had a strong impact even in more temperate regions such as western Australia (Byrne 2007; Byrne et al. 2008) and northern Australia. E. urophylla is endemic to islands in eastern Indonesia, which are believed to have never been in direct contact with continental Australia (Payn et al. 2007). In a cpDNA study, the islands nearest Australia harbored the greatest diversity while those further west were more depauperate, consistent with either an east-to-west colonization, or possibly, hybridization (Payn et al. 2007). Analysis of nuclear microsatellite could not resolve the issue (Payn et al. 2008).
Metrosideros is a genus of Myrtaceae found on oceanic islands across the Pacific from the Philippines, south to New Zealand, and north east to the Hawaiian Islands, which has also received attention in biogeographic studies. Chloroplast haplotype diversity was found to be low in extra-refugial areas, compared with a greater complexity in the vicinity of the putative glacial refugia. As seen in some eucalypts (McKinnon et al. 2001a, 2004a), the sharing of chloroplast haplotypes between different species of Metrosideros in New Zealand suggested a history of recurring hybridization and introgression, possibly initiated during periods of refugial confinement (Gardner et al. 2004). M. excelsa populations were analyzed using AFLP, and there was no evidence of greater genetic variation in areas of New Zealand that have been proposed to be glacial refugia in contrast to cpDNA studies (Broadhurst et al. 2008). Despite having small winged seeds potentially providing for some long distance dispersal, the general pattern of chloroplast variation in Metrosideros across the Hawaiian Islands suggests that islands on a chain were mostly colonized once, followed by in situ diversification before further colonization occurred down the chain (Percy et al. 2008). However, microsatellite analysis suggest that more complicated scenarios are more likely, with genetic and morphological diversity structured not simply by distance and species barriers, but also involving parallel evolution, in addition to hybridization (Harbaugh et al. 2009). In Metrosideros, greater diversity in chloroplast DNA does not imply greater diversity in nuclear markers. The same phenomenon has been found in Eucalyptus, and this may in part be due to greater pollen dispersal than seed dispersal, which may be especially effective in promoting genetic cohesion in widespread species (e.g., Shepherd et al. 2010).
Transcriptomics, proteomics, and metabolomics
Transcriptomics and EST databases
Genomic resources in the form of EST collections have been created for species of Eucalyptus. Initial EST sequencing efforts were based on cDNA libraries derived from different tissues, and developmental stages, and individuals and species have been partially sequenced, resulting in EST datasets. These datasets have enabled the identification of the DNA sequences of expressed genes, levels of gene expression, and alternative splicing forms from identical loci. The generation of such collections has now become easier through next-generation sequencing technologies, in which the cloning step is omitted and tens of thousands of ESTs can be sequenced simultaneously. Sequencing of ESTs has been carried out either randomly, to generate an index of expressed genes, or has been directed, to discover genes that are selectively expressed typically with a focus on wood formation or abiotic or biotic stresses. Whereas EST databases are a rich resource by themselves, they are even more important when training algorithms for genome-wide gene predictions. However, only a small fraction of the Myrtaceae-sequenced ESTs have been made public. Currently, there are 37,480 ESTs from the Myrtaceae family available in GenBank, 36,981 of which are from five Eucalyptus species and one Eucalyptus hybrid, 491 from Melaleuca alternifolia, and eight from Myrciaria dubia. Other private EST resources have been reported as of 2005 [e.g., Oji Paper Co. (Japan, 80,000 ESTs), Genolyptus (Brazil, 135,093 ESTs) (Grattapaglia et al. 2004a), and Arborgen Inc. (USA 218,000 ESTs), (Poke et al. 2005)]. A number of EST projects focused on cold stress and wood formation in Eucalyptus. Keller et al. (2009) sequenced 13,056 ESTs and annotated 11,303 of these from cold-acclimated leaves from E. gunnii to discover expressed genes involved in cold tolerance. Their data suggest that eucalypts utilize carbohydrate accumulation and membrane modification for cell protection in cold stress. Furthermore, they described 57 transcription factors that are expressed in cold acclimation (Keller et al. 2009). Rasmussen-Poblete et al. (2008) sequenced 9,913 ESTs from cold-stressed seedlings of E. globulus and discovered all known genes involved in lignin biosynthesis in their library, as well as 11 transcription factor families. An EST database from developing xylem and two subtractive libraries from mature vs. juvenile wood in E. grandis with 9,222 ESTs was also published (Rengel et al. 2009). The EST and unigene collection that resulted from this project is named EUCAWOOD and is enriched in genes involved in wood formation (www.polebio.scsv.ups-tlse.fr/Eucalyptus/eucawood/, accessed on 1/4/2011). It can be downloaded completely or used for BLAST searches and searches for tissue-specific expression. The database is a large resource for genes related to wood formation, and 141 transcription factors from 41 transcription factor families were discovered. A set of 639 putative EST-SSRs was identified from this database. These genetic markers are a potentially valuable resource for gene–trait association studies, especially for wood traits. The EUCAGEN (Eucalyptus Genome Network) database (web.up.ac.za/eucagen/default.aspx?a1 = 1) presents a summary of the EST databases described above, as well as a link to the current 8× draft assembly of the Eucalyptus grandis genome. A combination of lower sequencing costs and the availability of a reference genome will lead to an increase in the number and value of EST discovery projects.
Next-generation sequencing resources
The first study to use next-generation sequencing technologies to generate a large EST collection for an uncharacterized plant genome was published with E. grandis (Novaes et al. 2008). Over one million ESTs derived from three 454 runs of xylem RNA collected from 21 individuals sampled in seven unrelated open-pollinated families of E. grandis demonstrated the power of this approach to rapidly and inexpensively generate whole transcriptome information and reveal thousands of potential SNP markers. The sequence reads were assembled into 71,384 contigs, 5,838 of which were more than 500 bp long. Next, the 454 sequences were compared with 86,328 ESTs generated in the Genolyptus project by Sanger sequenced ESTs assembled in 21,432 contigs (Grattapaglia et al. 2004a). This enabled a detailed comparison of the two sequencing technologies. Whereas 84% of the Sanger contigs had a homolog in the 454 dataset, only 41% of the 454 contigs found a match in the Sanger sequences. This may be due to the combined effects of greater gene coverage in the 454 dataset together with shorter contigs within the same dataset that could not be matched to the longer Sanger contigs (Novaes et al. 2008). Estimates of the ratio of non-synonymous SNPs per non-synonymous site (Ka) to synonymous SNPs per synonymous site (Ks) revealed whether genes were under purifying selection. Within 2,001 contigs, the average ratio of Ka/Ks was 0.3, indicating purifying selection acting on most of the expressed genes. Most gene ontology (GO) categories that could be assigned from these contigs were under purifying selection as well (Novaes et al. 2008).
Külheim and co-workers used 454 sequencing to discover SNPs associated with plant defense traits in Eucalyptus. Twenty-three loci over approximately 50 kb were sequenced from bulk DNA from about 450 individuals of four species of Eucalyptus (Külheim et al. 2009). A total of 8,631 SNPs were discovered and, on average, the studied loci contained between one SNP per 16 bp for E. camaldulensis—the highest density of SNPs found in any forest tree so far—and one SNP per 33 bp for E. nitens. This was significantly greater than Novaes’ study where one SNP was found in every 192 bp, probably because Külheim et al. studied many more individuals derived from sampling a much wider geographical distribution and also sequenced both introns and exons at a greater depth. The density of SNPs in other forest trees are somewhere between these estimates with one in 25 bp for Quercus crispula (Quang et al. 2008) and one in every 60 bp for Populus tremula (Ingvarsson 2005). E. camaldulensis grows naturally across most of mainland Australia and has the largest natural distribution of all Eucalyptus species. The dataset from Külheim et al. contains 456 individuals from 93 populations across the geographic range (Külheim et al. 2009). There are natural barriers between populations that may have led to large genetic diversity within the species (Butcher et al. 2009; McDonald et al. 2009) and the occurrence of rare SNPs. Most studies that have genotyped individuals or estimated nucleotide diversity focus on a smaller number of population and individuals. This study shows that, to characterize these parameters, best results can be achieved when sampling across the geographic range. Also, with one SNP in every 16 bp on average, primers for regular PCR or SNP assays based on oligonucleotide hybridization followed by PCR may not work on all individuals of that species.
Recently, ultra-deep Illumina mRNA sequencing was used to de novo assemble an expressed gene catalog for a fast-growing E. grandis × E. urophylla F1 hybrid clone. The database contained 17,945 contigs larger than 200 bp (average length of 1,193 bp) and total size of 22 Mbp. Each contig was associated with annotations by homology to other angiosperm genes, as well as functional annotations using GO, KEGG, and Pfam terms. More than 80,000 polymorphic SNPs were identified with a minimum of 20× sequence coverage and average spacing of one SNP per 193 bp in the hybrid transcriptome (Mizrachi et al. 2010). A similar approach was used to generate more than 48,000 de novo contigs (ESTs) with an average length of 560 bp for E. camaldulensis seedlings subjected to water stress to specifically reveal gene transcripts related to this phenotype for association genetics studies. More than 250,000 SNPs from several thousand genes were identified (Thumma and Southerton, unpublished). All these large EST datasets, and additional ones currently in progress, together with the existing ones derived from Sanger sequencing, will be key elements to support the E. grandis genome annotation effort with transcriptional evidence.
Gene expression studies
Various methods with medium to high-throughput have been used to quantify transcript abundance in species of Eucalyptus. Several different platforms were employed, e.g., microarrays (Barros et al. 2009; Solomon et al. 2010), in silico analysis of EST databases (Vicentini et al. 2005), sequencing of cDNA libraries on next-generation sequencers (Külheim 2010), serial analysis of gene expression (De Carvalho et al. 2008; Moon et al. 2007) and cDNA-amplified fragment length polymorphism (Ranik et al. 2006). Several of these studies aimed at providing candidate genes to be tested in association genetics experiments (see Section “Gene discovery and association genetics”).
Solomon et al. (2010) investigated the temporal regulation of genes in developing xylem in Eucalyptus and found that 217 transcripts (8% of the transcripts on the microarray) were influenced by diurnal stimuli. Those genes were involved in carbon allocation, hormone signaling, stress response, and wood formation. Using a suppression subtractive hybridization (SSH) library between xylem and leaves of E. gunnii, Paux et al. (2004) discovered 181 transcripts which were confirmed by RT-PCR that were preferentially expressed in differentiating secondary xylem. Interestingly, many of these transcripts had either “no hit” (44%) or “unknown function” (17%) when compared with published databases, indicating the discovery of novel genes. Not surprisingly, a large proportion of the discovered transcripts belong to two cellular processes, cell signaling, and cell wall biogenesis (Paux et al. 2004). The same group also investigated which genes were involved in the formation of tension wood and found 196 genes with differential expression patterns (Paux et al. 2005). A cellulose synthase gene was found to play a key role in the formation of the G-layer in response to bending. Foucart et al. (2006) created a SSH library between xylem and phloem. Of the 263 differentially expressed genes, 87 were upregulated in xylem. These genes were involved in hormone signaling and metabolism, secondary cell wall synthesis, and proteolysis. Gene expression was studied in E. nitens branches oriented at 45 degrees using microarrays containing 4,900 xylem cDNAs. Wood fiber characteristics were analyzed by X-ray diffraction, and chemical and histochemical methods. Expression of two closely related fasciclin-like arabinogalactan proteins, and a beta-tubulin was inversely correlated with microfibril angle in upper and lower xylem from branches, and some important genes involved in responses to gravitational stress in eucalypt xylem were identified (Qiu et al. 2008).
The future of global transcript profiling will likely be via next-generation sequencing as the costs are comparable to those of microarrays but with a greater range and sensitivity (Shendure 2008). Two methods are currently applied to the analysis of global transcriptomes with next-generation sequencing: The first method is applicable for non-model species and assembles the reads into contigs, followed by annotation from public databases (Külheim 2010). The alternative approach aligns the reads directly to a reference genome. Whereas the major source of error in microarray approaches is cross-hybridization, in next-generation sequencing-based approaches, errors can arise during assembly of short reads against a reference genome, which can lead to false alignments. For species of Eucalyptus, this is now possible by using the 8× draft assembly, and possibly, an approximation could be attempted for other closely related Myrtaceae from the same tribe.
Proteomics
Proteomic analysis in eucalypts has thus far also focused on wood formation, but the field is in its infancy with just one paper published (Celedon et al. 2007). Proteomic studies benefit greatly from access to an annotated genome or to extensive EST resources from which a protein database can be created. This can be used with proteomics data derived from the detection of digested proteins via LC-MS/MS. This approach has been used successfully in other species including Sitka spruce (Lippert et al. 2007) and poplar (Plomion et al. 2006). In eucalypts, the increasingly large EST resources and the upcoming reference genome sequence of E. grandis will lead to new opportunities in the field of proteomics. Once a large protein database has been created, studies of the proteome involved in wood formation or the structural proteins responsible for secondary metabolites, two appealing targets in Eucalyptus, will become feasible.
Metabolomics
The family Myrtaceae is well known for a wealth of secondary metabolites. These include flavonoids and tannins, some of which have antimicrobial properties (Martos et al. 2000; Okamura et al. 1993), for example, terpenes (essential oils) (Keszei et al. 2008) and a group which is unique to eucalypts, the formylated phloroglucinol compounds (FPC) (Eschler et al. 2000). The FPCs are particularly important in ecological interactions as feeding deterrents of marsupial herbivores such as the koala (Moore et al. 2005), common brushtail possum (Scrivener et al. 2004), and to some insects (Andrew et al. 2007). Little is known about the biosynthesis of the FPCs, and the Eucalyptus genome sequence may direct us to the enzyme family responsible for their biosynthesis. There are few metabolomic studies of Eucalyptus with most focused on cataloging broad groups of secondary metabolites including essential oils (terpenes) and flavonoids and some wood chemicals. Tucker et al. (2010) have recently used 1H NMR as an unbiased approach for quantification of the eucalypt metabolome in ~130 species to identify new compounds associated with resistance to mammalian browsing. Metabolomic studies have also been developed with Melaleuca alternifolia, which is the source of medicinal tea tree oil. These studies include, gene discovery (Keszei et al. 2010a,b), gene expression and identification of quantitative trait nucleotides (QTN) in relation to foliar terpene yield (Webb, Külheim and Foley unpublished). A further species, M. quinquenervia, is highly invasive and is regarded as one of the world’s worst woody weeds. In the Florida Everglades a successful biological control program has been established that provides excellent opportunities for genomic studies of insect herbivory (Padovan et al. 2010).
Eucalypts may prove to have a greater number of secondary metabolism genes than other annotated genomes. Velasco et al. (2007) made an inter-species comparison of gene numbers within secondary metabolism pathways of Vitis vinifera, which, like Eucalyptus is rich in terpenes and flavonoids. They found that gene copy numbers in pathways of secondary metabolism were higher than those in poplar and much higher than those in Arabidopsis. Grape has 14 copies of the phenylalanine ammonia-lyase (PAL) and 10 copies of flavonoid 3’,5’-hydroxylase (F3’5’H), more than any other species investigated thus far (Velasco et al. 2007). In Arabidopsis, all but one gene in the flavonoid biosynthetic pathway are present as single copy.
To illustrate the potential richness of secondary metabolism genes in eucalypts, we have made a preliminary analysis of the terpene synthase (TPS) family, which is responsible for the diversity of terpenes in plants and which has been well characterized elsewhere (Bohlmann et al. 1998). Arabidopsis was reported to contain 32 TPS genes, plus 8 pseudogenes (Aubourg et al. 2002); P. trichocarpa has 47 TPS genes (Tuskan et al. 2006); Oryza sativa has 15 plus 2 pseudogenes (Goff et al. 2002); and Vitis vinifera 89 plus 27 pseudogenes (Jaillon et al. 2007) (Table 6). BLAST searches within each of these genomes revealed similar numbers except for rice, where we discovered 46 TPS genes. A BLAST search in the E. grandis genome revealed 120 putative TPS genes plus 17 pseudogenes (Table 6). In all TPS subfamilies, E. grandis has equal number of genes to grape or more, except for the diterpene synthases (diTPS), where there are only two copies in eucalypts. Such a preliminary analysis, however, requires due caution because over- or underestimates could arise from pseudogenes, heterozygosity of the genome, and lack of completeness of the genome. For all species, genes with a single stop codon were maintained in the functional group. Furthermore, plant genomes with low sequence coverage, such as grape and Eucalyptus, may still contain a number of sequencing errors leading to falsely assigned stop codons. Although heterozygosity could also lead to an increase in gene numbers, it should be noted that most TPS genes occur in large gene clusters of up to 11 copies in E. grandis and that most of these tandem repeats are more closely related to each other than to genes from other clusters. Nonetheless, this does hint at some of the interesting analyses that will be possible once the E. grandis genome will be fully annotated.
Genetic mapping, QTL, and eQTL identification
By far, the majority of growth, quality, and adaptive traits of interest to plant breeders is quantitative in nature and affected by genetic variation at many loci throughout the genome as well as by the environment and their interactions. Genome-wide dissection of quantitative traits require the construction of complete genetic linkage maps with sufficient genomic coverage and large segregating populations to allow detection of moderate to large effect QTLs. In this section, we review the status of genetic linkage map construction in species and genera of the Myrtaceae. We also review the use of those maps to analyze the genetic basis of quantitative trait variation at the level of the genome in approaches that have recently included quantitative data from high-throughput gene expression profiling experiments with the aim of identifying individual genes underlying trait variation.
Linkage mapping of Myrtaceae genomes
The first complete genetic linkage maps produced in the Myrtaceae, for E. grandis and E. urophylla (Grattapaglia and Sederoff 1994), were also some of the first produced for forest tree species and indeed for all woody plants. Linkage mapping efforts to date have been almost exclusively carried out in species of Eucalyptus (Table 7). Besides a few maps generated using RFLP technology (Byrne et al. 1995; Thamarus et al. 2002), most maps were enabled by PCR-based marker systems such as RAPD and AFLP analysis (Gan et al. 2003; Grattapaglia and Sederoff 1994; Marques et al. 1998; Myburg et al. 2003; Verhaegen and Plomion 1996). The availability of widely segregating intra- and interspecific crosses and the two-way pseudo-testcross mapping approach (Grattapaglia and Sederoff 1994) allowed the use of dominant markers and inbred line mapping models in outbred forest tree pedigrees. The anonymous nature of the RAPD and AFLP markers and low proportion of shared polymorphism limited the transfer of linkage information across mapping pedigrees. More informative, codominant markers such as isozymes, RFLPs, and ESTs were indeed successfully used to map eucalypt genomes (Byrne et al. 1995; Gion et al. 2000; Thamarus et al. 2002) but did not provide the levels of polymorphism and throughput required for routine mapping in multiple pedigrees. It was only with the more recent development of large numbers of highly polymorphic microsatellite (or SSR) markers for Eucalyptus (Brondani et al. 1998, 2002, 2006; Glaubitz et al. 2001; Ottewell et al. 2005; Steane et al. 2001) that comparative mapping could be performed and linkage map synteny established across multiple pedigrees (Brondani et al. 2006; Freeman et al. 2006; Marques et al. 2002; Hudson et al. 2011; Kullan et al. 2012).
More than 20 genetic linkage maps, generally comprising fewer than 400 dominant and/or codominant markers, have been produced in the Myrtaceae (Table 7) mainly for species of the genus Eucalyptus and, in some cases, for F1 hybrids of these species. Map lengths have ranged from slightly below 1,000 to 2,100 cM depending on the species, marker technology, map coverage, and mapping algorithms used for linkage map construction. Map coverage has generally been greater than 90% allowing efficient genome-wide detection of quantitative trait loci (see below). The focus of linkage mapping in commercially grown species and hybrids of Eucalyptus reflects the need for the concerted efforts of molecular geneticists and breeders to produce mapping pedigrees and construct linkage maps. Such interactions and marker resources have generally not been established for other Myrtaceae genera. Outside of Eucalyptus, genetic maps have only been constructed in the related genus Corymbia, which is also commercially important in tropical and subtropical regions. Shepherd et al. (2006) used a combination of microsatellite markers transferred from Eucalyptus as well as microsatellite markers developed de novo in Corymbia (Jones et al. 2001) to generate genetic linkage maps for the F1 (Corymbia torelliana × C. citriodora subsp. variegata) hybrid parents of a wide F2 hybrid pedigree.
Although highly informative, the relatively low multiplex ratio of microsatellites limits their use for rapid high-density genetic linkage mapping required for dissecting QTLs down to the candidate gene levels. In Eucalyptus this limitation was overcome by building a high-density transcript linkage map with 1,845 genes using an SFPs (Single Feature Polymorphisms) microarray (Neves et al. 2011), and with a 7,680 DArT marker array (Sansaloni et al. 2010) used in genetic linkage mapping efforts in South Africa, Brazil, and Australia (Hudson et al. 2011; Kullan et al. 2012; Petroli et al. 2011) (Table 7). Several high-density genetic linkage maps with up to 2,500 high-confidence (LOD > 3.0) DArT markers in consensus linkage maps of intra- or interspecific hybrid pedigrees have been reported with average marker spacing of ~0.5 cM (Fig. 5). Furthermore, DNA sequences were obtained for almost all of the cloned DArT marker fragments on the array and early indications are that 90% of the mapped markers can be placed uniquely in the draft E. grandis genome sequence. This finding is consistent with the nature of DArT marker analysis, which targets single-copy DNA through the use of a methylation-sensitive restriction enzyme.

Linkage maps for E. grandis (Group 6): (a) Microsatellite-only framework map; (b) Framework map of DArT + microsatellites at likelihood for marker ordering >3.0; (c) Full map fitting all markers linked at LOD>15 with a relaxed ordering threshold. The total recombination distance (~120 cM) for this linkage group remains constant with the increasing number of markers showing that DArT markers effectively increase map density. Microsatellites in red and DArT markers in black (Petroli et al. (2011)
High-density linkage maps produced with DArT markers have been applied to guide the assembly of the E. grandis genome sequence scaffolds into 11 superscaffolds, which putatively represent the 11 autosomal chromosomes. The initial 8× coverage assembly of the E. grandis genome comprised 691 Mbp of genome sequence in 6,043 scaffolds. A combination of high-density DArT marker placements and microsatellite loci was subsequently used to anchor 606 Mbp (88%) of the genome assembly to 11 chromosome models. The first map-based assembly of the E. grandis genome (V1.0, Myburg et al. unpublished) therefore contains 11 large scaffolds that range from 39 to 80 Mbp (average, 55 Mbp) and are cross-linked to the 11 main linkage groups of the E. grandis genome with more than 2,000 DArT and microsatellite markers. The remainder (12%) of the genome assembly is contained in 4,941 smaller, unanchored scaffolds, many of which will be anchored to the main genome scaffolds as additional DArT and microsatellite markers are mapped. The high density of DArT linkage maps and ability to anchor a large proportion of the markers to genome sequences will allow the establishment of a high-resolution genetic framework for the dissection of quantitative traits across eucalypt pedigrees via the anchored markers in the E. grandis reference genome sequence. However, as part of the DArT development process, the transferability of DArT markers between genera (Eucalyptus and Corymbia) was found to be low (Sansaloni et al. 2010). Dedicated DArT development efforts will therefore have to be initiated to make this technology available for high-density linkage mapping of other genera of the Myrtaceae.
Even higher density linkage maps will in future be possible in Eucalyptus with concerted efforts of genome-wide discovery of SNP markers enabled by next-generation DNA sequencing technologies. Nevertheless, the very high frequency of SNPs observed in Eucalyptus will challenge the commonly used SNP genotyping technologies, particularly for multi-species genotyping platforms where the combined SNP diversity of several species need to be (Grattapaglia et al. 2011a) accommodated (see Section “Molecular marker resources”). Ultimately, the extremely high-throughput recently achieved with next-generation DNA sequencing technologies will enable direct detection and genotyping of SNPs from short sequence tags at high coverage on reduced genomic representations of individual offspring sequenced in multiplexed barcoded pools. This approach, generally termed Genotyping-by-Sequencing (GbS) has been successfully applied to crop plants (Elshire et al. 2011; Poland et al. 2012). In Eucalyptus a recent test of the GbS protocol provided several thousand high-quality SNPs (Faria et al. 2012), while a similar DArT-based method of genome complexity reduction combined with massive Illumina short tag sequencing was used to build a high density linkage map with over 4,000 dominant and co-dominant SNP markers (Sansaloni et al. 2011).
QTL mapping in Myrtaceae genomes
QTL mapping studies have been performed in a relatively small number of Eucalyptus tree species in the subgenus Symphyomyrtus, mainly due the commercial importance of the species for plantation forestry and active breeding programs for these species and their hybrids. Linkage maps developed have been successfully used for genetic dissection of a variety of quantitative characters including growth and form, wood properties, vegetative propagation, flowering time, biotic and abiotic stress resistance, and traits related to secondary metabolism (Table 8). The development of similar genetic mapping resources in other subgenera and genera of the Myrtaceae will pave the way for investigation of the evolution of quantitative characters at the genus and family level.
QTL mapping studies performed in Eucalyptus have been reviewed (Grattapaglia and Kirst 2008; Myburg et al. 2007). Findings from QTL mapping experiments in Eucalyptus trees (Table 8), summarized here, are likely to be relevant for most genera and species in the Myrtaceae. Typically, fewer than ten major effect QTLs have been detected for most studied traits. These QTLs jointly explained up to 52% of phenotypic variance for traits related to vegetative propagation (Grattapaglia et al. 1995) and 81% for secondary metabolism (Shepherd et al. 1999), both estimates likely overestimated. Recent QTL studies, however, have reported a substantially larger number of QTLs for wood quality traits whose effects estimated rarely exceed 5%. Thumma et al. (2010) detected 36 QTLs for cellulose content, pulp yield, lignin content, density, and microfibril angle (MFA) in E. nitens. Gion et al. (2011) described a total of 117 QTLs for a number of wood and end-use related traits, including chemical, technological, physical, mechanical and anatomical properties. Most QTLs had effects below 5% and only 13 of them had major effects above 15%. In a recent multi-pedigree QTL study, Freeman et al. (2011) found 98 QTLs for a range of growth and wood quality traits. Substantial QTL x pedigree and QTL x environment interactions were observed corroborating the anticipated complications of applying these results for marker assisted selection. The high genetic diversity in Eucalyptus species has facilitated QTL detection in most intra- and interspecific crosses. QTL mapping has been particularly productive in interspecific hybrid pedigrees where large effect segregating alleles potentially represent major gene effects of species differences (Grattapaglia et al. 1995; Marques et al. 1999; Shepherd et al. 1999, 2008b; Verhaegen et al. 1997). In several cases (Freeman et al. 2008b; Mamani et al. 2010; Marques et al. 2002, 2005; O’Reilly-Wapstra et al. 2011; Thamarus et al. 2004), QTLs detected in one pedigree could be detected on homologous linkage groups in other, often unrelated, pedigrees validating the initial report and providing support for further investigation of key genomic loci. This was made possible by the development of transportable microsatellite markers that could be mapped in multiple pedigrees.
After an initial period when linkage group numbering was assigned ad hoc in different linkage maps that were mostly built with non-transferable dominant markers, Brondani et al. (2006) in a consolidation effort unified the existing linkage mapping data from several different maps using the few microsatellites that had been mapped to that point. This unification now widely adopted in more recent mapping reports has facilitated the continued addition of new markers and genes and expanded the prospects of making comparative analysis of putative QTL synteny.
The assembled meta-chromosomes of the E. grandis reference genome sequence are also being numbered and oriented according to this convention using a genome-wide framework of microsatellite and DArT markers. The general conservation of the base chromosome number (n = 11) and the strong co-linearity of genomes of Eucalyptus species and closely related Corymbia suggest that the numbering convention and orientation of linkage maps could find use at the family level. This should allow interesting evolutionary genomics questions to be addressed.
Despite the success of QTL mapping studies and the promise of marker-assisted breeding for quantitative traits, QTLs mapped in Eucalyptus have generally not been deployed in tree breeding programs (see Section “Molecular breeding”). Several major barriers have limited the routine application of QTL mapping information, and similar barriers will exist for other outbred genera in the Myrtaceae. Factors such as the high cost of genotyping and phenotyping have resulted in relatively small populations (generally fewer than 300 individuals, Table 8) being used for QTL analysis. This has limited the power to detect alleles segregating in these pedigrees and most likely resulted in overestimation of the effects of those that have been detected (Beavis 1994). The resolution of the QTL mapping experiments has been low. Observed QTL intervals typically span 10 to 20 cm, which likely comprise genomic regions harboring several hundred genes often including many genes that could be considered putative candidates based on their biochemical or cellular functions. Only a small proportion of the allelic variation that exist in a breeding populations is sampled in each QTL mapping pedigree and high levels of linkage equilibrium observed in tree species limit the application of the observed marker–trait linkages to within-pedigree selection of progeny with desirable QTL allele combinations. QTL validation in related pedigrees and different environments is expensive and time-consuming, especially for tree species with long generation times. There is still a lack of understanding of the population genetics and evolution of quantitative traits in most plant species. Knowledge is particularly lacking on the frequency of QTL alleles and distribution of effects in natural and breeding populations. Finally, long generation times and high rates of inbreeding depression in outbred eucalypt species (Costa e Silva et al. 2011; Griffin and Cotterill 1988) have precluded the development of inbred lines with contrasting alleles, which are needed to isolate and characterize (Mendelize) individual allele effects.
Several recommendations can be put forward that would increase the success and applicability of future QTL mapping efforts in the Myrtaceae. Dedicated QTL mapping pedigrees need to be developed that maximize segregation for traits of interest. Such pedigrees may include multiple families or crosses and that sample more genetic variation for traits of interest and allow estimation of QTL effects in multiple genetic backgrounds. QTL mapping populations should also be replicated in multiple sites to estimate genotype by environment interaction, information that has been largely lacking in Eucalyptus. Larger experimental populations (>500 individuals) together with the higher map density achieved by new marker systems like DArT will increase the statistical power and resolution of QTL mapping experiments. Key genetic loci that have been identified by comparative QTL mapping in multiple pedigrees can be tagged at high resolution with SNP markers developed from the underlying genome sequence. This might increase the utility of QTL markers for molecular breeding and provide an entry point for discovering underlying genes.
Genetical genomics for quantitative trait dissection
Many QTLs are expected to result from a polymorphism that affects the expression of an underlying gene. QTLs can be mapped for variation in transcript levels at thousands of genes using genomics tools such as microarray analysis. Overlap between the trait (phenotype) QTL and an expression QTL (eQTL) for a particular gene provides evidence that the gene may be contributing to quantitative trait variation at that locus. By measuring the expression levels of more than 2,600 genes in developing xylem tissues of an interspecific (E. grandis × E. globulus) backcross population, (Kirst et al. 2004) demonstrated the potential use of eQTLs to study the coordinated regulation of traits such as lignin biosynthesis and volume growth at the gene level. It was also demonstrated that more than 30% of genes had eQTLs that were clustered together in loci which may contain key regulators responsible for the coordination of many developmental and/or biosynthetic pathways (Kirst et al. 2005a). Such master regulatory loci may be associated with large effect QTLs in segregating populations and could, therefore, be targets for marker-aided breeding. The completion of the Eucalyptus genome sequence and anchoring of thousands of DArT and microsatellite markers from high-density genetic linkage maps will be an important enabling step for genetical genomics studies. It will allow discrimination among cis-acting eQTLs (where the eQTL maps to the gene locus) and trans-acting eQTLs (located elsewhere in the genome, presumably associated with transcriptional regulators of target genes). Furthermore, eQTL studies in hybrid pedigrees may allow identification of genomic loci and genes that are associated with differences among parental species, which will facilitate the tagging and introgression of such loci in hybrid breeding programs and provide valuable insights into the genetic basis of species differentiation.
Gene discovery and association genetics
Association studies are in progress in several forest tree species including Eucalyptus (Butcher and Southerton 2007). In contrast to genome-wide association studies in humans and model plant species (e.g., Arabidopsis thaliana), association studies in forest trees are currently focused on candidate genes implicated in the trait of interest. In this section, we briefly review the results from gene discovery studies in Eucalyptus, the only Myrtaceae genus where such efforts have been carried out, followed by results from association studies. As only a few association studies have been published in Eucalyptus, results from other forest trees will be mentioned for comparative purposes.
Candidate gene discovery
Identification of candidate genes controlling important traits such as wood quality has been the focus of a number of studies in forest trees. Gene discovery has been achieved by developing EST data base resources by a number of groups both public and private with most of the EST resources in Eucalyptus targeting wood traits and abiotic stress response (see Section “Transcriptomics, proteomics and metabolomics”). Several candidate genes affecting cell wall biosynthesis in wood experiencing tension forces have been identified in eucalypts using microarray-based global gene expression experiments (Paux et al. 2005; Qiu et al. 2008). Even though array-based studies are useful for discovering candidate genes, functional studies can confirm the role of a gene in controlling a trait. Goicoechea et al. (2005) characterized the role of EgMYB2, a transcriptional activator identified from differentiating xylem from Eucalyptus. Using protein-binding analyses, they showed that EgMYB2 specifically binds to the promoters of CCR and CAD, the two terminal genes in the lignin biosynthetic pathway, and regulates their expression. Transgenic tobacco plants over-expressing EgMYB2 showed significantly thicker cell wall walls and altered lignin profiles. Spokevicius et al. (2007) have analyzed the role of the E. grandis β-tubulin gene (EgrTUB1) in controlling microfibril orientation, a trait strongly correlated with wood stiffness. Using a novel transformation technique that produced somatic xylem sectors (Spokevicius et al. 2005), they showed significant changes in microfibril orientation in sectors over-expressing EgrTUB1 (Spokevicius et al. 2007). In a recent study, MacMillan et al. (2010) have implicated fasciclin-like arabinogalactan proteins (FLAs) in wood stiffness and chemical composition. Expression of FLA proteins was shown to be negatively correlated with microfibril angle in E. nitens (Qiu et al. 2008). Using knockout mutants and novel biomechanical tests, MacMillan et al. (2010) revealed that FLA proteins contribute to plant stem strength by affecting neutral sugar composition and cellulose deposition in secondary walls of stems.
In the absence of obvious candidates or to provide supportive evidence for the potential role of a candidate, a QTL approach may be useful. A recent example was the use of a QTL study to launch fine-scale mapping of functional genetic variation in a gene involved in cellulose deposition (COBRA-like gene) in E. nitens (Thumma et al. 2009). Positional information may be particularly valuable for candidate gene reduction, where complex biochemistry provides a plethora of candidates, or mechanisms are unclear such as in several recent studies of foliar chemistry and biotic stresses in eucalypts (Freeman et al. 2008a, b; Henery et al. 2007; O’Reilly-Wapstra et al. 2011).
Association genetics
While gene discovery studies and biochemical characterization are useful to understand how genes are involved in the control of important quantitative traits, for practical application in breeding, specific alleles at these genes have to be found. Dissection of the molecular basis of trait variation by QTL mapping revealed numerous loci controlling variation in growth and wood properties in Eucalyptus and Corymbia (see Section “Genetic mapping, QTL, and eQTL identification”). However, these marker–trait associations have not been useful in forest tree breeding programs due to the low extent of linkage disequilibrium (LD) in highly outbred populations (see Section “Molecular breeding”). To overcome this limitation, the focus of molecular marker research in forest trees shifted to population-based association studies (Neale and Savolainen 2004).
Association studies in natural tree populations will result in high resolution of marker–trait associations after due account for the existence of population structure (Neale and Savolainen 2004). Several statistical methods have been proposed to account for this structure when testing for association (Price et al. 2006; Pritchard et al. 2000; Yu et al. 2006). Unlike QTL studies where specialized controlled-cross families are required, existing populations can frequently be used in association studies. This is particularly attractive in forest trees as development of specialized crosses such as full sib families is slow and expensive. Existing breeding populations are suitable for association studies as they generally include several hundreds of unrelated families. As genetic variation in the whole population is studied and associations are detected using the entire population, markers identified in association studies can, in principle, be immediately applied in breeding programs.
The first published association study in a forest tree species was in Eucalyptus (Thumma et al. 2005). Two SNPs in the CCR gene associated with microfibril angle were found in a E. nitens population involving 290 unrelated individuals. These results were further validated in two full-sib families of E. nitens and the closely related species E. globulus. Functional studies in Arabidopsis have also implicated CCR in affecting cellulose MF orientation (Ruel et al. 2009). Since that first study, association studies analyzing wood traits, adaptation to drought, cold, aridity, and disease resistance have been reported mainly in pines and poplars (Dillon et al. 2010; Eckert et al. 2009,2010; Gonzalez-Martinez et al. 2007, 2008; Ingvarsson et al. 2008; Quesada et al. 2010). Results from an expanded association study with hundreds of SNPs in about 100 cell wall genes has revealed several SNPs significantly associated with different wood quality traits in E. nitens (Southerton et al. 2004; Thumma and Southerton unpublished). Some of these SNPs were further validated in two other large natural populations. Meta-analysis by combining genotype data from three populations with more than 1,500 trees revealed 13 SNPs significantly associated with cellulose and pulp yield (Thumma and Southerton, unpublished). In a first application of marker-assisted selection, 13 significant markers were genotyped in two breeding populations. Analysis of the data has shown that the frequencies of the favorable genotypes are, in general, higher in breeding populations compared with unselected base populations further validating the significance of these SNPs (Thumma and Southerton unpublished). Trees from the two breeding populations were ranked based on SNP genotypes, and these marker-based rankings along with breeding values will be used in selecting superior trees. These studies demonstrate the potential of association studies to yield useful markers for forest tree breeding programs.
The rapid decline in LD generally found in forest tree species offers unique opportunities for functional analysis of genes. Thumma et al. (2009) identified a synonymous SNP (SNP7) in a eucalypt COBRA-like (EniCOBL4) gene that is significantly associated with cellulose content and pulp yield in E. nitens. LD analysis revealed that SNP7 occurs in low LD with two small flanking haplotype blocks. This pattern of LD made it possible to test the functional significance of SNP7. Using different methods such as allele-specific expression, protein binding, and methylation analyses, they were able to show that SNP7 is a cis-acting regulatory variant influencing allelic expression. In a recent study Külheim et al (2011) used 195 SNPs in 24 candidate genes from known biosynthetic pathways to investigate associations for 33 traits related to plant secondary metabolites that defend eucalypt foliage against both vertebrate and invertebrate herbivores in E. globulus. For the 37 significant associations found across 11 candidate genes and 19 traits, the effects of SNPs on phenotypic variation were within the expected range (0.018 < r2 < 0.061) for forest trees. This study successfully linked allelic variants to ecologically important phenotypes which can have a large impact on the entire community. These studies demonstrate the power of association studies in populations with low LD to reveal functional variants affecting quantitative traits. While these results show that association studies are useful in identifying markers in pure species, their usefulness in hybrid breeding populations is less straightforward. Many advanced generation breeding programs in Eucalyptus are based on hybrid populations (Grattapaglia and Kirst 2008) that involve relatively small number of parental lines. As LD will be generally high in F1 populations and a few generations thereof, resolution of marker–trait associations from association studies using such populations will be low. Genome-wide association studies in such populations could, however, reveal important genomic segments that could be integrated into genomic selection models (see Section “Molecular breeding”).
Results from association studies in forest trees to date indicate that the effect of associated markers is typically very small, rarely exceeding 5%. These associations are similar to the magnitude of QTL effects found in forest trees. Large effect QTLs reported are most likely due to the small population sizes used (Brown et al. 2003; Grattapaglia et al. 2009), although some QTLs identified in crosses between species (Shepherd et al. 2008b) could be real as they may represent major gene effects of species differences. The small effect of markers generally found in forest trees is in agreement with findings from association studies in humans. Results from recent genome-wide association studies in humans suggest that common genetic variants are responsible for only a small fraction of trait variation. It was suggested that rare variants of large effect may not have been captured by using common SNPs in association studies (Goldstein 2009). Even though the effect of individual SNPs may be small, it should be possible to use combinations of SNPs to capture a large proportion of variation in traits. In loblolly pine (Pinus taeda), a substantial proportion of cumulative phenotype variance (20% of phenotypic variance and 40% of additive genetic variance in specific gravity) was explained by jointly analyzing a few significant SNPs (Gonzalez-Martinez et al. 2007).
The number of genes analyzed to date in association studies in forest trees is relatively small. Even highly heritable traits such as wood quality are expected to be influenced by variation in hundred of cell wall genes (Carpita et al. 2001). To capture a large proportion of trait variation, the number of candidate genes examined needs to be increased substantially. Consequently, selection of appropriate candidate genes will be crucial in future association studies. Developments in next-generation sequencing technology will have a major impact on candidate gene discovery. Deep sequencing of RNA with next-generation sequencing technology will help profile genome-wide gene expression patterns. RNA sequencing is useful not only for identification of candidate genes but also for identifying SNPs within the candidate genes. The availability of draft Eucalyptus genome will accelerate this process. The cost of genotyping large numbers of SNPs across large number of samples is rapidly falling based on novel Genotyping-by-Sequencing methods that have been successfully applied to Eucalyptus species (Sansaloni et al. 2011; Faria et al. 2012). As sequencing costs fall and throughput increases, it should soon be possible to sequence individual genomes. SNPs identified from these sequencing projects could then be directly tested for their effect on the trait without the need for genotyping individual SNPs. Using information from such projects, it should be possible to identify both rare and common SNPs controlling complex traits.
Molecular breeding
Systematic breeding efforts in the Myrtaceae have been essentially restricted to species of Eucalyptus. A few occasional studies have described genetic variance components of fruit traits in Psidium (guava) (Thaipong and Boonprakob 2005), or reported the intraspecific variation, genetic control, and gains from selection for oil content and composition in Melaleuca (Butcher et al. 1994; Doran et al. 2006; Shelton et al. 2002). Except for a few studies that have developed and used molecular markers to investigate diversity in Psidium (Prakash et al. 2002; Risterucci et al. 2005) and Melaleuca (Rossetto et al. 1999b), tangible applications of molecular tools to advance breeding populations have been carried out only in Eucalyptus, (Grattapaglia 2004; Grattapaglia and Kirst 2008; Myburg et al. 2007).
Eucalypts were introduced worldwide in the first quarter of the 1800s and quickly selected for plantations as their remarkable growth and adaptability were realized (Doughty 2000). The dawn of industrially oriented eucalypt plantations in the 1960s and 1970s led to a systematic approach to breeding in several countries like Brazil, South Africa, Portugal, Australia, and Chile (Eldridge et al. 1993). Main target traits for genetic improvement have been volume growth and wood density. Recently, pulp yield has received more attention while resistance to biotic and abiotic stresses is usually secondary. Large genetic gains have been obtained in the early stages of eucalypt breeding through species and provenance selection followed by individual selection for population improvement and establishment of seed orchards (Potts 2004). A major breakthrough in eucalypt plantation technology occurred in the 1970s with the plantation of the first commercial stands of selected clones derived from hardwood cuttings (Campinhos 1980; Martin and Quillet 1974). Since then, vegetative propagation coupled to hybrid breeding has become a powerful strategy for the improvement of productivity and wood quality. Eucalypt hybrid clones currently make up a significant component of commercial plantations worldwide in exotic environments (de Assis 2000) whereas, in their native range, eucalypt hybrids face multiple biotic challenges that can decrease yield (Potts and Dungey 2004). The prospects of molecular breeding are discussed in the framework of such specialized Eucalyptus breeding programs, bearing in mind that eucalypts are still in early stages of domestication, a fact that has important implications when planning the use of genomic approaches in operational breeding.
Marker-based management of breeding populations
Molecular markers have been used to answer questions related to the management of genetic variation, identity, and relationship in breeding and production populations. The correct identification of clones is currently the most widespread application of molecular technologies in operational Eucalyptus breeding and production forestry. Quality control of large-scale clonal plantations is essential, especially in vertically integrated production systems where the industry owns the forest plantation and therefore relies on the availability of wood from specific clones with particular wood properties. Because nursery propagation takes place several years before wood consumption, mislabeling can seriously affect the whole production process. Correct clonal identity also has important implications in breeding procedures where mislabeled elite clones used as parents in seed orchards can significantly affect the expected gains from breeding. RAPD and AFLP markers were initially used to this end (Gaiotto et al. 1997; Keil and Griffin 1994; Nesbitt et al. 1997) but quickly substituted by more powerful and accurate microsatellite markers (Kirst et al. 2005b). Recently, improved genotyping systems based on tetra, penta, and hexanucleotide repeats microsatellites and SNPs have been developed and successfully used for high resolution fingerprinting, inter-individual genetic distance estimation, species distinction, and assignment of hybrid individuals to their most likely ancestral species (Faria et al. 2011; Correia et al. 2011). Standard panels of informative microsatellites have been routinely used for varietal protection of elite clones in Brazil since 2002, following inclusion of molecular markers as supplementary descriptors in the national regulation (Grattapaglia 2008).
In addition to the use of markers for fundamental population genetics studies in Eucalyptus (see Section “Molecular population genetics”), DNA genotyping data have been valuable to assist in the design of seed collections (Nesbitt et al. 1995), improve the structure of breeding populations and seed orchards (Marcucci-Poltri et al. 2003; Zelener et al. 2005), and assess the levels of genetic diversity in national breeding programs. For example, microsatellite markers were used to compare the Australian National E. globulus Breeding Program (n = 140) to that observed in native trees (n = 340). While expected heterozygosity was high in the breeding population, and similar to that displayed in the native trees, allelic richness was lower, suggesting a loss of rare alleles during selection. Observed heterozygosity (individual heterozygosity), however, was consistently higher in the breeding population than in native trees, suggesting that heterozygotes have been preferentially selected within the program (Jones et al. 2006). Markers have been successfully used to estimate outcrossing rates, study parentage, and outside pollen contamination in seed orchards, parameters that have also been valuable to provide general guidelines for risk assessment of gene flow from plantation to natural stands (Barbour et al. 2010; Burczyk et al. 2002; Chaix et al. 2003; Gaiotto et al. 1997; Jones et al. 2008; Patterson et al. 2004; Rao et al. 2008). In breeding programs, DNA paternity testing was successfully employed to retrospectively select parents of higher specific combining ability and demonstrate realized gains in volume growth above 24% in commercial forest stands (Grattapaglia et al. 2004b). This DNA-based breeding tactic was later expanded and formally presented as the more general "breeding without breeding" strategy for forest trees (El-Kassaby and Lstiburek 2009). Finally, early attempts to use marker-based distance data among parents to predict performance of offspring were not very encouraging possibly due to the limited genome coverage. Only less than 5% of the variation in specific combing ability could be accounted for by RAPD markers (Vaillancourt et al. 1995), and similar results were obtained with microsatellites (de Aguiar et al. 2007), although Baril et al. (1997) suggested that RAPD-based distance could successfully predict the value of hybrid progenies.
Marker-assisted selection
A number of QTL mapping studies have been carried out in Eucalyptus with encouraging results with regard to the ability to detect large effect QTLs, although it is clear that most of them have been widely overestimated (see Section “QTL mapping in Myrtaceae genomes”). No report, however, exists so far on the actual use of such QTL data for operational breeding by marker-assisted selection (MAS). In view of the rapid decay of LD typically seen in Eucalyptus populations, marker–trait associations detected in a specific bi-parental segregating family in theory would hardly hold in unrelated pedigrees (Strauss et al. 1992). This was recently demonstrated for the association between microsatellite markers and a major effect QTL (Ppr1) for resistance to the Puccinia psiidi rust-causing fungus. Although Ppr1 was successfully validated in unrelated families, when the two microsatellites flanking Ppr1 were genotyped in a large breeding population, they were found to be in linkage equilibrium (Mamani et al. 2010).
This result confirms early expectations that long-range LD marker–trait association detected by QTL analysis in bi-parental pedigrees may only be useful for selection within the same or related families where the QTL was originally detected (Grattapaglia et al. 1995). This represents a considerable limitation for most Eucalyptus breeding programs that typically evaluate a broad diversity of unrelated families to capture valuable allelic combinations for multiple traits. Nonetheless, MAS has been proposed as a potential strategy to identify top individuals at early age within particular elite full-sib families of very large size by tracking a few large effect QTLs (Grattapaglia and Kirst 2008; Johnson et al. 2000; O'Malley et al. 1994). QTL mapping studies in forest trees with increased statistical power have shown that a larger number of QTL with relatively small and variable effects across backgrounds and environments typically underlie complex traits (Dillen et al. 2008; Ukrainetz et al. 2008; Freeman et al. 2011). Breeding for several traits simultaneously by QTL-based MAS in such a scenario is practically precluded while the potential for MAS-based on the cumulative effect of several marker–trait associations found in candidate genes could be realized (see Section “Gene discovery and association genetics”).
Genomic selection
Genomic selection (GS), an approach originally put forward for domestic animals (Meuwissen et al. 2001), was recently proposed as a promising approach for molecular breeding in forest trees (Grattapaglia et al. 2009; Grattapaglia and Resende 2011). GS involves selection decisions based on genomic breeding values estimated as the sum of the effects of genome-wide markers capturing most QTL for the target traits. Based on a set of deterministic simulations Grattapaglia and Resende (2011) initially proposed that GS could radically improve the efficiency of forest tree breeding in elite populations. Recent groundbreaking GS experiments in two contrasting Eucalyptus breeding populations totaling 1,700 individuals genotyped for > 3,000 DArT markers and phenotyped for growth and wood quality traits have confirmed those predictions. Realized selection accuracies between 55 and 88% were obtained, matching or surpassing the accuracies achieved by conventional phenotypic selection. Substantial proportions (74–97%) of trait heritability were captured by fitting over a thousand significant genome-wide markers simultaneously underlying an estimated 200 QTLs per trait. The location of genomic regions explaining trait variation largely coincided between populations, although GS models predicted poorly across populations, likely as a result of variable patterns of linkage disequilibrium, inconsistent allelic effects across genetic backgrounds and genotype X environment interaction (Resende et al. 2012). In this scenario, selection efficiency gain evaluated as the ratio of GS and phenotypic selection exceeds 350% by reducing breeding generation time from 8 to 2 years (Grattapaglia and Resende 2011). With the rapid technological advances and declining costs of genotyping, we anticipate that GS will rapidly receive increased attention by Eucalyptus breeders and soon become an effective operational tool in advanced breeding programs.
Transgenic technology
Despite the acknowledged economic importance of Eucalyptus in world forestry, very few public reports exist on transgenic experiments in species of the genus. Eucalyptus tissue can be transformed by Agrobacterium tumefaciens (Machado et al. 1997), but difficulties are faced for regeneration, typically found to be highly genotype-dependent. Attempts to transform guava (Psidium) using A. tumefaciens also faced difficulties in the regeneration step (Rai et al. 2010). Reports on Eucalyptus-transformed plants are scant and generally developed for easily regenerable genotypes (Ho et al. 1998; Tournier et al. 2003). Proprietary transformation protocols have been developed, however, that allegedly would work on different eucalypt genotypes (Kawazu et al. 2003; M. Hinchee personal communication).
Besides the potential economic impact of a genotype-independent transformation system, an efficient transgenic technology for Eucalyptus would represent a fundamental step to advance functional genomics studies. To mitigate the biological limitation for the study of wood formation using mutant phenotypes, in vitro wood formation systems have been employed to introduce transgenes transiently or stably into growing Eucalyptus wood-producing tissue (Spokevicius et al. 2005) and recently used to show that β-tubulin determines cellulose microfibril orientation during xylogenesis in E. globulus (Spokevicius et al. 2007).
Transgenic technology promises to be a powerful complementary tool available to the molecular breeders. Transgenic trait modification, once successfully proven, should be well adapted to the clonal propagation system used in industrial Eucalyptus forests. The introduction of genes that confer traits for which the existing natural variation in Eucalyptus is insufficient, limiting plantation establishment in exotic environments (e.g., pest and pathogen susceptibility and/or abiotic stress tolerance such as frost or drought), is likely to be the initial target. In fact, the tree biotechnology company ArborGen recently received approval from the US Department of Agriculture to plant a quarter million genetically modified freeze tolerant Eucalyptus trees in Southern USA to provide an economically viable hardwood option in the region (http://www.arborgen.us/uploads/press-releases/Dear%20BRS%20Stakeholder.pdf, accessed on 1/4/2011). In conclusion, the current information on Eucalyptus transgenesis points to an encouraging future concerning the possibility of generating stably transformed Eucalyptus plants (Labate et al. 2009). However, some strategic issues to be considered regarding the adoption of transgenic technology in Eucalyptus and forestry in general have been anticipated (Grattapaglia and Kirst 2008).
The status of the E. grandis genome
In July 2007, the US Department of Energy (DOE) Joint Genome Institute (JGI) announced that it would sequence the genome of E. grandis (rose gum or flooded gum), a widely grown subtropical plantation species listed by the US-DOE as a candidate biomass energy crop. A first-generation inbred (S1) clone of E. grandis (BRASUZ1, donated by the pulp and paper company Suzano in Brazil) (Fig. 3b) was selected by the Eucalyptus research community as the target for genome sequencing (Myburg et al. 2008). Following the breakthrough that the poplar genome sequence (Tuskan et al. 2006) triggered for the study of traits unique to woody plants, it is expected that a Eucalyptus reference genome will provide additional opportunities for comparative genomic analysis and shed light on the evolution of traits such as perennial growth, woody biomass production, and carbon sequestration.
Following a whole-genome shotgun sequencing approach very similar to that used for the poplar genome (Tuskan et al. 2006), the DOE-JGI has over the past two years sequenced the estimated 640 Mbp genome of E. grandis to an average Sanger sequence coverage approaching 8×. Approximately 7 million paired Sanger sequence reads were produced from 3 and 8 kb plasmid libraries, 40 kb fosmid libraries, and two bacterial artificial chromosome libraries of average 145-kb insert size. The shotgun sequences were assembled into 6,043 scaffolds totaling 691 Mbp, of which 643 Mbp (93%) is contained in contiguous sequences. Despite the selection of an inbred (S1) genotype of E. grandis (BRASUZ1), assembly of the two haplotypes of the clone into a single consensus sequence proved challenging. A substantial proportion of the genome (>25% of loci dispersed throughout the genome) assembled into two haplotypes of 3–4× coverage, while the remainder of the genome assembled into a single haplotype of 6 × to 7 × coverage. Currently, over 88% (605 Mbp) of the draft mapped assembly (V1.0) of the E. grandis genome is included in 11 large sequence scaffolds averaging 55 Mbp in size. Sequence similarity searches with 1.6 million ESTs from leaf and xylem tissues of BRASUZ1 revealed that 96% of expressed sequences map to the 11 superscaffolds suggesting that only a small proportion of genes are contained in smaller scaffolds that have yet to be anchored to the main chromosome assemblies. Additional marker development in these regions and linkage mapping will position these scaffolds in future updates of the E. grandis genome assembly. Ab initio and homology-based annotation of the E. grandis genome sequence will be supported by over 4 million ESTs produced by GS FLX Titanium sequencing (Roche/454 Life Sciences) from xylem and leaf tissues of E. grandis (BRASUZ1) and a clonal genotype of E. globulus (X46, Forestal Mininco, Chile). In addition, the several published and yet unpublished sets of Eucalyptus ESTs (see Section “Transcriptomics, proteomics and metabolomics”) have been used in parallel genome annotation efforts at the DOE-JGI and Ghent University. In addition to the E. grandis genome sequence, the DOE-JGI has performed genome-wide (30× Illumina paired-end) resequencing of E. globulus clone X46. The annotated E. grandis genome assembly, E. globulus resequencing data, and all supporting EST sequences have been released via the JGI's comparative plant genome database, Phytozome (http://www.phytozome.net/) at the beginning of 2011. The Kazusa DNA Institute in Japan has produced a draft assembly of the genome of E. camaldulensis and recently released the information (Hirakawa et al 2011).
Opportunities for advancing Myrtaceae genomics
The Eucalyptus reference sequence and associated genomic resources will have an important impact on basic as well as applied biological research in the Myrtaceae. As the pivotal representative of the family, the Eucalyptus genome will be the first reference genome in the Rosid order Myrtales, an important sister lineage to the Eurosids, which now include reference genomes for Arabidopsis, Populus, Medicago, Carica, Glycine, Ricinus, Cucumis, and Manihot. The Eucalyptus genome will therefore be informative for comparative genomic studies within the Rosids as well as the Eudicots. Together with the Vitis genome (Jaillon et al. 2007; Velasco et al. 2007) representing the order Vitales, an earlier diverging lineage in the Rosids (Jansen et al. 2006), it will serve to confirm the paleo-hexaploid nature of Rosid genomes.
A recent comparative genomic analysis of Coffea (representing the Asterids) with Vitis, Arabidopsis, and Populus (Rosids) indicated that the Rosids and Asterids shared a paleo-hexaploid ancestor (Cenci et al. 2010) as was previously suggested based on multiple alignment of tomato and Rosid genes (Tang et al. 2008). The Eucalyptus genome should therefore contain the ancient (γ) genome triplication, which has been detected in Vitis and the Eurosids (Jaillon et al. 2007). Analysis of the genome sequence will resolve whether additional lineage-specific genome-wide duplications like the one (ρ) in the Populus lineage and two (α and β) in the Arabidopsis lineage (Tang et al. 2008) also occurred in the Myrtales. No additional genome-wide duplications were observed in the Vitis (Jaillon et al. 2007) or Carica (Ming et al. 2008) genomes. Depending on the number of genome duplications that occurred in the Myrtales lineage after its divergence from other Rosid lineages and subsequent rates of gene loss, the number of protein coding genes in the E. grandis genome and likely in several other members of the Myrtaceae should be within the range of that observed for the Rosids (i.e., 27,000 to 41,000).
The vast majority of Myrtaceae species of potential genomic interest have relatively small genomes varying from 235 to 640 Mbp and possibly equally or less complex than the Eucalyptus genome. This fact, together with the rapidly evolving third-generation sequencing technologies (Rusk 2009), should contribute to the generation of high-quality reference genome sequences for several Myrtaceae species of interest in the not-so-distant future. Incredible opportunities for genomic research will then be driven by increasingly affordable high-throughput genome sequencing technologies that will be used not only for resequencing but also for genome-wide, high density genotyping.
Fundamental questions in comparative genome evolution, speciation, reproductive isolation, ecosystem, and population genomics will be approached at the genome-wide level. Standing phylogenetic questions or hypotheses at the global family level or tribe-specific will be settled, although new issues will probably emerge once much higher DNA sequence resolution will be achieved. For example, genome-wide studies focusing on the evolutionary relationships and the timings of diversification might provide further stronger evidences for the incredibly high species diversification proposed for the Syzygieae and Myrteae tribes (Biffin et al. 2010).
Genome-wide comparative studies between E. grandis and E. globulus will provide exciting opportunities to study genome evolution and facilitate genomic dissection of the superior wood quality of E. globulus, a premier pulping species, possibly identifying sequence polymorphisms to be used in molecular breeding practice. The same approach could be used to study particular characteristics of other species in the genus. The availability of multiple genomes for species of Myrtaceae will also motivate increased efforts in metabolomics surveys, exploring the extraordinary diversity of secondary metabolites in Myrtaceae and their role in complex interactions with herbivore’s life history, habitat selection, dietary constraints, fitness, competition, and coevolution. Finally, ecological and population genomics studies based on environmental association analysis will allow searching for correlations between climate variables and sequence variants at the whole-genome level across a range-wide sample of populations, a particularly exciting approach to understand Myrtaceae evolution.
References
Agrimonti C, Bianchi R, Bianchi A, Ballero M, Poli F, Marmiroli N (2007) Understanding biological conservation strategies: a molecular-genetic approach to the case of myrtle (Myrtus communis L.) in two Italian regions: Sardinia and Calabria. Conserv Genet 8:385–396
Albaladejo RG, Carrillo LF, Aparicio A, Fernandez-Manjarres JF, Gonzalez-Varo JP (2009) Population genetic structure in Myrtus communis L. in a chronically fragmented landscape in the Mediterranean: can gene flow counteract habitat perturbation? Plant Biol 11:442–453
Albaladejo RG, Sebastiani F, Gonzalez-Martinez SC, Gonzalez-Varo JP, Vendramin GG, Aparicio A (2010) Isolation of microsatellite markers for the common Mediterranean shrub Myrtus communis (Myrtaceae). Am J Bot 97:E23–E25
Alves A, Rosado C, Faria D, Guimarães L, Lau D, Brommonschenkel S, Grattapaglia D, Alfenas A (2012) Genetic mapping provides evidence for the role of additive and non-additive QTLs in the response of inter-specific hybrids of Eucalyptus to Puccinia psidii rust infection. Euphytica 183:27–38
Andrew RL, Wallis IR, Harwood CE, Henson M, Foley WJ (2007) Heritable variation in the foliar secondary metabolite sideroxylonal in Eucalyptus confers cross-resistance to herbivores. Oecologia 153:891–901
Arumuganathan K, Earle ED (1991) Nuclear DNA content of some important plant species. Plant Mol Biol Rep 9:208–218
Atchison E (1947) Chromosome numbers in the Myrtaceae. Am J Bot 34:159–164
Aubourg S, Lecharny A, Bohlmann J (2002) Genomic analysis of the terpenoid synthase (AtTPS) gene family of Arabidopsis thaliana. Mol Genet Genom 267:730–745
Bacles CFE, Brooks J, Lee DJ, Schenk PM, Lowe AJ, Kremer A (2009) Reproductive biology of Corymbia citriodora subsp variegata and effective pollination across its native range in Queensland, Australia. Southern Forests 71:125–132
Barbour RC, Crawford AC, Henson M, Lee DJ, Potts BM, Shepherd M (2008) The risk of pollen-mediated gene flow from exotic Corymbia plantations into native Corymbia populations in Australia. For Ecol Manag 256:1–19
Barbour RC, Wise SL, McKinnon GE, Vaillancourt RE, Williamson GJ, Potts BM (2010) The potential for gene flow from exotic eucalypt plantations into Australia's rare native eucalypts. For Ecol Manag 260:2079–2087
Baril CP, Verhaegen D, Vigneron P, Bouvet JM, Kremer A (1997) Structure of the specific combining ability between two species of Eucalyptus. I. RAPD data. Theor Appl Genet 94:796–803
Barros E, van Staden CA, Lezar S (2009) A microarray-based method for the parallel analysis of genotypes and expression profiles of wood-forming tissues in Eucalyptus grandis. BMC Biotechnol 9:12
Bayly MJ, Ladiges PY (2007) Divergent paralogues of ribosomal DNA in eucalypts (Myrtaceae). Mol Phylogenet Evol 44:346–356
Bayly MJ, Udovicic F, Gibbs AK, Parra-O C, Ladiges PY (2008) Ribosomal DNA pseudogenes are widespread in the eucalypt group (Myrtaceae): implications for phylogenetic analysis. Cladistics 24:131–146
Beavis W (1994) The power and deceit of QTL experiments: lessons from comparative QTL studies. In: Wilkinson D (ed) 49th Annual Corn and Sorghum Research Conference. IL, Chicago, pp 250–266
Beheregaray LB (2008) Twenty years of phylogeography: the state of the field and the challenges for the Southern Hemisphere. Mol Ecol 17:3754–3774
Bell CD, Soltis DE, Soltis PS (2010) The age and diversification of the angiosperms re-revisited. Am J Bot 97:1296–1303
Bennett MD, Leitch IJ (1997) Nuclear DNA amounts in angiosperms—583 new estimates. Ann Bot 80:169–196
Bennett MD, Leitch IJ (2005) Nuclear DNA amounts in angiosperms: progress, problems and prospects. Ann Bot 95:45–90
Bennett MD, Smith JB (1976) Nuclear-DNA amounts in angiosperms. Phil Trans Roy Soc London Sr B Biolo Sci 274:227–274
Biffin E, Craven LA, Crisp MD, Gadek PA (2006) Molecular systematics of Syzygium and allied genera (Myrtaceae): evidence from the chloroplast genome. Taxon 55:79–94
Biffin E, Harrington MG, Crisp MD, Craven LA, Gadek PA (2007) Structural partitioning, paired-sites models and evolution of the ITS transcript in Syzygium and Myrtaceae. Mol Phylogenet Evol 43:124–139
Biffin E, Lucas EJ, Craven LA, da Costa IR, Harrington MG, Crisp MD (2010) Evolution of exceptional species richness among lineages of fleshy-fruited Myrtaceae. Ann Bot 106:79–93
Bohlmann J, Meyer-Gauen G, Croteau R (1998) Plant terpenoid synthases: molecular biology and phytogenetic analysis. Proc Natl Acad Sci U S A 95:4126
Broadhurst L, Byrne M, Craven L, Lepschi B (2004) Genetic congruence with new species boundaries in the Melaleuca uncinata complex (Myrtaceae). Austr J Bot 52:729–737
Broadhurst LM, Young AG, Murray BG (2008) AFLPs reveal an absence of geographical genetic structure among remnant populations of pohutukawa (Metrosideros excelsa, Myrtaceae). N Z J Bot 46:13–21
Brondani RPV, Brondani C, Tarchini R, Grattapaglia D (1998) Development, characterization and mapping of microsatellite markers in Eucalyptus grandis and E. urophylla. Theor Appl Genet 97:816–827
Brondani RP, Brondani C, Grattapaglia D (2002) Towards a genus-wide reference linkage map for Eucalyptus based exclusively on highly informative microsatellite markers. Mol Genet Genom 267:338–347
Brondani RPV, Williams ER, Brondani C, Grattapaglia D (2006) A microsatellite-based consensus linkage map for species of Eucalyptus and a novel set of 230 microsatellite markers for the genus. BMC Plant Biol 6:1–16
Brooker MIH (2000) A new classification of the genus Eucalyptus L'Her. (Myrtaceae). Aust Syst Bot 13:79–148
Brown GK, Udovicic F, Ladiges PY (2001) Molecular phylogeny and biogeography of Melaleuca, Callistemon and related genera (Myrtaceae). Aust Syst Bot 14:565–585
Brown GR, Bassoni DL, Gill GP, Fontana JR, Wheeler NC, Megraw RA, Davis MF, Sewell MM, Tuskan GA, Neale DB (2003) Identification of Quantitative Trait Loci Influencing Wood Property Traits in Loblolly Pine (Pinus taeda L.). III. QTL Verification and Candidate Gene Mapping. Genetics 164:1537–1546
Bruna S, Portis E, Cervelli C, De Benedetti L, Schiva T, Mercuri A (2007) AFLP-based genetic relationships in the Mediterranean myrtle (Myrtus communis L.). Sci Hortic 113:370–375
Bundock PC, Hayden M, Vaillancourt RE (2000) Linkage maps of Eucalyptus globulus using RAPD and microsatellite markers. Silvae Genet 49:223–232
Bundock PC, Potts BM, Vaillancourt RE (2008) Detection and stability of quantitative trait loci (QTL) in Eucalyptus globulus. Tree Genet Genomes 4:85–95
Burczyk J, Adams WT, Moran GF, Griffin AR (2002) Complex patterns of mating revealed in a Eucalyptus regnans seed orchard using allozyme markers and the neighbourhood model. Mol Ecol 11:2379–2391
Butcher PA, Southerton SG (2007) MAS in forestry species. Marker-assisted selection (MAS) in crops, livestock, forestry and fish: current status and the way forward. FAO, Rome, pp 283–305
Butcher PA, Doran JC, Slee MU (1994) Intraspecific variation in leaf oils of Melaleuca-Alternifolia (Myrtaceae). Biochem Syst Ecol 22:419–430
Butcher PA, McDonald MW, Bell JC (2009) Congruence between environmental parameters, morphology and genetic structure in Australia's most widely distributed eucalypt, Eucalyptus camaldulensis. Tree Genet Genomes 5:189–210
Byrne M (2007) Phylogeography provides an evolutionary context for the conservation of a diverse and ancient flora. Austr J Bot 55:316–325
Byrne M (2008) Phylogeny, diversity and evolution of eucalypts. In: Sharma A, Sharma A (eds) Plant genome: biodiversity and evolution volume 1, part E, phanerogams-angiosperm. Science Publishers Enfield, pp 303–346
Byrne M, Hopper SD (2008) Granite outcrops as ancient islands in old landscapes: evidence from the phylogeography and population genetics of Eucalyptus caesia (Myrtaceae) in western Australia. Biol J Linn Soc 93:177–188
Byrne M, Macdonald B (2000) Phylogeography and conservation of three oil mallee taxa, Eucalyptus kochii ssp. kochii, ssp. plenissima and E. horistes. Austr J Bot 48:305–312
Byrne M, Moran GF (1994) Population divergence in the chloroplast genome of Eucalyptus nitens. Heredity 73:18–28
Byrne M, Moran GF, Tibbits WN (1993) Restriction map and maternal inheritance of chloroplast DNA in Eucalyptus nitens. J Heredity 84:218–220
Byrne M, Murrell JC, Allen B, Moran G (1995) An integrated genetic linkage map for eucalypts using RFLP, RAPD and isozyme markers. Theor Appl Genet 91:869–875
Byrne M, Marquez-Garcia MI, Uren T, Smith DS, Moran GF (1996) Conservation and genetic diversity of microsatellite loci in the genus Eucalyptus. Austr J Bot 44:331–341
Byrne M, Murrell JC, Owen JV, Kriedemann P, Williams ER, Moran GF (1997a) Identification and mode of action of quantitative trait loci affecting seedling height and leaf area in Eucalyptus nitens. Theor Appl Genet 94:674–681
Byrne M, Murrell JC, Owen JV, Williams ER, Moran GF (1997b) Mapping of quantitative trait loci influencing frost tolerance in Eucalyptus nitens. Theor Appl Genet 95:975–979
Byrne M, Elliott CP, Yates C, Coates DJ (2007) Extensive pollen dispersal in a bird-pollinated shrub, Calothamnus quadrifidus, in a fragmented landscape. Mol Ecol 16:1303–1314
Byrne M, Yeates DK, Joseph L, Kearney M, Bowler J, Williams MAJ, Cooper S, Donnellan SC, Keogh JS, Leys R, Melville J, Murphy DJ, Porch N, Wyrwoll KH (2008) Birth of a biome: insights into the assembly and maintenance of the Australian arid zone biota. Mol Ecol 17:4398–4417
Caldiz MS, Premoli AC (2005) Isozyme diversity in large and isolated populations of Luma apiculata (Myrtaceae) in north-western Patagonia, Argentina. Austr J Bot 53:781–787
Calonje M, Martin-Bravo S, Dobes C, Gong W, Jordon-Thaden I, Kiefer C, Kiefer M, Paule J, Schmickl R, Koch MA (2009) Non-coding nuclear DNA markers in phylogenetic reconstruction. Plant Syst Evol 282:257–280
Campinhos EN (1980) More wood of better quality through intensive silviculture with rapid growth improved Brazilian Eucalyptus. Tappi 63:145–147
Carpita N, Tierney M, Campbell M (2001) Molecular biology of the plant cell wall: searching for the genes that define structure, architecture and dynamics. Plant Mol Biol 47:1–5
Celedon PAF, de Andrade A, Xavier Meireles KG, de Carvalho M, Gomes Caldas DG, Moon DH, Carneiro RT, Franceschini LM, Oda S, Labate CA (2007) Proteomic analysis of the cambial region in juvenile Eucalyptus grandis at three ages. Proteomics 7:2258–2274
Cenci A, Combes MC, Lashermes P (2010) Comparative sequence analyses indicate that Coffea (Asterids) and Vitis (Rosids) derive from the same paleo-hexaploid ancestral genome. Mol Genet Genom 283:493–501
Chaix G, Gerber S, Razafimaharo V, Vigneron P, Verhaegen D, Hamon S (2003) Gene flow estimation with microsatellites in a Malagasy seed orchard of Eucalyptus grandis. Theor Appl Genet 107:705–712
Coates DJ, Sampson JF, Yates CJ (2007) Plant mating systems and assessing population persistence in fragmented landscapes. Austr J Bot 55:239–249
Conti E, Litt A, Wilson PG, Graham SA, Briggs BG, Johnson LAS, Sytsma KJ (1997) Interfamilial relationships in Myrtales: molecular phylogeny and patterns of morphological evolution. Syst Bot 22:629–647
Cook LG, Morris DC, Edwards RD, Crisp MD (2008) Reticulate evolution in the natural range of the invasive wetland tree species Melaleuca quinquenervia. Mol Phylogenet Evol 47:506–522
Copeland LM, Bruhl JJ, Craven LA, Brubaker CL (2008) New chromosome numbers in Homoranthus (Myrtaceae: Chamelaucieae) and notes on their taxonomic utility. Aust Syst Bot 21:443–447
Correia L, Faria D, Grattapaglia D (2011) Comparative assessment of SNPs and microsatellites for fingerprinting, parentage and assignment testing in species of Eucalyptus. BMC Proceedings 5:P41
Costa e Silva J, Hardner C, Tilyard P, Potts BM (2011) The effects of age and environment on the expression of inbreeding depression in Eucalyptus globulus. Heredity 107:50–60
Coutinho TA, Wingfield MJ, Alfenas AC, Crous PW (1998) Eucalyptus rust: a disease with the potential for serious international implications. Plant Dis 82:819–825
Craven LA (2006) New combinations in Melaleuca for Australian species of Callistemon (Myrtaceae). Novon 16:468–475
Craven LA, Biffin E (2005) Anetholea anisata transferred to, and two new Australian taxa of Syzygium (Myrtaceae). Blumea 50:157–162
Craven LA, Biffin E (2010) An infrageneric classification of Syzygium (Myrtaceae). Blumea 55:94–99
Craven LA, Biffin E, Ashton PS (2006) Acmena, Acmenosperma, Cleistocalyx, Piliocalyx and Waterhousea formally transferred to Syzygium (Myrtaceae). Blumea 51:131–142
Crawford NG, Hagen C, Sahli HF, Stacy EA, Glenn TSC (2008) Fifteen polymorphic microsatellite DNA loci from Hawaii’s Metrosideros polymorpha (Myrtaceae: Myrtales), a model species for ecology and evolution. Mol Ecol Res 8(2):308–310
Crisp M, Cook L, Steane D (2004) Radiation of the Australian flora: what can comparisons of molecular phylogenies across multiple taxa tell us about the evolution of diversity in present-day communities? Phil Trans Roy Soc London Sr Biol Sci 359:1551–1571
Crisp MD, Cook LG, Steane DA (2005) Molecular dating and eucalypts: reply to Ladiges and Udovicic. Aust Syst Bot 18:295–296
Crisp MD, Burrows GE, Cook LG, Thornhill AH, Bowman DMJS (2011) Flammable biomes dominated by eucalypts originated at the Cretaceous-Palaeogene boundary. Nature Communications 2:193
da Costa IR, Forni-Martins ER (2006) Chromosome studies in species of Eugenia, Myrciaria and Plinia (Myrtaceae) from south-eastern Brazil. Austr J Bot 54:409–415
Da Costa IR, Forni-Martins ER (2007) Karyotype analysis in South American species of Myrtaceae. Bot J Linn Soc 155:571–580
da Costa IR, Dornelas MC, Forni-Martins ER (2008) Nuclear genome size variation in fleshy-fruited neotropical Myrtaceae. Plant Syst Evol 276:209–217
Dawson MI (1987) Contributions to a chromosome atlas of the New-Zealand flora—29 Myrtaceae. N Z J Bot 25:367–369
de Aguiar MS, Ferreira DF, Aguiar AM, Bison O, Rezende GDP, Grattapaglia D (2007) Potential of hybrids among elite clones of eucalypt by microsatellite markers. Pesqui Agropecu Bras 42:1007–1012
de Assis TF (2000) Production and use of Eucalyptus hybrids for industrial purposes. In: Nikles DG (ed) Hybrid breeding and genetics of forest trees. Proceedings of QFRI/CRC-SPF Symposium, 9-14th April 2000 Noosa, Queensland, Australia. Department of Primary Industries, Brisbane, pp 63–74
De Carvalho M, Gomes Caldas DG, Carneiro RT, Moon DH, Salvatierra GR, Franceschini LM, De Andrade A, Fiorani Celedon PA, Oda S, Labate CA (2008) SAGE transcript profiling of the juvenile cambial region of Eucalyptus grandis. Tree Physiol 28:905–919
De Lange PJ, Datson PM, Murray BG, Toelken HR (2005) Hybridism in the Kunzea ericoides complex (Myrtaceae): an analysis of artificial crosses. Aust Syst Bot 18:117–131
Dillen S, Storme V, Marron N, Bastien C, Neyrinck S, Steenackers M, Ceulemans R, Boerjan W (2008) Genomic regions involved in productivity of two interspecific poplar families in Europe. 1. Stem height, circumference and volume. Tree Genet Genomes 5:147–164
Dillon SK, Nolan M, Li W, Bell C, Wu HX, Southerton SG (2010) Allelic variation in cell wall candidate genes affecting solid wood properties in natural populations and land races of Pinus radiata. Genetics 185:1477–1487
Doran JC, Baker GR, Williams ER, Southwell IA (2006) Genetic gains in oil yields after nine years of breeding Melaleuca alternifolia (Myrtaceae). Aust J Exp Ag 46:1521–1527
Doughty RW (2000) The Eucalyptus. The Johns Hopkins University Press, Baltimore and London, A natural and commercial history of the gum tree
Dray FA Jr, Hale RE, Madeira PT, Bennett BC, Center TD (2009) Concordance between life history traits, invasion history, and allozyme diversity of the Everglades invader Melaleuca quinquenervia. Aquat Bot 90:296–302
Drummond RSM, Keeling DJ, Richardson TE, Gardner RC, Wright SD (2000) Genetic analysis and conservation of 31 surviving individuals of a rare New Zealand tree, Metrosideros bartlettii (Myrtaceae). Mol Ecol 9:1149–1157
Eckert AJ, Bower AD, Wegrzyn JL, Pande B, Jermstad KD, Krutovsky KV ST, Clair JB, Neale DB (2009) Association genetics of coastal douglas fir (Pseudotsuga menziesii var. menziesii, pinaceae). i. cold-hardiness related traits. Genetics 182:1289–1302
Eckert AJ, van Heerwaarden J, Wegrzyn JL, Nelson CD, Ross-Ibarra J, Gonzalez-Martinez SC, Neale DB (2010) Patterns of population structure and environmental associations to aridity across the range of Loblolly Pine (Pinus taeda L., Pinaceae). Genetics 185:969–982
Edwards RD, Craven LA, Crisp MD, Cook LG (2010) Melaleuca revisited: cpDNA and morphological data confirm that Melaleuca L. (Myrtaceae) is not monophyletic. Taxon 59:744–754
Eldridge K, Davidson J, Harwood C, van Wyk G (1993) Eucalypt domestication and breeding. Clarendon Press, Oxford, UK
El-Kassaby YA, Lstiburek M (2009) Breeding without breeding. Genet Res 91:111–120
Elliott CP, Byrne M (2004) Phylogenetics and the conservation of rare taxa in the Eucalyptus angustissima complex in western Australia. Conserv Genet 5:39–47
Elliott CP, Byrne M (2005) Isolation and characterization of microsatellites in the woody shrub, Calothamnus quadrifidus (Myrtaceae). Mol Ecol Notes 5:757–759
Ellis MF, Sedgley M, Gardner JA (1991) Interspecific pollen pistil interaction in Eucalyptus L’Her (Myrtaceae)—the effect of taxonomic distance. Ann Bot 68:185–194
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. Plos One 6
Eschler BM, Pass DM, Willis R, Foley WJ (2000) Distribution of foliar formylated phloroglucinol derivatives amongst Eucalyptus species. Biochem Syst Ecol 28:813–824
Faria DA, Mamani EMC, Pappas GJ, Grattapaglia D (2011) Genotyping systems for Eucalyptus based on tetra-, penta-, and hexanucleotide repeat EST microsatellites and their use for individual fingerprinting and assignment tests. Tree Genetics & Genomes 7:63–77
Faria DA, Tanno P, Reis A, Martins A, Ferreira ME, Grattapaglia D (2012) Genotyping-by-Sequencing (GbS) the highly heterozygous genome of Eucalyptus provides large numbers of high quality genome-wide SNPs. Plant & Animal Genomes XX Conference, San Diego, p Abstract P0521
Ferreira-Ramos R, Laborda PR, Santos MD, Mayor MS, Mestriner MA, de Souza AP, Alzate-Marin AL (2008) Genetic analysis of forest species Eugenia uniflora L. through of newly developed SSR markers. Conserv Genet 9:1281–1285
Field DL, Ayre DJ, Whelan RJ, Young AG (2008) Relative frequency of sympatric species influences rates of interspecific hybridization, seed production and seedling performance in the uncommon Eucalyptus aggregata. J Ecol 96:1198–1210
Field DL, Ayre DJ, Whelan RJ, Young AG (2009) Molecular and morphological evidence of natural interspecific hybridization between the uncommon Eucalyptus aggregata and the widespread E. rubida and E. viminalis. Conserv Genet 10:881–896
Foster SA, McKinnon GE, Steane DA, Potts BM, Vaillancourt RE (2007) Parallel evolution of dwarf ecotypes in the forest tree Eucalyptus globulus. New Phytol 175:370–380
Foucart C, Paux E, Ladouce N, San-Clemente H, Grima-Pettenati J, Sivadon P (2006) Transcript profiling of a xylem vs phloem cDNA subtractive library identifies new genes expressed during xylogenesis in Eucalyptus. New Phytol 170:739–752
Franceschinelli EV, Vasconcelos GMP, Landau EC, Ono KY, Santos FAM (2007) The genetic diversity of Myrciaria floribunda (Myrtaceae) in Atlantic forest fragments of different sizes. J Trop Ecol 23:361–367
Franzon RC, Castro CM, Raseira MDB (2010) Genetic variability in Surinam cherry populations originated from self-pollination and cross-pollination, estimated by AFLP. Rev Bras Frutic 32:240–250
Freeman JS, Jackson HD, Steane DA, McKinnon GE, Dutkowski GW, Potts BM, Vaillancourt RE (2001) Chloroplast DNA phylogeography of Eucalyptus globulus. Austr J Bot 49:585–596
Freeman JS, Potts BM, Shepherd M, Vaillancourt RE (2006) Parental and consensus linkage maps of Eucalyptus globulus using AFLP and microsatellite markers. Silvae Genet 55:202–217
Freeman JS, Marques CMP, Carocha V, Borralho N, Potts BM, Vaillancourt RE (2007) Origins and diversity of the Portuguese landrace of Eucalyptus globulus. Ann Forest Sci 64:639–647
Freeman JS, O'Reilly-Wapstra M, Vaillancourt RE, Wiggins N, Potts BM (2008a) Quantitative trait loci for key defensive compounds affecting herbivory of eucalypts in Australia. New Phytol 178:846–851
Freeman JS, Potts BM, Vaillancourt RE (2008b) Few Mendelian genes underlie the quantitative response of a forest tree, Eucalyptus globulus, to a natural fungal epidemic. Genetics 178:563–571
Freeman JS, Whittock SP, Potts BM, Vaillancourt RE (2009) QTL influencing growth and wood properties in Eucalyptus globulus. Tree Genet Genomes 5:713–722
Freeman J, Potts B, Downes G, Thavamanikumar S, Pilbeam D, Hudson C, Vaillancourt R (2011) QTL analysis for growth and wood properties across multiple pedigrees and sites in Eucalyptus globulus. BMC Proceedings 5:O8
Gadek PA, Wilson PG, Quinn CJ (1996) Phylogenetic reconstruction in Myrtaceae using matK, with particular reference to the position of Psiloxylon and Heteropyxis. Aust Syst Bot 9:283–290
Gaiotto FA, Bramucci M, Grattapaglia D (1997) Estimation of outcrossing rate in a breeding population of Eucalyptus urophylla with dominant RAPD and AFLP markers. Theor Appl Genet 95:842–849
Gamage HK, Schmidt S (2009) A robust method for chromosome quantification and ploidy determination in woody species. Austr J Bot 57:87–93
Gan S, Shi J, Li M, Wu K, Wu J, Bai J (2003) Moderate-density molecular maps of Eucalyptus urophylla S. T. Blake and E. tereticornis Smith genomes based on RAPD markers. Genetica 118:59–67
Gardner RC, De Lange PJ, Keeling DJ, Bowala T, Brown HA, Wright SD (2004) A late Quaternary phylogeography for Metrosideros (Myrtaceae) in New Zealand inferred from chloroplast DNA haplotypes. Biol J Linn Soc 83:399–412
Gibbs AK, Udovicic F, Drinnan AN, Ladiges PY (2009) Phylogeny and classification of Eucalyptus subgenus Eudesmia (Myrtaceae) based on nuclear ribosomal DNA, chloroplast DNA and morphology. Aust Syst Bot 22:158–179
Gion JM, Rech P, Grima-Pettenati J, Verhaegen D, Plomion C (2000) Mapping candidate genes in Eucalyptus with emphasis on lignification genes. Mol Breed 6:441–449
Gion JM, Carouche A, Deweer S, Bedon F, Pichavant F, Charpentier JP, Bailleres H, Rozenberg P, Carocha V, Ognouabi N, Verhaegen D, Grima-Pettenati J, Vigneron P, Plomion C (2011) Comprehensive genetic dissection of wood properties in a widely-grown tropical tree: Eucalyptus. Bmc Genomics 12:301
Glaubitz JC, Emebiri LC, Moran GF (2001) Dinucleotide microsatellites from Eucalyptus sieberi: inheritance, diversity, and improved scoring of single-base differences. Genome 44:1041–1045
Godinez-Alvarez H (2004) Pollination and seed dispersal by lizards: a review. Rev Chil Hist Nat 77:569–577
Goff SA, Ricke D, Lan T-H, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchison D, Martin C, Katagiri F, Lange BM, Moughamer T, Xia Y, Budworth P, Zhong J, Miguel T, Paszkowski U, Zhang S, Colbert M, W-l S, Chen L, Cooper B, Park S, Wood TC, Mao L, Quail P, Wing R, Dean R, Yu Y, Zharkikh A, Shen R, Sahasrabudhe S, Thomas A, Cannings R, Gutin A, Pruss D, Reid J, Tavtigian S, Mitchell J, Eldredge G, Scholl T, Miller RM, Bhatnagar S, Adey N, Rubano T, Tusneem N, Robinson R, Feldhaus J, Macalma T, Oliphant A, Briggs S (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296:92–100
Goicoechea M, Lacombe E, Legay S, Mihaljevic S, Rech P, Jauneau A, Lapierre C, Pollet B, Verhaegen D, Chaubet-Gigot N, Grima-Pettenati J (2005) EgMYB2, a new transcriptional activator from Eucalyptus xylem, regulates secondary cell wall formation and lignin biosynthesis. Plant J 43:553–567
Goldstein DB (2009) Common Genetic Variation and Human Traits. New England Journal of Medicine 360:1696–1698
Gonzalez-Martinez SC, Krutovsky KV, Neale DN (2006) Forest tree population genomics and adaptive evolution. New Phytol 170:227–238
Gonzalez-Martinez SC, Wheeler NC, Ersoz E, Nelson CD, Neale DB (2007) Association genetics in Pinus taeda L. I. wood property traits. Genetics 175:399–409
Gonzalez-Martinez SC, Huber D, Ersoz E, Davis JM, Neale DB (2008) Association genetics in Pinus taeda L. II. Carbon isotope discrimination. Heredity 101:19–26
Gonzalez-Varo JP, Albaladejo RG, Aparicio A (2009) Mating patterns and spatial distribution of conspecific neighbours in the Mediterranean shrub Myrtus communis (Myrtaceae). Plant Ecol 203:207–215
Govaerts R, Sobral M, Ashton P, Barrie F, Holst BK, Landrum LR, Matsumoto K, Mazine FF, Nic Lughadha E, Proença C, Soares-Silva LH, Wilson PG, Lucas EJ (2008) World checklist of Myrtaceae. Kew Publishing, Royal Botanic Gardens, Kew
Grattapaglia D (2004) Integrating genomics into Eucalyptus breeding. Genet Mol Res 3:369–379
Grattapaglia D (2008) Genomics of Eucalyptus, a global tree for energy, paper and wood. In: Moore P, Ming R (eds) Genomics of tropical crop plants. Springer Sciences, pp 257-295
Grattapaglia D, Bradshaw HD (1994) Nuclear DNA content of commercially important Eucalyptus species and hybrids. Can J Forest Res 24:1074–1078
Grattapaglia D, Kirst M (2008) Eucalyptus applied genomics: from gene sequences to breeding tools. New Phytol 179:911–929
Grattapaglia D, Resende MDV (2011) Genomic selection in forest tree breeding. Tree Genet Genomes 7:241–255
Grattapaglia D, Sederoff R (1994) Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics 137:1121–1137
Grattapaglia D, Bertolucci FL, Sederoff RR (1995) Genetic mapping of QTLs controlling vegetative propagation in Eucalyptus grandis and E. urophylla using a pseudo-testcross strategy and RAPD markers. Theor Appl Genet 90:933–947
Grattapaglia D, Bertolucci FL, Penchel R, Sederoff RR (1996) Genetic mapping of quantitative trait loci controlling growth and wood quality traits in Eucalyptus grandis using a maternal half-sib family and RAPD markers. Genetics 144:1205–1214
Grattapaglia D, Alfenas AC, Coelho ASG, Bearzoti E, Pappas GJ, Pasquali G, Pereira G, Colodette J, Gomide JL, Bueno J, Cascardo JC, Brondani RPV, Brommonschenkel SH (2004a) Building resources for molecular breeding of Eucalyptus. In: Borralho NMG, Pereira JS, Marques C, Coutinho J, Madeira M, Tomé M (eds) International IUFRO Conference: Eucalyptus in a changing world. RAIZ, Instituto Investigação da Floresta e Papel, Portugal, Aveiro Portugal, pp 20–32
Grattapaglia D, Ribeiro VJ, Rezende G (2004b) Retrospective selection of elite parent trees using paternity testing with microsatellite markers: an alternative short term breeding tactic for Eucalyptus. Theor Appl Genet 109:192–199
Grattapaglia D, Plomion C, Kirst M, Sederoff RR (2009) Genomics of growth traits in forest trees. Curr Opin Plant Biol 12:148–156
Grattapaglia D, Silva Junior OB, Kirst M, Lima BM, Faria DA, Pappas GJ Jr (2011a) High-throughput SNP genotyping in the highly heterozygous genome of Eucalyptus: assay success, polymorphism and transferability across species. BMC Plant Biol 11:65
Grattapaglia D, de Alencar S, Pappas G (2011b). Genome-wide genotyping and SNP discovery by ultra-deep Restriction-Associated DNA (RAD) tag sequencing of pooled samples of E. grandis and E. globulus. BMC Proceedings 5:P45
Greilhuber J (2008) Cytochemistry and C-values: the less-well-known world of nuclear DNA amounts. Ann Bot 101:791–804
Griffin AR, Cotterill PP (1988) Genetic-variation in growth of outcrossed, selfed and open-pollinated progenies of Eucalyptus regnans and some implications for breeding strategy. Silvae Genet 37:124–131
Griffin AR, Burgess IP, Wolf L (1988) Patterns of hybridization and manipulated hybridization in the genus Eucalyptus L'Herit—a review. Austr J Bot 36:41–66
Grosser C, Potts BM, Vaillancourt RE (2010) Microsatellite based paternity analysis in a clonal Eucalyptus nitens seed orchard. Silvae Genet 59:57–62
Harbaugh DT, Wagner WL, Percy DM, James HF, Fleischer RC (2009) Genetic structure of the polymorphic Metrosideros (Myrtaceae) complex in the Hawaiian Islands using nuclear microsatellite data. PLos One 4
Harrington MG, Gadek PA (2004) Molecular systematics of the Acmena alliance (Myrtaceae): phylogenetic analyses and evolutionary implications with reference to Australian taxa. Aust Syst Bot 17:63–72
Harwood C (2011) Introductions: doing it right. In: Walker J (ed) Developing a eucalypt resource: learning from Australia and elsewhere. Wood Technology Research Centre, University of Canterbury, Christchurch, New Zealand, pp 43–54
He Q, Liang G, Xie J, Li W (2006) Genetic diversity analysis of wax apple germplasm by ISSR markers. Acta Hort Sin 33:392–394
Henery ML, Moran GF, Wallis IR, Foley WJ (2007) Identification of quantitative trait loci influencing foliar concentrations of terpenes and formylated phloroglucinol compounds in Eucalyptus nitens. New Phytol 176:82–95
Heywood VH (1996) Flowering plants of the world. B.T. Batsford Ltd., London
Hill KD, Johnson LAS (1995) Systematic studies in the eucalypts 7. A revision of the bloodwoods, genus Corymbia (Myrtaceae). Telopea 6:185–504
Hillyer MJ, Boulter SL, Kitching R, Huges JM (2007) Isolation and characterisation of eight polymorphic microsatellite loci in the rainforest canopy tree, Syzgium sayeri (Myrtaceae). Mol Ecol Notes 7:1199–1120
Hirakawa H, Nakamura Y, Kaneko T, Isobe S, Sakai H, Kato T, Hibino T, Sasamoto S, Watanabe A, Yamada M, Nakayama S, Fujishiro T, Kishida Y, Kohara M, Tabata S, Sato S (2011) Survey of the genetic information carried in the genome of Eucalyptus camaldulensis. Plant Biotechnology 28:471–480
Ho CK, Chang SH, Tsay JY, Tsai CJ, Chiang VL, Chen ZZ (1998) Agrobacterium tumefaciens-mediated transformation of Eucalyptus camaldulensis and production of transgenic plants. Plant Cell Reports 17:675–680
Holman JE, Hughes JM, Fensham RJ (2003) A morphological cline in Eucalyptus: a genetic perspective. Mol Ecol 12:3013–3025
Hudson CJ (2007) Genetic studies of the Eucalyptus gunnii–archeri complex and related species on the central plateau. Honours thesis, University of Tasmania
Hudson CJ, Kullan ARK, Freeman JS, Faria DA, Grattapaglia D, Kilian A, Myburg AA, Potts BM, Vaillancourt RE (2011) High synteny and colinearity among Eucalyptus genomes revealed by high-density comparative genetic mapping. Tree Genetics & Genomes 8:339–352
Ingvarsson PK (2005) Nucleotide polymorphism and linkage disequilbrium within and among natural populations of European Aspen (Populus tremula L., Salicaceae). Genetics 169:945–953
Ingvarsson PK, Garcia MV, Luquez V, Hall D, Jansson S (2008) Nucleotide polymorphism and phenotypic associations within and around the phytochrome b2 locus in European aspen (Populus tremula, Salicaceae). Genetics 178:2217–2226
Jackson HD, Steane DA, Potts BM, Vaillancourt RE (1999) Chloroplast DNA evidence for reticulate evolution in Eucalyptus (Myrtaceae). Mol Ecol 8:739–751
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyere C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, Le Clainche I, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezzotti M, Lecharny A, Scarpelli C, Artiguenave F, Pe ME, Valle G, Morgante M, Caboche M, Adam-Blondon AF, Weissenbach J, Quetier F, Wincker P (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449:463–U465
James EA, McDougall KL (2007) Extent of clonality, genetic diversity and decline in the endangered mallee Eucalyptus imlayensis. Austr J Bot 55:548–553
James SA, Puttock CF, Cordell S, Adams RP (2004) Morphological and genetic variation within Metrosideros polymorpha (Myrtaceae) on Hawai'i. N Z J Bot 42:263–270
Janaki-Ammal EK, Khosla SN (1969) Breaking barrier to polyploidy in genus Eucalyptus. Proc In Acad Sci Sect B 70:248
Jansen RK, Kaittanis C, Saski C, Lee SB, Tomkins J, Alverson AJ, Daniell H (2006) Phylogenetic analyses of Vitis (Vitaceae) based on complete chloroplast genome sequences: effects of taxon sampling and phylogenetic methods on resolving relationships among rosids. BMC Evol Biol 6:32
Jensen RJ, Schwoyer M, Crawford DJ, Stuessy TF, Anderson GJ, Baeza CM OMS, Ruiz E (2002) Patterns of morphological and genetic variation among populations of Myrceugenia fernandeziana (Myrtaceae) on Masatierra island: implications for conservation. Syst Bot 27:534–547
Johnson LAS, Briggs BG (1984) Myrtales and Myrtaceae—a phylogenetic analysis. Ann MO Bot Gard 71:700–756
Johnson GR, Wheeler NC, Strauss SS (2000) Financial feasibility of marker-aided selection in Douglas-fir. Can J Forest Res 30:1942–1952
Jones R (2009) Molecular evolution and genetic control of flowering in the Eucalyptus globulus species complex. School of Plant Science. University of Tasmania, Hobart, p 221
Jones ME, Stokoe RL, Cross MJ, Scott LJ, Maguire TL, Shepherd M (2001) Isolation of microsatellite loci from spotted gum (Corymbia variegata), and cross-species amplification in Corymbia and Eucalyptus. Mol Ecol Notes 1:276–278
Jones R, Steane DA, Potts BM, Vaillancourt RE (2002) Microsatellite and morphological analysis of Eucalyptus globulus populations. Can J For Res 32:59–66
Jones RC, McKinnon GE, Potts BM, Vaillancourt RE (2005) Genetic diversity and mating system of an endangered tree Eucalyptus morrisbyi. Austr J Bot 53:367–377
Jones TH, Steane DA, Jones RC, Pilbeam D, Vaillancourt RE, Potts BM (2006) Effects of domestication on genetic diversity in Eucalyptus globulus. For Ecol Manag 234:78–84
Jones TH, Vaillancourt RE, Potts BM (2007) Detection and visualization of spatial genetic structure in continuous Eucalyptus globulus forest. Mol Ecol 16:697–707
Jones M, Shepherd M, Henry R, Delves A (2008) Pollen flow in Eucalyptus grandis determined by paternity analysis using microsatellite markers. Tree Genet Genomes 4:37–47
Junghans DT, Alfenas AC, Brommonschenkel SH, Oda S, Mello EJ, Grattapaglia D (2003) Resistance to rust (Puccinia psidii Winter) in Eucalyptus: mode of inheritance and mapping of a major gene with RAPD markers. Theor Appl Genet 108:175–180
Kaneko S, Isagi Y, Nobushima F (2007) Development of microsatellite markers for Metrosideros boninensis (Myrtaceae), an endangered endemic plant species from the Bonin Islands, Japan. Conservation Genetics 8(3):753–755
Kaneko S, Isagi Y, Nobushima F (2008) Genetic differentiation among populations of an oceanic island: the case of Metrosideros boninensis, an endangered endemic tree species in the Bonin Islands. Plant Spec Biol 23:119–128
Kawazu T, Doi K, Kondo K (2003) Process for transformation of mature trees of Eucalyptus plants. US Patent 6,563,024 B1
Keil M, Griffin AR (1994) Use of random amplified polymorphic DNA (RAPD) markers in the discrimination and verification of genotypes in Eucalyptus. Theor Appl Genet 89:442–450
Keller G, Marchal T, SanClemente H, Navarro M, Ladouce N, Wincker P, Couloux A, Teulières C, Marque C (2009) Development and functional annotation of an 11,303-EST collection from Eucalyptus for studies of cold tolerance. Tree Genet Genomes 5:317–327
Keszei A, Brubaker CL, Foley WJ (2008) A molecular perspective on terpene variation in Australian Myrtaceae. Austr J Bot 56:197–213
Keszei A, Brubaker CL, Carter R, Kollner T, Degenhardt J, Foley WJ (2010a) Functional and evolutionary relationships between terpene synthases from Australian Myrtaceae. Phytochemistry 71:844–852
Keszei A, Hassan Y, Foley WJ (2010b) A biochemical interpretation of terpene chemotypes in Melaleuca alternifolia. J Chem Ecol 36:652–661
Kirst M, Brondani RPV, Brondani C, Grattapaglia D (1997) Screening of designed primer pairs for recovery of microsatellite markers and their transferability among species of Eucalyptus. In: EMBRAPA (ed) IUFRO Conference on Eucalyptus Genetics and Silviculture. EMBRAPA CNPF, Salvador, BA, Brazil, pp 167–171
Kirst M, Myburg AA, De Leon JPG, Kirst ME, Scott J, Sederoff R (2004) Coordinated genetic regulation of growth and lignin revealed by quantitative trait locus analysis of cDNA microarray data in an interspecific backcross of Eucalyptus. Plant Phys 135:2368–2378
Kirst M, Basten CJ, Myburg AA, Zeng ZB, Sederoff RR (2005a) Genetic architecture of transcript-level variation in differentiating xylem of a Eucalyptus hybrid. Genetics 169:2295–2303
Kirst M, Cordeiro CM, Rezende G, Grattapaglia D (2005b) Power of microsatellite markers for fingerprinting and parentage analysis in Eucalyptus grandis breeding populations. J Heredity 96:161–166
Külheim C (2010) Applying second-generation sequencing to non-model species. Aust Biochemist 41:10–13
Külheim C, Yeoh SH, Maintz J, Foley WJ, Moran GF (2009) Comparative SNP diversity among four Eucalyptus species for genes from secondary metabolite biosynthetic pathways. BMC Genomics 10:1–11
Külheim C, Yeoh SH, Wallis IR, Laffan S, Moran GF, Foley WJ (2011) The molecular basis of quantitative variation in foliar secondary metabolites in Eucalyptus globulus. New Phytologist 191:1041–1053
Kullan ARK, Van Dyk MM, Jones N, Kanzler A, Bayley A, Myburg AA (2012) High-density genetic linkage maps with over 2,400 sequence-anchored DArT markers for genetic dissection in an F2 pseudo-backcross of Eucalyptus grandis × E. urophylla. Tree Genet Genomes 8:163–175
Labate CA, de Assis TF, Oda S, de Mello EJ, González ER, Zauza EAV, Mori ES, de Moraes MLT, Cid LPB, Alfenas AC, Foelkel C, Moon DH, de Carvalho MCdCG, Caldas DGG, Carneiro RT, de Andrade A, Salvatierra GR (2009) Eucalyptus. In: Kole C, Hall TC (eds) Compendium of transgenic crop plants, pp 35-108
Ladiges PY (1997) Phylogenetic history and classification of eucalypts. In: Woinarski JCZ (ed) Eucalypt ecology: individuals to ecosystems. Cambridge University Press, Cambridge, pp 16–29
Ladiges PY, Udovicic F (2000) Comment on a new classification of the eucalypts. Aust Syst Bot 13:149–152
Ladiges PY, Udovicic F (2005) Comment on molecular dating of the age of eucalypts. Aust Syst Bot 18:291–293
Ladiges PY, Udovic F, Drinnan AN (1995) Eucalypt phylogeny—molecules and morphology. Aust Syst Bot 8:483–497
Ladiges PY, McFadden GI, Middleton N, Orlovich DA, Treloar N, Udovicic F (1999) Phylogeny of Melaleuca, Callistemon, and related genera of the Beaufortia suballiance (Myrtaceae) based on 5S and ITS-1 spacer regions of nrDNA. Cladistics 15:151–172
Ladiges PY, Udovicic F, Nelson G (2003) Australian biogeographical connections and the phylogeny of large genera in the plant family Myrtaceae. J Biogeogr 30:989–998
Lam N, Wilson PG, Heslewood MM, Quinn CJ (2002) A phylogenetic analysis of the Chamelaucium alliance (Myrtaceae). Aust Syst Bot 15:535–543
Le S, Nock C, Henson M, Shepherd M (2009) Genetic differentiation among and within three red mahoganies (series Annulares), Eucalyptus pellita, E. resinifera and E. scias (Myrtaceae). Aust Syst Bot 22:332–343
Leitch IJ, Bennett MD (2004) Genome downsizing in polyploid plants. Biol J Linn Soc 82:651–663
Lin H, Jian M, Liang LY, Pei WJ, Liu XZ, Zhang HY (2010) Production of polyploids from cultured shoot tips of Eucalyptus globulus Labill by treatment with colchicine. Afr J Biotechnol 9:2252–2255
Lippert D, Chowrira S, Ralph SG, Zhuang J, Aeschliman D, Ritland C, Ritland K, Bohlmann J (2007) Conifer defense against insects: proteome analysis of Sitka spruce (Picea sitchensis) bark induced by mechanical wounding or feeding by white pine weevils (Pissodes strobi). Proteomics 7:248–270
Lucas EJ, Belsham SR, Lughadha EMN, Orlovich DA, Sakuragui CM, Chase MW, Wilson PG (2005) Phylogenetic patterns in the fleshy-fruited Myrtaceae—preliminary molecular evidence. Plant Syst Evol 251:35–51
Lucas EJ, Harris SA, Mazine FF, Bellsham SR, Lughadha EMN, Telford A, Gasson PE, Chase MW (2007) Suprageneric phylogenetics of Myrteae, the generically richest tribe in Myrtaceae (Myrtales). Taxon 56:1105–1128
Lughadha EN, Proenca C (1996) A survey of the reproductive biology of the Myrtoideae (Myrtaceae). Ann MO Bot Gard 83:480–503
Luikart G, England P, Tallmon DA, Jordan S, Taberlet P (2003) The power and promise of population genomics: from genotyping to genome typing. Nat Rev Genet 4:981–994
Ma XL, Considine JA, Yan GJ (2004) Chloroplast DNA variation and inheritance in waxflowers (Myrtaceae). Austr J Bot 52:55–61
Machado LDR, deAndrade GM, Cid LPB, Penchel RM, Brasileiro ACM (1997) Agrobacterium strain specificity and shooty tumour formation in eucalypt (Eucalyptus grandis x E. urophylla). Plant Cell Rep 16:299–303
MacMillan CP, Mansfield SD, Stachurski ZH, Evans R, Southerton SG (2010) Fasciclin-like arabinogalactan proteins: specialization for stem biomechanics and cell wall architecture in Arabidopsis and Eucalyptus. Plant J 62:689–703
Mamani EMC, Bueno NW, Faria DA, Guimarães LMS, Lau D, Alfenas ACA, Grattapaglia D (2010) Positioning of the major locus for Puccinia psidii rust resistance (Ppr1) on the Eucalyptus reference map and its validation across unrelated pedigrees. Tree Genet Genomes 6:953–962
Marcucci-Poltri SN, Zelener N, Rodriguez Traverso J, Gelid P, Hopp HE (2003) Selection of a seed orchard of Eucalyptus dunnii based on genetic diversity criteria calculated using molecular markers. Tree Physiol 23:625–632
Margis R, Felix D, Caldas JF, Salgueiro F, De Araujo DSD, Breyne P, Van Montagu M, De Oliveira D, Margis-Pinheiro M (2002) Genetic differentiation among three neighboring Brazil-cherry (Eugenia uniflora L.) populations within the Brazilian Atlantic rain forest. Biodivers Conserv 11:149–163
Marie D, Brown SC (1993) A cytometric exercise in plant DNA histograms, with 2c-values for 70 species. Biol Cell 78:41–51
Marques CM, Araujo JA, Ferreira JG, Whetten R, O'Malley DM, Liu BH, Sederoff R (1998) AFLP genetic maps of Eucalyptus globulus and E. tereticornis. Theor Appl Genet 96:727–737
Marques CM, Vasquez-Kool J, Carocha VJ, Ferreira JG, O'Malley DM, Liu BH, Sederoff R (1999) Genetic dissection of vegetative propagation traits in Eucalyptus tereticornis and E. globulus. Theor Appl Genet 99:936–946
Marques CM, Brondani RPV, Grattapaglia D, Sederoff R (2002) Conservation and synteny of SSR loci and QTLs for vegetative propagation in four Eucalyptus species. Theor Appl Genet 105:474–478
Marques CM, Carocha VJ, de Sa ARP, Oliveira MR, Pires AM, Sederoff R, Borralho NMG (2005) Verification of QTL linked markers for propagation traits in Eucalyptus. Tree Genet Genomes 1:103–108
Martin B, Quillet J (1974) The propogation by cuttings of forest trees in the Congo. Bois et Forets des Tropiques 155:15–33
Martos I, Ferreres F, Tomas-Barberan FA (2000) Identification of flavonoid markers for the botanical origin of Eucalyptus honey. J Agric Food Chem 48:1498–1502
McDonald MW, Butcher PA, Larmour JS, Bell JC (2003) Intra- and interspecific allozyme variation in eucalypts from the spotted gum group, Corymbia, section 'Politaria' (Myrtaceae) (vol 13, pg 491, 2000). Aust Syst Bot 16:643–U617
McDonald MW, Brooker MIH, Butcher PA (2009) A taxonomic revision of Eucalyptus camaldulensis (Myrtaceae). Aust Syst Bot 22:257–285
McGowen MH, Wiltshire RJE, Potts BM, Vaillancourt RE (2001) The origin of Eucalyptus vernicosa, a unique shrub eucalypt. Biol J Linn Soc 74:397–405
McKinnon GE, Steane DA, Potts BM, Vaillancourt RE (1999) Incongruence between chloroplast and species phylogenies in Eucalyptus subgenus Monocalyptus. Am J Bot 86:1038–1046
McKinnon GE, Vaillancourt RE, Jackson HD, Potts BM (2001a) Chloroplast sharing in the Tasmanian eucalypts. Evolution 55:703–711
McKinnon GE, Vaillancourt RE, Tilyard PA, Potts BM (2001b) Maternal inheritance of the chloroplast genome in Eucalyptus globulus and interspecific hybrids. Genome 44:831–835
McKinnon GE, Jordan GJ, Vaillancourt RE ASD, Potts BM (2004a) Glacial refugia and reticulate evolution: the case of the Tasmanian eucalypts. Phil Trans Roy Soc London Sr B Biol Sci 359:275–284
McKinnon GE, Vaillancourt RE, Steane DA, Potts BM (2004b) The rare silver gum, Eucalyptus cordata, is leaving its trace in the organellar gene pool of Eucalyptus globulus. Mol Ecol 13:3751–3762
McKinnon GE, Potts BM, Steane DA, Vaillancourt RE (2005) Population and phylogenetic analysis of the cinnamoyl coA reductase gene in Eucalyptus globulus (Myrtaceae). Austr J Bot 53:827–838
McKinnon GE, Vaillancourt RE, Steane DA, Potts BM (2008) An AFLP marker approach to lower-level systematics in Eucalyptus (Myrtaceae). Am J Bot 95:368–380
McKinnon GE, Smith JJ, Potts BM (2010) Recurrent nuclear DNA introgression accompanies chloroplast DNA exchange between two eucalypt species. Mol Ecol 19:1367–1380
Messaoud C, Afif M, Boulila A, Rejeb MN, Boussaid M (2007) Genetic variation of Tunisian Myrtus communis L. (Myrtaceae) populations assessed by isozymes and RAPDs. Ann For Sci 64:845–853
Meuwissen TH, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Mimura M, Barbour RC, Potts BM, Vaillancourt RE, Watanabe KN (2009) Comparison of contemporary mating patterns in continuous and fragmented Eucalyptus globulus native forests. Mol Ecol 18:4180–4192
Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, Senin P, Wang W, Ly BV, Lewis KL, Salzberg SL, Feng L, Jones MR, Skelton RL, Murray JE, Chen C, Qian W, Shen J, Du P, Eustice M, Tong E, Tang H, Lyons E, Paull RE, Michael TP, Wall K, Rice DW, Albert H, Wang ML, Zhu YJ, Schatz M, Nagarajan N, Acob RA, Guan P, Blas A, Wai CM, Ackerman CM, Ren Y, Liu C, Wang J, Wang J, Na JK, Shakirov EV, Haas B, Thimmapuram J, Nelson D, Wang X, Bowers JE, Gschwend AR, Delcher AL, Singh R, Suzuki JY, Tripathi S, Neupane K, Wei H, Irikura B, Paidi M, Jiang N, Zhang W, Presting G, Windsor A, Navajas-Perez R, Torres MJ, Feltus FA, Porter B, Li Y, Burroughs AM, Luo MC, Liu L, Christopher DA, Mount SM, Moore PH, Sugimura T, Jiang J, Schuler MA, Friedman V, Mitchell-Olds T, Shippen DE, dePamphilis CW, Palmer JD, Freeling M, Paterson AH, Gonsalves D, Wang L, Alam M (2008) The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452:991–996
Missiaggia AA, Piacezzi AL, Grattapaglia D (2005) Genetic mapping of Eef1, a major effect QTL for early flowering in Eucalyptus grandis. Tree Genet Genomes 1:79–84
Miwa M, Tanaka R, Shinone M, Kojima K, Hogetsu T (2000) Development of polymorphic microsatellite markers in a tropical tree species, Melaleuca cajuputi (Myrtaceae). Mol Ecol 9:639–640
Mizrachi E, Hefer C, Ranik M, Joubert F, Myburg AA (2010) Prelude to a genome: de novo assembly, annotation and profiling of an expressed gene catalog of a fast-growing Eucalyptus hybrid clone using Illumina mRNA-seq. XVIII Plant & Animal Genomes Conference, San Diego, abstract W233
Moon DH, Salvatierra GR, Caldas DGG, de Carvalho M, Carneiro RT, Franceschini LM, Oda S, Labate CA (2007) Comparison of the expression profiles of susceptible and resistant Eucalyptus grandis exposed to Puccinia psidii Winter using SAGE. Funct Plant Biol 34:1010–1018
Moore BD, Foley WJ, Wallis IR, Cowling A, Handasyde KA (2005) Eucalyptus foliar chemistry explains selective feeding by koalas. Biol Lett 1:64–67
Moran GF (1992) Patterns of genetic diversity in Australian tree species. New Forest 6:49–66
Mori SA, Boom BM, Decarvalino AM, Dossantos TS (1983) Ecological importance of Myrtaceae in an eastern Brazilian wet forest. Biotropica 15:68–70
Myburg AA, Griffin RA, Sederoff RR, Whetten RW (2003) Comparative genetic linkage maps of Eucalyptus grandis, Eucalyptus globulus and their F1 hybrid based on a double pseudo-backcross mapping approach. Theor Appl Genet 107:1028–1042
Myburg AA, Vogl C, Griffin RA, Sederoff RR, Whetten RW (2004) Genetics of postzygotic isolation in Eucalyptus: whole-genome analysis of barriers to introgression in a wide interspecific cross of Eucalyptus grandis and E. globulus. Genetics 166:1405–1418
Myburg AA, Potts BM, Marques CM, Kirst M, Gion JM, Grattapaglia D, Grima-Pettenati J (2007) Eucalyptus. In: Kole C (ed) Genome mapping and molecular breeding in plants Springer. NY, USA, New York, pp 115–160
Myburg AA, Grattapaglia D, Tuskan GA, Schmutz J, Barry K, Bristow J (2008) The Eucalyptus Genome Network Sequencing the Eucalyptus genome: genomic resources for renewable energy and fiber production. XVI Plant & Animal Genome Conference, San Diego, CA
Neale DB, Savolainen O (2004) Association genetics of complex traits in conifers. Trends Plant Sci 9:325–330
Nesbitt KA, Potts BM, Vaillancourt RE, West AK, Reid JB (1995) Partitioning and distribution of RAPD variation in a forest tree species, Eucalyptus globulus (Myrtaceae). Heredity 74:628–637
Nesbitt KA, Potts BM, Vaillancourt RE, Reid JB (1997) Fingerprinting and pedigree analysis in Eucalyptus globulus using RAPDs. Silvae Genet 46:6–11
Neves LG, Mamani EMC, Alfenas AC, Kirst M, Grattapaglia D (2011) A high-density transcript linkage map with 1,845 expressed genes positioned by microarray-based Single Feature Polymorphisms (SFP) in Eucalyptus. Bmc Genomics 12:189
Nevill PG, Bossinger G, Ades PK (2008) Genome sharing patterns in south-east Australian eucalypts. Proceedings of 'Old Forests, New Management' Sir Mark Oliphant Conference, Hobart, Tasmania, Australia, abstract 158
Nevill PG, Bossinger G, Ades PK (2010) Phylogeography of the world's tallest angiosperm, Eucalyptus regnans: evidence for multiple isolated Quaternary refugia. J Biogeogr 37:179–192
Niedenzu F (1893) Myrtaceae. In: Engler A, Prantl K (eds) Die Natürlichen Pflanzenfamilien III, pp 57–107
Nodari RO, Guerra MP, Meler K, Ducroquet JP (1997) Genetic variability of Feijoa sellowiana germplasm. Acta Hort (ISHS) 452:41–46
Novaes E, Drost DR, Farmerie WG, Pappas GJ, Grattapaglia D, Sederoff R, Kirst M (2008) High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics 9:312–326
O’Reilly-Wapstra JM, Freeman JS, Davies NW, Vaillancourt RE, Fitzgerald H, Potts BM (2011) Quantitative trait loci for foliar terpenes in a global eucalypt species. Tree Genetics & Genomes 7:485–498
O'Brien MM, Quinn CJ, Wilson PG (2000) Molecular systematics of the Leptospermum suballiance (Myrtaceae). Austr J Bot 48:621–628
Ochieng J, Steane DA, Ladiges PY, Baverstock PR, Henry RJ, Shepherd M (2007a) Microsatellites retain phylogenetic signals across genera in eucalypts (Myrtaceae). Genet Mol Biol 30:1125–1134
Ochieng JW, Henry RJ, Baverstock PR, Steane DA, Shepherd M (2007b) Nuclear ribosomal pseudogenes resolve a corroborated monophyly of the eucalypt genus Corymbia despite misleading hypotheses at functional ITS paralogs. Mol Phylogenet Evol 44:752–764
Ochieng J, Shepherd M, Baverstock P, Nikles G, Lee DJ, Henry RJ (2008) Genetic variation within two sympatric spotted gum eucalypts exceeds between taxa variation. Silvae Genet 57:249–256
Ochieng JW, Shepherd M, Baverstock PR, Nikles G, Lee DJ, Henry RJ (2010) Two sympatric spotted gum species are molecularly homogeneous. Conserv Genet 11:45–56
Ohri D (2002) Genome size variation in some tropical hardwoods. Biol Plantarum 45:455–457
Ohri D, Bhargava A, Chatterjee A (2004) Nuclear DNA amounts in 112 species of tropical hardwoods - New estimates. Plant Biology 6:555–561
Okamura H, Mimura A, Yakou Y, Niwano M, Takahara Y (1993) Antioxidant activity of tannins and flavonoids in Eucalyptus rostrata. Phytochemistry 33:557–561
Okun DO, Kenya EU, Oballa PO, Odee DW, Muluvi GM (2008) Analysis of genetic diversity in Eucalyptus grandis (Hill ex Maiden) seed sources using inter simple sequence repeats (ISSR) molecular markers. Afr J Biotechnol 7:2119–2123
O'Malley DM, Sederoff RR, Grattapaglia D (1994) Methods for within family selection in woody perennials using genetic markers. USPTO Patent Number 6054634
Ottewell KM, Donnellan SC, Moran GF, Paton DC (2005) Multiplexed microsatellite markers for the genetic analysis of Eucalyptus leucoxylon (Myrtaceae) and their utility for ecological and breeding studies in other Eucalyptus species. J Heredity 96:445–451
Oudjehih B, Bentouati A (2006) Chromosome numbers of the 59 species of Eucalyptus L'Herit. (Myrtaceae). Caryologia 59:207–212
Padovan A, Keszei A, Köllner TG, Degenhardt J, Foley WJ (2010) The molecular basis of host plant selection in Melaleuca quinquenervia by a successful biological control agent. Phytochemistry 71:1237–1244
Paiva JA, Prat E, Vautrin S, Santos MD, San-Clemente H, Brommonschenkel S, Fonseca PG, Grattapaglia D, Song X, Ammiraju JS, Kudrna D, Wing RA, Freitas AT, Bergès H, Grima-Pettenati J (2011) Advancing Eucalyptus genomics: identification and sequencing of lignin biosynthesis genes from deep-coverage BAC libraries. BMC Genomics 12:137
Parnell JAN, Craven LA, Biffin E (2007) Matters of scale: Dealing with one of the largest genera of angiosperms. In: Hodkinson TR, Parnell JAN (eds) Reconstructing the tree of life: taxonomy and systematics of species rich taxa. The Systematics Association Special Volume Series 72, Boca Raton CRC Press, pp 251–273
Parra-O C, Bayly UF, Ladiges PY (2006) ETS sequences support the monophyly of the eucalypt genus Corymbia (Myrtaceae). Taxon 55:653–663
Parra-O C, Bayly MJ, Drinnan A, Udovicic F, Ladiges P (2009) Phylogeny, major clades and infrageneric classification of Corymbia (Myrtaceae), based on nuclear ribosomal DNA and morphology. Aust Syst Bot 22:384–399
Parra-O C, Bayly MJ, Drinnan A, Udovicic F, Ladiges P (2010) Phylogeny, major clades and infrageneric classification of Corymbia (Myrtaceae), based on nuclear ribosomal DNA and morphology (vol 22, pg 384, 2009). Aust Syst Bot 23:141-+
Patterson B, Gore P, Potts BM, Vaillancourt RE (2004) Advances in pollination techniques for large-scale seed production in Eucalyptus globulus. Austr J Bot 52:781–788
Paux E, Tamasloukht M, Ladouce N, Sivadon P, Grima-Pettenati J (2004) Identification of genes preferentially expressed during wood formation in Eucalyptus. Plant Mol Biol 55:263–280
Paux E, Carocha V, Marques C, de Sousa AM, Borralho N, Sivadon P, Grima-Pettenati J (2005) Transcript profiling of Eucalyptus xylem genes during tension wood formation. New Phytol 167:89–100
Payn KG, Dvorak WS, Myburg AA (2007) Chloroplast DNA phylogeography reveals the island colonisation route of Eucalyptus urophylla (Myrtaceae). Austr J Bot 55:673–683
Payn KG, Dvorak WS, Janse BJH, Myburg AA (2008) Microsatellite diversity and genetic structure of the commercially important tropical tree species Eucalyptus urophylla, endemic to seven islands in eastern Indonesia. Tree Genet Genomes 4:519–530
Percy DM, Garver AM, Wagner WL, James HF, Cunningham CW, Miller SE, Fleischer RC (2008) Progressive island colonization and ancient origin of Hawaiian Metrosideros (Myrtaceae). Proc R Soc B Biol Sci 275:1479–1490
Pereira M, Oliveira ALd, Pereira REdA, Sena JAD, Costa JRVd, Almeida Md, Goncalves AN (2005) Morphologic and molecular characterization of Myrciaria spp species. Rev Bras Frutic 27:507–510
Petroli C, Sansaloni C, Carling J, Mamani E, Steane D, Myburg A, Vaillancourt R, Kilian A, Pappas G, Bonfim da Silva O, Grattapaglia D (2011) Genomic characterization, high-density mapping and anchoring of DArT markers to the reference genome of Eucalyptus. BMC Proceedings 5:P35
Pinto G, Loureiro J, Lopes T, Santos C (2004) Analysis of the genetic stability of Eucalyptus globulus Labill. somatic embryos by flow cytometry. Theor Appl Genet 109:580–587
Plomion C, Lalanne C, Claverol S, Meddour H, Kohler A, Bogeat-Triboulot MB, Barre A, Le Provost G, Dumazet H, Jacob D, Bastien C, Dreyer E, de Daruvar A, Guehl JM, Schmitter JM, Martin F, Bonneu M (2006) Mapping the proteome of poplar and application to the discovery of drought-stress responsive proteins. Proteomics 6:6509–6527
Poke FS, Vaillancourt RE, Elliott RC, Reid JB (2003) Sequence variation in two lignin biosynthesis genes, cinnamoyl CoA reductase (CCR) and cinnamyl alcohol dehydrogenase 2 (CAD2). Mol Breeding 12:107–118
Poke FS, Vaillancourt RE, Potts BM, Reid JB (2005) Genomic research in Eucalyptus. Genetica 125:79–101
Poke FS, Martin DP, Steane DA, Vaillancourt RE, Reid JB (2006) The impact of intragenic recombination on phylogenetic reconstruction at the sectional level in Eucalyptus when using a single copy nuclear gene (cinnamoyl CoA reductase). Mol Phylogenet Evol 39:160–170
Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Development of High-Density Genetic Maps for Barley and Wheat Using a Novel Two-Enzyme Genotyping-by-Sequencing Approach. PLoS One 7:e32253
Potts BM (2004) Genetic improvement of eucalypts. In: Burley J, Evans J, Youngquist JA (eds) Encyclopedia of forest science. Elsevier Science, Oxford, pp 1480–1490
Potts BM, Dungey HS (2004) Interspecific hybridization of Eucalyptus: key issues for breeders and geneticists. New Forest 27:115–138
Potts BM, Reid JB (1990) The evolutionary significance of hybridization in Eucalyptus. Evolution 44:2151–2152
Potts BM, Wiltshire RJE (1997) Eucalypt genetics and genecology. In: Williams J, Woinarski J (eds) Eucalypt ecology: individuals to ecosystems. Cambridge University Press, Cambridge, pp 56–91
Potts BM, Barbour RC, Hingston AB, Vaillancourt RE (2003) Turner review no. 6: genetic pollution of native eucalypt gene pools—identifying the risks. Austr J Bot 51:1–25
Praca MM, Carvalho CR, Ribeiro C, Novaes DB (2009) Nuclear DNA content of three Eucalyptus species estimated by flow and image cytometry. Austr J Bot 57:524–531
Prakash DP, Narayanaswamy P, Sondur SN (2002) Analysis of molecular diversity in guava using RAPD markers. J Hortic Sci Biotech 77:287–293
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pryor LD, Johnson LAS (1971) A classification of the eucalypts. Australian National University Press, Canberra
Pryor LD, Johnson LAS (1981) Eucalyptus, the universal Australian. In: Keast A (ed) Ecological biogeography of Australia. W. Junk, The Hague pp 499–536.
Qiu D, Wilson IW, Gan S, Washusen R, Moran GF, Southerton SG (2008) Gene expression in Eucalyptus branch wood with marked variation in cellulose microfibril orientation and lacking G-layers. New Phytol 179:94–103
Quang ND, Ikeda S, Harada K (2008) Nucleotide variation in Quercus crispula Blume. Heredity 101:166–174
Quesada T, Gopal V, Cumbie WP, Eckert AJ, Wegrzyn JL, Neale DB, Goldfarb B, Huber DA, Casella G, Davis JM (2010) Association mapping of quantitative disease resistance in a natural population of loblolly pine (Pinus taeda L.). Genetics 186:677–686
Rai MK, Asthana P, Jaiswal VS, Jaiswal U (2010) Biotechnological advances in guava (Psidium guajava L.): recent developments and prospects for further research. Trees-Struct Funct 24:1–12
Ranik M, Creux NM, Myburg AA (2006) Within-tree transcriptome profiling in wood-forming tissues of a fast-growing Eucalyptus tree. Tree Physiol 26:365–375
Rao H, Patterson B, Potts B, Vaillancourt R (2008) A microsatellite study on outcrossing rates and contamination in an Eucalyptus globulus breeding arboretum. J Forestry Res 19:136–140
Rasmussen-Poblete S, Valdes J, Gamboa MC, Valenzuela PDT, Krauskopf E (2008) Generation and analysis of an Eucalyptus globulus cDNA library constructed from seedlings subjected to low temperature conditions. Electron J Biotechnol 11:13
Rathbone DA, McKinnon GE, Potts BM, Steane DA, Vaillancourt RE (2007) Microsatellite and cpDNA variation in island and mainland populations of a regionally rare eucalypt, Eucalyptus perriniana (Myrtaceae). Austr J Bot 55:513–520
Rengel D, San Clemente H, Servant F, Ladouce N, Paux E, Wincker P, Couloux A, Sivadon P, Grima-Pettenati J (2009) A new genomic resource dedicated to wood formation in Eucalyptus. BMC Plant Biol 9:36
Resende MD, Resende MF, Jr., Sansaloni CP, Petroli CD, Missiaggia AA, Aguiar AM, Abad JM, Takahashi EK, Rosado AM, Faria DA, Pappas GJ Jr, Kilian A, Grattapaglia D (2012) Genomic selection for growth and wood quality in Eucalyptus: capturing the missing heritability and accelerating breeding for complex traits in forest trees. New Phytol 194:116–128
Risterucci AM, Duval MF, Rohde W, Billotte N (2005) Isolation and characterization of microsatellite loci from Psidium guajava L. Mol Ecol Notes 5:745–748
Rossetto M, McLauchlan A, Harriss FCL, Henry RJ, Baverstock PR, Lee LS, Maguire TL, Edwards KJ (1999a) Abundance and polymorphism of microsatellite markers in the tea tree Melaleuca alternifolia Myrtaceae. Theor Appl Genet 98:1091–1098
Rossetto M, Slade RW, Baverstock PR, Henry RJ, Lee LS (1999b) Microsatellite variation and assessment of genetic structure in tea tree (Melaleuca alternifolia Myrtaceae). Mol Ecol 8:633–643
Rossetto M, Harriss FCL, McLauchlan A, Henry RJ, Baverstock PR, Lee LS (2000) Interspecific amplification of Tea Tree (Melaleuca alternifolia–Myrtaceae) microsatellite loci—potential implications for conservation studies. Austr J Bot 48:367–373
Ruel K, Berrio-Sierra J, Derikvand MM, Pollet B, Thévenin J, Lapierre C, Jouanin L, Joseleau JP (2009) Impact of CCR1 silencing on the assembly of lignified secondary walls in Arabidopsis thaliana. New Phytol 184:99–113
Rusk N (2009) Cheap third-generation sequencing. Nat Methods 6:244–245
Rye BL (1979) Chromosome-number variation in the Myrtaceae and its taxonomic implications. Austr J Bot 27:547–573
Ryyanen HJ, Tonteri A, Vasemagi A, Primmer CR (2007) A comparison of biallelic markers and microsatellites for the estimation of population and conservation genetic parameters in Atlantic salmon (Salmo salar). J Hered 98:692–704
Sale MM, Potts BM, West AK, Reid JB (1993) Relationships within Eucalyptus using chloroplast DNA. Aust J Sys Bot 6:127–138
Sale M, Potts BM, West A, Reid J (1996) Molecular differentiation within and between Eucalyptus risdonii. E. amygdalina and their hybrids using RAPD markers. Austr J Bot 44:559–569
Salgueiro F, Felix D, Caldas JF, Margis-Pinheiro M, Margis R (2004) Even population differentiation for maternal and biparental gene markers in Eugenia uniflora, a widely distributed species from the Brazilian coastal Atlantic rain forest. Divers Distrib 10:201–210
Sampson JF, Byrne M (2008) Outcrossing between an agroforestry plantation and remnant native populations of Eucalyptus loxophleba. Mol Ecol 17:2769–2781
Sansaloni CP, Petroli CD, Carling J, Hudson CJ, Steane DA, Myburg AA, Grattapaglia D, Vaillancourt RE, Kilian A (2010) A high-density diversity arrays technology (DArT) microarray for genome-wide genotyping in Eucalyptus. Plant Methods 6:16
Sansaloni C, Petroli C, Jaccoud D, Carling J, Detering F, Grattapaglia D, Kilian A (2011) Diversity Arrays Technology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proceedings 5:P54
Santos KL, Santos MO, Laborda PR, Souza AP, Peroni N, Nodari RO (2008) Isolation and characterization of microsatellite markers in Acca sellowiana (Berg) Burret. Mol Ecol Res 8:998–1000
Schmidt-Adam G, Young AG, Murray BG (2000) Low outcrossing rates and shift in pollinators in New Zealand pohutukawa (Metrosideros excelsa; Myrtaceae). Am J Bot 87:1265–1271
Scrivener NJ, Johnson CN, Wallis IR, Takasaki M, Foley WJ, Krockenberger AK (2004) Which trees do wild common brushtail possums (Trichosurus vulpecula) prefer? Problems and solutions in scaling laboratory findings to diet selection in the field. Evol Ecol Res 6:77–87
Seguel BI, Penaloza HE, Gaete CN, Montenegro BA, Torres PA (2000) Collecting and molecular characterisation murta (Ugni molinae Turcz.) germplasm in Chile. Agro Sur 28:32–41
Serce S, Simsek O, Gunduz K, Aka-Kacar Y, Ercisli S (2008) Relationships among myrtle accessions from Turkey as revealed by fruit characteristics and RAPD. Rom Biotechn Lett 13:4054–4065
Sexton T, Henry R, McManus LJ, Thomas D, Shepherd M (2010a) Conservation of single nucleotide polymorphisms in 44 wood quality candidate genes of Eucalyptus globulus, Eucalyptus pyrocarpa and Eucalyptus pilularis. XVIII Plant and Animal Genome Conference, San Diego, CA, abstract W238
Sexton T, Henry RJ, McManus LJ, Bowen S, Shepherd M (2010b) Capture of assay template by multiplex PCR of long amplicons for genotyping SNPs and InDels with MALDI-TOF mass spectrometry. Mol Breeding 25:471–480
Shelton D, Aitken K, Doimo L, Leach D, Baverstock P, Henry R (2002) Genetic control of monoterpene composition in the essential oil of Melaleuca alternifolia (Cheel). Theor Appl Genet 105:377–383
Shendure J (2008) The beginning of the end for microarrays? Nat Methods 5:585–587
Shepherd M, Jones M (2004) Molecular markers in tree improvement: characterisation and use in Eucalyptus. In: Lörz H, Wenzel G (eds) Biotechnology in Agriculture and Forestry, vol 55, Molecular marker systems in plant breeding and crop improvement. Springer-Verlag, Heidelberg, pp 399–409
Shepherd M, Raymond CA (2010) Species delineation and gene flow in the Blackbutts (genus Eucalyptus subgenus Eucalyptus section Pseudophloius). Conserv Genet 11:1965–1978
Shepherd M, Chaparro JX, Teasdale R (1999) Genetic mapping of monoterpene composition in an interspecific eucalypt hybrid. Theor Appl Genet 99:1207–1215
Shepherd M, Kasem S, Lee D, Henry R (2006) Construction of microsatellite linkage maps for Corymbia. Silvae Genet 55:228–238
Shepherd M, Kasam S, Ablett G, Ochieng JW, Crawford A (2008a) Genetic structuring of the spotted gum complex (genus Corymbia section Politaria). Aust Syst Bot 21:15–25
Shepherd M, Kasem S, Lee D, Henry R (2008b) Mapping species differences for adventitious rooting in a Corymbia torelliana × Corymbia citriodora subspecies variegata hybrid. Tree Genet Genomes 4:715–725
Shepherd M, Sexton TR, Thomas D, Henson M, Henry RJ (2010) Geographical and historical determinants of microsatellite variation in Eucalyptus pilularis. Can J Forest Res 40:1051–1063
Sinclair EA, Bussell JD, Krauss SL, Hobbs R, Dixon KW (2006) Molecular markers detect multiple origins of Agonis flexuosa (Myrtaceae) plants used in urban bushland restoration. Ecol Manag Rest 7:234–235
Slee AV, Brooker H, Duffy M, West JG (2006) EUCLID eucalypts of Australia. 3 rd edition DVD edn. Centre for Plant Biodiversity Research–CSIRO publishing Canberra
Solomon OL, Berger DK, Myburg AA (2010) Diurnal and circadian patterns of gene expression in the developing xylem of Eucalyptus trees. S Afr J Bot 76:425–439
Soltis DE, Soltis PS, Bennett MD, Leitch IJ (2003) Evolution of genome size in the angiosperms. Am J Bot 90:1596–1603
Southerton SG, Birt P, Porter J, Ford HA (2004) Review of gene movement by bats and birds and its potential significance for eucalypt plantation forestry. Aust Forestry 67:44–53
Spokevicius AV, Van Beveren K, Leitch MM, Bossinger G (2005) Agrobacterium-mediated in vitro transformation of wood-producing stem segments in eucalypts. Plant Cell Rep 23:617–624
Spokevicius AV, Southerton SG, MacMillan CP, Qiu D, Gan S, Tibbits JFG, Moran GF, Bossinger B (2007) Beta-tubulin affects cellulose microfibril orientation in plant secondary fibre cell walls. Plant J 51:717–726
Steane DA (2005) Complete nucleotide sequence of the chloroplast genome from the Tasmanian blue gum, Eucalyptus globulus (Myrtaceae). DNA Res 12:215–220
Steane DA, Byrne M, Vaillancourt RE, Potts BM (1998) Chloroplast DNA polymorphism signals complex interspecific interactions in Eucalyptus (Myrtaceae). Aust Syst Bot 11:25–40
Steane DA, McKinnon GE, Vaillancourt RE, Potts BM (1999) ITS sequence data resolve higher level relationships among the eucalypts. Mol Phylogenet Evol 12:215–223
Steane D, Vaillancourt R, Russell J, Powell W, Marshall D, Potts B (2001) Development and characterisation of microsatellite loci in Eucalyptus globulus (Myrtaceae). Silvae Genet v 50:89–91
Steane DA, Nicolle D, McKinnon GE, Vaillancourt RE, Potts BM (2002) Higher-level relationships among the eucalypts are resolved by ITS-sequence data. Aust Syst Bot 15:49–62
Steane DA, Jones RC, Vaillancourt RE (2005) A set of chloroplast microsatellite primers for Eucalyptus (Myrtaceae). Mol Ecol Notes 5:538–541
Steane DA, Conod N, Jones RC, Vaillancourt RE, Potts BM (2006) A comparative analysis of population structure of a forest tree, Eucalyptus globulus (Myrtaceae), using microsatellite markers and quantitative traits. Tree Genet Genomes 2:30–38
Steane DA, Nicolle D, Potts BM (2007) Phylogenetic positioning of anomalous eucalypts by using ITS sequence data. Aust Syst Bot 20:402–408
Steane DA, Nicolle D, Sansaloni CP, Petroli CD, Carling J, Kilian A, Myburg AA, Grattapaglia D, Vaillancourt RE (2011) Population genetic analysis and phylogeny reconstruction in Eucalyptus (Myrtaceae) using high-throughput, genome-wide genotyping. Mol Phylogenet Evol 59:206–224
Stevens PF (2008) Angiosperm phylogeny website. Version 9, June 2008 [and more or less continuously updated since]." http://www.mobot.org/MOBOT/research/APweb/. (accessed on 1/4/2011).
Strauss SH, Lande R, Namkoong G (1992) Limitations of molecular-marker-aided selection in forest tree breeding. Can J Forest Res 22:1050–1061
Sytsma KJ, Litt A, Zjhra ML, Pires JC, Nepokroeff M, Conti E, Walker J, Wilson PG (2004) Clades, clocks, and continents: historical and biogeographical analysis of Myrtaceae, Vochysiaceae, and relatives in the southern hemisphere. Int J Plant Sci 165:S85–S105
Tang H, Wang X, Bowers JE, Ming R, Alam M, Paterson AH (2008) Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res 18:1944–1954
Telles MPD, Coelho ASG, Chaves LJ, Diniz JAF, Valva FD (2003) Genetic diversity and population structure of Eugenia dysenterica DC. ("cagaiteira"-Myrtaceae) in Central Brazil: spatial analysis and implications for conservation and management. Conserv Genet 4:685–695
Thaipong K, Boonprakob U (2005) Genetic and environmental variance components in guava fruit qualities. Sci Hortic 104:37–47
Thamarus K, Groom K, Murrell J, Byrne M, Moran G (2002) A genetic linkage map for Eucalyptus globulus with candidate loci for wood, fibre and floral traits. Theor Appl Genet 104:379–387
Thamarus K, Groom K, Bradley A, Raymond CA, Schimleck LR, Williams ER, Moran GF (2004) Identification of quantitative trait loci for wood and fibre properties in two full-sib pedigrees of Eucalyptus globulus. Theor Appl Genet 109:856–864
Thumma BR, Nolan MR, Evans R, Moran GF (2005) Polymorphisms in cinnamoyl CoA reductase (CCR) are associated with variation in microfibril angle in Eucalyptus spp. Genetics 171:1257–1265
Thumma BR, Matheson BA, Zhang DQ, Meeske C, Meder R, Downes GM, Southerton SG (2009) Identification of a cis-acting regulatory polymorphism in a eucalypt cobra-like gene affecting cellulose content. Genetics 183:1153–1164
Thumma BR, Southerton SG, Bell JC, Owen JV, Henery ML, Moran GF (2010) Quantitative trait locus (QTL) analysis of wood quality traits in Eucalyptus nitens. Tree Genet Genomes 6:305–317
Tierney DA, Wardle GM (2008) The relative fitness of parental and hybrid Kunzea (Myrtaceae): the interaction of reproductive traits and ecological selection. Am J Bot 95:146–155
Tournier V, Grat S, Marque C, El Kayal W, Penchel R, de Andrade G, Boudet AM, Teulieres C (2003) An efficient procedure to stably introduce genes into an economically important pulp tree (Eucalyptus grandis x Eucalyptus urophylla). Transgenic Res 12:403–411
Trindade MD, Chaves LJ (2005) Genetic structure of natural Eugenia dysenterica DC (Myrtaceae) populations in northeastern Goias, Brazil, accessed by morphological traits and RAPD markers. Genet Mol Biol 28:407–413
Tucker DJ, Wallis IR, Bolton JM, Marsh KJ, Rosser AA, Brereton IM, Nicolle D, Foley WJ (2010) A metabolomic approach to identifying chemical mediators of mammal–plant interactions. J Chem Ecol 36:727–735
Turner C, Wiltshire RJE, Potts BM, Vaillancourt RE (2000) Allozyme variation and conservation of the Tasmanian endemics, Eucalyptus risdonii. E. tenuiramis and E. coccifera. Conserv Genet 1:209–216
Turner TL, Bourne EC, Von Wettberg EJ, Hu TT, Nuzhdin SV (2010) Population resequencing reveals local adaptation of Arabidopsis lyrata to serpentine soils. Nat Genet 42:260–263
Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J, Sterck L, Aerts A, Bhalerao RR, Bhalerao RP, Blaudez D, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M, Chapman J, Chen GL, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q, Cunningham R, Davis J, Degroeve S, Dejardin A, dePamphilis C, Detter J, Dirks B, Dubchak I, Duplessis S, Ehlting J, Ellis B, Gendler K, Goodstein D, Gribskov M, Grimwood J, Groover A, Gunter L, Hamberger B, Heinze B, Helariutta Y, Henrissat B, Holligan D, Holt R, Huang W, Islam-Faridi N, Jones S, Jones-Rhoades M, Jorgensen R, Joshi C, Kangasjarvi J, Karlsson J, Kelleher C, Kirkpatrick R, Kirst M, Kohler A, Kalluri U, Larimer F, Leebens-Mack J, Leple JC, Locascio P, Lou Y, Lucas S, Martin F, Montanini B, Napoli C, Nelson DR, Nelson C, Nieminen K, Nilsson O, Pereda V, Peter G, Philippe R, Pilate G, Poliakov A, Razumovskaya J, Richardson P, Rinaldi C, Ritland K, Rouze P, Ryaboy D, Schmutz J, Schrader J, Segerman B, Shin H, Siddiqui A, Sterky F, Terry A, Tsai CJ, Uberbacher E, Unneberg P, Vahala J, Wall K, Wessler S, Yang G, Yin T, Douglas C, Marra M, Sandberg G, Van de Peer Y, Rokhsar D (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604
Udovicic F, Ladiges PY (2000) Informativeness of nuclear and chloroplast DNA regions and the phylogeny of the eucalypts and related genera (Myrtaceae). Kew Bull 55:633–645
Udovicic F, McFadden GI, Ladiges PY (1995) Phylogeny of Eucalyptus and Angophora based on 5S rDNA spacer sequence data. Mol Phylogenet Evol 4:247–256
Ukrainetz NK, Ritland K, Mansfield SD (2008) Identification of quantitative trait loci for wood quality and growth across eight full-sib coastal Douglas-fir families. Tree Genet Genomes 4:159–170
Vaillancourt RE, Jackson HD (2000) A chloroplast DNA hypervariable region in eucalypts. Theor Appl Genet 101:473–477
Vaillancourt RE, Potts BM, Watson M, Volker PW, Hodges G, Reid JB, West A (1995) Detection and prediction of heterosis in Eucalyptus globulus. Forest Genet 2:11–19
Vaillancourt RE, Petty A, McKinnon GE (2004) Maternal inheritance of mitochondria in Eucalyptus globulus. J Heredity 95:353–355
Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, Pindo M, FitzGerald LM, Vezzulli S, Reid J, Malacarne G, Iliev D, Coppola G, Wardell B, Micheletti D, Macalma T, Facci M, Mitchell JT, Perazzolli M, Eldredge G, Gatto P, Oyzerski R, Moretto M, Gutin N, Stefanini M, Chen Y, Segala C, Davenport C, Demattà L, Mraz A, Battilana J, Stormo K, Costa F, Tao Q, Si-Ammour A, Harkins T, Lackey A, Perbost C, Taillon B, Stella A, Solovyev V, Fawcett JA, Sterck L, Vandepoele K, Grando SM, Toppo S, Moser C, Lanchbury J, Bogden R, Skolnick M, Sgaramella V, Bhatnagar SK, Fontana P, Gutin A, Van de Peer Y, Salamini F, Viola R (2007) A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One 2:e1326
Verhaegen D, Plomion C (1996) Genetic mapping in Eucalyptus urophylla and Eucalyptus grandis using RAPD markers. Genome 39:1051–1061
Verhaegen D, Plomion C, Gion J-M, Poitel M, Costa P, Kremer A (1997) Quantitative trait dissection analysis in Eucalyptus using RAPD markers: 1.Detection of QTL in interspecific hybrid progeny, stability of QTL expression across different ages. Theor Appl Genet 95:597–608
Vicentini R, Sassaki FT, Gimenes MA, Maia IG, Menossi M (2005) In silico evaluation of the Eucalyptus transcriptome. Genet Mol Biol 28:487–495
Whittock S, Steane DA, Vaillancourt RE, Potts BM (2003) Molecular evidence shows that the tropical boxes (Eucalyptus subgenus Minutifructus) are over-ranked. Trans Roy Soc of South Aust 127:27–32
Wilf P, Cuneo NR, Johnson KR, Hicks JF, Wing SL, Obradovich JD (2003) High plant diversity in eocene south america: evidence from Patagonia. Science 300:122–125
Williams JE, Brooker MIH (1997) Eucalypts: an introduction. In: Williams JE, Woinarski JCZ (eds) Eucalypt ecology: individuals to ecosystems. Cambridge University Press, Cambridge, pp 1–15
Williams JE, Woinarski JCZ (1997) Eucalypt ecology: individuals to ecosystems. Cambridge University Press, Cambridge
Wilson PG, O'Brien MM, Gadek PA, Quinn CJ (2001) Myrtaceae revisited: a reassessment of infrafamilial groups. Am J Bot 88:2013–2025
Wilson PG, O'Brien MM, Heslewood MM, Quinn CJ (2005) Relationships within Myrtaceae sensu lato based on a matK phylogeny. Plant Syst Evol 251:3–19
Wilson PG, Heslewood MM, Quinn CJ (2007) Re-evaluation of the genus Babingtonia (Myrtaceae) in eastern Australia and New Caledonia. Aust Syst Bot 20:302–318
Wiltshire RJE (2004) Eucalypts. In: Burley J, Evans J, Youngquist JA (eds) Encyclopedia of forest science. Elsevier Science, Oxford, pp 1687–1699
Wright ME, Ranker TA (2010) Dispersal and habitat fidelity of bog and forest growth forms of Hawaiian Metrosideros (Myrtaceae). Bot J Linn Soc 162:558–571
Wright SD, Yong CG, Dawson JW, Whittaker DJ, Gardner RC (2000) Riding the ice age El Nino? Pacific biogeography and evolution of Metrosideros subg. Metrosideros (Myrtaceae) inferred from nuclear ribosomal DNA. Proc Natl Acad Sci USA 97:4118–4123
Wright SD, Yong CG, Wichman SR, Dawson JW, Gardner RC (2001) Stepping stones to Hawaii: a trans-equatorial dispersal pathway for Metrosideros (Myrtaceae) inferred from nrDNA (ITS plus ETS). J Biogeogr 28:769–774
Yates CJ, Ladd PG, Coates DJ, McArthur S (2007) Hierarchies of cause: understanding rarity in an endemic shrub Verticordia staminosa (Myrtaceae) with a highly restricted distribution. Austr J Bot 55:194–205
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zauza EAV, Alfenas AC, Old K, Couto MMF, Graca RN, Maffia LA (2010) Myrtaceae species resistance to rust caused by Puccinia psidii. Aust Plant Path 39:406–411
Zelener N, Poltri SN, Bartoloni N, Lopez CR, Hopp HE (2005) Selection strategy for a seedling seed orchard design based on trait selection index and genomic analysis by molecular markers: a case study for Eucalyptus dunnii. Tree Physiol 25:1457–1467
Zucchi MI, Brondani RPV, Pinheiro JB, Brondani C, Vencovsky R (2002) Transferability of microsatellite markers from Eucalyptus, spp. to Eugenia dysenterica (Myrtaceae family). Mol Ecol Notes 2:512–513
Zucchi MI, Brondani RPV, Pinheiro JB, Chaves LJ, Coelho ASG, Vencovsky R (2003) Genetic structure and gene flow in Eugenia dysenterica DC in the Brazilian Cerrado utilizing SSR markers. Genet Mol Biol 26:449–457
Zucchi MI, Pinheirol JB, Aguiar AV, Chaves LJ, Coelho ASG, Vencovsky R (2004) Spatial autocorrelation of divergence in populations of Eugenia dysenterica DC using SSR markers. Floresta Amb 11:29–38
Acknowledgments
This work was supported by the following grants: (1) Brazilian Ministry of Science and Technology CNPq research grant 577047/2008-6, EMBRAPA grant 03.08.01.010, FAP-DF/CNPq Grant NEXTREE 193.000.570/2009 and CNPq research productivity fellowship awarded to DG; (2) Australian Research Council (DP0770506); (3) Mondi and Sappi through the Forest Molecular Genetics Programme (University of Pretoria, South Africa); (4) CRC for Forestry (Australia); (5) Australian Research Council's Linkage Projects funding scheme to WJF (project number LP0667708); (6) CRC for Sustainable Forestry.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Abbott
A contribution to the Special Issue “The genomes of the giants: a walk through the forest of tree genomes”
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 134 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Grattapaglia, D., Vaillancourt, R.E., Shepherd, M. et al. Progress in Myrtaceae genetics and genomics: Eucalyptus as the pivotal genus. Tree Genetics & Genomes 8, 463–508 (2012). https://doi.org/10.1007/s11295-012-0491-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-012-0491-x