
Abstract
Multiple sclerosis (MS) is an autoimmune demyelinating neurodegenerative disease of the central nervous system (CNS) due to injury of the myelin sheath by immune cells. The clotting factor fibrinogen is involved in the pathogenesis of MS by triggering microglia and the progress of neuroinflammation. Fibrinogen level is correlated with MS severity; consequently, inhibition of the fibrinogen cascade may reduce MS neuropathology. Thus, this review aimed to clarify the potential role of fibrinogen in the pathogenesis of MS and how targeting of fibrinogen affects MS neuropathology. Accumulation of fibrinogen in the CNS may occur independently or due to disruption of blood–brain barrier (BBB) integrity in MS. Fibrinogen acts as transduction and increases microglia activation which induces the progression of inflammation, oxidative stress, and neuronal injury. Besides, brain fibrinogen impairs the remyelination process by inhibiting the differentiation of oligodendrocyte precursor cells. These findings proposed that fibrinogen is associated with MS neuropathology through interruption of BBB integrity, induction of neuroinflammation, and demyelination with inhibition of the remyelination process by suppressing oligodendrocytes. Therefore, targeting of fibrinogen and/or CD11b/CD18 receptors by metformin and statins might decrease MS neuropathology. In conclusion, inhibiting the expression of CD11b/CD18 receptors by metformin and statins may decrease the pro-inflammatory effect of fibrinogen on microglia which is involved in the progression of MS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Most of neurodegenerative diseases including Alzheimer’s disease (AD) and Parkinson’s disease (PD) are age-related disorders that are most common in elderly subjects [1]. However, multiple sclerosis (MS) is the most common demyelinating neurodegenerative disease of the central nervous system (CNS) in young adults [2]. Of note, MS was initially described by Jean-Martin Charcot a French neurologist in 1868 who illustrated multiple scars in the brain and spinal cord [3]. It has been shown that MS interrupts motor and sensory neuronal signal transmission leading to motor and sensory deficits [4]. MS is characterized by specific symptoms including vision loss in one eye, double vision, muscle weakness, and motor-sensory incoordination [5]. Notably, MS may be progressive or relapsing forms in which the symptoms disappear and return [5]. Of interest, MS affects about 2.8 million people globally with the difference among many populations; MS is more common in women at 20–50 years [6]. Remarkably, MS is not a curable disease, and 85% of MS cases presented as an isolated clinical syndrome, 45% have motor or sensory dysfunctions, 20% of MS patients have optic neuritis, and 10% presented with brainstem disorders [7]. Particularly, 85% of MS patients presented with acute exacerbations followed by improvement [8]. Nevertheless, 15% of MS patients presented with gradual motor-sensory dysfunction without a period of recovery [8]. However, a combination of these two forms may occur, and relapse is triggered by several factors including viral infections and stress [7, 8]. Symptoms of MS are increased during exposure to high temperatures [9]. An acute attack of MS is treated by corticosteroids and/or plasmapheresis [10]. However, chronic MS is managed by disease-modifying treatments like interferons, glatiramer, and mitoxantrone [11].
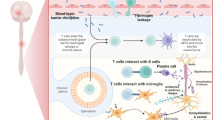
Furthermore, MS is regarded as an autoimmune disease causing the injury of myelin sheath by immune cells and inhibiting the production of myelin [12]. Oligodendrocytes which are concerned with the synthesis of the myelin sheath are mostly affected in MS [13, 14]. Progressive loss of myelin sheath with axonal injury leads to neuronal dysfunction [15]. Partial remyelination in the remission state and demyelination in the relapse phase lead to sclerotic lesions in the CNS [16]. Besides, reactive astrocytosis in response to neuronal injury promotes plaque formation [8]. Moreover, MS plaques are numerous focal areas of demyelination scattered in the brain’s white matter and spinal cords as well as in the deep grey matter and cerebral cortex [17]. In the MS plaques, there is a large infiltration of immune cells including T lymphocytes, monocytes, B cells, and plasma cells [18]. It has been shown that inflammation acts as a central role in the pathogenesis of MS due to the uninhibited activation of T lymphocytes [19]. Peripheral auto-reactive T lymphocytes prompt inflammatory changes in the MS [20]. However, the underlying mechanism for the activation of peripheral auto-reactive T lymphocytes is not fully elucidated. Polyclonal activation of peripheral auto-reactive T lymphocytes by viral antigens or molecular mimicry may be the convincing mechanism [21]. Peripheral auto-reactive T lymphocytes can cross the blood–brain barrier (BBB) by binding integrin on the immune cells and vascular cell adhesion protein 1 (VCAM-1) on the endothelial cells [21]. Expression of integrin and VCAM-1 are increased by inflammation and pro-inflammatory cytokines [22, 23]. T-cell-induces the expression and release of matrix metalloproteinase (MMPs) which augment the entry of T cell which is also involved in the degeneration of myelin components [24]. After the entry of peripherally auto-reactive T lymphocytes, they bind to MHCII expressed by dendritic and antigen-presenting cells leading to the reactivation of T cells toward pro-inflammatory phenotype [25]. These changes provoke disruption of myelin components and the release of other CNS antigens with subsequent recruitment of other immune cells and production of specific myelin auto-antibodies which support further injury and loss of myelin sheath [26]. These immune-inflammatory reactions cause further injury of BBB that promotes entry of auto-reactive T lymphocytes and generation of soluble factors which attack synaptic regions causing neuronal dysfunction [27, 28] (Fig. 1).

Pathophysiology of multiple sclerosis (MS): entry of autoreactive T cells which interact with microglia and active release of pro-inflammatory cytokines and osteopontin. Besides, autoreactive T cells antigen-presenting cells (APCs) expressing major histocompatibility II (MHCII) lead to injury of oligodendrocytes and disruption of neuronal myelin sheath with the development of multiple sclerosis (MS)
It has been reported that fibrinogen is involved in the pathogenesis of MS by activating microglia and the development of neuroinflammation [29]. Fibrinogen also called factor I is a 340 kDa glycoprotein produced by hepatocytes and circulates in the blood [30]. The main function of fibrinogen is controlling of blood homeostasis during vascular injury and tissue damage [30]. Fibrinogen was initially discovered by Paul Morawitz in 1905. The normal circulating fibrinogen level is 3 g/L which increases to 4.5 g/L during pregnancy [31]. Fibrinogen is converted by thrombin to fibrin and the formation of blood clots during vascular injury to prevent bleeding [31, 32] (Fig. 2).

Fibrinogen pathway: prothrombin is converted with the assistance of calcium by prothrombin activator to thrombin which enhances the conversion of fibrinogen to fibrinogen monomer. The formed fibers are cross-linked with the assistance of an activated fibrin stabilizing factor
Acquired and congenital hypofibrinogenemia is associated with bleeding tendency; however, hyperfibrinogenemia is linked with thrombotic disorders [33]. Fibrinogen is regarded as an acute-phase protein in response to systemic inflammation in different inflammatory disorders [34]. Hyperfibrinogenemia in response to inflammation and malignancy induces thromboembolic disorders [35]. In addition, fibrinogen forms a bridge between platelets via binding fibrinogen receptors (GpIIb/IIIa) on platelets [36]. Acquired dysfibrinogenemia is developed due to dysfunction of fibrinogen that developed in chronic disorders including cirrhosis and chronic hepatitis [37]. Diseased liver produced dysfunctional fibrinogen due to a defect in the glycosylation process of amino acids [38]. Furthermore, circulating autoantibodies in autoimmune disorders interferes with fibrinogen causing the development of dysfunctional fibrinogen [39]. As well, some medications like glucocorticoids and isotretinoin interfere with fibrinogen function [40]. Mutation of fibrinogen induces the production of abnormal fibrinogen involved in the development of familial renal amyloidosis [40]. However, fibrinogen in familial renal amyloidosis is not associated with bleeding or thrombosis [40]. It has been revealed that fibrinogen level is correlated with MS severity [41]. Therefore, inhibition of the fibrinogen cascade may reduce MS neuropathology. Thus, this review aimed to clarify the potential role of fibrinogen in the pathogenesis of MS and how targeting of fibrinogen affects MS neuropathology.
Fibrinogen and MS
Fibrinogen has pleiotropic effects and plays a critical role in the pathogenesis of MS by induction of inflammatory process and neuroinflammation [11]. Deposition of fibrinogen in the CNS precedes neuroinflammation in MS [41]. However, accumulation of fibrinogen in the CNS may occur due to disruption of BBB integrity in MS [29, 41]. Disturbance of BBB integrity in MS may precede the development of brain lesions that may promote the entrance of plasma proteins including fibrinogen [29, 41]. It has been demonstrated that extracellular vesicles from blood plasma can induce experimental autoimmune encephalomyelitis in mice due to the activation of CD8 T cells by fibrinogen [42]. Likewise, analysis of extracellular vesicles from blood plasma from MS patients showed a higher concentration of fibrinogen [42]. Therefore, fibrinogen is implicated in the development and progression of MS through the induction of neuroinflammation and disease relapse. Fibrinogen is not merely the indicator of BBB injury in MS but acts as transduction increases microglia activation via triggering expression of αvβ3 integrin on CD11b/CD18 [43]. Likewise, brain fibrinogen induces the progression of inflammation, oxidative stress, and neuronal injury [44]. Interestingly, brain fibrinogen impairs the remyelination process by inhibiting the differentiation of oligodendrocyte precursor cells [45]. In vivo study using photon microscopy demonstrated that the clustering of microglia in the perivascular space induced by fibrinogen is developed before the progression of demyelination in MS [46]. Ghorbani and Yong [47] observed that the extracellular matrix acts as a possible modifier of remyelination and neuroinflammation. Furthermore, fibrinogen and fibrinogen-like 1,2 are associated with neuroinflammation and MS neuropathology [48]. Furthermore, fibrinogen can act as an immunomodulatory in the CNS, triggering neuroinflammation and demyelination in MS [49]. A case-control study included 119 MS patients and 68 healthy controls observed that single gene polymorphism 455G/A and VH1299R variant in the fibrinogen gene was associated with higher MS risk [49]. Davalos et al. [50] revealed that deposition of fibrinogen in the CNS triggers the pathogenesis of MS. Many postmortem studies revealed that deposition of fibrinogen in the brain perivascular regions was observed not only in the active sclerotic lesions but also in the pre-active brain lesions mainly in the white and gray matter of cerebral cortex. It has been observed that fibrinogen plasma level was increased in MS patients [40]. An observational cohort study involving 58 MS patients showed that fibrinogen plasma level was increased in patients with active MS and positively correlated with CNS lesions [40]. This study indicated that fibrinogen plasma level is a sensitive biomarker for the early detection of MS.
Of interest, the deposition of fibrinogen in the perivascular space activates microglia and releases pro-inflammatory cytokines causing BBB dysfunction and the progression of MS [40]. A cohort study illustrated that CSF fibrinogen was low in chronic MS patients as compared to other inflammatory disorders involving the CNS [51]. However, in acute MS with injury of BBB integrity, plasma fibrinogen level is increased [51]. Furthermore, increased fibrinogen level in the proteome of platelets was observed in patients with progressive MS [52]. This finding indicated that platelet hyper-reactivity is augmented in MS patients due to overexpression of fibrinogen. Notably, platelets play a critical role in the progression of inflammation in MS [53]. Platelet hyper-reactivity is implicated in the progression of autoimmunity and inflammation in the early phase of MS neuropathology [53]. Therefore, the pro-thrombotic state in MS increases the risk of stroke and cardiovascular complications [54]. Besides, fibrinogen interacts with other coagulation factors in the pathogenesis of MS [54]. A case-control study observed that MS patients were associated with platelet hyperactivity compared to controls [55]. Platelet hyperactivity in MS could be due to endothelial injury and the release of endothelial microparticles which reflect activated T cells in the endothelium [56]. In MS, there is a profound platelet hyper-responsiveness to the different physiological activators leading to intravascular thrombosis [57]. Platelet hyper-responsiveness in MS is also due to hyperinflammation and oxidative stress. Pro-oxidant and pro-inflammatory state in MS also promotes platelet aggregation [57]. It has been shown that activated platelets are highly abundant in MS lesions [53]. Markedly, platelets play a role in the activation of the coagulation cascade through the induction of local generation of thrombin and release of stored clotting factors [58]. In addition, platelets can synthesize fibrinogen, von-Willebrand factor, thrombosthenin, thrombospondin, and membrane glycoproteins [59]. Of note, fibrinogen binds platelets GPIIb–IIIa receptors leading to platelet aggregation and thrombosis [60]. Therefore, activated platelets in MS are highly susceptible to the effect of fibrinogen leading to platelet aggregation.
These findings proposed that fibrinogen is associated with MS neuropathology through interruption of BBB integrity, induction of neuroinflammation, and demyelination with inhibition of the remyelination process by suppressing oligodendrocytes (Fig. 3).

Role of fibrinogen in MS: disruption of the blood–brain barrier (BBB) enhances leakage of fibrinogen from blood vessels. Deposited fibrinogen in the multiple sclerotic lesions leads to the activation of microglia and the release of pro-inflammatory cytokines. As well, fibrinogen triggers demyelination and inhibits remyelination
Oxidative Stress and Fibrinogen
Oxidative stress plays an essential role in the pathogenesis of MS through the induction of the demyelination process [61]. Reactive oxygen species (ROS) trigger peripheral activation of T cells and the development of autoreactive T cells. ROS triggers the activation of microglia and induction of neuronal apoptosis [61]. Inflammatory reactions in MS can aggravate oxidative stress burst in the activated macrophages and microglia leading to neuronal demyelination. In sequence, oxidative stress augments the propagation of inflammation in MS [62]. Consequently, there is positive feedback activation between oxidative stress and inflammation in MS. A case–control study showed that biomarkers of oxidative stress were increased in patients with relapsing–remitting MS compared to healthy controls [63]. These findings proposed that oxidative stress can aggravate inflammatory reactions and contribute to more neuronal injury and progression of MS (Fig. 4).

Oxidative stress/mitochondrial dysfunction/fibrinogen in multiple sclerosis (MS): fibrinogen binds the CD11b receptor in microglia leading to the activation of NADPH oxidase and the release of Reactive oxygen species (ROS) leading to mitochondrial dysfunction and injury of oligodendrocytes. Additionally, oxidative stress induces structural and functional changes in fibrinogen leading to thrombosis. ROS trigger peripheral activation of T cells and the development of autoreactive T cells. ROS triggers the activation of microglia and induction of neuronal apoptosis
On the other hand, oxidative stress induces structural and functional changes in fibrinogen leading to more risk of thrombosis [64]. In addition, oxidative modification of fibrinogen increases its propensity for spontaneous activation and progression of thrombosis [65]. Chronic oxidative stress and inflammation predispose to the development of venous thromboembolism [66]. A cohort study that included patients with suspected venous thromboembolism illustrated that nitrated fibrinogen level was higher in patients who develop venous thromboembolism compared to patients that were not developed venous thromboembolism [66]. This finding suggests that nitrated fibrinogen could be a possible biomarker of venous thromboembolism. However, mitochondrial superoxide enhances oxidative modification and fibrinogen proteolysis leading to coagulopathy during chronic inflammatory conditions [67]. Interestingly, leaky BBB in AD enhances fibrinogen in the brain leading to the activation of inflammation and oxidative stress through the stimulation of microglia [68]. Further, fibrinogen binds the CD11b receptor in microglia leading to the activation of NADPH oxidase and the release of ROS [69]. Therefore, selective deletion of the brain CD11b receptor prevents fibrinogen-induced microglia activation and attenuates ROS-mediated neurotoxicity in mice [70]. These verdicts indicated a potential interaction between oxidative stress and fibrinogen in MS neuropathology. Thus, higher oxidative stress in MS provokes oxidative modification of fibrinogen which in turn triggers oxidative stress and inflammation in MS.
Mitochondrial Dysfunction and Fibrinogen
It has been reported that mitochondrial dysfunction is involved in neuronal loss in MS due to unrestrained activation of microglia and associated neuronal injury [71]. Of note, the impairment of mitochondrial permeability transition pore by Ca2+ dyshomeostasis and ROS is the central mechanism for the progress of mitochondrial dysfunction in MS [72]. In addition, the pathological opening of mitochondrial permeability transition pores in response to nitrogen species, Ca2+ and ROS, provoke an influx of many solutes into the mitochondrial matrix causing matrix expansion and mitochondrial rupture, and cell deaths [72]. Furthermore, mitochondrial dysfunction is regarded as a vital trigger of programmed axon death in MS [73]. Uric acid and serum lactate are considered potential biomarkers of mitochondrial dysfunction [74]. A case-control study included 32 MS patients and 20 healthy controls revealed that lactate serum level but not serum uric acid was augmented in MS patients compared to the controls [74]. It has been anticipated that mitochondrial dysfunction modifies lymphocyte homeostasis causing a defective apoptotic process of auto-reactive T cells to allocate them to perpetuate within the CNS and maintain an inflammatory cycle in MS patients [75]. Thus, activation of Th1 cells and their lymphokines like interferon-gamma (INF-γ) and IL-2 which induce the transformation of B-lymphocyte to plasma cells generate autoantibodies against myelin antigens [75]. As a result, mitochondrial dysfunction might be a primary cause for MS progression via alteration of lymphocyte activity, or a secondary result due to oxidative stress caused by MS (Fig. 4).
Notably, mitochondrial dysfunction and the release of mitochondrial DNA induce the release of ROS which causes the oxidation of lipoprotein and plasma proteins like fibrinogen leading to the development of atherosclerosis [76]. An experimental study illustrated that fibrinogen interrupts mitochondrial membrane potential and the development of mitochondrial dysfunction in mice with burn injury [77]. In a cross-sectional study involving patients with sarcopenia which developed due to mitochondrial dysfunction, plasma fibrinogen level and its products were increased compared to healthy controls [78]. This observation suggests that increasing fibrinogen levels is associated with the development and progression of mitochondrial dysfunction. In addition, higher fibrinogen level in preeclampsia is correlated with disease severity due to the induction of mitochondrial dysfunction and inflammation [53]. Of note, chronic inflammation is associated with the development of mitochondrial dysfunction [79]. In this state, fibrinogen acts as a link between immunoinflammatory response and the development of mitochondrial dysfunction. Herein, fibrinogen is linked with the direct development of mitochondrial dysfunction or indirectly through the induction of inflammation.
Inflammatory Signaling Pathways and Fibrinogen
Diverse types of inflammatory signaling pathways and receptors including nuclear factor kappa B (NF-κB), nod-like receptor pyrin 3 receptor (NLRP3) inflammasome, and toll-like receptor 4 (TLR4) are implicated in the pathogenesis of MS [80].
Toll-Like Receptor 4
Toll-like receptor 4 (TLR4) is an innate immune sensor aware of the immune system to the presence of external pathogens [81]. Activation of TLR4 triggers the release of pro-inflammatory cytokines and activation of adaptive immune response to eliminate invading pathogens [81]. TLR4 detects danger signals which are products of inflammation and tissue injury. TLR4 is highly expressed by immune cells in the CNS and is involved in MS neuropathology [82]. Importantly, TLR4 agonists participate in the amplification of harmful inflammatory responses. Fibrinogen activates the immune response by stimulating TLR4 which is involved in immune activation [79]. Fibrinogen triggers the release of chemokines like macrophage inflammatory protein 1 alpha (MIP-1α) from macrophages by activating TLR4 [83]. In addition, the fibrinogen-TLR4 complex promotes the release of pro-inflammatory cytokines leading to the propagation of inflammatory disorders [84]. Fibrinogen can act synergistically with MIP-1β in the propagation of inflammation in patients with acute coronary syndrome [85]. Notably, MIP-1α is augmented in MS lesions and correlated with disease severity [86]. Therefore, fibrinogen is regarded as a component of damage-associated molecular patterns involved in the early immune response [87]. The findings of these studies suggest that the fibrinogen-TLR4 complex plays a critical role in the pathogenesis of MS.
NF-κB
NF-κB is a DNA-binding protein necessary for the transcription of chemokines and pro-inflammatory cytokines. Predominantly, immune deregulation and increased expression of NF-κB are linked with the progression of neuronal injury, neuroinflammation, and the development of neurodegeneration [88]. NF-κB is overstated in MS leading to immune dysregulation and induction of the release of pro-inflammatory cytokines. Inhibition of the NF-κB signaling pathway by dimethyl fumarate may reduce MS severity [89]. It has been observed that native and memory B cells from MS patients have a higher level of phosphorylated NF-κB which was inhibited by mycophenolate [90]. In addition, glatiramer attenuates the activation of NF-κB by inhibiting CD40 which is over-activated in MS [90].
Fibrinogen plays an integral role in the regulation of immune response and release of pro-inflammatory cytokines through NF-κB activation [91]. In vitro study demonstrated that fibrinogen promotes the release of IL-8 and monocyte chemoattractant protein 1 (MCP-1) in a concentration-dependent manner by activating the NF-κB signaling pathway [91]. Inhibition of IκB kinase by herbal parthenolide prevents expression and the release of MCP-1 under the effect of fibrinogen. Guo et al. [91] suggest a role of NF-κB in mediating the inflammatory effect of fibrinogen. Rubel et al. [92] revealed that fibrinogen through interaction with CD11b/CD18 can activate the apoptotic pathway in human neutrophils. Furthermore, fibrinogen triggers the development of endothelial dysfunction by increasing the expression of adhesion molecules like VCAM-1 and intercellular adhesion molecule 1 (ICAM-1) which promote the expression of NF-κB [93]. These verdicts indicated that fibrinogen triggers NF-κB activation, and accordingly may increase inflammatory reactions in MS.
NLRP3 Inflammasome
One of the most important inflammatory signaling pathways is the NLRP3 inflammasome which is concerned in the activation of caspase-1, and maturation of IL-1β and IL-18 [94]. NLRP3 inflammasome is triggered by miscellaneous stimuli and inflammatory signaling pathways like NF-κB [94]. NLRP3 inflammasome is intricate in the pathogenesis of neuroinflammation, and the development of neurodegeneration [95]. NLRP3 inflammasome is also exaggerated and linked with the severity of MS [96]. NLRP3 inflammasome within activated microglia promotes the expression and the release of IL-1β and IL-18. Confirmation from preclinical and clinical findings illustrated that aberrant activation of NLRP3 inflammasome is associated with the pathogenesis of MS. Over-activation of NLRP3 inflammasome in MS is apparent by increasing IL-1β CSF levels in severely affected patients [97]. Targeting of NLRP3 inflammasome by specific inhibitors can reduce MS severity [97].
It has been shown that fibrinogen and fibrinogen-like protein 2 through activation of TLR4 promotes the expression of NLRP3 inflammasome and the release of pro-inflammatory cytokines [98]. Roseborough et al. [99] showed that fibrinogen propagates pro-inflammatory signaling by priming microglial NLRP3 inflammasome in a dose-dependent manner. In addition, extracellular vesicles released activated microglia by fibrinogen priming signaling to naïve cells. These extracellular vesicles from injured microglia are increased in the peripheral circulation and correlated with increasing IL-6 and IL-1β which are a biomarker of NLRP3 inflammasome activity [99]. Therefore, a higher level of fibrinogen level in MS induces more inflammatory changes and the development of neuroinflammation.
Neuroinflammation
It has been reported by different studies that neuroinflammation is connected with the progression of diverse neurodegenerative disorders [96, 100]. Lymphocytes in the CNS activate inflammatory disorders and the progress of neuroinflammation [94]. Neuroinflammation in the early stage of MS can cause synaptopathy independent of the demyelination process, and this may explain cognitive dysfunction in the early phase of MS patients. However, in the late phase of MS, overstatement of immune disturbance and progress neuroinflammation stimulate MS pathogenesis [101]. Notably, cholinergic activity is impaired in MS patients that control the activity and response of immune cells. A decrease in the acetylcholine level in the immune cells promotes the release of pro-inflammatory cytokines with the progress of neuroinflammation [102, 103]. Of interest, fibrinogen is regarded as a potent inducer of neuroinflammation in different neurodegenerative disorders including AD, MS, and traumatic brain injury [104]. Hyperfibrinogenemia and fibrin deposits are associated with memory deficit and cognitive dysfunction in AD. Fibrinogen activates astrocytes, microglia, and neurons leading to neuroinflammation and neuronal injury [104]. Fibrinogen binds CD11b/CD18 which is also called Mac-1 on monocytes, macrophages, and microglia leading to the release of ROS in experimental autoimmune encephalomyelitis [105]. In addition, fibrinogen triggers neuroinflammation through the activation of platelets [106]. Remarkably, fibrinogen enhances the recruitment of peripheral monocytes and interaction with myelin-specific antigens [107]. Therefore, genetic deletion of the fibrinogen gene reduces neuroinflammation and demyelination in transgenic mice with MS [108]. Likewise, genetic deletion or use of specific inhibitors against CD11b/CD18 attenuates the development of experimental autoimmune encephalomyelitis in mice [109]. According to these findings, inhibition of fibrinogen could be a novel therapeutic strategy against neuroinflammation in MS.
Brain-Derived Neurotrophic Factor
Brain-derived neurotrophic factor (BDNF) also called abrineurin is a protein encoded by the BDNF gene [110]. BDNF is a member of neurotrophin growth factors found in the brain and periphery. BDNF acts on tropomyosin receptor kinase B (TrkB) receptors which are catalytic receptors for different neurotrophins involved in the growth and differentiation of cells [111]. Neuronal TrkB is highly active in the hippocampus, basal forebrain, and cerebral cortex. In addition, BDNF also activates low-affinity nerve growth factor receptor (LNGFR) whose function is not fully elucidated [112].
BDNF levels may decrease in MS due to the advanced neurodegeneration process [113]. A case-control study on 22 MS patients compared to 19 healthy controls exposed that BDNF serum level was decreased in MS patients compared to the controls [113]. Though, a recent study detected that BDNF serum level was not considerably reduced in MS patients compared to healthy controls [114]. In addition, a systemic review comprising 30 studies (689 MS patients and 583 healthy controls) discovered that BDNF serum level was decreased in MS patients compared to healthy controls [115].
It has been shown that fibrinogen increases the expression of TrkB in astrocytes in a dose-dependent manner [104]. However, over-expression of astrocytes TrkB is linked with morphological changes of astrocytes [116] with subsequent over-production of nitric oxide (NO), the release of nitrotyrosine and ROS which promote neurodegeneration [117]. These findings proposed that overexpression of TrkB by fibrinogen induces neurodegeneration in MS (Fig. 5).

Tropomyosin receptor kinase B (TrkB) in multiple sclerosis (MS): Fibrinogen increases the expression of tropomyosin receptor kinase B (TrkB) receptors in astrocytes. Over-expression of astrocytes TrkB is linked with morphological changes of astrocytes with subsequent over-production of nitric oxide (NO), the release of nitrotyrosine and reactive oxygen species (ROS) which promote MS
Targeting of Fibrinogen in MS
It has been shown platelet activity is increased in MS patients due to the over-expression of platelet GPIIb/IIIa receptors which are activated by fibrinogen. Platelet over-activity in MS promotes the formation of platelet microparticles and platelet aggregate which increase cardiovascular complications [118]. Platelet abnormality is developed in MS that causes more inflammation in the neurovascular unit. Platelets are the main source of IL-1α which affect brain endothelium and enhance entry of immune cells into the CNS causing cerebrovascular inflammation [119]. In addition, platelets contribute to the progression of inflammation in the early stage of MS [120]. Therefore, GPIIb/IIIa receptor antagonists may reduce thrombotic events in MS patients.
Of note, fibrinogen binds CD11b/CD18 also called complement receptor type 3 (CR3) which is also called Mac-1 on monocytes, macrophages and microglia to induce the release of ROS in MS [105]. Thus, inhibition of CD11b/CD18 receptors by specific antagonists may attenuate fibrinogen-induced neuroinflammation and demyelination. However, most CD11b/CD18 receptors were used in experimental studies but are not confirmed clinically. Therefore, the repurposing of clinical approval drugs which affect the expression of CD11b/CD18 is promising.
Metformin
Metformin is an insulin-sensitizing drug that improves peripheral insulin sensitivity and reduces hepatic glucose uptake. It is used as a first line in the management of type 2 diabetes mellitus (T2DM) [121, 122]. In addition, metformin has a neuroprotective effect against the development and progression of AD [123]. As well, metformin reduces neuroinflammation and demyelination in MS [124]. Different preclinical and clinical studies (Table 1) indicated that metformin is effective in the management of MS. Metformin attenuates the induction of experimental autoimmune encephalomyelitis by restricting the infiltration of mononuclear cells into the CNS, down-regulating the expression of pro-inflammatory cytokines [gamma interferone (IFN-γ), tumor necrosis factor alpha (TNF-α), IL-6, IL-17, and inducible NO synthase (iNOS)], cell adhesion molecules, matrix metalloproteinase 9, and chemokine [125]. Metformin inhibited T cell-mediated immune responses including Ag-specific recall responses and production of Th1 or Th17 cytokines, while it induced the generation of IL-10 in spleen cells of treated experimental autoimmune encephalomyelitis animals [125]. Metformin reduced Th17 and increased Treg cell percentages along with the levels of associated cytokines. Molecules involved in cellular metabolism were altered in mice with experimental autoimmune encephalomyelitis. Suppressed activation of the mechanistic target of rapamycin (mTOR) and its downstream target, hypoxia-inducible factor 1 α (HIF-1α), likely mediated the protective effects of metformin [126]. Treatment with metformin has beneficial anti-inflammatory effects in patients with MS by a significant increase in the number and regulatory functions of CD4+ and CD25+, and regulatory T cells compared with controls [127].
A previous comparative study illustrated that metformin therapy reduced fibrinogen levels in obese patients with T2DM [128]. Baptista and colleagues observed that metformin therapy reduced fibrinogen levels and inflammatory biomarkers in olanzapine-induced metabolic dysfunction in patients with schizophrenia [129]. However, a systematic review involving 9 randomized, placebo-controlled trials of 2302 patients showed that metformin therapy did not reduce fibrinogen levels significantly [130]. Though, this systematic review did not involve clinical studies regarding fibrinogen levels and the effects of metformin in T2DM patients. In progressive MS, fibrinogen level is progressively increased in the cerebral cortex and CSF in patients with MS [131]. Fibrinogen not only induces demyelination but also inhibits remyelination and neurogenesis by inducing the expression of bone morphogenic protein (BMP) receptors which inhibit remyelination [132]. Therefore, inhibiting the expression of BMP may prevent the inhibitory effect of fibrinogen on the remyelination process. It has been shown that metformin inhibits the expression of BMP and its signaling in trauma-induced ossification [133]. In addition, metformin through an AMP-activated protein kinase (AMPK)-dependent pathway negatively regulates BMP signaling [134]. Therefore, metformin through inhibition of fibrinogen and related BMP signaling may attenuate the development and progression of MS.
Statins
Statins are a class of lipid-lowering agents used in the management of hypercholesterolemia in patients with cardiovascular complications [134]. Statins act by inhibiting hyroxymethylglutaryl-CoA (HMG-CoA) a rate-limiting enzyme in denovo cholesterol biosynthesis [135]. Of note, statins have neuroprotective effects against different neurodegenerative diseases [1]. In addition, different studies revealed that statins are effective against MS neuropathology (Table 2) [136, 137]. However, there is a conflict regarding the beneficial effects of statins in MS [138]. Statins are safe drugs, widely used in patients with cardiovascular complications, and have pleiotropic effects. Therefore, statins may affect fibrinogen which is implicated in MS neuropathology. It has been reported that atorvastatin reduced fibrinogen levels in patients with hyperlipidemia [139]. However, fluvastatin increases fibrinogen levels [140]. Therefore, there is a controversy regarding the effect of statins on fibrinogen level. Recently, a cohort study illustrated that rosuvastatin fibrinogen level had significant fibrinolytic effects [141].
Despite these controversies, statins reduce the expression of CD11b/CD18 thereby reducing monocyte activation [142]. In addition, statins can mitigate neuroinflammation in different neurological disorders by inhibiting the expression of CD11b/CD18 [143]. Therefore, statins seem to not affect fibrinogen levels but decrease its effect on the brain by reducing CD11b/CD18.
Taken together, both statins and metformin which modulate the fibrinogen pathway and inflammatory reactions in MS could be adjuvant treatments with immunomodulatory agents in the management of MS. In this bargain, preclinical and large-scale prospective studies are recommended in this regard.
Conclusions
MS is an autoimmune demyelinating neurodegenerative disease of the CNS due to injury of myelin sheath by immune cells and inhibition of the production of myelin. The clotting factor fibrinogen is intricate in the pathogenesis of MS by triggering microglia and the progress of neuroinflammation. Fibrinogen level is correlated with MS severity; therefore, inhibition of the fibrinogen cascade may reduce MS neuropathology.
Deposition of fibrinogen in the CNS precedes neuroinflammation in MS. Accumulation of fibrinogen in the CNS may occur due to disruption of BBB integrity in MS. Disturbance of BBB integrity in MS may precede the development of brain lesions that may promote the entrance of plasma proteins including fibrinogen. Fibrinogen acts as transduction and increases microglia activation via triggering expression of CD11b/CD18. Likewise, brain fibrinogen induces the progression of inflammation, oxidative stress, and neuronal injury. Interestingly, brain fibrinogen impairs the remyelination process by inhibiting the differentiation of oligodendrocyte precursor cells. Moreover, increased fibrinogen level in the proteome of platelets is correlated with the progression of MS. These findings proposed that fibrinogen is associated with MS neuropathology through interruption of BBB integrity, induction of neuroinflammation, and demyelination with inhibition of the remyelination process by suppressing oligodendrocytes. Therefore, targeting of fibrinogen and/or CD11b/CD18 receptors by metformin and statins may reduce MS neuropathology.
Taken together, inhibition expression of CD11b/CD18 receptors by metformin and statins decreases the pro-inflammatory effect of fibrinogen on microglia which is involved in the progression of MS.
Data Availability
All data supporting the findings of this study are available in the paper.
References
Alsubaie N, Al-Kuraishy HM, Al-Gareeb AI, Alharbi B, De Waard M, Sabatier J-M et al (2022) Statins use in Alzheimer disease: bane or boon from frantic search and narrative review. Brain Sci 12(10):1290
Dobson R, Giovannoni G (2019) Multiple sclerosis—a review. Eur J Neurol 26(1):27–40
Kumar DR, Aslinia F, Yale SH, Mazza JJ (2011) Jean-Martin Charcot: the father of neurology. Clin Med Res 9(1):46–49
Khare M, Singh A, Zamboni P (2014) Prospect of brain machine interface in motor disabilities: the future support for multiple sclerosis patient to improve quality of life. Ann Med Health Sci Res 4(3):305–312
Ben-Zacharia AB (2011) Therapeutics for multiple sclerosis symptoms. Mount Sinai J Med 78(2):176–191
Wijeratne T, Carroll W (2021) World Brain Day 2021: global campaign to stop multiple sclerosis. SAGE Publications, London, pp 1318–1319
Sandi D, Kokas Z, Biernacki T, Bencsik K, Klivényi P, Vécsei L (2022) Proteomics in multiple sclerosis: the perspective of the clinician. Int J Mol Sci 23(9):5162
Bethoux F, Miller DM, Kinkel RP (2001) Recovery following acute exacerbations of multiple sclerosis: from impairment to quality of life. Mult Scler J 7(2):137–142
Sumowski JF, Leavitt VM (2014) Body temperature is elevated and linked to fatigue in relapsing-remitting multiple sclerosis, even without heat exposure. Arch Phys Med Rehabil 95(7):1298–1302
Lipphardt M, Mühlhausen J, Kitze B, Heigl F, Mauch E, Helms HJ et al (2019) Immunoadsorption or plasma exchange in steroid-refractory multiple sclerosis and neuromyelitis optica. J Clin Apher 34(4):381–391
Neuhaus O, Kieseier BC, Hartung H-P (2007) Pharmacokinetics and pharmacodynamics of the interferon-betas, glatiramer acetate, and mitoxantrone in multiple sclerosis. J Neurol Sci 259(1–2):27–37
Feinstein A (2004) The neuropsychiatry of multiple sclerosis. Can J Psychiatry 49(3):157–163
Stys PK (2010) Multiple sclerosis: autoimmune disease or autoimmune reaction? Can J Neurol Sci 37(S2):S16–S23
Prineas JW, Parratt JD (2012) Oligodendrocytes and the early multiple sclerosis lesion. Ann Neurol 72(1):18–31
Sedel F, Bernard D, Mock DM, Tourbah A (2016) Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology 110:644–653
Patani R, Balaratnam M, Vora A, Reynolds R (2007) Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol Appl Neurobiol 33(3):277–287
Frohman EM, Racke MK, Raine CS (2006) Multiple sclerosis—the plaque and its pathogenesis. N Engl J Med 354(9):942–955
Brownell B, Hughes JT (1962) The distribution of plaques in the cerebrum in multiple sclerosis. J Neurol Neurosurg Psychiatry 25(4):315
Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M et al (2009) The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132(5):1175–1189
Haase S, Linker RA (2021) Inflammation in multiple sclerosis. Ther Adv Neurol Disord 14:17562864211007688
Puthenparampil M, Zito A, Pantano G, Federle L, Stropparo E, Miante S et al (2019) Peripheral imbalanced TFH/TFR ratio correlates with intrathecal IgG synthesis in multiple sclerosis at clinical onset. Mult Scler J 25(7):918–926
Peterson JW, Bö L, Mörk S, Chang A, Ransohoff RM, Trapp BD (2002) VCAM-1-positive microglia target oligodendrocytes at the border of multiple sclerosis lesions. J Neuropathol Exp Neurol 61(6):539–546
Rice GP, Hartung H-P, Calabresi PA (2005) Anti-α4 integrin therapy for multiple sclerosis: mechanisms and rationale. Neurology 64(8):1336–1342
Yong VW, Zabad RK, Agrawal S, DaSilva AG, Metz LM (2007) Elevation of matrix metalloproteinases (MMPs) in multiple sclerosis and impact of immunomodulators. J Neurol Sci 259(1–2):79–84
Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T et al (2005) Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med 11(3):328–334
Parker Harp CR, Archambault AS, Sim J, Ferris ST, Mikesell RJ, Koni PA et al (2015) B cell antigen presentation is sufficient to drive neuroinflammation in an animal model of multiple sclerosis. J Immunol 194(11):5077–5084
Morris G, Reiche EMV, Murru A, Carvalho AF, Maes M, Berk M et al (2018) Multiple immune-inflammatory and oxidative and nitrosative stress pathways explain the frequent presence of depression in multiple sclerosis. Mol Neurobiol 55:6282–6306
Al-Kuraishy HM, Al-Gareeb AI, Saad HM, Batiha GE-S (2023) The potential therapeutic effect of statins in multiple sclerosis: beneficial or detrimental effects. Inflammopharmacology. https://doi.org/10.1007/s10787-023-01240-x
Adams R, Schachtrup C, Davalos D, Tsigelny I, Akassoglou K (2007) Fibrinogen signal transduction as a mediator and therapeutic target in inflammation: lessons from multiple sclerosis. Curr Med Chem 14(27):2925–2936
Lowe GD, Rumley A, Mackie IJ (2004) Plasma fibrinogen. Ann Clin Biochem 41(6):430–440
Neerman-Arbez M (2006) Molecular basis of fibrinogen deficiency. Pathophysiol Haemost Thromb 35(1–2):187–198
Doolittle RF (2017) The conversion of fibrinogen to fibrin: a brief history of some key events. Matrix Biol 60:5–7
Besser MW, MacDonald SG (2016) Acquired hypofibrinogenemia: current perspectives. J Blood Med 7:217–225
Fuller GM (2020) Fibrinogen: a multifunctional acute phase protein. Acute phase proteins. CRC Press, Boca Raton, pp 169–183
Yamashita H, Kitayama J, Taguri M, Nagawa H (2009) Effect of preoperative hyperfibrinogenemia on recurrence of colorectal cancer without a systemic inflammatory response. World J Surg 33:1298–1305
Jastrzebska M, Lisman D, Szelepajlo A, Oledzki S, Chelstowski K, Clark J et al (2019) Evaluation of platelet reactivity during combined antiplatelet therapy in patients with stable coronary artery disease in relation to diabetes type 2 and the GPIIB/IIIA receptor gene polymorphism. J Physiol Pharmacol 70(2):175–185
Arai S, Kamijo T, Takezawa Y, Sugano M, Nakazawa H, Yanagisawa R et al (2020) Acquired dysfibrinogenemia: monoclonal λ-type IgA binding to fibrinogen caused lower functional plasma fibrinogen level and abnormal clot formation. Int J Hematol 112:96–104
Dumitrescu G (2020) Coagulation in liver failure: the role of thromboelastometry and fibrinogen. Karolinska Institutet, Solna
Perween S, Abidi M, Faizy AF (2022) Biophysical changes in methylglyoxal modified fibrinogen and its role in the immunopathology of type 2 diabetes mellitus. Int J Biol Macromol 202:199–214
Acuña JM, de la Cruz MH, Ros AL, Tapia SP, Ginés MLM, de Andrés Frutos CD (2017) Elevated plasma fibrinogen levels in multiple sclerosis patients during relapse. Mult Scler Relat Disord 18:157–160
Ahmad U, Frederiksen JL (2020) Fibrinogen: a potential biomarker for predicting disease severity in multiple sclerosis. Mult Scler Relat Disord 46:102509
Willis CM, Nicaise AM, Menoret A, Ryu JK, Mendiola AS, Jellison ER et al (2019) Extracellular vesicle fibrinogen induces encephalitogenic CD8+ T cells in a mouse model of multiple sclerosis. Proc Natl Acad Sci 116(21):10488–10493
Ryu JK, Davalos D, Akassoglou K (2009) Fibrinogen signal transduction in the nervous system. J Thromb Haemost 7:151–154
Yates RL, Esiri MM, Palace J, Jacobs B, Perera R, DeLuca GC (2017) Fibrin (ogen) and neurodegeneration in the progressive multiple sclerosis cortex. Ann Neurol 82(2):259–270
Petersen MA, Ryu JK, Akassoglou K (2018) Fibrinogen in neurological diseases: mechanisms, imaging and therapeutics. Nat Rev Neurosci 19(5):283–301
Davalos D, Kyu Ryu J, Merlini M, Baeten KM, Le Moan N, Petersen MA et al (2012) Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 3(1):1227
Ghorbani S, Yong VW (2021) The extracellular matrix as modifier of neuroinflammation and remyelination in multiple sclerosis. Brain 144(7):1958–1973
Sulimai NH, Brown J, Lominadze D (2022) Fibrinogen, fibrinogen-like 1 and fibrinogen-like 2 proteins, and their effects. Biomedicines 10(7):1712
Abbadessa G, Miele G, Di Pietro A, Sparaco M, Palladino R, Armetta I et al (2022) Multiple sclerosis and genetic polymorphisms in fibrinogen-mediated hemostatic pathways: a case–control study. Neurol Sci 43:1–9
Davalos D, Mahajan KR, Trapp BD (2019) Brain fibrinogen deposition plays a key role in MS pathophysiology–yes. Mult Scler J 25(11):1434–1435
Ehling R, Pauli FD, Lackner P, Kuenz B, Santner W, Lutterotti A et al (2011) Fibrinogen is not elevated in the cerebrospinal fluid of patients with multiple sclerosis. Fluids Barriers CNS 8(1):1–5
Bijak M, Olejnik A, Rokita B, Morel A, Dziedzic A, Miller E et al (2019) Increased level of fibrinogen chains in the proteome of blood platelets in secondary progressive multiple sclerosis patients. J Cell Mol Med 23(5):3476–3482
Langer HF, Chavakis T (2013) Platelets and neurovascular inflammation. Thromb Haemost 110(11):888–893
Christensen S, Farkas DK, Pedersen L, Miret M, Christiansen CF, Sørensen HT (2012) Multiple sclerosis and risk of venous thromboembolism: a population-based cohort study. Neuroepidemiology 38(2):76–83
Sheremata WA, Jy W, Horstman LL, Ahn YS, Alexander JS, Minagar A (2008) Evidence of platelet activation in multiple sclerosis. J Neuroinflammation 5:1–6
Minagar A, Jy W, Jimenez J, Sheremata W, Mauro L, Mao W et al (2001) Elevated plasma endothelial microparticles in multiple sclerosis. Neurology 56(10):1319–1324
Saluk-Bijak J, Dziedzic A, Bijak M (2019) Pro-thrombotic activity of blood platelets in multiple sclerosis. Cells 8(2):110
Strukova S (2004) Role of platelets and serine proteinases in coupling of blood coagulation and inflammation. Biochem Mosc 69:1067–1081
Bijak M, Saluk J, Ponczek MBP, Nowak P, Wachowicz B (2013) The synthesis of proteins in unnucleated blood platelets. Adv Hygiene Exp Med 67:672–679
Bennett JS (2001) Platelet-fibrinogen interactions. Ann N Y Acad Sci 936(1):340–354
Ohl K, Tenbrock K, Kipp M (2016) Oxidative stress in multiple sclerosis: central and peripheral mode of action. Exp Neurol 277:58–67
Ortiz GG, Pacheco-Moisés FP, Bitzer-Quintero OK, Ramírez-Anguiano AC, Flores-Alvarado LJ, Ramírez-Ramírez V et al (2013) Immunology and oxidative stress in multiple sclerosis: clinical and basic approach. Clin Dev Immunol 2013:1–14
Padureanu R, Albu CV, Mititelu RR, Bacanoiu MV, Docea AO, Calina D et al (2019) Oxidative stress and inflammation interdependence in multiple sclerosis. J Clin Med 8:1815
Wang L, Li L, Wang H, Liu J (2016) Study on the influence of oxidative stress on the fibrillization of fibrinogen. Biochem J 473(23):4373–4384
Štikarová J, Kotlín R, Riedel T, Suttnar J, Pimková K, Chrastinová L et al (2013) The effect of reagents mimicking oxidative stress on fibrinogen function. Sci World J 2013:1–8
Martinez M, Cuker A, Mills A, Lightfoot R, Fan Y, Tang WW et al (2012) Nitrated fibrinogen is a biomarker of oxidative stress in venous thromboembolism. Free Radic Biol Med 53(2):230–236
Han CY, Pichon TJ, Wang X, Ringgold KM, St John AE, Stern SA et al (2022) Leukocyte activation primes fibrinogen for proteolysis by mitochondrial oxidative stress. Redox Biol 51:102263
McLarnon JG (2021) A leaky blood–brain barrier to fibrinogen contributes to oxidative damage in Alzheimer’s disease. Antioxidants 11(1):102
Husemann J, Obstfeld A, Febbraio M, Kodama T, Silverstein SC (2001) CD11b/CD18 mediates production of reactive oxygen species by mouse and human macrophages adherent to matrixes containing oxidized LDL. Arterioscler Thromb Vasc Biol 21(8):1301–1305
Ryu JK, Rafalski VA, Meyer-Franke A, Adams RA, Poda SB, Rios Coronado PE et al (2018) Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat Immunol 19(11):1212–1223
Witte ME, Mahad DJ, Lassmann H, van Horssen J (2014) Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol Med 20(3):179–187
Su K, Bourdette D, Forte M (2013) Mitochondrial dysfunction and neurodegeneration in multiple sclerosis. Front Physiol 4:169
Merlini E, Coleman MP, Loreto A (2022) Mitochondrial dysfunction as a trigger of programmed axon death. Trends Neurosci 45(1):53–63
Hewedi K, Abd El Aziz AF, Essmat A, Faheem M (2020) Laboratory evaluation for mitochondrial dysfunction in multiple sclerosis patients. Al-Azhar Int Med J 1(11):190–192
Signorile A, Ferretta A, Ruggieri M, Paolicelli D, Lattanzio P, Trojano M et al (2020) Mitochondria, oxidative stress, cAMP signalling and apoptosis: a crossroads in lymphocytes of multiple sclerosis, a possible role of nutraceutics. Antioxidants 10(1):21
Shemiakova T, Ivanova E, Grechko AV, Gerasimova EV, Sobenin IA, Orekhov AN (2020) Mitochondrial dysfunction and DNA damage in the context of pathogenesis of atherosclerosis. Biomedicines 8(6):166
Ueki R, Liu L, Kashiwagi S, Kaneki M, Khan M, Hirose M et al (2016) Role of elevated fibrinogen in burn-induced mitochondrial dysfunction: protective effects of glycyrrhizin. Shock (Augusta, GA) 46(4):382
Chen J-L, Chen D-M, Luo C, Sun Y, Zhao Y-X, Huang C-Q et al (2021) Fibrinogen, fibrin degradation products and risk of sarcopenia. Clin Nutr 40(8):4830–4837
Cruz CSD, Kang M-J (2018) Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 41:37–44
Zhou Y, Fang L, Peng L, Qiu W (2017) TLR9 and its signaling pathway in multiple sclerosis. J Neurol Sci 373:95–99
Gran B, Nyirenda MH, Crooks J (2013) The role of Toll-like receptors in multiple sclerosis and experimental autoimmune encephalomyelitis. In: Yamamura T, Gran B (eds) Multiple sclerosis immunology: a foundation for current and future treatments. Springer, New York, pp 149–176
Marta M, Meier UC, Lobell A (2009) Regulation of autoimmune encephalomyelitis by toll-like receptors. Autoimmun Rev 8(6):506–509
Smiley ST, King JA, Hancock WW (2001) Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol 167(5):2887–2894
Hodgkinson CP, Patel K, Ye S (2008) Functional Toll-like receptor 4 mutations modulate the response to fibrinogen. Thromb Haemost 100(2):301–307
Xu F, Lv S, Chen Y, Song X, Jin Z, Yuan F et al (2012) Macrophage inflammatory protein-1β and fibrinogen are synergistic predictive markers of prognosis of intermediate coronary artery lesions. Cardiology 121(1):12–19
Bayat P, Farshchi M, Yousefian M, Mahmoudi M, Yazdian-Robati R (2021) Flavonoids, the compounds with anti-inflammatory and immunomodulatory properties, as promising tools in multiple sclerosis (MS) therapy: a systematic review of preclinical evidence. Int Immunopharmacol 95:107562
Rosin DL, Okusa MD (2011) Dangers within: DAMP responses to damage and cell death in kidney disease. J Am Soc Nephrol 22(3):416–425
Al-Kuraishy HM, Al-Gareeb AI, Fageyinbo MS, Batiha GE-S (2022) Vinpocetine is the forthcoming adjuvant agent in the management of COVID-19. Future Sci 8(5):FSO797
Leibowitz SM, Yan J (2016) NF-κB pathways in the pathogenesis of multiple sclerosis and the therapeutic implications. Front Mol Neurosci 9:84
Chen D, Ireland SJ, Remington G, Alvarez E, Racke MK, Greenberg B et al (2016) CD40-mediated NF-κB activation in B cells is increased in multiple sclerosis and modulated by therapeutics. J Immunol 197(11):4257–4265
Guo M, Sahni SK, Sahni A, Francis CW (2004) Fibrinogen regulates the expression of inflammatory chemokines through NF-κB activation of endothelial cells. Thromb Haemost 92(10):858–866
Rubel C, Gómez S, Fernández GC, Isturiz MA, Caamaño J, Palermo MS (2003) Fibrinogen-CD11b/CD18 interaction activates the NF-κB pathway and delays apoptosis in human neutrophils. Eur J Immunol 33(5):1429–1438
Zadeh FJ, Mohammadtaghizadeh M, Bahadori H, Saki N, Rezaeeyan H (2020) The role of exogenous fibrinogen in cardiac surgery: stop bleeding or induce cardiovascular disease. Mol Biol Rep 47(10):8189–8198
Batiha GE, Al-Gareeb AI, Rotimi D, Adeyemi OS, Al-Kuraishy HM (2022) Common NLRP3 inflammasome inhibitors and Covid-19: divide and conquer. Sci Afr 18:e01407
Holbrook JA, Jarosz-Griffiths HH, Caseley E, Lara-Reyna S, Poulter JA, Williams-Gray CH et al (2021) Neurodegenerative disease and the NLRP3 inflammasome. Front Pharmacol 12:643254
Garcia-Ramírez M, Hernández C, Palomer X, Vázquez-Carrera M, Simó R (2016) Fenofibrate prevents the disruption of the outer blood retinal barrier through downregulation of NF-κB activity. Acta Diabetol 53:109–118
Shao S, Chen C, Shi G, Zhou Y, Wei Y, Fan N et al (2021) Therapeutic potential of the target on NLRP3 inflammasome in multiple sclerosis. Pharmacol Ther 227:107880
Hu J, Wang H, Li X, Liu Y, Mi Y, Kong H et al (2020) Fibrinogen-like protein 2 aggravates nonalcoholic steatohepatitis via interaction with TLR4, eliciting inflammation in macrophages and inducing hepatic lipid metabolism disorder. Theranostics 10(21):9702
Roseborough A, Zhu Y, Zhao L, Laviolette S, Pasternak S, Whitehead S (2023) Fibrinogen primes the microglial NLRP3 inflammasome and propagates pro-inflammatory signaling via extracellular vesicles: Implications for blood–brain barrier dysfunction. Neurobiol Dis 177:106001
Liu Q, Zhang F, Zhang X, Cheng R, Ma J-x, Yi J et al (2018) Fenofibrate ameliorates diabetic retinopathy by modulating Nrf2 signaling and NLRP3 inflammasome activation. Mol Cell Biochem 445:105–115
Naegele M, Martin R (2014) The good and the bad of neuroinflammation in multiple sclerosis. Handb Clin Neurol 122:59–87
Musella A, Gentile A, Rizzo FR, De Vito F, Fresegna D, Bullitta S et al (2018) Interplay between age and neuroinflammation in multiple sclerosis: effects on motor and cognitive functions. Front Aging Neurosci 10:238
Gatta V, Mengod G, Reale M, Tata AM (2020) Possible correlation between cholinergic system alterations and neuro/inflammation in multiple sclerosis. Biomedicines 8(6):153
Sulimai N, Lominadze D (2020) Fibrinogen and neuroinflammation during traumatic brain injury. Mol Neurobiol 57(11):4692–4703
Solovjov DA, Pluskota E, Plow EF (2005) Distinct roles for the α and β subunits in the functions of integrin αMβ2. J Biol Chem 280(2):1336–1345
Langer HF, Choi EY, Zhou H, Schleicher R, Chung K-J, Tang Z et al (2012) Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ Res 110(9):1202–1210
Ryu JK, Petersen MA, Murray SG, Baeten KM, Meyer-Franke A, Chan JP et al (2015) Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat Commun 6(1):8164
Akassoglou K, Adams RA, Bauer J, Mercado P, Tseveleki V, Lassmann H et al (2004) Fibrin depletion decreases inflammation and delays the onset of demyelination in a tumor necrosis factor transgenic mouse model for multiple sclerosis. Proc Natl Acad Sci 101(17):6698–6703
Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H et al (2007) The fibrin-derived γ377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med 204(3):571–582
Castrén E, Monteggia LM (2021) Brain-derived neurotrophic factor signaling in depression and antidepressant action. Biol Psychiatry 90(2):128–136
Li Y, Wei C, Wang W, Li Q, Wang ZC (2023) Tropomyosin receptor kinase B (TrkB) signalling: targeted therapy in neurogenic tumours. J Pathol 9(2):89–99
Kendall A, Ekman S, Skiöldebrand E (2023) Nerve growth factor receptors in equine synovial membranes vary with osteoarthritic disease severity. J Orthop Res 41(2):316–324
Wens I, Keytsman C, Deckx N, Cools N, Dalgas U, Eijnde B (2016) Brain derived neurotrophic factor in multiple sclerosis: effect of 24 weeks endurance and resistance training. Eur J Neurol 23(6):1028–1035
Naegelin Y, Saeuberli K, Schaedelin S, Dingsdale H, Magon S, Baranzini S et al (2020) Levels of brain-derived neurotrophic factor in patients with multiple sclerosis. Ann Clin Transl Neurol 7(11):2251–2261
Karimi N, Ashourizadeh H, Pasha BA, Haghshomar M, Jouzdani T, Shobeiri P et al (2022) Blood levels of brain-derived neurotrophic factor (BDNF) in people with multiple sclerosis (MS): a systematic review and meta-analysis. Mult Scler Relat Disorders 65:103984
Fahimi A, Baktir MA, Moghadam S, Mojabi FS, Sumanth K, McNerney MW et al (2017) Physical exercise induces structural alterations in the hippocampal astrocytes: exploring the role of BDNF-TrkB signaling. Brain Struct Funct 222:1797–1808
Colombo E, Cordiglieri C, Melli G, Newcombe J, Krumbholz M, Parada LF et al (2012) Stimulation of the neurotrophin receptor TrkB on astrocytes drives nitric oxide production and neurodegeneration. J Exp Med 209(3):521–535
Morel A, Rywaniak J, Bijak M, Miller E, Niwald M, Saluk J (2017) Flow cytometric analysis reveals the high levels of platelet activation parameters in circulation of multiple sclerosis patients. Mol Cell Biochem 430:69–80
Thornton P, McColl BW, Greenhalgh A, Denes A, Allan SM, Rothwell NJ (2010) Platelet interleukin-1α drives cerebrovascular inflammation. Blood 115(17):3632–3639
Han MH, Hwang S-I, Roy DB, Lundgren DH, Price JV, Ousman SS et al (2008) Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451(7182):1076–1081
Al-Kuraishy HM, Al-Gareeb AI, Waheed HJ, Al-Maiahy TJ (2018) Differential effect of metformin and/or glyburide on apelin serum levels in patients with type 2 diabetes mellitus: concepts and clinical practice. J Adv Pharm Technol Res 9(3):80
Al-Kuraishy HM, Sami OM, Hussain NR, Al-Gareeb AI (2020) Metformin and/or vildagliptin mitigate type II diabetes mellitus induced-oxidative stress: the intriguing effect. J Adv Pharm Technol Res 11(3):142
Al-Kuraishy HM, Al-Gareeb AI, Saad HM, Batiha GE-S (2023) Long-term use of metformin and Alzheimer’s disease: beneficial or detrimental effects. Inflammopharmacology 31:1–9
Dziedzic A, Saluk-Bijak J, Miller E, Bijak M (2020) Metformin as a potential agent in the treatment of multiple sclerosis. Int J Mol Sci 21(17):5957
Nath N, Khan M, Paintlia MK, Hoda MN, Giri S (2009) Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J Immunol 182(12):8005–8014
Sun Y, Tian T, Gao J, Liu X, Hou H, Cao R et al (2016) Metformin ameliorates the development of experimental autoimmune encephalomyelitis by regulating T helper 17 and regulatory T cells in mice. J Neuroimmunol 292:58–67
Negrotto L, Farez MF, Correale J (2016) Immunologic effects of metformin and pioglitazone treatment on metabolic syndrome and multiple sclerosis. JAMA Neurol 73(5):520–528
Fanghänel G, Silva U, Sanchez-Reyes L, Sisson D, Sotres D, Torres EM (1998) Effects of metformin on fibrinogen levels in obese patients with type 2 diabetes. Revista de Investigacion Clinica; Organo del Hospital de Enfermedades de la Nutricion 50(5):389–394
Baptista T, Sandia I, Lacruz A, Rangel N, de Mendoza S, Beaulieu S et al (2007) Insulin counter-regulatory factors, fibrinogen and C-reactive protein during olanzapine administration: effects of the antidiabetic metformin. Int Clin Psychopharmacol 22(2):69–76
Simental-Mendia LE, Pirro M, Atkin SL, Banach M, Mikhailidis DP, Sahebkar A (2018) Effect of metformin on plasma fibrinogen concentrations: a systematic review and meta-analysis of randomized placebo-controlled trials. Curr Pharm Des 24(9):1034–1040
Magliozzi R, Hametner S, Facchiano F, Marastoni D, Rossi S, Castellaro M et al (2019) Iron homeostasis, complement, and coagulation cascade as CSF signature of cortical lesions in early multiple sclerosis. Ann Clin Transl Neurol 6(11):2150–2163
Pous L, Deshpande SS, Nath S, Mezey S, Malik SC, Schildge S et al (2020) Fibrinogen induces neural stem cell differentiation into astrocytes in the subventricular zone via BMP signaling. Nat Commun 11(1):630
Lin H, Shi F, Jiang S, Wang Y, Zou J, Ying Y et al (2020) Metformin attenuates trauma-induced heterotopic ossification via inhibition of bone morphogenetic protein signalling. J Cell Mol Med 24(24):14491–14501
Al-Kuraishy HM, Al-Gareeb AI, Hussien NR, Al-Naimi MS, Rasheed HA (2019) Statins an oft-prescribed drug is implicated in peripheral neuropathy: the time to know more. JPMA 69(8):S108–S112
Al-Kuraishy HM, Al-Gareeb AI (2017) Acylation-stimulating protein is a surrogate biomarker for acute myocardial infarction: role of statins. J Lab Phys 9(03):163–169
Stüve O, Prod’homme T, Slavin A, Youssef S, Dunn S, Steinman L et al (2003) Statins and their potential targets in multiple sclerosis therapy. Expert Opin Ther Targets 7(5):613–622
Wang J, Xiao Y, Luo M, Luo H (2011) Statins for multiple sclerosis. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD008386.pub3
Feng X, Han D, Kilaru BK, Franek BS, Niewold TB, Reder AT (2012) Inhibition of interferon-beta responses in multiple sclerosis immune cells associated with high-dose statins. Arch Neurol 69(10):1303–1309
Eyal Leibovitz M, Neli Hazanov M, Angela Frieman M, Itzhak Elly M, Dov Gavish M (2004) Atorvastatin reduces fibrinogen levels in patients with severe hypercholesterolemia: additional evidence to support the anti-inflammatory effects of statins. Isr Med Assoc J 6(8):456–459
Haverkate F, Koopman J, de Maat M (1998) Statins and fibrinogen. Lancet 351(9113):1430–1431
Schol-Gelok S, de Maat MP, Biedermann JS, van Gelder T, Leebeek FW, Lijfering WM et al (2020) Rosuvastatin use increases plasma fibrinolytic potential: a randomised clinical trial. Br J Haematol 190(6):916–922
Stavroulopoulos A, Petras D, Kakavas I, Agroyannis I, Stamatelou K, Vyssoulis G et al (2010) Monocyte expression of adhesion molecules during low-and high-flux polysulfone hemodialysis and the effect of atorvastatin administration. Blood Purif 29(3):274–279
Cucchiara B, Kasner SE (2001) Use of statins in CNS disorders. J Neurol Sci 187(1–2):81–89
Charles MA, Morange P, Eschwège E, André P, Vague P, Juhan-Vague I (1998) Effect of weight change and metformin on fibrinolysis and the von Willebrand factor in obese nondiabetic subjects: the BIGPRO1 Study. Biguanides and the prevention of the risk of obesity. Diabetes Care 21(11):1967–1972
Abdi M, Pasbakhsh P, Shabani M, Nekoonam S, Sadeghi A, Fathi F et al (2021) Metformin therapy attenuates pro-inflammatory microglia by inhibiting NF-κB in cuprizone demyelinating mouse model of multiple sclerosis. Neurotox Res 39:1732–1746
Xu D, Wang M (2022) Research progress of statins on immune regulation of multiple sclerosis and experimental allergic encephalomyelitis. Allergol Immunopathol (Madr) 50(6):76–83
Ghasami K, Faraji F, Fazeli M, Ghazavi A, Mosayebi G (2016) Interferon β-1a and atorvastatin in the treatment of multiple sclerosis. Iran J Immunol 13(1):16–26
Chataway J, Schuerer N, Alsanousi A, Chan D, MacManus D, Hunter K et al (2014) Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet 383(9936):2213–2221
Stefanou M-I, Palaiodimou L, Katsanos AH, Milionis H, Kosmidou M, Lambadiari V et al (2022) The effects of HMG-CoA reductase inhibitors on disease activity in multiple sclerosis: a systematic review and meta-analysis. Mult Scler Relat Disorders 58:103395
Acknowledgements
No.
Funding
Open Access funding enabled and organized by Projekt DEAL. Open Access funding enabled and organized by Projekt DEAL. This work was supported by the University of Witten-Herdecke Germany.
Author information
Authors and Affiliations
Contributions
MA, HMA-K and BMA conceptualized the manuscript, wrote, edited and reviewed the main text and approved the final edition of the manuscript. AA, MP, OE, HMS and GE-SB prepared the figures, wrote, corrected, amended and approved the final edition of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Institutional Review Board Statement
Nil.
Informed Consent Statement
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alruwaili, M., Al-kuraishy, H.M., Alexiou, A. et al. Pathogenic Role of Fibrinogen in the Neuropathology of Multiple Sclerosis: A Tale of Sorrows and Fears. Neurochem Res 48, 3255–3269 (2023). https://doi.org/10.1007/s11064-023-03981-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-023-03981-1