
Abstract
While methane-powered vehicles produce fewer greenhouse gas emissions in comparison to conventional fuel vehicles, there is a significant amount of methane slip in their exhaust that needs to be treated. This study investigates non-thermal plasma (NTP) assisted catalytic methane oxidation as an alternative method for the low temperature methane slip abatement applicable to the exhaust of biogas methane-powered vehicles. It is concluded that high CH4 conversion and CO2 selectivity can be obtained using NTP-catalysis at low temperature with Pd/Al2O3 found to be the most promising candidate among all catalysts tested. In addition, it was found that CH4 conversion efficiency was dependent on the feed gas components and gas hourly space velocity as well as how the activation energy is introduced. For example, a combination of plasma and external heat supply provides advantages in terms of CH4 conversion along with lower plasma energy consumption. The presence of N2 and O2 in the feed gas during NTP-catalytic methane oxidation results in unfavourable NOX formation which linearly increases with CH4 conversion. These results conclude that the most suitable aftertreatment option involves the combination of an oxidation catalyst with plasma to target the hydrocarbon and CH4 oxidation, followed by an ammonia-SCR system to convert the NOX formed in plasma assisted zone.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Methane-powered vehicles have gained recent interest, with currently 23 million vehicles worldwide, due to their life cycle greenhouse gas (GHG) emissions benefits over conventional gasoline- and diesel- powered vehicles [1,2,3]. The use of methane as a fuel, either from fossil derived natural gas or renewable natural gas (biomethane), can reduce GHG emissions as well as other air pollutants. However, these benefits are mitigated by the presence of a significant amount of biogas derived-unburned methane (methane slip) in the engine exhaust [4], in particular during cold-start. Methane is a strong GHG with global warming potential which is 21 times higher than that of carbon dioxide. The low gas temperatures and the presence of impurities in the vehicle exhaust make the current catalytic converters inefficient for the methane removal and new and novel exhaust aftertreatment systems are required.
The thermal catalytic oxidation of methane has been extensively investigated [5,6,7,8]. However, even for the most active catalysts, 100% conversion cannot be achieved at temperatures below 400 °C, especially when water is present. An alternative approach to activate CH4 at low temperatures is to utilize non-thermal plasma (NTP). Typically, gas phase NTP reactions are nonselective; however, combining an NTP with catalyst can improve the system performance in terms of conversion and product selectivity [9,10,11]. Most plasma-catalyst studies have used a dielectric barrier discharge (DBD) reactor [12,13,14] with one or more dielectric material and a catalyst that either filled the gap between the plasma electrodes or placed after the discharge zone. NTP-assisted catalytic systems, using different reactor configurations, have received a lot of attentions in the literature for various reactions including dry reforming of methane [15, 16], CO2 hydrogenation [17, 18], NOX reduction [19] and the water–gas shift reaction [20]. Although there are a number of studies that have examined the NTP-assisted catalytic methane oxidation, few have studied it under conditions relevant to methane-powered vehicles exhaust applications [21, 22].
One of the concerns with plasma-catalyst systems for the methane oxidation is the benefits brought by the hybridization of the plasma and the catalyst. For example, the synergistic effect between the plasma and the catalyst for this reaction depends on several factors including the nature of power supply [21] and the reaction conditions [21,22,23]. While some researchers questioned such synergy and attributed the catalytic effect mostly to the subsequent CO oxidation [22], others reported a true synergistic effect on the CH4 conversion [23]. An in-situ study of the role of the NTP in the hybrid catalytic reaction under CH4 and CH4 + O2 conditions has provided evidence that no significant structural changes occurred within the catalyst on application of the NTP and that the temperature of the Pd nanoparticles was lower than that necessary to activate the thermal CH4 oxidation reaction [24]. Under similar conditions, using a diffuse reflectance infra-red Fourier transform spectroscopy coupled with mass spectrometer (DRIFTS-MS) cell, an enhancement in the formation of surface species on the Pd catalyst was reported [25]. Upon review of the body of work reported to date, the role of the catalyst during methane oxidation in the plasma-catalyst systems remains unclear. In addition, the effect of the components in a gas mixture that mimics the exhaust gas conditions, on the methane conversion and product selectivity needs to be clarified.
While the high endothermicity of the methane dissociation (434 kJ/mol) plays a crucial role in the plasma assisted reaction, the impact of high concentrations of H2O, CO2, O2 and N2/NOX has not been explored in detail. Baylet et al. [22] showed that an understanding of the undesirable side reactions in the plasma-only system, the role of CO2 and water in the plasma assisted catalytic process needs to be clarified. Moreover, the presence of N2 and O2 in the feed gas during plasma-assisted methane oxidation has been reported to result in NOX formation [22] and there are no reported investigation on the relationship between methane conversion and NOX formation in complex feed gas mixtures with the plasma-catalyst systems. In terms of temperature dependence, an increase in the reactor temperature to 200 °C was shown to have a positive effect on the CH4 conversion [22]. Nozaki et al. [26] discussed that the dissociation of methane by electron impact was almost independent of the reaction temperature and that the vibrationally excited methane and water were the activated species at low temperature. Considering that the range of temperature experienced by the exhaust gas is wide, e.g. up to 300 °C when considering cold start conditions, the effect of temperature should be examined in more detail.
Previously, the NTP-assisted catalytic methane oxidation has only been explored using Pd/Al2O3, Al2O3, Ru/TiO2 and Au/Al2O3 [27,28,29]. In order to investigate how the type of catalyst affects the performance of the plasma-catalyst and identify the best catalyst formulation for this reacting system, several supported metal catalysts were prepared and tested under the same reaction conditions. The selected catalysts in this study were among the ones reported for CH4 activation for a wide range of reactions [30,31,32,33]. The aim of this study is to investigate the effect of the nature of the catalyst, the feed gas components as well as operating temperature conditions on the plasma-catalyst system performance in the methane oxidation reaction. Herein, an investigation of NOX production due to uncontrolled plasma reactions between N2 and O2 present in the gas stream and the link to methane conversion is established as well as strategies to mitigate NOX slip.
Experimental and Methods
Catalyst Preparation
The catalysts were prepared by incipient wetness impregnation. A NH4-ZSM-5 (Zeolyst International, molar SiO2/Al2O3 ratio of 30 and 80, approximately 0.17 cm3 g−1 pore volume) was calcined in air at 550 °C for 8 h to obtain the H-ZSM-5 form (denoted ZSM-5). γ-Al2O3 was obtained from Sasol which had a pore volume of 0.5 cm3 g−1 and this was dried in ambient air at 110 °C for 8 h. Both the ZSM-5 and γ-Al2O3 powders were pressed into pellets before being ground and sieved between 250 and 425 µm mesh sizes particles. The ZSM-5 or γ-Al2O3 supports were impregnated with solutions made by dissolving an appropriate amount of Co(NO3)2·6H2O (99.999%, Sigma-Aldrich), Cu(NO3)2·xH2O (99.999%, Sigma-Aldrich), Fe(NO3)3·9H2O (≥ 98%, Sigma-Aldrich), LiNO3 (99.99%, Sigma-Aldrich), (NH4)6Mo7O24·4H2O (99.98%, Sigma-Aldrich), Ni(NO3)2·6H2O (99.999%, Sigma-Aldrich) or Pd(NO3)2·2H2O (99%, Sigma-Aldrich) in 10 wt% HNO3 (prepared from concentrated HNO3 (69–70%, BDH)) or diluting an appropriate amount of ruthenium (III) nitrosyl nitrate solution (1.5 wt% Ru, Ru(NO)(NO3)x(OH)y x + y = 3, Sigma-Aldrich) or rhodium (III) nitrate solution (10 wt% Rh, Rh(NO3)3, Sigma-Aldrich) in deionized water. After 48 h impregnation, the impregnated samples were dried in air at 110 °C for 8 h. The dried precursors were subsequently calcined in flowing air at 500 °C for 8 h following a temperature ramp of 10 °C min−1 and then, cooled to room temperature before testing. The alumina-based catalysts were prepared to obtain 2 wt% nominal loading of Ru, Co, Fe, Cu, Rh, Pt, Ni or Li and 10 wt% nominal loading of Pd. A 2 wt% Pd/Al2O3 was also tested and was obtained from Johnson Matthey.
Experimental Setup
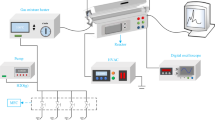
A schematic representation of the experimental setup is shown in Fig. 1. It consisted of a quartz tubular reactor (outside diameter (O.D.): 6 mm; inside diameter (I.D.): 4 mm) packed with different catalysts with a particle size in the range of 250–425 µm, operated in a plug flow mode. The NTP was generated in a DBD reactor configuration via a tungsten wire placed in the centre of the quartz tube as the ground electrode, insulated within a sealed quartz capillary tube (O.D.: 1.8 mm) to prevent arcing, and the power electrode (aluminium foil) wrapped on the outer surface of the reactor. The length of the discharge was determined by the length of the foil and was kept constant for all the experiments at 2 cm. This ensured that the plasma region entirely covered the packed bed, irrespective of the materials used.

Schematic of experimental setup
An alternating current power supply (PVM500 model) was used to ignite the plasma. The electrical parameters were monitored using an oscilloscope (Tektronix MDO3022) connected to the reactor using a high voltage probe (Tektronix P6015A) to measure the applied voltage and a probe to measure the plasma current using a 10 Ω resistor. An example of a voltage-current profile associated to the plasma is shown in Fig. 2. An electric air blower heater (LE Mini Sensor Kit) with controllable temperature and air pressure was used to supply the external heat to the DBD reactor when plasma reactions at elevated temperatures were required. The reactor temperature as a function of the heater temperature at a constant air pressure was calibrated by replacing the ground electrode with a K-type thermocouple inside the DBD reactor in the absence of plasma.

a An example of voltage-current profile. b A zoomed-in plot of current in the range of − 30 µs–30 µs. Conditions: 7 kV, 30 kHz, 0.5 kPa CH4, 10 kPa O2 in N2, gas hourly space velocity (GHSV) = 171,429 mL g−1 h−1
The reactor feed containing the desired gas mixtures of CH4, O2, CO, CO2, NO, NH3 and Ar/N2 (all gas cylinders were supplied by BOC, 99.99% purity) was supplied using mass flow controllers (Bronkhorst El flow). The H2O vapor was added to the feed gas stream by passing Ar/N2 as a carrier gas through a custom-made saturator at specific temperature that was controlled using a Grant™ GD120 thermostatic bath. The produced water was removed from the gas stream after the reactor using a condenser, kept at 0 °C. All inlet and outlet gas lines were heat traced and maintained at 110 °C to prevent condensation. A gas chromatograph (GC; Agilent 7820A) was used to analyse the reactants and products and was equipped with a Porapak Q packed column and a flame ionization detector fitted with a methaniser to detect CO and CO2. Propene, added post reactor and before the condenser to the effluent, was used as an internal standard. The concentration of NOx was measured with a Signal 4000 VM series analyser equipped with a chemiluminescence detector. When NH3 was added to the feed gas, a Bruker Vertex 70 FTIR spectrometer fitted with a gas cell with a volume of 190 cm3 was used for the analysis of reactants and products. An additional catalyst bed for NH3-SCR, consisting of Cu-CHA provided by Johnson Matthey Technology was used in these experiments.
Parameter Calculations
The average power consumed during the plasma process was calculated by Eq. (1). The instantaneous current was estimated by measuring the voltage across the resistor (Vr), placed on the ground region (see Fig. 1) and having the resistance (R), i.e. 10 Ω, according to Eq. (2).
where I was the current, Pave was the average power, t was time, T was a period, and V was the applied peak voltage. The specific input energy (SIE), representing the discharge energy density was calculated as the ratio of the average plasma power and the volumetric total gas flowrate at inlet.
In order to facilitate a comparison between plasma and thermal effects on the process an equivalent SIE for thermal systems was defined as the ratio of the thermal process heat duty (Q) in J s−1 and the volumetric total flowrate at inlet [20, 21].
The heat duty for the thermal systems was assumed to be the sensible heat needed to bring the temperature of the gaseous reactants to the final reaction temperature. This assumption was based on the fact that the discharge plasma power was measured according to the amount of energy injected to the plasma system without considering the energy of reactions [21]. The heat duty was calculated using the Eq. (5).
where Tr was the room temperature (RT), Tf was the final reaction temperature and CP was the heat capacity. The heat capacities for the gas components were calculated using equations from the literature (e.g. [34]) for an ideal gas state as a function of temperature.
The CH4 conversion and product selectivity were calculated using Eqs. 6 and 7, respectively:
Given the atmospheric conditions in this study, the reactant and product gases were assumed to follow ideal gas law. The carbon balance (CH4 + CO + CO2) was calculated and found to be close to 100% for all experiments. Only CO and CO2 were observed as the carbon-based products for all the conditions investigated, therefore, the sum of CO and CO2 selectivities were considered to be 100% to exclude small deviations. On the same basis, the CO2 selectivity for experiments with CO2 present in the feed was calculated using [CO2 selectivity] = 100 − [CO selectivity].
The errors associated with CH4 conversion, CO/CO2 selectivity and NOX formation in this study were ± 5%, ± 4% and ± 91 ppmv, respectively. The thermodynamic calculations to assess the stable reaction products at various conditions were performed in Aspen Plus V9 using the built-in Gibbs reactor package.
Results and Discussion
Catalyst Screening
The CH4 conversion, CO/CO2 selectivity and SIE results for the different catalysts prepared are reported in Table 1. Note that, while the catalysts reported in Table 1 are screened using one set of reaction conditions, the effect of varying the reaction conditions of the plasma-catalyst system is also further investigated for the best performing catalysts (see Sects. 3.2 and 3.3).
With zeolite-based materials, an increase in the molar SiO2/Al2O3 ratio from 30 to 80 led to an increase in both CH4 conversion and CO2 selectivity from 48 to 65% and from 40 to 50%, respectively. The addition of Mo to both ZSM-5 (SiO2/Al2O3 = 30 mol/mol) and ZSM-5 (SiO2/Al2O3 = 80 mol/mol) enhanced the CH4 conversion by 33% and 12% (up to 77% and 81%), respectively. The CO2 selectivity stayed constant for the Mo/ZSM-5 (SiO2/Al2O3 = 80 mol/mol) while for Mo/ZSM-5 (SiO2/Al2O3 = 30 mol/mol), the CO2 selectivity increased from 43 to 57%. The observed enhancement in conversion when Mo was added to the catalyst formulation could be related to the previously reported reactivity of molybdenum towards CH4 activation [32, 35,36,37,38,39,40]. Mo/ZSM-5 has been applied to the methane aromatization reaction. This catalyst is bi-functional—methane is activated on the transition metal sites (Mo) to form intermediates which are further converted to aromatic compounds e.g. benzene on zeolite acidic sites [32]. As Mo is also active in partial oxidation reactions, e.g. oxidation of methanol to formaldehyde [36, 37], it is likely that in the presence of plasma, the methane could be partially oxidized and this could initiate a water gas shift-NTP reaction.
Over Al2O3, the NTP resulted in 79% CH4 conversion and 86% CO2 selectivity, a better performance when compared to the zeolite-based catalysts. Impregnating Pd on the Al2O3 support enhanced both the CH4 conversion (up to 94–99%) and CO2 selectivity (> 99%) with increasing precious metal loading (from 2 to 10 wt%). All other metals explored led to lower methane conversions than the pure alumina. The addition of Ru, Co, Cu, Rh and Pt led to a CO2 selectivity similar or better than alumina; however, impregnation with Li, Fe or Ni resulted in significantly lower performance. It is likely that several reactions e.g. steam CH4 reforming, partial oxidation, dry CH4 reforming, CO2 methanation and H2O-gas shift reaction occur simultaneously (with different rates) under the plasma-catalyst condition of this study, besides the CH4 oxidation reaction (equations (S24) to (S35)). The active metals might promote side reactions resulting in an overall decrease in CO2 selectivity and CH4 conversion. For example, Gao et al. [41] have recently reported on the high-performance plasma enabled CO2 methanation over bimetallic Ni–Fe catalysts using a nanosecond pulsed power supply. The low methane conversion and CO2 selectivity observed with Fe, Li and Ni could indeed indicate that despite the known activity of these catalysts towards CO oxidation, in the presence of plasma, a number of side reactions take place.
As reported in Table 1, Pd/Al2O3 catalysts had the highest activity and selectivity in comparison to other catalysts tested. The Brunauer–Emmett–Teller (BET) surface area and X-ray diffraction (XRD) patterns for the Pd catalysts are reported in the ESI. It is noteworthy that Pd-based catalysts are known to have the best performance in thermal catalytic CH4 oxidation [7, 42, 43]. Lee et al. [23] examined a 2 wt% Pd/Al2O3 powder catalyst in a fixed-bed reactor design during NTP-catalytic CH4 oxidation at ambient temperature and obtained a significant increase in CH4 conversion and CO2 selectivity when adding the catalyst in the plasma zone, in line with the results of this study. Baylet et al. [22] compared the performance of post-plasma and in-plasma catalyst systems with plasma-only system for CH4 combustion over a Pd/Al2O3/cordierite catalyst and reported that while CH4 conversion remained the same for post-plasma catalyst and plasma-only systems, there was a significant decrease in CH4 conversion for in-plasma catalyst system, attributed mainly to the lower residence time of the gas components when the catalyst was loaded in the discharge area. De Rosa et al. [21] reported a plasma-only, NTP-catalytic and thermal catalytic methane oxidation over a 2 wt% Pd/Al2O3 in 0.5 kPa CH4, 10 kPa O2, 5.5 kPa CO2, 7 kPa H2O in N2 using a different reactor configuration, i.e. an alumina tube reactor with a stainless steel wire as the internal electrode, in comparison to the reactor configuration of this study. No synergistic effect on CH4 conversion was observed when combining plasma and catalyst with an AC power supply although there was a synergistic effect on CH4 conversion using a nanopulse power supply [21] and similar findings were reported by Nozaki et al. [26].
In-situ DRIFTS-MS studies revealed that the formation of surface species, e.g. carbonate and formate, was significantly affected by the plasma in the methane oxidation over a Pd catalyst in comparison to the thermal catalytic reactions [25]. In addition, temperature increases in the NTP-catalyst system have been observed and it was suggested that this process was not isothermal especially at high plasma SIEs [21, 24, 44]. De Rosa et al. [21] reported a linear relationship between the reactor temperature (up to 300 °C) and the SIE in the plasma-only and plasma catalyst systems. Molteni et al. [44] reported that the temperature rise induced by the plasma was significant during CH4 oxidation reaction and this might lead to increased methane conversion. However, Gibson et al. [24] reported that the temperature rise in the bed was insufficient to lead to the thermal catalytic CH4 conversion. Using an IR camera and EXAFS, it was found that during the CH4 oxidation over a Pd/Al2O3 catalyst packed in the NTP zone, the temperature of the Pd nanoparticles was around 162 °C, i.e. below the temperature required for the activation of CH4 (> 200 °C) [24]. The heat generated during plasma-induced CH4 oxidation reaction, on the other hand, might promote the further oxidation of CO to form CO2 over the catalyst, because temperatures below 200 °C are sufficient for CO oxidation over Pd/Al2O3 catalysts [45, 46].
The calculated SIE results, shown in Table 1, varied in the range of 4.3–6.9 J mL−1 for different catalyst system even though the experiments in Table 1 were conducted under apparently identical reaction conditions (e.g. constant applied voltage and frequency). The metals impregnated on the catalyst surface might be the source of variations in SIEs by changing the dielectric constant in the discharge zone [47].
Effect of Gas Components and GHSV
The effect of the reactant feed gas composition and GHSVs on the CH4 conversion and CO/CO2 selectivity over Al2O3 and 10 wt% Pd/Al2O3 catalyst is reported in Fig. 3. To further differentiate the effect of Pd metal on Al2O3 support, a catalyst with a higher loading of Pd (10 wt% Pd/Al2O3) was selected for the rest of study. The gas compositions consisted of:
-
Condition (a): 0.5 kPa CH4, 10 kPa O2 in Ar;
-
Condition (b): 0.5 kPa CH4, 10 kPa O2 in N2;
-
Condition (c): 0.5 kPa CH4, 10 kPa O2, 7 kPa H2O, 5.5 kPa CO2 and 0.1 kPa NO, in N2.

Effect of feed gas composition and GHSV on NTP-catalytic CH4 oxidation over 10 wt% Pd/Al2O3 at 7 kV, 30 kHz. (a and d) Condition (a): 0.5 kPa CH4, 10 kPa O2 in Ar; (b and e) Condition (b): 0.5 kPa CH4, 10 kPa O2 in N2; (c and f) Condition (c): 0.5 kPa CH4, 10 kPa O2, 7 kPa H2O, 5.5 kPa CO2 and 0.1 kPa NO in N2
With Ar as the balance gas, the conversion of CH4 over the 10 wt% Pd/Al2O3 is high, e.g. 98% at GHSV = 171,429 mL g−1 h−1, and significantly higher than over Al2O3 at the same GHSV, e.g. 59% at GHSV = 171,429 mL g−1 h−1. An increase in the GHSV in the plasma-catalyst system reduced the overall residence time of the gas components, and the energy density, resulting in, as expected, lower conversions. As observed in Fig. 3, doubling the GHSV from 171,429 to 342,858 mL g−1 h−1 decreased the CH4 conversion slightly for the 10 wt% Pd/Al2O3 from 98 to 96% and, significantly for Al2O3 from 59 to 43%. The CO2 selectivity remained around 100% over the 10 wt% Pd/Al2O3 and decreased from 89 to 78% for Al2O3. It is believed that besides the electron-impact dissociation of CH4 and O2 in the plasma, the excited Ar species can dissociate CH4 and O2 through a “Penning dissociation” phenomenon which correlates to an energy transfer from excited species to other molecules [15, 48, 49]. The relatively small decrease in conversion of methane when increasing the GHSV could be attributed to the kinetically enhanced collisions between the excited noble gas atoms and methane molecules. As shown in Fig. 3b, if the GHSV is further increased, the positive effect of the gas phase chemistry is superseded by the reaction kinetics over the catalyst, represented by a ~ tenfold reduction in CH4 conversion at the highest space velocity.
The impact of using N2 as the balance gas on the CH4 conversion was assessed with condition (b). At a similar GHSV, there was a decrease in the CH4 conversion for both 10 wt% Pd/Al2O3 and Al2O3 catalysts when using N2 instead of Ar as balance. The effect of N2 was more pronounced at high GHSVs, e.g. at 342,858 mL g−1 h−1, the CH4 conversion decreased from 96% for condition (a) to almost 10% for condition (b) over the 10 wt% Pd/Al2O3 catalyst. As discussed above, the Penning effect can account, to a certain extent, for the higher CH4 conversion in Ar. While CO2 selectivity remained above 90% over the 10 wt% Pd/Al2O3 for both conditions (a) and (b), it decreased significantly over the Al2O3 catalyst when replacing Ar with N2 at a constant GHSV. For example, the CO2 selectivity reduced from 89% at condition (a) to 58% at condition (b) when GHSV was 171,429 mL g−1 h−1 and Al2O3 was used as the catalyst.
Modeling studies for a mixture of CO2/CH4/N2 (1:1:8) have shown that the dissociation of CH4 and CO2 splitting to CO and O are promoted by the metastable singlet and triplet states generated by electron impact excitation of N2, which enhances CH4 and CO2 conversions [50] (equations listed in ESI, Section S2). This effect is not observed for CH4 conversion in this study but the decrease in CO2 selectivity over Al2O3 by switching to N2 balance could be attributed to the collision of some of the generated CO2 molecules with metastable N2.
When a complex gas mixture containing CH4, O2, CO2, H2O, NO and N2 balance [condition (c)] was tested over the 10 wt% Pd/Al2O3 catalyst, the methane conversion remained high, with less than 10% decrease compared to the simpler gas mixtures used in conditions (a) and (b). For example, at a GHSV of 171,429 mL g−1 h−1 when changing the feed composition from condition (a) to (b) and (c), the methane conversion was 97.7, 96.1 and 89.7%, respectively, while the CO2 selectivity remained very high (above 98%) in all cases. In contrast, when using Al2O3, the CH4 conversion increased in condition (c) compared with condition (b), from 25 to 43% at GHSV of 171,429 mL g−1 h−1. In line with the Al2O3 results, an improvement in CH4 conversion using condition (c) gas was also observed in comparison to condition (b). However, the CO2 selectivity reduced significantly when switching to condition (c) over Al2O3 compared with condition (b), from 58% to − 11% at GHSV of 171,429 mL g−1 h−1. Note that the negative CO2 selectivity indicates that some of the inlet CO2 was converted during the plasma reaction, for example through methane reforming, as discussed in [22]. This would be consistent with the observed increase in CO selectivity.
Under thermal conditions, it is known that the Pd catalyst activity is inhibited by the presence of combustion products e.g. H2O or CO2 [6, 42] which may also be the reason for the small decrease in conversion under NTP conditions observed with the 10 wt% Pd/Al2O3 catalyst. Similarly, the addition of H2O to the plasma-assisted catalytic toluene oxidation was shown to have a negative effect on catalyst activity, and was attributed to the competitive adsorption of water and also to a change in the plasma discharge behavior through quenching energetic electrons and reactive species [51, 52]. However, a previous study observed a promotional effect of H2O addition to the plasma-assisted catalytic CO oxidation through facilitating the decomposition of carbonate species on the Cu/Ce catalyst and enhancing catalyst stability [53].
When considering different feed compositions and reaction conditions, it can be concluded that, for the current study, a promotional effect of the addition of Pd compared to the support alone was also observed, both in terms of CH4 conversion and CO2 selectivity.
Effect of External Heat Supply and Plasma Energy
To evaluate the effect of combining an external heat source with the NTP to activate the catalyst for the CH4 oxidation reaction, a heat blower was used to control the reactor temperature up to 300 °C. The performance of the NTP-catalytic CH4 oxidation over the 10 wt% Pd/Al2O3 with a feed composition of 0.5 kPa CH4, 10 kPa O2, 7 kPa H2O, 5.5 kPa CO2 and 0.1 kPa NO in N2 as a function of reactor temperature and plasma SIE is reported in Fig. 4. The variation in plasma SIE from 1.1 to around 4.9 J mL−1 is the result of a change in the applied peak voltage from 5 to 7 kV at a constant frequency of 30 kHz. The plasma SIE followed a linear relationship with the applied voltage as shown in Fig. S2. As the applied voltage increased there was also an increase in the reactor temperature which contributed to a small increase in the plasma SIE, presented in Fig. S3. For example, at 7 kV and 30 kHz, the plasma SIE increased from around 3.7 to 4.7 J mL−1 when the temperature increased from RT to 300 °C.

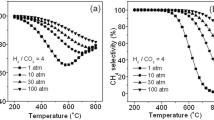
CH4 conversion (a) and CO2 selectivity (b) as a function of temperature and plasma SIE in NTP-catalytic CH4 oxidation (the lines are only to guide the eye). Reaction conditions: 10 wt% Pd/Al2O3, 0.5 kPa CH4, 10 kPa O2, 7 kPa H2O, 5.5 kPa CO2 and 0.1 kPa NO in N2, 171,429 mL g−1 h−1 GHSV
Because CH4 is a more stable molecule compared to other hydrocarbons, due to the strength of the C–H bonds, it requires high temperatures or energies to be activated. Typically, the thermal catalytic CH4 oxidation over supported Pd catalysts is initiated at temperatures above 200 °C and often only reaches 100% CH4 conversion at 400 °C or above [24, 42]. Examining CH4 oxidation reaction over the 10 w% Pd/Al2O3 at the operating conditions shown in Fig. 4, in the thermal mode (no plasma), no CH4 conversion from RT to 250 °C took place and only 4% CH4 conversion was observed at 300 °C. The limited CH4 conversion at 300 °C in this study may be attributed to the presence of H2O and CO2 in the gas feed which act as poisons [6, 42]. From Fig. 4a it is observed that an increase in plasma SIE and temperature led to an enhancement of the CH4 conversion. For example, at a constant plasma SIE of 1.5 J mL−1, an increase in temperature from RT to 300 °C increased the CH4 conversion from 0 to 98%. Similarly, without external heat supply (at RT in Fig. 4a), increasing the plasma SIE from 1.2 to 3.7 J mL−1 improved the CH4 conversion from almost 0 to 90%. The plasma-assisted catalytic process without external heat contribution can activate CH4 at RT and very high conversion of CH4 to products can be achieved with an increase in plasma SIE. Similar effect of the plasma SIE on CH4 conversion during plasma-only CH4 oxidation was reported [23].
Increasing the plasma SIE and temperature also enhanced the CO2 selectivity over the 10 wt% Pd/Al2O3 catalyst as shown in Fig. 4b. The CO2 selectivity was above 90% once the CH4 conversion was greater than 10%. Once the Pd catalyst became activated in the reaction, through heat supply or plasma, the CO2 selectivity reached around 100%. Conversely, at low temperatures (< 150 °C) and plasma SIEs (e.g. 1.1 J mL−1) where the CH4 conversion is below 10%, other reactions such as steam reforming of methane or dry reforming of methane govern the conversion, as also discussed in Sect. 3.2, and resulted in negative or low CO2 selectivities. In agreement with these results, Pham Huu et al. [28] reported a shift in CH4 conversion towards lower light-off temperatures when using plasma-assisted catalytic CH4 oxidation with in-plasma and post-plasma configurations in comparison to thermal catalytic reaction and plasma alone.
To achieve the same level of CH4 conversion in a plasma-catalyst system, the applied peak voltage and reactor temperature can be tuned, as shown in Table 2. From an energy perspective, increasing the external heat supply significantly reduced the plasma energy and total energy required to obtain the same CH4 conversion. For example, the plasma SIE required to obtain 50% CH4 conversion, reduced from 3.1 to 1.3 J mL−1 resulting in the total SIE reducing from 3.1 to 1.6 J mL−1 when the temperature increased from RT to 260 °C, respectively. Similarly, a decrease in plasma SIE from 3.7 to 1.6 J mL−1 required an increase in the temperature from RT to 293 °C to obtain 90% CH4 conversion at the reactor outlet, leading to a decrease in total SIE from 3.7 to 1.9 J mL−1. In summary, a combination of plasma and external heat supply in a plasma-catalyst gives access to high CH4 conversion with lower energy consumption.
The results in Table 2 also indicate that the external heat is more efficient than the plasma at initiating the methane oxidation reaction, in line with the results reported by [21]. However, it is worth noting that, in a practical application, the heat is dependent on the exhaust gas temperature, which leads to inefficient cold start emission control using traditional approaches. Under such conditions, where the heat supply is low, plasma could be used as it is almost instantaneous in its onset. As shown in Fig. S4, a process control with time dependent regulation (decrease) of the plasma SIE could guarantee methane emission abatement while minimizing the energy penalty by tuning the SIE from the NTP.
NOX Production and Mitigation Strategies
Together with the CH4 conversion and CO2 selectivity, the effect of plasma on NOX formation/conversion needs to be considered in after-treatment applications. The presence of N2 as the balance gas resulted in the production of a combination of NO and NO2 in the plasma discharge zone (Table 3). The amount of NOX produced was 836 ppmv in the presence of the Pd/Al2O3 catalyst while the use of Al2O3 support in the plasma zone resulted in a decrease in NOX formation to 644 ppmv. These changes also reflected changes in the CH4 conversion with high NOx formation occurring at high CH4 conversions, as a result of the increase in electric field intensity. This consequently lead to an increase in the rate of formation of active species (such as N, O, CH3) through electron impact reactions, and the effective collisions between them (equations listed in ESI, section S2). Moreover, the NO/NO2 ratio also varied and increased in the following order: Al2O3 < Pd/Al2O3. Under the conditions of this study, thermodynamic calculations suggest that only NO is a stable product of N2 and O2 reaction, especially at high SIEs. This indicates that the plasma provides non-equilibrium conditions with generation of a mixture of NO and NO2.
The effect of the reaction conditions on the CH4 conversion and NOX formation over the 10 wt% Pd/Al2O3 is reported in Table 4. Under condition (c), the feed gas contained 1000 ppmv NOX (corresponding to 0.1 kPa NO) and the effluent data indicated the formation of an additional 224 ppmv NOX during the plasma-catalytic reaction. This reflected a decrease in the amount of NOX formed in the plasma in comparison to condition (b) due to the addition of NO in the feed gas. These preliminary results seemed to indicate that the presence of NO in the feed gas led to reduced formation of NOX. However, when the reactor temperature was increased to 300 °C (condition (d)), the formation of NOX in the DBD increased to 714 ppmv, to a total of 1714 ppmv outlet concentration. This is consistent with the reported formation of NOX (mainly from plasma gas phase reactions via Eqs. (8, 9, 10, 11, 12, and 13)) being enhanced even if NO is already present in the feed. These results are in good agreement with those reported in [54], which attributed this behaviour to the fact that a 10% oxygen concentration in the feed is above the critical oxygen concentration range, leading to an increase in the NOX concentration with the energy density. The present results also indicate that the energy density dependence is driven by both the plasma and heat. The general trend reported in Fig. 5 appears to indicate a good correlation between the CH4 conversion and the effluent NOX concentration, for all the catalysts tested in this study, irrespective of reaction conditions. The results, herein, also indicate that the simultaneous NOX reduction and methane oxidation is not possible within the conditions investigated due to the electron impact dissociation of N2 and O2 (Eqs. (8, 9, and 10)) and the subsequent reactions that occur between the active species, some of which are listed in ESI Section S2 [22, 50,51,52,53,54,55,56].

CH4 conversion as a function of NOX formation over all catalysts tested in this study at different reaction conditions. Note NO is not added to the gas feed in any of the tests in this figure. (Green Inverted triangle) ZSM-5 (SiO2/Al2O3 = 30); (Blue triangle) ZSM-5 (SiO2/Al2O3 = 80); (Golden brown Left pointer triangle) 2 wt% Mo/ZSM-5 (SiO2/Al2O3 = 30); (Diamond) 2 wt% Mo/ZSM-5 (SiO2/Al2O3 = 80); (Red circle) Al2O3; (Brown Hexagon) 2 wt% Co/Al2O3; (Orange pentagon) 2 wt% Cu/Al2O3; (Star) 2 wt% Fe/Al2O3; (Orange asterisk) 2 wt% Li/Al2O3; (Multiplication symbol) 2 wt% Ni/Al2O3; (Black square box) 2 wt% Pd/Al2O3; (Minus symbol) 10 wt% Pd/Al2O3; (Plus symbol) 2 wt% Pt/Al2O3; (Vertical blue line) 2 wt% Rh/Al2O3; (Cyan right pointer triangle) 2 wt% Ru/Al2O3 (Color figure online)
To address this challenge and to mitigate for the NOX formation, a different approach was explored, whereby the addition of ammonia and a suitable SCR catalyst was considered. While NH3-SCR (Eqs. (14, 15, and 16)) is a mature technology, it is not suitable for simultaneous conversion of methane and NOX under realistic conditions during cold start [57].
The catalyst of choice for the SCR process is a commercially available system, Cu-CHA from Johnson Matthey Technology. A typical thermal activation light-off experiment is reported in Fig. 6. As expected and reported in the literature [58], the activity of the Cu-CHA catalyst for NOX reduction was found to be high, with 100% conversion achieved at 190 °C under standard thermal activation. As noted earlier, the conversion of methane under these conditions is negligible below 400 °C, as illustrated in Fig. 6. Thus, an assessment of the effect of plasma on the simultaneous reduction of NOX and CH4 oxidation during cold start using NH3 as a reducing agent was examined.

NO (red) and CH4 conversions (blue) as function of reactor temperature using a commercial Cu-CHA catalyst. Reaction conditions: 0.5 kPa CH4, 10 kPa O2, 0.1 kPa NO, and 0.1 kPa NH3 in Ar, GHSV = 85,714 mL g−1 h−1 (Color figure online)
For a better understanding of the formation and evolution of the reaction products/intermediates during the plasma exposure, without the interference from high concentrations of H2O and CO2, the inlet feed was simplified (CH4, O2, NO and NH3 with N2 balance) and the outlet line of the reactor was connected directly to an FTIR. The following vibrational bands were used as diagnostics for the presence of different species: CH4 at 3016 and 1304 cm−1, CO2 at 2360 cm−1, CO at 2143 cm−1, NO at 1883 cm−1, NO2 at 1614 cm−1, N2O at 2223 cm−1, HNO3 at 1325 and 1718 cm−1 [56].
Figure 7 reports the FTIR spectra of the reactants and products from the DBD reactor with the addition of Cu-CHA to the discharge, at 2.1–2.3 J mL−1. When the plasma is ignited, a 40% reduction in the methane concentration was observed and a decrease in the intensity of NO band. However, since the NO2 concentration simultaneously increased, very little change was observed in the overall NOX conversion indicating that the NO was oxidised to NO2. This is in good agreement with [54]. Nitrous oxide was also formed in the plasma discharge (equations (S6) to (S9)), with concentration of approximately 430 ppmv N2O. Wang and Tokunaga [54, 59] reported that in the presence of a high concentration of O radicals and O2, both NOX and N2O are formed in plasma (equations (S1) to (S25)). It should, however, be noted that the increase of the peaks at 2210–2250 cm−1 might not be entirely due to N2O formation, but also to other -NCO species.

FTIR spectra for inlet feed and final reactor products with Cu-CHA catalyst in the DBD. Reaction conditions: 0.5 kPa CH4/10 O2/0.1 NO/0.1 NH3 in N2
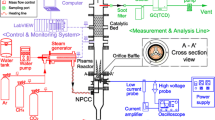
To prevent the NH3 from being oxidised in the plasma instead of being utilised as a reducing agent, a hybrid solution was further explored. It made use of a hybrid dual-stage reactor where the oxidation reactions took place upstream in the plasma discharge (in-plasma catalysis (IPC)) and the SCR catalyst and NH3 injection were located downstream, i.e. in a post-plasma catalysis (PPC) configuration, as shown in Fig. 8 (with further details in Fig. S5). This configuration benefited from the supply of a small amount of heat from the methane oxidation zone to the SCR region.

Experimental setup for CH4 oxidation using plasma-catalyst and thermal NH3-SCR post-plasma treatment
The dual stage configuration resulted in a full methane conversion with > 99% CO2 selectivity, as evidenced in Fig. 9 from the disappearance of the bands at 3016 and 1304 cm−1, and the increase of the CO2 feature at 2360 cm−1, respectively. Once NH3 was injected downstream of the plasma reactor, the NOX were fully converted, as shown by the disappearance of the NO and NO2 bands. No significant change was observed in the intensity of the band at 2223 cm−1 when compared to the one in Fig. 7 (417 ppmv N2O), which indicated that this species is produced in the plasma discharge and the catalysts used herein did not promote the formation nor the removal of this product. Zhao et al. [60] have shown a strong correlation between the SIE and N2O formation when a V2O5-WO3/TiO2 catalyst was used. In our previous study on HC-SCR [19] we have shown that the N2O selectivity is dependent on the applied frequency, temperature and NOx conversion and we have proposed that the duration of the pulse can be of high importance, as also discussed by de Rosa et al. [21] While N2O slip is highly undesirable, the current study was not intended for exhaustive mitigation strategies but rather to put forward a possible solution for cold-start CH4 and NOX emission control for diesel engines.

FTIR spectra for final reaction products of the dual stage reactor. 0.5% CH4/10% O2/0.1% NO/0 (blue)−0.1% (red) NH3/N2; GHSV = 85,714 mL g−1 h−1; 70 mg 10% Pd/Al2O3; 70 mg Cu-CHA catalyst downstream plasma discharge, 190 °C reaction temperature (Color figure online)
As demonstrated in the present study, the main disadvantage of using an in-plasma catalyst discharge to reach complete oxidation of the methane under realistic exhaust conditions was the formation or low conversion of NOX (Eqs. (8, 9, 10, 11, 12, and 13) and (S1) to (S25)).
It was found that the concentration of oxygen in the feed had a critical role on the NTP denitration performance [54] and, while high conversions of NOX can be achieved with O2 concentration lower than 1%, this is not realistic for automotive exhaust conditions. It is important to note that the addition of water and ammonia is beneficial towards the NOX removal efficiency even with O2 concentrations > 10% as long as the SIE is kept below 2 J mL−1, as shown by Wang et al. It is suggested that, as soon as the SIE increases, allowing for more reactive oxygen species (ROS) to be formed, the removal efficiency becomes negative, with reactions (Eqs. (12), (13)) more prone to take place. Furthermore, the interaction between the excited NO* and other N-containing species formed in the plasma and on the oxide ions at the catalyst surface, gives rise to –NOX and –NO2 species, which could lead to an increase in the overall NOX concentration, resulting therefore in a negative NOX removal efficiency [54, 61]. The ease of –N radicals formation is of paramount importance in terms of NOX concentration and it might be that a different way of generating plasma instead of the DBD method could indeed lead to significant differences in NOX concentration, as seen in [54].
The properties observed for the combined hybrid system indicate that the use of the NTP + catalyst closely followed by an ammonia-SCR system may hold the solution to diesel cold start emission control, as proposed in Fig. 10.

Proposed solution for simultaneous CH4 and NOX conversion for Diesel cold start
Conclusions
This paper reports on the NTP-catalytic methane oxidation as an aftertreatment technology to remove methane emissions in the exhaust of methane-powered vehicles. High CH4 conversion and CO2 selectivities can be achieved with the presence of a catalyst in a DBD reactor. The CO2 selectivity during NTP-catalytic CH4 oxidation is related to the CH4 conversion and increases linearly with the CH4 conversion. In agreement with thermal catalysis, Pd/Al2O3 catalysts are shown to have the best performance for the NTP-catalytic methane oxidation in terms of CH4 conversion and CO2 selectivity among all catalysts tested. The performance of the plasma-catalyst system is found to depend on feed gas components and GHSVs. Complex gas mixtures that simulate real exhaust conditions can reduce methane conversion when using Pd/Al2O3 catalysts. A combination of plasma and external heat supply in a plasma-catalyst system can achieve high CH4 conversion and CO2 selectivity with lower energy consumption. This is an approach that appears particularly suitable to cold-start conditions using biogas as a fuel. The presence of N2 and O2 in the plasma system can lead to unfavorable NOX formation, which is not preventable as long as the plasma exists due to electron-impact dissociation reaction of these molecules. The NOX formation has a linear dependence with CH4 conversion during NTP-catalytic CH4 oxidation. The addition of a second catalyst bed, post-plasma, where NH3 can be injected for selectively reduce the nitrogen oxides at temperatures below 200 °C was shown to be an efficient solution in this case.
It is concluded that the most suitable aftertreatment option involves the combination of an oxidation catalyst with plasma to target the hydrocarbon and CH4 oxidation, followed by the NH3-SCR system to convert the NOX.
References
Curran SJ, Wagner RM, Graves RL et al (2014) Well-to-wheel analysis of direct and indirect use of natural gas in passenger vehicles. Energy 75:194–203. https://doi.org/10.1016/j.energy.2014.07.035
Torchio MF, Santarelli MG (2010) Energy, environmental and economic comparison of different powertrain/fuel options using well-to-wheels assessment, energy and external costs–European market analysis. Energy 35:4156–4171. https://doi.org/10.1016/j.energy.2010.06.037
U.S. Department of Energy (2021) Alternative fuels data center: natural gas vehicles. https://afdc.energy.gov/vehicles/natural_gas.html
U.K. Department of Transport (2015) RICARDO-AEA provision of HGV emissions testing, RM4470-SB2925. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/468172/hgv-emissions-testing.pdf
Gélin P, Urfels L, Primet M, Tena E (2003) Complete oxidation of methane at low temperature over Pt and Pd catalysts for the abatement of lean-burn natural gas fuelled vehicles emissions: influence of water and sulphur containing compounds. Catal Today 83:45–57. https://doi.org/10.1016/S0920-5861(03)00215-3
Gholami R, Alyani M, Smith K (2015) Deactivation of Pd catalysts by water during low temperature methane oxidation relevant to natural gas vehicle converters. Catalysts 5:561–594. https://doi.org/10.3390/catal5020561
Cargnello M, Jaén JJD, Garrido JCH et al (2012) Exceptional activity for methane combustion over modular Pd@CeO2 subunits on functionalized Al2O3. Science 337:713–717. https://doi.org/10.1126/science.1222887
Coney C, Stere C, Millington P et al (2020) Spatially-resolved investigation of the water inhibition of methane oxidation over palladium. Catal Sci Technol 10:1858–1874. https://doi.org/10.1039/D0CY00154F
Neyts EC, Ostrikov K, SunkaraBogaerts MKA (2015) Plasma catalysis: synergistic effects at the nanoscale. Chem Rev 115:13408–13446. https://doi.org/10.1021/acs.chemrev.5b00362
Azzolina-Jury F, Bento D, Henriques C, Thibault-Starzyk F (2017) Chemical engineering aspects of plasma-assisted CO2 hydrogenation over nickel zeolites under partial vacuum. J CO2 Util 22:97–109. https://doi.org/10.1016/j.jcou.2017.09.017
Patil BS, Cherkasov N, Lang J et al (2016) Low temperature plasma-catalytic NOX synthesis in a packed DBD reactor: effect of support materials and supported active metal oxides. Appl Catal B Environ 194:123–133. https://doi.org/10.1016/j.apcatb.2016.04.055
Uytdenhouwen Y, Bal KM, Neyts EC et al (2021) On the kinetics and equilibria of plasma-based dry reforming of methane. Chem Eng J 405:126630. https://doi.org/10.1016/j.cej.2020.126630
Khoja AH, Tahir M, Amin NAS (2019) Recent developments in non-thermal catalytic DBD plasma reactor for dry reforming of methane. Energy Convers Manag 183:529–560. https://doi.org/10.1016/j.enconman.2018.12.112
Ahmad F, Lovell EC, Masood H et al (2020) Low-temperature CO2 methanation: synergistic effects in plasma-Ni hybrid catalytic system. ACS Sustain Chem Eng 8:1888–1898. https://doi.org/10.1021/acssuschemeng.9b06180
Vakili R, Gholami R, Stere CE et al (2020) Plasma-assisted catalytic dry reforming of methane (DRM) over metal-organic frameworks (MOFs)-based catalysts. Appl Catal B Environ 260:118195. https://doi.org/10.1016/j.apcatb.2019.118195
Wang H, Zhao B, Qin L et al (2020) Non-thermal plasma-enhanced dry reforming of methane and CO2 over Ce-promoted Ni/C catalysts. Mol Catal 485:110821. https://doi.org/10.1016/j.mcat.2020.110821
Chen H, Goodarzi F, Mu Y et al (2020) Effect of metal dispersion and support structure of Ni/silicalite-1 catalysts on non-thermal plasma (NTP) activated CO2 hydrogenation. Appl Catal B Environ 272:119013. https://doi.org/10.1016/j.apcatb.2020.119013
Chen H, Mu Y, Xu S et al (2020) Recent advances in non-thermal plasma (NTP) catalysis towards C1 chemistry. Chin J Chem Eng 28:2010–2021. https://doi.org/10.1016/j.cjche.2020.05.027
Stere CE, Adress W, Burch R et al (2014) Ambient temperature hydrocarbon selective catalytic reduction of NOX using atmospheric pressure nonthermal plasma activation of a Ag/Al2O3 catalyst. ACS Catal 4:666–673. https://doi.org/10.1021/cs4009286
Wangkawong K, Phanichphant S, Inceesungvorn B et al (2020) Kinetics of water gas shift reaction on Au/CeZrO4: a comparison between conventional heating and dielectric barrier discharge (DBD) plasma activation. Top Catal 63:363–369. https://doi.org/10.1007/s11244-020-01245-8
De Rosa F, Hardacre C, Graham WG et al (2021) Comparison between the thermal and plasma (NTP) assisted palladium catalyzed oxidation of CH4 using AC or nanopulse power supply. Catal Today 384:177–186. https://doi.org/10.1016/j.cattod.2021.04.015
Baylet A, Marécot P, Duprez D et al (2012) Synergetic effect of plasma/catalysis hybrid system for CH4 removal. Appl Catal B Environ 113–114:31–36. https://doi.org/10.1016/j.apcatb.2011.10.026
Lee H, Lee DH, Song YH et al (2015) Synergistic effect of non-thermal plasma-catalysis hybrid system on methane complete oxidation over Pd-based catalysts. Chem Eng J 259:761–770. https://doi.org/10.1016/j.cej.2014.07.128
Gibson EK, Stere CE, Curran-McAteer B et al (2017) Probing the role of a non-thermal plasma (NTP) in the hybrid NTP catalytic oxidation of methane. Angew Chem Int Edit 56:9351–9355. https://doi.org/10.1002/anie.201703550
Stere C, Chansai S, Gholami R et al (2020) A design of a fixed bed plasma DRIFTS cell for studying the NTP-assisted heterogeneously catalysed reactions. Catal Sci Technol 10:1458–1466. https://doi.org/10.1039/D0CY00036A
Nozaki T, Okazaki K (2013) Non-thermal plasma catalysis of methane: principles, energy efficiency, and applications. Catal Today 211:29–38. https://doi.org/10.1016/j.cattod.2013.04.002
Yao S, Chen Z, Weng S et al (2019) Mechanism of CO2-formation promotion by Au in plasma-catalytic oxidation of CH4 over Au/γ-Al2O3 at room temperature. J Hazard Mater 373:698–704. https://doi.org/10.1016/j.jhazmat.2019.04.003
Pham Huu T, Gil S, Da Costa P et al (2015) Plasma-catalytic hybrid reactor: application to methane removal. Catal Today 257:86–92. https://doi.org/10.1016/j.cattod.2015.03.001
Kim S-S, Kwon B, Kim J (2007) Plasma catalytic methane conversion over sol–gel derived Ru/TiO2 catalyst in a dielectric-barrier discharge reactor. Catal Commun 8:2204–2207. https://doi.org/10.1016/j.catcom.2007.04.029
Kiani D, Sourav S, Tang Y et al (2021) Methane activation by ZSM-5-supported transition metal centers. Chem Soc Rev. https://doi.org/10.1039/D0CS01016B
Tang P, Zhu Q, Wu Z, Ma D (2014) Methane activation: the past and future. Energy Environ Sci 7:2580–2591. https://doi.org/10.1039/C4EE00604F
Horn R, Schlögl R (2015) Methane activation by heterogeneous catalysis. Catal Lett 145:23–39. https://doi.org/10.1007/s10562-014-1417-z
Hu X, Liu Y, Dou L et al (2022) Plasma enhanced anti-coking performance of Pd/CeO2 catalysts for the conversion of methane. Sustain Energy Fuels 6:98–109. https://doi.org/10.1039/D1SE01441B
Smith JM, Van Ness HC, Abbott MM (2001) Introduction to chemical engineering thermodynamics, Sixth. McGraw-Hill, New York
Liu HF, Liu RS, Liew KY et al (1984) Partial oxidation of methane by nitrous oxide over molybdenum on silica. J Am Chem Soc 106:4117–4121. https://doi.org/10.1021/ja00327a009
Louis C, Tatibouët J-M, Che M (1988) Catalytic properties of silica-supported molybdenum catalysts in methanol oxidation: the influence of molybdenum dispersion. J Catal 109:354–366. https://doi.org/10.1016/0021-9517(88)90218-7
Chen Y, Zhang X, Wang X et al (2021) Insights into the structure-activity relationship in aerobic alcohol oxidation over a metal–organic-framework-supported molybdenum(VI) catalyst. J Am Chem Soc 143:4302–4310. https://doi.org/10.1021/jacs.0c12963
Kurlov A, Deeva EB, Abdala PM et al (2020) Exploiting two-dimensional morphology of molybdenum oxycarbide to enable efficient catalytic dry reforming of methane. Nat Commun 11:4920. https://doi.org/10.1038/s41467-020-18721-0
Suzuki K, Hayakawa T, Shimizu M, Takehira K (1994) Partial oxidation of methane over silica supported molybdenum oxide catalysts. Catal Lett 30:159–169. https://doi.org/10.1007/BF00813682
Xiao T, Hanif A, York APE et al (2002) Study on the mechanism of partial oxidation of methane to synthesis gas over molybdenum carbide catalyst. Phys Chem Chem Phys 4:4549–4554. https://doi.org/10.1039/B204347E
Gao Y, Dou L, Zhang S et al (2021) Coupling bimetallic Ni-Fe catalysts and nanosecond pulsed plasma for synergistic low-temperature CO2 methanation. Chem Eng J 420:127693. https://doi.org/10.1016/j.cej.2020.127693
Gholami R, Smith KJ (2015) Activity of PdO/SiO2 catalysts for CH4 oxidation following thermal treatments. Appl Catal B Environ 168–169:156–163. https://doi.org/10.1016/j.apcatb.2014.12.037
Danielis M, Betancourt LE, Orozco I et al (2021) Methane oxidation activity and nanoscale characterization of Pd/CeO2 catalysts prepared by dry milling Pd acetate and ceria. Appl Catal B Environ 282:119567. https://doi.org/10.1016/j.apcatb.2020.119567
Molteni M, Donazzi A (2020) Model analysis of atmospheric non-thermal plasma for methane abatement in a gas phase dielectric barrier discharge reactor. Chem Eng Sci 212:115340. https://doi.org/10.1016/j.ces.2019.115340
Phan DQ, Kureti S (2017) CO oxidation on Pd/Al2O3 catalysts under stoichiometric conditions. Top Catal 60:260–265. https://doi.org/10.1007/s11244-016-0608-9
Xu W, Zhan Z, Di L, Zhang X (2015) Enhanced activity for CO oxidation over Pd/Al2O3 catalysts prepared by atmospheric-pressure cold plasma. Catal Today 256:148–152. https://doi.org/10.1016/j.cattod.2015.01.017
Taheraslani M, Gardeniers H (2020) Coupling of CH4 to C2 hydrocarbons in a packed bed DBD plasma reactor: the effect of dielectric constant and porosity of the packing. Energies 13:468. https://doi.org/10.3390/en13020468
Goujard V, Tatibouët J-M, Batiot-Dupeyrat C (2011) Carbon dioxide reforming of methane using a dielectric barrier discharge reactor: effect of helium dilution and kinetic model. Plasma Chem Plasma Process 31:315–325. https://doi.org/10.1007/s11090-010-9283-y
Jo S, Hoon Lee D, Seok Kang W, Song Y-H (2013) Methane activation using noble gases in a dielectric barrier discharge reactor. Phys Plasmas 20:083509. https://doi.org/10.1063/1.4818795
Wang W, Snoeckx R, Zhang X et al (2018) Modeling plasma-based CO2 and CH4 conversion in mixtures with N2, O2, and H2O: the bigger plasma chemistry picture. J Phys Chem C 122:8704–8723. https://doi.org/10.1021/acs.jpcc.7b10619
Wu J, Xia Q, Wang H, Li Z (2014) Catalytic performance of plasma catalysis system with nickel oxide catalysts on different supports for toluene removal: effect of water vapor. Appl Catal B Environ 156–157:265–272. https://doi.org/10.1016/j.apcatb.2014.03.017
Falkenstein Z, Coogan JJ (1997) Microdischarge behaviour in the silent discharge of nitrogen–oxygen and water–air mixtures. J Phys D Appl Phys 30:817–825. https://doi.org/10.1088/0022-3727/30/5/015
Zhang J, Liu Y, Yao X et al (2020) Enhanced moisture resistance of Cu/Ce catalysts for CO oxidation via plasma−catalyst interactions. Chemosphere 261:127739. https://doi.org/10.1016/j.chemosphere.2020.127739
Wang Z, Kuang H, Zhang J et al (2019) Nitrogen oxide removal by non-thermal plasma for marine diesel engines. RSC Adv 9:5402–5416. https://doi.org/10.1039/C8RA09217F
Snoeckx R, Heijkers S, Van Wesenbeeck K et al (2016) CO2 conversion in a dielectric barrier discharge plasma: N2 in the mix as a helping hand or problematic impurity? Energy Environ Sci 9:999–1011. https://doi.org/10.1039/C5EE03304G
Pavlovich MJ, Ono T, Galleher C et al (2014) Air spark-like plasma source for antimicrobial NOX generation. J Phys D Appl Phys 47:505202. https://doi.org/10.1088/0022-3727/47/50/505202
Nova I, Tronconi E (2014) Urea-SCR technology for deNOX after treatment of diesel exhausts, 1st edn. Springer, New York
Villamaina R, Nova I, Tronconi E et al (2019) Effect of the NH4NO3 addition on the Low-T NH3-SCR performances of individual and combined Fe- and Cu-zeolite catalysts. Emiss Control Sci Technol 5:290–296. https://doi.org/10.1007/s40825-019-00140-3
Tokunaga O, Suzuki N (1984) Radiation chemical reactions in NOX and SO2 removals from flue gas. Radiat Phys Chem 24:145–165. https://doi.org/10.1016/0146-5724(84)90013-X
Zhao W, Liu Y, Wei H et al (2019) NO removal by plasma-enhanced NH3-SCR using methane as an assistant reduction agent at low temperature. Appl Sci 9:2751. https://doi.org/10.3390/app9132751
Bogaerts A, Tu X, Whitehead JC et al (2020) The 2020 plasma catalysis roadmap. J Phys D Appl Phys 53:443001. https://doi.org/10.1088/1361-6463/ab9048
Acknowledgements
The authors acknowledge funding from Innovate UK under grant ref: 102661. Open access data can be found via the University of Manchester research portal.
Author information
Authors and Affiliations
Contributions
RG: Investigation, Writing-Original draft preparation, Writing-Reviewing and editing; CS: Investigation, Methodology, Writing-Reviewing and editing; SC: Validation, Writing-Reviewing and editing; AS: Investigation, Validation; AG: Conceptualization, Funding acquisition, Writing-Reviewing and editing; PH: Supervision, Funding acquisition; PM: Funding acquisition, Project administration; CH: Conceptualization, Funding acquisition, Project administration, Supervision, Validation, Writing-Reviewing and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors report no declaration of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gholami, R., Stere, C., Chansai, S. et al. Optimization of Non-thermal Plasma-Assisted Catalytic Oxidation for Methane Emissions Abatement as an Exhaust Aftertreatment Technology. Plasma Chem Plasma Process 42, 709–730 (2022). https://doi.org/10.1007/s11090-022-10253-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11090-022-10253-3