
Abstract
The F11 Receptor (F11R), also called Junctional Adhesion Molecule-A (JAM-A) (F11R/JAM-A), is a transmembrane glycoprotein of the immunoglobulin superfamily, which is mainly located in epithelial and endothelial cell tight junctions and also expressed on circulating platelets and leukocytes. It participates in the regulation of various biological processes, as diverse as paracellular permeability, tight junction formation and maintenance, leukocyte transendothelial migration, epithelial-to-mesenchymal transition, angiogenesis, reovirus binding, and platelet activation. Dysregulation of F11R/JAM-A may result in pathological consequences and disorders in normal cell function. A growing body of evidence points to its role in carcinogenesis and invasiveness, but its tissue-specific pro- or anti-tumorigenic role remains a debated issue. The following review focuses on the F11R/JAM-A tissue-dependent manner in tumorigenesis and metastasis and also discusses the correlation between poor patient clinical outcomes and its aberrant expression. In the future, it will be required to clarify the signaling pathways that are activated or suppressed via the F11R/JAM-A protein in various cancer types to understand its multiple roles in cancer progression and further use it as a novel direct target for cancer treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Intercellular interactions are fundamental for the development and function maintenance of multicellular organisms through the regulation of physiological events, such as tissue barrier formation, tissue integrity regulation, or inflammatory cell recruitment regulation [1,2,3]. Cells accommodate to changes occurring in their microenvironment due to the information transmitted by adhesion receptors on neighboring cells, which regulate receptor-mediated cellular interactions through participation in the signal transduction pathways [1, 4, 5]. These receptors use their cytoplasmic domains that often contain specific motifs (including proline-rich motifs, phosphorylation sites, FERM and PDZ domain-binding motifs) to interact with appropriate specific interaction motifs in cytoplasmic proteins (SH3, Src-homology-3; SH2, Src-homology-2; FERM or PDZ domains, respectively) [6,7,8,9].
Cell–cell adhesion and contact between adjacent cells take place through multiprotein complexes located in intercellular junctions. In vertebrates, these cellular structures mostly comprise adherens junctions (AJs), gap junctions (GJs), desmosomes, and tight junctions (TJs) [10,11,12,13,14,15]. The latter ones are dynamic structures that are distributed at the most apical end of the lateral endothelial and epithelial cell membrane and establish two barrier types, such as a paracellular diffusion barrier (the ‘gate’ function) and an intramembrane diffusion barrier (the ‘fence’ function). TJs as paracellular gates are responsible for the selective permeability of water, solutes, ions, and small molecules that perform a critical role in the tissue and organ homeostasis maintenance. The second type of barrier limits the membrane component replacement among the apical and basolateral cell surface domains that have an impact on apicobasal cell polarity regulation [3, 10, 12, 14, 16]. Furthermore, TJs also play an essential role as a bidirectional signaling platform [12, 16] that regulates various cellular processes, such as proliferation, differentiation and polarization of cells [17,18,19,20,21,22], cellular stress response [23], gene expression [24], and the cytoskeleton [14, 25, 26]. Structurally, TJs mainly consist of adaptor proteins, transcriptional and post-transcriptional regulators, transmembrane and signaling proteins [10, 12, 16, 17, 27,28,29,30,31,32] (Fig. 1 summarizes the key tight junction-associated proteins). In numerous human diseases, particularly in inflammatory disorders, tumor progression, and cancer metastasis, TJ disturbances are observed [13, 30, 33,34,35,36,37,38,39,40,41,42,43,44,45,46,47].

Key components of tight junctions (TJs). Tight junctions are composed of four main protein classes: transmembrane, adaptor, signaling, and transcriptional and post-transcriptional regulators. Diagram illustrates key proteins associated with TJs; it is not a full list of proteins presented in this cell structure. AMOT angiomotin, AP1 activator protein 1, aPKC atypical protein kinase C, BVES blood vessel epicardial substance, CAR coxsackievirus and adenovirus receptor, CDK4 cyclin-dependent kinase 4, CLMP CAR-like membrane protein, Crb3 protein crumbs homolog 3, ESAM endothelial cell-selective adhesion molecule, GEF-H1, guanine nucleotide exchange factor H1, huASH1 human absent small and homeotic discs protein 1 homolog, JACOP junction-associated coiled-coil protein, JAMs junctional adhesion molecules, JEAP junction-enriched and -associated protein, LYRIC lysine-rich CEACAM1 co-isolated protein, MAGI membrane-associated guanylate kinase with inverted orientation, MarcelD3 MARVEL domain-containing protein 3, MUPP1 multi-PDZ domain protein-1, Pals1 MAGUK p55 subfamily member 5, PAR partitioning defective, PATJ Pals1-associated tight junction protein, PILT protein incorporated later into tight junctions, PP protein phosphatase, PTEN phosphatase and tensin homolog, Rab Ras-related protein Rab, RPTPβ receptor-type tyrosine-protein phosphatase β, TAZ transcriptional coactivator with PDZ-binding motif, Tuba tubulin alpha-1A chain, WNK4 serine/threonine-protein kinase WNK4, YAP Yes-associated protein, Yes tyrosine-protein kinase Yes, ZO zonula occludens, ZONAB ZO-1-associated nucleic-acid binding protein
Junctional adhesion molecules (JAMs) are cell adhesion molecules (CAMs) of the immunoglobulin superfamily (IgSF), which are mainly located in epithelial and endothelial cell intercellular junctions [48] and also expressed on circulating platelets [49] and leukocytes [49, 50]. A variety of biological processes are regulated by the JAMs’ ability to trigger intracellular cascades of signals at intercellular contact sites, including the regulation of leukocyte diapedesis, TJ formation and maintenance, paracellular permeability, cell polarization, barrier function regulation, cell migration, and platelet activation [1, 4, 27, 51,52,53]. The following review focuses on the current knowledge about the F11R/JAM-A protein, especially its tissue-specific involvement in tumorigenesis and metastasis. We also discuss the correlation between poor cancer patient clinical outcomes and aberrant F11R/JAM-A expression.
F11R/JAM-A structure, localization, and function
The first discovered member of the junctional adhesion molecule family was F11R/JAM-A (currently also known as JAM, JAM-1, F11R, CD321) [27, 48, 54,55,56,57]. Initially, this protein was described as the human platelet F11 receptor for a monoclonal antibody called mAb.F11, which induces platelet activation [54, 55] and later as an adherens and tight junction protein [48, 58]. F11R/JAM-A is a transmembrane glycoprotein that consists of a short C-terminal cytosolic tail, a single transmembrane segment, and an extracellular N-terminal region that contains two Ig-like domains (Fig. 2a) [27, 29, 48, 52, 54, 58, 59]. The cytoplasmic tail is composed of 40 amino acid residues, contains phosphorylation sites and the C-terminal PDZ (PSD-95/Discs-large/ZO-1 [15, 31, 60,61,62]) domain-binding motif (-SSFLVCOOH) which mediates direct protein interactions [57]. F11R/JAM-A is expressed by various cell types and tissues, among other endothelial and epithelial cells [48], platelets [55], leukocytes [49, 50], hematopoietic stem cells (HSC) [63], glial cells [64], spermatozoa and Sertoli cells [65, 66], heart [58], intestine [67], kidney [58], liver [58], lung [58], and lymphoid organs [68, 69] (distribution and functions are summarized in Table 1). Of note, its functions depend on sequence motifs, i.e., adhesive activity is regulated by extracellular domain motifs, whereas the cytoplasmic domain motifs adjust scaffolding and signaling protein interactions. Predictably, F11R/JAM-A contributes to the regulation of the variety of cellular processes, as diverse as epithelial/endothelial barrier function [30, 67, 70, 71], transendothelial migration of leukocytes (TEM) [48, 72,73,74,75,76], hemostasis [77,78,79], angiogenesis [80,81,82], hematopoiesis [83], the male germ cell [65, 84] and central nervous system [85] development, immune homeostasis and inflammation [86], the epithelial-to-mesenchymal transition (EMT) [87, 88], intercellular junction assembly [89, 90], cell migration regulation [57, 61, 81, 91], platelet aggregation [54, 92] and adhesion [92, 93], cell adhesion [30], and reovirus binding [94, 95]. Furthermore, it was proven that F11R/JAM-A is involved in the development of several pathologies, such as cardiovascular diseases [96,97,98,99], inflammatory bowel disease [100], rheumatoid arthritis [101], neurological disorders [72, 102], reovirus infection [94, 103, 104], and various cancer types [66, 87, 88, 105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142].

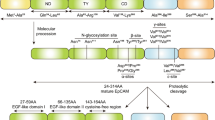
F11R/JAM-A structure and its homophilic interactions. a F11R/JAM-A consists of a short C-terminal cytosolic tail, a single transmembrane segment, and an extracellular N-terminal region. The extracellular segment has a membrane-distal VH-type Ig-like domain (D1, S28-K125) and membrane-proximal C2-type Ig-like domain (D2, P135-R228). The D1 contains cis-dimerization motif (R59V60E61) and trans-dimerization motif (N43N44P45), and the D2 has a single N-glycan at N185 residue. The cytoplasmic region includes phosphorylation sites (Y280, S284) and the type II PDZ domain-binding motif (–F297L298V299–COOH). b In the cis-dimerization process, two F11R/JAM-A molecules on the same cell form an inverted U-shaped homodimer by salt bridges between two oppositely charged amino acid residues (E61⋯R59; R59⋯E61). c The N43N44P45 motif participates in the trans-homophilic binding of two F11R/JAM-A cis-dimers on opposing cells. Likely, uncharged, polar residues (N43⋯N44; N44⋯N43) mostly mediate in trans-dimerization
F11R/JAM-A extracellular domain
The extracellular domain of F11R/JAM-A consists of the distal VH-type Ig-like domain (D1 domain, S28-K125) involved in homophilic binding and membrane-proximal C2-type Ig-like domain (D2 domain, P135-R228) participating in heterophilic interactions with the leukocyte αLß2 integrin (LFA-1, leukocyte function-associated antigen-1; CD11a/CD18 [143]) and reovirus protein σ1 (σ1 ligand) [27, 29, 52, 59, 73, 92,93,94, 144, 145]. The D1 domain has two structural motifs, i.e., the cis-dimerization motif (R59V60E61) and trans-dimerization motif (N43N44P45), both are involved in F11R/JAM-A adhesive interactions (Fig. 2a). In the cis-dimerization process, two F11R/JAM-A molecules on the same cell form an inverted U-shaped homodimer by salt bridges between two oppositely charged amino acid residues (E61⋯R59; R59⋯E61) (Fig. 2b). The previously mentioned N43N44P45 motif (Fig. 2c) participates in the trans-homophilic binding of two F11R/JAM-A cis-dimers on opposing cells [29, 51, 52, 59,60,61, 90, 93, 144, 146,147,148]. The D2 domain contains a single N-glycan at N185 residue, which stabilizes the F11R/JAM-A homodimers [148, 149]. The membrane-proximal Ig-like domain does not participate directly in the connection of F11R/JAM-A monomers, but it was proved that N-glycan at position N185 is a dimerization regulator [59, 146, 149]. However, the N-glycosylation of F11R/JAM-A protein is fundamental for its function, such as the reduction in cell migration, increased activity of Rap1, barrier function intensification, and leukocyte adhesion regulation [149]. Epitopes present in the extracellular region of F11R/JAM-A play a crucial role in the cellular processes, such as platelet aggregation [54, 92], TJ formation [89, 90], leukocyte integrin αLβ2 binding [73], and reovirus protein σ-1 attachment [94, 95].
F11R/JAM-A cytoplasmic domain
F11R/JAM-A directly associates with proteins through the C-terminal type II PDZ domain-binding motif (–F297L298V299–COOH), which is localized in the cytoplasmic domain (Fig. 2a) [4, 57, 150]. All proteins called direct binding partners such as zonula occludens-1 (ZO-1) [151,152,153], ZO-2 [154], afadin (also known as AF-6) [61, 151, 154], partitioning-defective 3 homolog (PAR-3) [155, 156], multi-PDZ domain protein 1 (MUPP1) [113], protein interacting with C kinase 1 (PICK-1) [157], calcium/calmodulin-dependent serine protein kinase (CASK) [158, 159], Rap guanine nucleotide exchange factor 6 (RAPGEF6/PDZ-GEF2) [61], and factor 2 (RAPGEF2/PDZ-GEF1) [154] belong to the group of PDZ domain-containing proteins that bind directly with F11R/JAM-A. Furthermore, in endothelial cells, monomeric F11R/JAM-A is incorporated by tetraspanin CD9 to F11R/JAM-A-CD9-αvβ3 integrin complex [160]. After the basic fibroblast growth factor (bFGF) stimulation, F11R/JAM-A is released from the developed complex and acts as a regulator of angiogenesis, endothelial cell migration, and MAPK (mitogen-activated protein kinase) activation. It is suggested that an unidentified cytoplasmic PDZ domain protein mediates the interaction of F11R/JAM-A with CD9 [160]. Besides, in different types of cells, four phosphorylation sites (Y280, S284, S287, S296) in the human F11R/JAM-A cytoplasmic domain have been hitherto identified experimentally [161,162,163,164], but only two of them (Tyr280, Ser284) have a known function (Fig. 2a) [77, 81, 165, 166]. To sum up, the cytoplasmic region of F11R/JAM-A participates in the TJ assembly [28], intracellular signaling pathways [30, 57, 62, 167], and cell polarity regulation [30, 168].
F11R/JAM-A expression in cancer
Around 90% of human carcinomas originate from epithelial tissues. For many years, it was thought that the loss of proteins associated with TJs is required in the early stages of the cancer metastasis (epithelial intercellular adhesion disturbances) [34, 169,170,171,172,173]. Meanwhile, in some cases, overexpression of TJ proteins is related to the regulation of intracellular signaling cascades responsible for tumorigenesis and metastasis [15]. Therefore, an imbalance in the F11R/JAM-A expression may result in pathological consequences and disorders in the normal cell function. F11R/JAM-A participation in cancer progression and invasiveness is still a debated issue. Hitherto, the effects of aberrant F11R/JAM-A expression and its potential mechanisms in breast cancer have been best studied [131,132,133,134,135,136,137,138,139,140,141,142]. Of note, there have also been reports of its contribution to the development of many other malignancies [66, 87, 88, 105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130]. In this review, we focus on the dysregulation of a TJ protein, namely F11R/JAM-A, and its contribution to human cancer progression and metastasis. Figure 3 illustrates current knowledge about the mechanisms by which F11R/JAM-A affects tumorigenesis. The correlation between the F11R/JAM-A expression level in different carcinomas and poor prognosis in patients is summarized in Table 2.

Schematic overview of the signaling pathways by which F11R/JAM-A affects tumorigenesis. The F11R/JAM-A function in cancer progression is not only associated with the regulation of cell migration but also with an influence on apoptosis, epithelial-to-mesenchymal transition (EMT), cancer stem cell maintenance (self-renewal and pro-survival factor), and cell proliferation. AF-6 afadin, Akt, protein kinase B, EMT epithelial-to-mesenchymal transition, ERK extracellular signal-regulated kinase, F11R/JAM-A F11 receptor/junctional adhesion molecule-A, GSK3 α/β glycogen synthase kinase 3, α and β isoforms, HER2, human epidermal growth factor receptor-2, JNK, c-Jun N-terminal kinase, MAPK, mitogen-activated protein kinase, MEK serine/tyrosine/threonine kinase, NFκB nuclear factor kappa B, PDZ-GEF2 Rap guanine nucleotide exchange factor 6, PI3K phosphoinositide 3-kinase, RAF serine/threonine-protein kinase, RAS small GTPase, TGF-β1 transforming growth factor-β1
Breast cancer
The F11R/JAM-A role in breast cancer progression and metastasis remains a controversial issue. In 2008, the first published evidence indicated that in breast tumor metastases, F11R/JAM-A expression is decreased versus in normal human mammary epithelium [135]. According to Naik et al. [135], in breast cancer cell lines, downregulation of F11R/JAM-A protein correlates with an increased ability to cell migration, which is required for tumor cell invasion and metastasis. High F11R/JAM-A amounts were observed on the surface of cells showing less ability to migrate (T47D, MCF-7, and MDA-MB-468, respectively), whereas low F11R/JAM-A level was found on MDA-MB-231 cells, which is marked by high migratory potential. In MDA-MB-231 cells, migration and invasion were suppressed by F11R/JAM-A overexpression. Additionally, enhanced invasiveness of highly migratory cells was observed after F11R/JAM-A knockdown through the use of short interfering RNAs. Authors provide evidence that a possible mechanism responsible for cell migration blockade via F11R/JAM-A is associated with the cytoskeletal rearrangement, directly with an increased stable focal adhesion formation [135].
Shortly afterward, two research groups showed contradictory results. Their studies demonstrated that poor prognosis in breast cancer patients correlates with F11R/JAM-A overexpression [131, 132, 134]. Indeed, recurrence (within five years) occurs in patients whose tumors had high F11R/JAM-A expression levels [132]. McSherry et al. [132] and Murakami et al. [131] observed a strong correlation between high F11R/JAM-A level and reduced patient survival after the analysis of 270 and 444 breast tumor samples. The divergence of data may be related to the fact that Naik et al. [135], in their research, used only 62 cases of commercial tumor tissue microarrays (including 12 low-grade tumors, and 50 malignant primary breast tumors). F11R/JAM-A upregulation in tumor tissues from various origins (including breast cancer) versus normal tissues was also revealed by Goetsch et al. [137].
Moreover, McSherry et al. [132] emphasized that the possible mechanism of attenuated breast cancer cell motility after F11R/JAM-A loss is associated with a β1-integrin level reduction. Based on the invasive breast cancer tissue microarray analysis, it was shown that the increase in β1-integrin expression, one of the proteins required for cell migration, was associated with poor clinical outcomes in breast cancer patients [132, 141]. In mouse breast cancer models, proliferation and formation of metastases were also associated with the β1-integrin level [174, 175]. In further studies, identification of the F11R/JAM-A signaling events regulating β1-integrin-dependent migratory activity has shown that F11R/JAM-A could indirectly activate Rap1 GTPase via AF-6 and PDZ-GEF2 proteins through complex formation [133]. Rap1 GTPase is an activator of β1-integrins and a regulator of breast tumorigenesis. These findings have confirmed the F11R/JAM-A role in the cancer cell migration enhancement through Rap1 GTPase and β1-integrin activation and provide the evidence that it could be a novel therapeutic target in breast cancer patients for the development of anti-migratory cancer therapies and a negative prognostic factor for murine and human mammary tumor growth [131, 133].
The F11R/JAM-A function in cancer progression is not only associated with the regulation of cell migration but also with an influence on apoptosis and proliferation [131, 134, 137, 140]. Wang and Liu [140] revealed the link between the F11R/JAM-A expression and transforming growth factor-β1 (TGF-β1) signaling in the regulation of breast cancer cell proliferation and invasion. They proposed the following mechanisms by which TGF-β1 controls F11R/JAM-A expression by distinct signaling pathways: the TGF-β1-stimulated F11R/JAM-A gene transcription via Smad-mediated signaling cascade and activation of p54 JNK (c-Jun N-terminal kinase signaling pathways) post-translational regulation of F11R/JAM-A protein degradation (through clathrin-mediated endocytosis) [140]. Another mechanism of action is associated with the fact that F11R/JAM-A acts as a survival factor for mammary carcinoma cells by protecting tumor cells from apoptosis. The F11R/JAM-A-mediated anti-apoptotic activity may correlate with changes in β1-integrin levels, which can also transmit anti-apoptotic signals. Increasing breast cancer cell susceptibility to apoptosis and reduction of aggressive tumor behavior are associated with F11R/JAM-A loss [131].
Interestingly, subsequent investigations revealed that aggressive breast cancer phenotypes are characterized by F11R/JAM-A and human epidermal growth factor receptor-2 (HER2) overexpression. Brennan et al. [134] speculate that F11R/JAM-A regulates HER2 proteasomal degradation and activity in vitro and may potentially be an oncotarget in HER2-positive breast cancers. In line with this model, the F11R/JAM-A upregulation enhances proliferation and reduces apoptosis through HER2 signaling by the PI3K (phosphoinositide 3-kinase) and MAPK pathways [137]. A recent study showed that the F11R/JAM-A extracellular domain cleavage co-occurring with the F11R/JAM-A overexpression correlates with the induction of resistance to HER2-targeted drugs. In semi in vivo and in vitro breast cancer models, invasive potential of cancer cells was intensified by cleaved F11R/JAM-A [139]. Moreover, Cruz et al. [142] studies showed that F11R/JAM-A also regulates HER3 expression through a pathway involving the transcription factors β-catenin and FOXA1. In vivo murine studies have confirmed the F11R/JAM-A role in apoptosis and breast tumor proliferation [131, 137]. Increased apoptosis in mice without F11R/JAM-A was associated with smaller tumors [131]. Another investigation elucidated that a specific anti-F11R/JAM-A monoclonal antibody reduced murine breast tumor xenograft growth [137]. Bednarek et al. demonstrated that the F11R/JAM-A protein can be considered as a novel target in the treatment of breast cancer metastasis and suggested the role of F11R/JAM-A-derived peptide as a possible anti-metastatic drug [136].
F11R/JAM-A expression is regulated by microRNAs. Reduced F11R/JAM-A expression and breast cancer cell motility as well as invasiveness are modulated by the enhanced miR-145 expression [138]. Of note, breast cancer cell lines and clinical samples characterize miR-145 downregulation [138]. Conversely, miR-495 upregulation in breast cancer tissue specimens and F11R/JAM-A as its potential target were proved. Migration of cancer cells induced by miR-495 is associated with high F11R/JAM-A level [176].
To sum up, despite a great body of evidence on the F11R/JAM-A functional role in breast cancer progression and metastasis, its mechanisms of action in this malignancy have not been fully elucidated. Initially, the invasion of breast cancer cells was shown to be induced by the F11R/JAM-A downregulation [135, 176]. However, breast cancer clinical datasets analysis demonstrated that its overexpression is strongly correlated with cancer patients' poor prognosis [131, 132, 134, 139, 142].
Gastric cancer
In 2020, gastric cancer was the fourth leading cause of cancer-related death and the fifth most prevalent malignancy worldwide [177]. The multipronged analysis showed F11R/JAM-A underexpression as a prognostic factor predicting poor clinical outcomes and enhanced tumor aggressiveness in gastric cancer patients [114]. Low F11R/JAM-A expression level in gastric cancer promotes tumor cell invasion and migration, but not proliferation, and contributes to large tumor size, lymphatic vessel invasion, lymph node metastasis, and advanced TNM Classification of Malignant Tumors stage. Unfortunately, the molecular mechanism involved in the described F11R/JAM-A biological actions was not defined [114]. A year later, Ikeo et al. [115] demonstrated attenuated proliferation and invasion of a rat gastric cancer-like cell line (RGK1) after F11R/JAM-A dampening. Similar results were obtained in a human gastric cancer cell line (NCI-N87); an additional observation was a diminished anti-apoptotic protein Bcl-xL expression. F11R/JAM-A knockdown did not change the AKT and Mcl-1 protein expression. Conclusively, F11R/JAM-A plays an essential role in gastric cancer progression through suppressed apoptosis and enhanced proliferation of cancer cells [115].
Pancreatic cancer
Pancreatic cancer is a highly aggressive tumor. F11R/JAM-A could be a prognostic value in cancer progression because its low expression level negatively correlates with the presence of distant metastasis, histologic grade, and positive lymph node status. F11R/JAM-A depletion in pancreatic cancer tissue specimens related to poor overall patient survival calculated by the Kaplan–Meier method. Because the F11R/JAM-A expression level was determined retrospectively by immunohistochemistry, more detailed studies should be performed to confirm the data mentioned above [111].
Nasopharyngeal carcinoma
Metastasis and invasiveness of various cancers depend on the conversion of epithelial cells into mesenchymal cells. In human nasopharyngeal carcinoma (NPC), the F11R/JAM-A overexpression acts as an epithelial–mesenchymal transition inducer by the PI3K/Akt pathway activation. Additionally, the analysis of 172 patients with NPC showed that the F11R/JAM-A upregulation correlates with poor prognosis and metastasis [88]. Based on the findings that the F11R/JAM-A expression could be regulated via microRNAs, Jiang et al. [117] recently reported that miR-543 is significantly higher expressed in NPC cell lines and tissues, and its upregulation enhances proliferation and invasion of cancer cells. The overexpression of F11R/JAM-A impeded the miR-543-induced proliferation of NPC cells suggesting that miR-543/F11R/JAM-A signaling plays a critical role in progression. Moreover, the results showed that the F11R/JAM-A underexpression elevated migration. In NPC tissues and cell lines, a reduced F11R/JAM-A expression was indicated in comparison to the normal control group [117]. The obtained data were not consistent with earlier studies conducted by Tian et al. [88]. In parallel, it was shown that the miR-124 overexpression intensified radiosensitivity and suppressed stem-like properties of cancer cells through F11R/JAM-A targeting. In NPC tissues, miR-124 downregulation and correlation with patient poor overall survival were revealed [125].
Lung cancer
In patients with non-small cell lung cancer (NSCLC), F11R/JAM-A is mainly expressed in cell membranes. Its overexpression occurred in 37% of lung tumor specimens and correlated with advanced TNM stage, lymph node metastasis, and diminished NSCLC patient survival. In some types of NSCLC cell lines, a high F11R/JAM-A amount was also observed. It induced NSCLC cell proliferation through cell cycle regulation. F11R/JAM-A suppression promotes the cell cycle arrest at the G1/S boundary, thereby decreases cancer cell proliferation and tumor growth. Its loss was accompanied by depletion in the amount of cell cycle-related proteins, such as cyclin D1, CDK4, 6, and P-Rb. F11R/JAM-A overexpression positively correlated with tumor aggressiveness and NSCLC progression [129].
F11R/JAM-A upregulation was also observed in lung adenocarcinoma and atypical adenomatous hyperplasia of lung. Knockdown of F11R/JAM-A was associated with intensified apoptosis of cancer cells, reduced in vivo tumorigenicity, attenuated colony-forming ability, decreased motility and invasiveness of cells, while increased expression was related to neoplasia occurrence [121]. Consistently, in lung adenocarcinoma patients cohort investigated by Zhao et al. [130], the upregulation of F11R/JAM-A and its influence on poor overall survival and high mortality rate were also indicated.
Glioblastoma
Glioblastomas are highly resistant to radiotherapy and chemotherapy. In glioblastomas (GBM), F11R/JAM-A overexpression correlates with shorter patient survival and overall poor outcome [120]. Further, F11R/JAM-A could be involved in cancer aggressiveness, in low-grade gliomas versus glioblastomas, and its level was significantly lower. The connection between patient survival and the F11R/JAM-A level in grade II and III gliomas was not detected, maybe because of a small patient number used in studies [123]. In previous research, it was proved that F11R/JAM-A is a prognostic factor in glioblastomas [120].
Ovarian cancer
Recently, the clinical significance of F11R/JAM-A gene expression was assessed by Boljevic et al. [116] in epithelial ovarian cancer (EOC), the most aggressive and frequent ovarian cancer type. Obtained results showed that patients with F11R/JAM-A overexpression tend to have worse overall survival versus patients with F11R/JAM-A depletion. Furthermore, an unfavorable clinicopathological feature in EOC is related to advanced International Federation of Gynecologists and Obstetricians (FIGO) stage, peritoneal metastasis, residual tumor, and high F11R/JAM-A expression, suggesting the F11R/JAM-A involvement in tumor aggressiveness. Further studies are required to elucidate molecular mechanisms responsible for the observed F11R/JAM-A role in EOC [116].
Endometrial carcinoma
In human endometrial carcinoma, the F11R/JAM-A expression is negatively correlated with poor patient prognosis (histologic grade, myometrial invasion, and stage). F11R/JAM-A underexpression in high-grade and advanced endometrial cancers is associated with invasiveness and low overall patient survival and progression-free survival rates. Furthermore, in studies on 3D-cultured endometrial carcinoma cells, the reduced F11R/JAM-A expression in poorly differentiated (KLE cell line) versus well-differentiated adenocarcinoma (Ishikawa cell line) was confirmed [118].
Uterine adnexa carcinoma
High-grade serous carcinoma of uterine adnexa (HGSC) is an epithelial ovarian malignancy histotype. Poorer patient clinical outcome (shorter progression-free survival and overall survival) correlated with F11R/JAM-A underexpression in all tested cohorts composed of 1526 cases. The data reliability is confirmed by a large number of cases studied and the observed reproducibility of obtained results. Furthermore, flow cytometric analysis of twenty-six human uterine cancer-derived cell lines determined F11R/JAM-A as a potential new prognostic biomarker. In this study, intensified EMT was demonstrated in tumors with the low F11R/JAM-A expression, which means that the EMT-dependent mechanism could be responsible for this effect [87]. Cancer progression via EMT initiation has been observed also in nasopharyngeal cancer and head and neck squamous cell carcinoma.
Multiple myeloma
Multiple myeloma (MM) is a hematological malignancy, which is characterized by the uncontrolled clonal plasma cell proliferation. One of the mechanisms of cancer cell survival and drug resistance development is cell adhesion. First published studies concerning the F11R/JAM-A expression in MM patients reported high F11R/JAM-A expression in primary cells and cancer cell lines (RPMI-8226, U266, NCI-H929, LP-1, KMS-12-BM, SKMM-2, OPM-2). Patients with F11R/JAM-A upregulation were characterized by worse outcomes within six years [178]. Three years later, Solimando et al. [124] confirmed the overexpression of F11R/JAM-A in several MM cell lines (RPMI 8226, U266, OPM-2, NCI-H929) and 147 biopsies and bone marrow from cancer patients. Poor clinical prognosis correlated with elevated F11R/JAM-A levels in the MM plasma cell surface. Also, the concentration of circulating soluble F11R/JAM-A in serum was intensified in comparison with healthy individuals. In vitro studies revealed that F11R/JAM-A blocking disturbed MM cell migration, proliferation, viability, colony formation, and chemotaxis. Besides, an anti-F11R/JAM-A monoclonal antibody treatment inhibited tumor progression in vivo in MM-bearing mice. Taken together, F11R/JAM-A was proposed as a potential novel therapeutic target against MM [124]. Up-to-date data showed that F11R/JAM-A directly mediates MM progression via angiogenesis enhancement. Furthermore, in newly diagnosed and relapsed MM patients, high F11R/JAM-A surface expression on bone marrow-derived endothelial cells (BM-ECs) associates with poor clinical outcomes and survival. Diminished MM progression and vascularity were attained after F11R/JAM-A blocking on BM-ECs [179].
Lymphoma
In diffuse large B-cell lymphoma patients with multiple extranodal lesions, F11R/JAM-A overexpression relation to EMT and cancer cell invasion in vitro and also in vivo were indicated. Regarding the underlying mechanism, the high F11R/JAM-A level initiated the TGF-β/NODAL signaling, whereby prompted increased cancer cell aggressiveness. Furthermore, patients with F11R/JAM-A upregulation had a poor prognosis, including shorter progression-free survival and lower complete remission rate [128]. Interestingly, in breast cancer, TGF-β signaling is also involved in cancer progression through F11R/JAM-A participation.
Oral cancer
Recently, the F11R/JAM-A protein expression was investigated by Upadhaya et al. [127] in oral epithelial dysplasia (OED) and oral squamous cell carcinoma (OSCC). Revealed F11R/JAM-A overexpression was related to cancer cell perineural invasion, aggressive histological tumor grades, and correlated with a low survival rate in comparison to cancer patients with F11R/JAM-A underexpression. The authors suggested that the dimerization mechanism is responsible for the high F11R/JAM-A level. In most OSCC tissues, F11R/JAM-A was delocalized to the cytoplasm from the cell membrane [127].
Renal cell carcinoma
In the human kidney, F11R/JAM-A is expressed in the distal convoluted tubule, connecting tubule, collecting duct cells, and weakly expressed in proximal tubule cells. In 282 biopsies from renal cell carcinoma (RCC) patients, the F11R/JAM-A underexpression and enhanced cancer cell migration were demonstrated, which means that this protein contributed to RCC progression. The RCC4 cell line migration was promoted by the F11R/JAM-A inhibition. Metalloproteinase-mediated F11R/JAM-A downregulation in HK-2 cells was induced after the pro-inflammatory cytokine (interferon-γ and tumor necrosis factor-α) treatment. The downregulation of F11R/JAM-A in RCC could be caused by enhanced metalloproteinase shedding. Any correlation with poor prognosis in RCC patients was not demonstrated. The performed analysis confirmed only a trend to a positive correlation with tumor grade. Nonetheless, F11R/JAM-A expression in biopsies of patients with clear cell renal cell carcinoma is associated with primary tumor category and tumor grade [112].
Cervical adenocarcinoma
The expression of F11R/JAM-A and claudin-1, 4, 7 proteins is significantly upregulated in patients with cervical adenocarcinoma and adenocarcinoma in situ (AIS). Based on immunoreactivity, Akimoto et al. [108] indicated that F11R/JAM-A or claudin-1 could be used as biomarkers for distinguishing cervical adenocarcinoma from non-neoplastic glands with high sensitivity (F11R/JAM-A with a higher rate) and specificity (both at the same rate) [108].
Head and neck squamous cell carcinoma
In head and neck squamous cell carcinoma (HNSCC), the F11R/JAM-A overexpression was revealed. All HNSCC differentiated stages (well, moderately, poorly) are characterized by high F11R/JAM-A mRNA expression. The plasma-soluble F11R/JAM-A level in HNSCC patients' serum was also high, which means that it could be a serum diagnostic marker of HNSCC [105, 119]. In further in vitro studies, the F11R/JAM-A dysregulation mechanism through the p63/GATA-3 was proved. In the Detroit562 cell line, F11R/JAM-A overexpression is also observed. Knockdown of F11R/JAM-A suppressed proliferation, migration, and invasion of cells [105].
Thyroid carcinoma
In anaplastic thyroid carcinoma (ATC) versus tissues from papillary thyroid cancer and normal thyroid, F11R/JAM-A underexpression was proved by the EMT-PCR array of 84 EMT-related genes. The observed downregulation was associated with tumor size, extrathyroid infiltration, and ATC histological type. In in vitro studies, cancer cell proliferation, transendothelial migration, and motility were dampened after the restoration of the F11R/JAM-A protein level. Noteworthy, the F11R/JAM-A overexpression was associated with an increased level of p53 and GSK3 α/β proteins phosphorylation. Regulation of GSK3 α/β and p53 signaling pathways through the F11R/JAM-A protein attenuates thyroid cancer cell aggressiveness [122]. Enhanced cancer cell proliferation and apoptosis inhibition by PI3K/Akt signaling pathway were determined also in breast cancer.
Testicular cancer
The F11R/JAM-A protein is present in spermatogonia and spermatocytes in the normal human testis and at inter-Sertoli cell tight junctions. It is overexpressed in seminoma cells, which may suggest that it promotes cancer cell migration and infiltration. In tubules with testicular carcinoma in situ, the F11R/JAM-A location is disorganized [66].
Salivary gland tumor
Salivary gland tumors (SGTs) are a comparatively rare disease, approx. 75% of which are not malignant tumors. Immunohistochemistry of 77 specimens of human SGTs and 40 non-tumorous tissues revealed the high F11R/JAM-A expression in ductal epithelium tumor cells compared to normal tissues. In malignant SGTs, claudin-4 and F11R/JAM-A are potential targets for molecular therapy [109].
Colorectal cancer
Severson’s et al. [61] findings demonstrate that F11R/JAM-A regulates human colonic epithelial cell migration through the F11R/JAM-A dimerization-mediated signaling. F11R/JAM-A dimerization facilitates the formation of a signaling complex (containing AF-6 and PDZ-GEF2), which in turn activates Rap1-GTPase, thereby elevating β1-integrin levels and enhancing epithelial cell migration [61]. Nava et al. [180] observed in their in vitro and in vivo studies that the F11R/JAM-A loss correlates with intensified intestinal epithelial cell proliferation in a dimerization-dependent manner, through the enhancement of Akt-dependent β-catenin activation. Whereas, in F11R/JAM-deficient mice, Akt inhibition reversed intestinal hyperproliferation. The authors reported that PI3K- and PTEN-dependent, Akt-mediated β-catenin transcriptional activation are required for increased cell proliferation [180]. Similar signaling pathways are involved in breast cancer progression through F11R/JAM-A.
Colorectal cancer is one of the deadliest carcinoma types worldwide. Recent research of the involvement of F11R/JAM-A and LFA-1 genetic variants in colorectal cancer development and metastasis was the first investigation of these gene variations in patients [110]. The authors revealed that the F11R/JAM-A rs790056 variation could influence the development of colorectal carcinoma (CC genotype has a threefold increased risk of colorectal cancer occurrence) and suggested that this variation could be evaluated as a potential predictive biomarker of this cancer type. Unfortunately, they did not define the relationship between genotypes and F11R/JAM-A expression (protein expression and soluble F11R/JAM-A levels or mRNA were not measured) [110].
Melanoma
Malignant melanoma cells have high metastatic potential. Ghislin et al. [107] revealed that F11R/JAM-A has an inhibitory role in melanoma transendothelial migration in vitro. The authors also showed that JAM-C plays an opposite role in melanoma A375 cells. F11R/JAM-A inhibition leads to intensified SLM8 cell migration through endothelial cells. Conversely, JAM-C impaired this efficiency of A375 cells [107]. F11R/JAM-A and JAM-C expression in the skin was previously approved by other research groups [48, 181]. To date, the F11R/JAM-A expression level in normal versus melanoma tissues is still undefined.
F11R/JAM-A immunological role in cancer
F11R/JAM-A affects immune-mediated processes because it is also expressed on immune cells, such as lymphocytes, polymorphonuclear neutrophils (PMNs), monocytes, and dendritic cells [50, 182, 183]. Notably, its ability to influence leukocyte trafficking is important in the context of its therapeutic potential in several pathological conditions, including cancer. In vivo, F11R/JAM-A-null mice demonstrated enhanced dendritic cell migration to lymph nodes and activation of specific immunity [182]. However, the lack of F11R/JAM-A negatively regulates polarized PMNs trafficking [50]. For polarized migration, F11R/JAM-A expression on PMN is essential [50, 75]. In a recent study, Bonilha et al. [184] showed that F11R/JAM-A present on the surface of dendritic cells regulates Th1 differentiation via its influence on CD4+ T cell-DC interactions during T cell priming. These findings demonstrated an F11R/JAM-A importance in the regulation of immune responses in pathological conditions, such as cancer, autoimmune diseases, and inflammation, in which CD4+ T cells play dominant roles [184].
The above-mentioned mechanisms may play an important role in cancer surveillance through the immune system [183]. Murakami et al. [185] demonstrated intensified antitumoral immune response, in the pancreatic islet cell carcinoma induced by SV40 T-antigen expression in β cells (Rip1Tag2 mice), after genetic-mediated F11R/JAM-A depletion. A decrease in cancer growth and invasiveness was associated with reduced angiogenesis and also enhanced infiltration of dendritic cells (CD11c+ and MHC-II+) and CD4+ and CD8+ T lymphocytes [185]. Therefore, it is suggested that F11R/JAM-A-mediated dendritic cell migration plays a role in cancer progression by indirect influence on the immune response. Impaired dendritic cell infiltration in tumors contributes to neoplasm development. Reduced number of dendritic cells leads to lack of tumor antigens which is directly associated with a decrease of tumor-specific T cell activation in the lymph nodes, consequently reduced adaptive immune response against cancer cells [186].
F11R/JAM-A may be a monocyte prognostic marker of glioblastoma (GBM). Its high expression in mononuclear cells is associated with high-grade GBM occurrence, which means that this protein is a prognostic factor predicting poor clinical outcomes in GBM cancer patients, independently of its molecular subtype [187]. F11R/JAM-A expression is different in bone marrow-derived monocytes than in microglia (significantly higher). Additionally, it was proved that brain-infiltrating macrophages acquire F11R/JAM-A expression in high-grade GBM (in vitro) and after bone marrow transplantation [187]. In the tumor microenvironment (TME), F11R/JAM-A expression is also high in cells like macrophages and microglia. Currently, there has been little literature concerning F11R/JAM-A protein in the immune TME [126, 187]. One recent study demonstrated a sex-specific F11R/JAM-A role in the GBM microenvironment [126]. Intensified tumor growth, cancer cell proliferation, and microglia activation (Fizz1 and Ifi202b anti-inflammatory gene overexpression) were observed in female mice with F11R/JAM-A deficiency. The crucial role of F11R/JAM-A in the female tumor microenvironment is to diminish microglial activation [126]. Taking into consideration F11R/JAM-A’s pivotal role in leukocyte trafficking, it is important to determine its significance in leukocyte infiltration into the tumor microenvironment, which may be used in cancer immunotherapy.
F11R/JAM-A on cancer stem cells
Cancer stem cells (CSCs) are tumor cells with stem cell-like attributes and intensified ability to self-renewal and tumor-initiation [188, 189]. Glioblastoma (GBM) was one of the first tumors on which the CSC role was investigated. In patient-derived GBM cells, it was proven that F11R/JAM-A protein is crucial for CSC maintenance (Fig. 3) [120]. F11R/JAM-A was identified on stem cell-like brain tumor-initiating cells, which means that it probably acts as a niche adhesion factor in glioblastoma and affects brain tumor-initiating cells’ oncogenic potential [123]. Moreover, Alvarado et al. [188] showed high expression of F11R/JAM-A in GBM CSCs in vitro (self-renewal and proliferation) as well as in vivo (tumor-initiation). MicroRNA-145 (miR-145) binds directly to F11R/JAM-A, suppressed it, and as a result attenuates self-renewal. Whereas, miR-145 is downregulated in GBM CSCs, which means that it is a negative regulator of F11R/JAM-A-mediated CSC maintenance. Reduced self-renewal after miR-145 implementation was associated with a decrease in stem cell markers expression (including NANOG, OCT4, and SOX2) and impaired Akt signaling. Additionally, high F11R/JAM-A levels in combination with low miR-145 levels were a prognostic factor of poor clinical outcome for GBM patients [188].
Similarly, in triple-negative breast cancer (TNBC) cells, F11R/JAM-A is significant in CSC self-renewal [190]. Its high expression was observed in the population of green fluorescence protein-positive (GFP+) MDA-MB-231 and HCC70 breast cancer cells and TNBC patient-derived xenograft aldehyde dehydrogenase-positive (ALDH+) CSCs in comparison to control cells (GFP− and ALDH−, respectively). Attenuated self-renewal was associated with F11R/JAM-A absence [190].
Taken together, the literature highlight an F11R/JAM-A pro-tumorigenic role in self-renewal and cellular differentiation stimulation.
Concluding remarks and perspectives
More than 20 years have passed since the discovery of F11R/JAM-A, even though this protein is still the subject of research interest. A growing body of evidence points to its role in tumorigenesis and metastasis. In the literature, the F11R/JAM-A implication in cancer progression remains a controversial issue. Its overexpression is demonstrated in breast cancer, glioblastoma, oral squamous cell carcinoma, ovarian cancer, head and neck squamous cell carcinoma, cervical adenocarcinoma, salivary gland tumor, testicular cancer, lymphoma, non-small cell lung cancer, and in multiple myeloma, oppositely, low expression levels are revealed in pancreatic, thyroid, endometrial, uterine adnexa, renal, and gastric cancers (Table 2). At the same time, inconsistent data have been published on the F11R/JAM-A expression in nasopharyngeal cancer. Above-mentioned reports confirm the tissue‐specific regulation of F11R/JAM-A expression. Indeed, low as well as its high levels have been correlated with poor clinical outcome prognosis of patients with different cancer types, which revealed that the prognostic value is also tissue-specific (Table 2). The poor outcome has been correlated with high expression in breast, glioblastoma, multiple myeloma, and lung cancer, whereas low expression in pancreatic, gastric, and endometrial carcinomas. Pro-tumorigenic or anti-tumorigenic F11R/JAM-A function depends on carcinoma type and should be further clarified. Its role seems to be complex and remains unclear. Also, the mechanisms responsible for its aberrant expression in tumor tissues remain to be defined. Of note, the F11R/JAM-A ability to interact with several proteins through its PDZ domain-binding motif in the cytoplasmic tail could probably be responsible for its different function in carcinogenesis through its involvement in various cell signaling pathways (Fig. 3). It is suggested that cancer progression is intensified by F11R/JAM-A upregulation. Probably, enhanced integrin-mediated migratory events associate with the F11R/JAM-A involvement in the epithelial-to-mesenchymal transition. However, its downregulation could intensify cancer initiation process through cell polarity loss and impair TJs structure. To understand its role in cancer, it will be essential to identify signaling pathways that are activated or suppressed via the F11R/JAM-A protein.
Conclusively, TJ proteins, namely F11R/JAM-A, could be a potential molecular marker of several human carcinomas because of its aberrant expression involved in cancer progression. F11R/JAM-A identification as a direct target for therapeutic antibodies or peptides causes that it could be considered as a potential new target for cancer treatment.
Data availability
Not applicable.
Code availability
Not applicable.
Abbreviations
- AF-6:
-
Afadin
- AJ(s):
-
Adherens junction(s)
- AMOT:
-
Angiomotin
- AP1:
-
Activator protein 1
- aPKC:
-
Atypical protein kinase C
- ATC:
-
Anaplastic thyroid carcinoma
- bFGF:
-
Fibroblast growth factor
- BM-EC(s):
-
Bone marrow-derived endothelial cell(s)
- BVES:
-
Blood vessel epicardial substance
- CAM(s):
-
Cell adhesion molecule(s)
- CAR:
-
Coxsackievirus and adenovirus receptor
- CASK:
-
Calcium/calmodulin-dependent serine protein kinase
- CDK4:
-
Cyclin-dependent kinase 4
- CLMP:
-
CAR-like membrane protein
- Crb3:
-
Protein crumbs homolog 3
- EMT:
-
Epithelial-to-mesenchymal transition
- EOC:
-
Epithelial ovarian cancer
- ERK:
-
Extracellular signal-regulated kinase
- ESAM:
-
Endothelial cell-selective adhesion molecule
- F11R:
-
F11-receptor
- GBM:
-
Glioblastomas
- GEF-H1:
-
Guanine nucleotide exchange factor H1
- GJ(s):
-
Gap junction(s)
- GSK3 α/β:
-
Glycogen synthase kinase 3, α and β isoforms
- HER2:
-
Human epidermal growth factor receptor-2
- HGSC:
-
High-grade serous carcinoma of uterine adnexa
- HNSCC:
-
Head and neck squamous cell carcinoma
- HSC(s):
-
Hematopoietic stem cells
- huASH1:
-
Human absent small and homeotic discs protein 1 homolog
- IgSF:
-
Immunoglobulin superfamily
- JACOP:
-
Junction-associated coiled-coil protein
- JAM(s):
-
Junctional adhesion molecule(s)
- JAM-A:
-
Junctional adhesion molecule-A
- JEAP:
-
Junction-enriched and -associated protein
- JNK:
-
C-Jun N-terminal kinase
- LFA-1:
-
Leukocyte function-associated antigen-1
- LYRIC:
-
Lysine-rich CEACAM1 co-isolated protein
- MAGI:
-
Membrane-associated guanylate kinase with inverted orientation
- MAPK:
-
Mitogen-activated protein kinase
- MarcelD3:
-
MARVEL domain-containing protein 3
- MEK:
-
Serine/tyrosine/threonine kinase
- MM:
-
Multiple myeloma
- MUPP1:
-
Multi-PDZ domain protein 1
- NFκB:
-
Nuclear factor kappa B
- NPC:
-
Nasopharyngeal carcinoma
- NSCLC:
-
Non-small cell lung cancer
- OED:
-
Oral epithelial dysplasia
- OSCC:
-
Oral squamous cell carcinoma
- Pals1:
-
MAGUK p55 subfamily member 5
- PAR-3:
-
Partitioning-defective 3 homolog
- PATJ:
-
Pals1-associated tight junction protein
- PI3K/Akt:
-
Phosphoinositide 3-kinase/protein kinase B
- PICK-1:
-
Protein interacting with C kinase 1
- PILT:
-
Protein incorporated later into tight junctions
- PP:
-
Protein phosphatase
- PTEN:
-
Phosphatase and tensin homolog
- Rab:
-
Ras-related protein Rab
- RAF:
-
Serine/threonine-protein kinase
- RAPGEF2/PDZ-GEF1:
-
Rap guanine nucleotide exchange factor 2
- RAPGEF6/PDZ-GEF2:
-
Rap guanine nucleotide exchange factor 6
- RAS:
-
Small GTPase
- RCC:
-
Renal cell carcinoma
- RPTPβ:
-
Receptor-type tyrosine-protein phosphatase β
- SGT(s):
-
Salivary gland tumor(s)
- TAZ:
-
Transcriptional coactivator with PDZ-binding motif
- Tuba:
-
Tubulin alpha-1A chain
- TEM:
-
Transendothelial migration
- TGF-β:
-
Transforming growth factor-β
- TJ(s):
-
Tight junction(s)
- WNK4:
-
Serine/threonine-protein kinase WNK4
- YAP:
-
Yes-associated protein
- Yes:
-
Tyrosine-protein kinase Yes
- ZO:
-
Zonula occludens
- ZONAB:
-
ZO-1-associated nucleic-acid binding protein
References
Keiper T, Santoso S, Nawroth PP, Orlova V, Chavakis T (2005) The role of junctional adhesion molecules in cell-cell interactions. Histol Histopathol 20:197–203
Friedl P, Mayor R (2017) Tuning collective cell migration by cell-cell junction regulation. Cold Spring Harb Perspect Biol 9:1–17. https://doi.org/10.1101/cshperspect.a029199
Tokuda S, Yu ASL (2019) Regulation of epithelial cell functions by the osmolality and hydrostatic pressure gradients: a possible role of the tight junction as a sensor. Int J Mol Sci 20:1–23. https://doi.org/10.3390/ijms20143513
Steinbacher T, Kummer D, Ebnet K (2018) Junctional adhesion molecule-A: functional diversity through molecular promiscuity. Cell Mol Life Sci 75:1393–1409. https://doi.org/10.1007/s00018-017-2729-0
Collins C, Nelson WJ (2015) Running with neighbors: coordinating cell migration and cell–cell adhesion. Curr Opin Cell Biol 36:62–70. https://doi.org/10.1016/j.ceb.2015.07.004
Pawson T, Nash P (2003) Assembly of cell regulatory systems through protein interaction domains. Science 300:445–452. https://doi.org/10.1126/science.1083653
Pawson T, Scott JD (1997) Signaling through scaffold, anchoring, and adaptor proteins. Science 278:2075–2080
Ball LJ, Kuhne R, Schneider-Mergener J, Oschkinat H (2005) Recognition of proline-rich motifs by protein-protein-interaction domains. Angew Chem Int Ed Engl 44:2852–2869. https://doi.org/10.1002/anie.200400618
Manjunath GP, Ramanujam PL, Galande S (2017) Structure function relations in PDZ-domain-containing proteins: implications for protein networks in cellular signalling. J Biosci 43:155–171. https://doi.org/10.1007/s12038-017-9727-0
Zihni C, Mills C, Matter K, Balda MS (2016) Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol 17:564–580. https://doi.org/10.1038/nrm.2016.80
Van Itallie CM, Tietgens AJ, Aponte A, Gucek M, Cartagena-Rivera AX, Chadwick RS, Anderson JM (2018) MARCKS-related protein regulates cytoskeletal organization at cell-cell and cell-substrate contacts in epithelial cells. J Cell Sci 131:1–14. https://doi.org/10.1242/jcs.210237
Matter K, Balda MS (2003) Signalling to and from tight junctions. Nat Rev Mol Cell Biol 4:225–236. https://doi.org/10.1038/nrm1055
Sugita K, Kabashima K (2020) Tight junctions in the development of asthma, chronic rhinosinusitis, atopic dermatitis, eosinophilic esophagitis, and inflammatory bowel diseases. J Leukoc Biol 107:749–762. https://doi.org/10.1002/JLB.5MR0120-230R
Rusu AD, Georgiou M (2020) The multifarious regulation of the apical junctional complex. Open Biol 10:190278. https://doi.org/10.1098/rsob.190278
Leech AO, Cruz RG, Hill AD, Hopkins AM (2015) Paradigms lost-an emerging role for over-expression of tight junction adhesion proteins in cancer pathogenesis. Ann Transl Med 3:184. https://doi.org/10.3978/j.issn.2305-5839.2015.08.01
Takano K, Kojima T, Sawada N, Himi T (2014) Role of tight junctions in signal transduction: an update. EXCLI J 13:1145–1162
Zihni C, Balda MS, Matter K (2014) Signalling at tight junctions during epithelial differentiation and microbial pathogenesis. J Cell Sci 127:3401–3413. https://doi.org/10.1242/jcs.145029
Balda MS, Garrett MD, Matter K (2003) The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J Cell Biol 160:423–432. https://doi.org/10.1083/jcb.200210020
Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q, Guan KL (2011) Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev 25:51–63. https://doi.org/10.1101/gad.2000111
Ikari A, Watanabe R, Sato T, Taga S, Shimobaba S, Yamaguchi M, Yamazaki Y, Endo S, Matsunaga T, Sugatani J (2014) Nuclear distribution of claudin-2 increases cell proliferation in human lung adenocarcinoma cells. Biochim Biophys Acta 1843:2079–2088. https://doi.org/10.1016/j.bbamcr.2014.05.017
Karaman R, Halder G (2018) Cell junctions in hippo signaling. Cold Spring Harb Perspect Biol 10:a028753. https://doi.org/10.1101/cshperspect.a028753
Diaz-Coranguez M, Liu X, Antonetti DA (2019) Tight junctions in cell proliferation. Int J Mol Sci 20:5972. https://doi.org/10.3390/ijms20235972
Lockwood C, Zaidel-Bar R, Hardin J (2008) The C. elegans zonula occludens ortholog cooperates with the cadherin complex to recruit actin during morphogenesis. Curr Biol 18:1333–1337. https://doi.org/10.1016/j.cub.2008.07.086
Gonzalez-Mariscal L, Dominguez-Calderon A, Raya-Sandino A, Ortega-Olvera JM, Vargas-Sierra O, Martinez-Revollar G (2014) Tight junctions and the regulation of gene expression. Semin Cell Dev Biol 36:213–223. https://doi.org/10.1016/j.semcdb.2014.08.009
Gonzalez-Mariscal L, Bautista P, Lechuga S, Quiros M (2012) ZO-2, a tight junction scaffold protein involved in the regulation of cell proliferation and apoptosis. Ann N Y Acad Sci 1257:133–141. https://doi.org/10.1111/j.1749-6632.2012.06537.x
Mattagajasingh SN, Huang SC, Hartenstein JS, Benz EJ Jr (2000) Characterization of the interaction between protein 4.1R and ZO-2. A possible link between the tight junction and the actin cytoskeleton. J Biol Chem 275:30573–30585. https://doi.org/10.1074/jbc.M004578200
Bazzoni G (2006) Endothelial tight junctions: permeable barriers of the vessel wall. Thromb Haemost 95:36–42. https://doi.org/10.1160/th05-07-0488
Rehder D, Iden S, Nasdala I, Wegener J, Brickwedde MK, Vestweber D, Ebnet K (2006) Junctional adhesion molecule-a participates in the formation of apico-basal polarity through different domains. Exp Cell Res 312:3389–3403. https://doi.org/10.1016/j.yexcr.2006.07.004
Mandell KJ, McCall IC, Parkos CA (2004) Involvement of the junctional adhesion molecule-1 (JAM1) homodimer interface in regulation of epithelial barrier function. J Biol Chem 279:16254–16262. https://doi.org/10.1074/jbc.M309483200
Mandell KJ, Babbin BA, Nusrat A, Parkos CA (2005) Junctional adhesion molecule 1 regulates epithelial cell morphology through effects on 1 integrins and rap1 activity. J Biol Chem 280:11665–11674. https://doi.org/10.1074/jbc.M412650200
Kojima T, Go M, Takano K, Kurose M, Ohkuni T, Koizumi J, Kamekura R, Ogasawara N, Masaki T, Fuchimoto J, Obata K, Hirakawa S, Nomura K, Keira T, Miyata R, Fujii N, Tsutsumi H, Himi T, Sawada N (2013) Regulation of tight junctions in upper airway epithelium. Biomed Res Int 2013:947072. https://doi.org/10.1155/2013/947072
Balda MS, Matter K (2016) Tight junctions as regulators of tissue remodelling. Curr Opin Cell Biol 42:94–101. https://doi.org/10.1016/j.ceb.2016.05.006
Claverie-Martin F (2015) Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis: clinical and molecular characteristics. Clin Kidney J 8:656–664. https://doi.org/10.1093/ckj/sfv081
Martin TA, Jiang WG (2009) Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta 1788:872–891. https://doi.org/10.1016/j.bbamem.2008.11.005
Wittekindt OH (2017) Tight junctions in pulmonary epithelia during lung inflammation. Pflugers Arch 469:135–147. https://doi.org/10.1007/s00424-016-1917-3
Kabashima K, Honda T, Ginhoux F, Egawa G (2019) The immunological anatomy of the skin. Nat Rev Immunol 19:19–30. https://doi.org/10.1038/s41577-018-0084-5
Soyka MB, Wawrzyniak P, Eiwegger T, Holzmann D, Treis A, Wanke K, Kast JI, Akdis CA (2012) Defective epithelial barrier in chronic rhinosinusitis: the regulation of tight junctions by IFN-gamma and IL-4. J Allergy Clin Immunol 130(1087–1096):e10. https://doi.org/10.1016/j.jaci.2012.05.052
Rerknimitr P, Otsuka A, Nakashima C, Kabashima K (2017) The etiopathogenesis of atopic dermatitis: barrier disruption, immunological derangement, and pruritus. Inflamm Regen 37:14. https://doi.org/10.1186/s41232-017-0044-7
Samadi N, Klems M, Untersmayr E (2018) The role of gastrointestinal permeability in food allergy. Ann Allergy Asthma Immunol 121:168–173. https://doi.org/10.1016/j.anai.2018.05.010
Anbazhagan AN, Priyamvada S, Alrefai WA, Dudeja PK (2018) Pathophysiology of IBD associated diarrhea. Tissue Barriers 6:e1463897. https://doi.org/10.1080/21688370.2018.1463897
de Boer WI, Sharma HS, Baelemans SM, Hoogsteden HC, Lambrecht BN, Braunstahl GJ (2008) Altered expression of epithelial junctional proteins in atopic asthma: possible role in inflammation. Can J Physiol Pharmacol 86:105–112. https://doi.org/10.1139/y08-004
Landy J, Ronde E, English N, Clark SK, Hart AL, Knight SC, Ciclitira PJ, Al-Hassi HO (2016) Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J Gastroenterol 22:3117–3126. https://doi.org/10.3748/wjg.v22.i11.3117
Sato T, Shibata W, Maeda S (2019) Adhesion molecules and pancreatitis. J Gastroenterol 54:99–107. https://doi.org/10.1007/s00535-018-1500-0
Kong Y, Naggert JK, Nishina PM (2018) The impact of adherens and tight junctions on physiological function and pathological changes in the retina. Adv Exp Med Biol 1074:545–551. https://doi.org/10.1007/978-3-319-75402-4_66
Oshima T, Miwa H (2016) Gastrointestinal mucosal barrier function and diseases. J Gastroenterol 51:768–778. https://doi.org/10.1007/s00535-016-1207-z
Reinhold AK, Rittner HL (2017) Barrier function in the peripheral and central nervous system—a review. Pflugers Arch 469:123–134. https://doi.org/10.1007/s00424-016-1920-8
Gon Y, Hashimoto S (2018) Role of airway epithelial barrier dysfunction in pathogenesis of asthma. Allergol Int 67:12–17. https://doi.org/10.1016/j.alit.2017.08.011
Martìn-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, Simmons D, E. D, (1998) Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol 142:117–127
Williams LA, Martin-Padura I, Dejana E, Hogg N, Simmons DL (1999) Identification and characterisation of human junctional adhesion molecule (JAM). Mol Immunol 36:1175–1188
Cera MR, Fabbri M, Molendini C, Corada M, Orsenigo F, Rehberg M, Reichel CA, Krombach F, Pardi R, Dejana E (2009) JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J Cell Sci 122:268–277. https://doi.org/10.1242/jcs.037127
Ebnet K (2017) Junctional adhesion molecules (JAMs): cell adhesion receptors with pleiotropic functions in cell physiology and development. Physiol Rev 97:1529–1554. https://doi.org/10.1152/physrev.00004.2017
Bazzoni G, Martinez-Estrada OM, Mueller F, Nelboeck P, Schmid G, Bartfai T, Dejana E, Brockhaus M (2000) Homophilic interaction of junctional adhesion molecule. J Biol Chem 275:30970–30976. https://doi.org/10.1074/jbc.M003946200
Cerutti C, Ridley AJ (2017) Endothelial cell-cell adhesion and signaling. Exp Cell Res 358:31–38. https://doi.org/10.1016/j.yexcr.2017.06.003
Sobocka MB, Sobocki T, Banerjee P, Weiss C, Rushbrook JI, Norin AJ, Hartwig J, Salifu MO, Markell MS, Babinska A, Ehrlich YH, Kornecki E (2000) Cloning ofthe human platelet F11 receptor: a cell adhesion molecule member ofthe immunoglobulin superfamily involved in platelet aggregation. Blood 95:2600–2609
Kornecki E, Walkowiak B, Naik UP, Ehrlich YH (1990) Activation of human platelets by a stimulatory monoclonal antibody. J Biol Chem 265:10042–10048
Sobocki T, Sobocka MB, Babinska A, Ehrlich YH, Banerjee P, Kornecki E (2006) Genomic structure, organization and promoter analysis of the human F11R/F11 receptor/junctional adhesion molecule-1/JAM-A. Gene 366:128–144. https://doi.org/10.1016/j.gene.2005.08.025
Bazzoni G, Tonetti P, Manzi L, Cera MR, Balconi G, Dejana E (2005) Expression of junctional adhesion molecule-A prevents spontaneous and random motility. J Cell Sci 118:623–632. https://doi.org/10.1242/jcs.01661
Gupta SK, Pillarisetti K, Ohlstein EH (2000) Platelet agonist F11 receptor is a member of the immunoglobulin superfamily and identical with junctional adhesion molecule (JAM): regulation of expression in human endothelial cells and macrophages. IUBMB Life 50:51–56
Prota AE, Campbell JA, Schelling P, Forrest JC, Watson MJ, Peters TR, Aurrand-Lions M, Imhof BA, Dermody TS, Stehle T (2003) Crystal structure of human junctional adhesion molecule 1: implications for reovirus binding. PNAS 100:5366–5371
Severson EA, Jiang L, Ivanov AI, Mandell KJ, Nusrat A, Parkos CA (2008) Cis-dimerization mediates function of junctional adhesion molecule A. Mol Biol Cell 19:1862–1872. https://doi.org/10.1091/mbc.E07-09-0869
Severson EA, Lee WY, Capaldo CT, Nusrat A, Parkos CA (2009) Junctional adhesion molecule A interacts with Afadin and PDZ-GEF2 to activate Rap1A, regulate beta1 integrin levels, and enhance cell migration. Mol Biol Cell 20:1916–1925. https://doi.org/10.1091/mbc.E08-10-1014
Sobocka MB, Sobocki T, Babinska A, Hartwig JH, Li M, Ehrlich YH, Kornecki E (2004) Signaling pathways of the F11 receptor (F11R; a.k.a. JAM-1, JAM-A) in human platelets: F11R dimerization, phosphorylation and complex formation with the integrin GPIIIa. J Recept Signal Transduct Res 24:85–105. https://doi.org/10.1081/rrs-120034252
Sugano Y, Takeuchi M, Hirata A, Matsushita H, Kitamura T, Tanaka M, Miyajima A (2008) Junctional adhesion molecule-A, JAM-A, is a novel cell-surface marker for long-term repopulating hematopoietic stem cells. Blood 111:1167–1172. https://doi.org/10.1182/blood-2007-03-081554
Stelzer S, Ebnet K, Schwamborn JC (2010) JAM-A is a novel surface marker for NG2-Glia in the adult mouse brain. BMC Neurosci 11:27. https://doi.org/10.1186/1471-2202-11-27
Shao M, Ghosh A, Cooke VG, Naik UP, Martin-DeLeon PA (2008) JAM-A is present in mammalian spermatozoa where it is essential for normal motility. Dev Biol 313:246–255
Tarulli GA, Stanton PG, Loveland KL, Rajpert-De Meyts E, McLachlan RI, Meachem SJ (2013) A survey of sertoli cell differentiation in men after gonadotropin suppression and in testicular cancer. Spermatogenesis 3:e24014. https://doi.org/10.4161/spmg.24014
Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, Dermody TS, Nusrat A, Parkos CA (2007) JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med 204:3067–3076. https://doi.org/10.1084/jem.20071416
Malergue F, Galland F, Martin F, Mansuelle P, Aurran-Lions M, Naqueta P (1998) A novel immunoglobulin superfamily junctional molecule expressed by antigen presenting cells endothelial cells and platelets. Mol Immunol 35:1111–1119
Cera MR, Del Prete A, Vecchi A, Corada M, Martin-Padura I, Motoike T, Tonetti P, Bazzoni G, Vermi W, Gentili F, Bernasconi S, Sato TN, Mantovani A, Dejana E (2004) Increased DC trafficking to lymph nodes and contact hypersensitivity in junctional adhesion molecule-A-deficient mice. J Clin Invest 114:729–738. https://doi.org/10.1172/JCI21231
Mitchell LA, Ward C, Kwon M, Mitchell PO, Quintero DA, Nusrat A, Parkos CA, Koval M (2015) Junctional adhesion molecule A promotes epithelial tight junction assembly to augment lung barrier function. Am J Pathol 185:372–386. https://doi.org/10.1016/j.ajpath.2014.10.010
Khounlotham M, Kim W, Peatman E, Nava P, Medina-Contreras O, Addis C, Koch S, Fournier B, Nusrat A, Denning TL, Parkos CA (2012) Compromised intestinal epithelial barrier induces adaptive immune compensation that protects from colitis. Immunity 37:563–573. https://doi.org/10.1016/j.immuni.2012.06.017
Del Maschio BA, De Luigi A, Martin-Padura I, Brockhaus M, Bartfai T, Fruscella P, Adorini L, Martino GV, Furlan R, De Simoni MG, Dejana E (1999) Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM). J Exp Med 190:1351–1356
Ostermann G, Weber KS, Zernecke A, Schroder A, Weber C (2002) JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol 3:151–158. https://doi.org/10.1038/ni755
Ostermann G, Fraemohs L, Baltus T, Schober A, Lietz M, Zernecke A, Liehn EA, Weber C (2005) Involvement of JAM-A in mononuclear cell recruitment on inflamed or atherosclerotic endothelium. Arterioscler Thromb Vasc Biol 25:729–735. https://doi.org/10.1161/01.ATV.0000157154.14474.3b
Corada M, Chimenti S, Cera MR, Vinci M, Salio M, Fiordaliso F, De Angelis N, Villa A, Bossi M, Staszewsky LI, Vecchi A, Parazzoli D, Motoike T, Latini R, Dejana E (2005) Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia–reperfusion injury. Proc Natl Acad Sci USA 102(30):10634–10639. https://doi.org/10.1073/pnas.0500147102
Khandoga A, Kessler JS, Meissner H, Hanschen M, Corada M, Motoike T, Enders G, Dejana E, Krombach F (2005) Junctional adhesion molecule-A deficiency increases hepatic ischemia-reperfusion injury despite reduction of neutrophil transendothelial migration. Blood 106:725–733. https://doi.org/10.1182/blood-2004-11-4416
Naik MU, Caplan JL, Naik UP (2014) Junctional adhesion molecule-A suppresses platelet integrin alphaIIbbeta3 signaling by recruiting Csk to the integrin-c-Src complex. Blood 123:1393–1402. https://doi.org/10.1182/blood-2013-04-496232
Karshovska E, Zhao Z, Blanchet X, Schmitt MM, Bidzhekov K, Soehnlein O, von Hundelshausen P, Mattheij NJ, Cosemans JM, Megens RT, Koeppel TA, Schober A, Hackeng TM, Weber C, Koenen RR (2015) Hyperreactivity of junctional adhesion molecule A-deficient platelets accelerates atherosclerosis in hyperlipidemic mice. Circ Res 116:587–599. https://doi.org/10.1161/CIRCRESAHA.116.304035
Naik MU, Stalker TJ, Brass LF, Naik UP (2012) JAM-A protects from thrombosis by suppressing integrin alphaIIbbeta3-dependent outside-in signaling in platelets. Blood 119:3352–3360. https://doi.org/10.1182/blood-2011-12-397398
Cooke VG, Naik MU, Naik UP (2006) Fibroblast growth factor-2 failed to induce angiogenesis in junctional adhesion molecule-A-deficient mice. Arterioscler Thromb Vasc Biol 26:2005–2011. https://doi.org/10.1161/01.ATV.0000234923.79173.99
Naik MU, Mousa SA, Parkos CA, Naik UP (2003) Signaling through JAM-1 and alphavbeta3 is required for the angiogenic action of bFGF: dissociation of the JAM-1 and alphavbeta3 complex. Blood 102:2108–2114. https://doi.org/10.1182/blood-2003-04-1114
Naik TU, Naik MU, Naik UP (2008) Junctional adhesion molecules in angiogenesis. Front Biosci 13:258–262. https://doi.org/10.2741/2676
Kobayashi I, Kobayashi-Sun J, Kim AD, Pouget C, Fujita N, Suda T, Traver D (2014) Jam1a-Jam2a interactions regulate haematopoietic stem cell fate through Notch signalling. Nature 512:319–323. https://doi.org/10.1038/nature13623
Wu KZ, Li K, Galileo DS, Martin-DeLeon PA (2017) Junctional adhesion molecule A: expression in the murine epididymal tract and accessory organs and acquisition by maturing sperm. Mol Hum Reprod 23:132–140. https://doi.org/10.1093/molehr/gaw082
Fededa JP, Esk C, Mierzwa B, Stanyte R, Yuan S, Zheng H, Ebnet K, Yan W, Knoblich JA, Gerlich DW (2016) MicroRNA-34/449 controls mitotic spindle orientation during mammalian cortex development. EMBO J 35:2386–2398. https://doi.org/10.15252/embj.201694056
Lakshmi SP, Reddy AT, Naik MU, Naik UP, Reddy RC (2012) Effects of JAM-A deficiency or blocking antibodies on neutrophil migration and lung injury in a murine model of ALI. Am J Physiol Lung Cell Mol Physiol 303:L758–L766. https://doi.org/10.1152/ajplung.00107.2012
Communal L, Medrano M, Sircoulomb F, Paterson J, Kobel M, Rahimi K, Hoskins P, Tu D, Lheureux S, Oza A, Ailles L, Provencher D, Rottapel R, Mes-Masson AM (2020) Low junctional adhesion molecule-A expression is associated with an epithelial to mesenchymal transition and poorer outcomes in high-grade serous carcinoma of uterine adnexa. Mod Pathol. https://doi.org/10.1038/s41379-020-0586-0
Tian Y, Tian Y, Zhang W, Wei F, Yang J, Luo X, Zhou T, Hou B, Qian S, Deng X, Qiu Y, Yao K (2015) Junctional adhesion molecule-A, an epithelial-mesenchymal transition inducer, correlates with metastasis and poor prognosis in human nasopharyngeal cancer. Carcinogenesis 36:41–48. https://doi.org/10.1093/carcin/bgu230
Liu Y, Nusrat A, Schnell FJ, Reaves TA, Walsh S, Pochet M, Parkos CA (2000) Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci 113:2363–2374
Liang TW, Demarco RA, Mrsny RJ, Gurney A, Gray A, Hooley J, Aaron HL, Huang A, Klassen T, Tumas DB, Fong S (2000) Characterization of huJAM: evidence for involvement in cell-cell contact and tight junction regulation. Am J Physiol Cell Physiol 279:C1733–C1743
Azari BM, Marmur JD, Salifu MO, Cavusoglu E, Ehrlich YH, Kornecki E, Babinska A (2010) Silencing of the F11R gene reveals a role for F11R/JAM-A in the migration of inflamed vascular smooth muscle cells and in atherosclerosis. Atherosclerosis 212:197–205. https://doi.org/10.1016/j.atherosclerosis.2010.05.014
Babinska A, Kedees MH, Athar H, Sobocki T, Sobocka MB, Ahmed T, Ehrlich YH, Hussain MM, Kornecki E (2002) Two regions of the human platelet F11-receptor (F11R) are critical for platelet aggregation, potentiation and adhesion. Thromb Haemost 87:712–721
Babinska A, Kedees M, Athar H, Ahmed T, Batuman O, Ehrlich YH, Hussain MM, Kornecki E (2002) F11-receptor (F11R/JAM) mediates platelet adhesion to endothelial cells: role in inflammatory thrombosis. Thromb Haemost 88:843–850. https://doi.org/10.1267/th02110843
Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS (2001) Junction adhesion molecule is a receptor for reovirus. Cell 104:441–451
Torres-Flores JM, Arias CF (2015) Tight junctions go viral! Viruses 7:5145–5154. https://doi.org/10.3390/v7092865
Schmitt MM, Megens RT, Zernecke A, Bidzhekov K, van den Akker NM, Rademakers T, van Zandvoort MA, Hackeng TM, Koenen RR, Weber C (2014) Endothelial junctional adhesion molecule-a guides monocytes into flow-dependent predilection sites of atherosclerosis. Circulation 129:66–76. https://doi.org/10.1161/CIRCULATIONAHA.113.004149
Waki H, Liu B, Miyake M, Katahira K, Murphy D, Kasparov S, Paton JF (2007) Junctional adhesion molecule-1 is upregulated in spontaneously hypertensive rats: evidence for a prohypertensive role within the brain stem. Hypertension 49:1321–1327. https://doi.org/10.1161/HYPERTENSIONAHA.106.085589
Babinska A, Azari BM, Salifu MO, Liu R, Jiang XC, Sobocka MB, Boo D, Al Khoury G, Deitch JS, Marmur JD, Ehrlich YH, Kornecki E (2007) The F11 receptor (F11R/JAM-A) in atherothrombosis: overexpression of F11R in atherosclerotic plaques. Thromb Haemost 97:272–281
Babinska A, Clement CC, Przygodzki T, Talar M, Li Y, Braun M, Wzorek J, Swiatkowska M, Ehrlich YH, Kornecki E, Watala C, Salifu MO (2019) A peptide antagonist of F11R/JAM-A reduces plaque formation and prolongs survival in an animal model of atherosclerosis. Atherosclerosis 284:92–101. https://doi.org/10.1016/j.atherosclerosis.2019.02.014
Vetrano S, Danese S (2009) The role of JAM-A in inflammatory bowel disease: unrevealing the ties that bind. Ann N Y Acad Sci 1165:308–313. https://doi.org/10.1111/j.1749-6632.2009.04045.x
Fang TJLCH, Lin YZ, Li RN, Ou TT, Wu CC, Tsai WC, Yen JH (2016) F11R mRNA expression and promoter polymorphisms in patients with rheumatoid arthritis. Int J Rheum Dis 19:127–133. https://doi.org/10.1111/1756-185X.12663
Padden M, Leech S, Craig B, Kirk J, Brankin B, McQuaid S (2007) Differences in expression of junctional adhesion molecule-A and beta-catenin in multiple sclerosis brain tissue: increasing evidence for the role of tight junction pathology. Acta Neuropathol 113:177–186. https://doi.org/10.1007/s00401-006-0145-x
Antar AA, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS (2009) Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59–71. https://doi.org/10.1016/j.chom.2008.12.001
Lai CM, Boehme KW, Pruijssers AJ, Parekh VV, Van Kaer L, Parkos CA, Dermody TS (2015) Endothelial JAM-A promotes reovirus viremia and bloodstream dissemination. J Infect Dis 211:383–393. https://doi.org/10.1093/infdis/jiu476
Kakuki T, Kurose M, Takano K, Kondoh A, Obata K (2016) Dysregulation of junctional adhesion molecule-A via p63/GATA-3 in head and neck squamous cell carcinoma. Oncotarget 7:33887–33900
Kelly K, Espitia CM, Zhao W, Wendlandt E (2015) Junctional adhesion molecule-A is overexpressed in advanced multiple myeloma and determines response to oncolytic reovirus. Oncotarget 6:41275–41289
Ghislin S, Obino D, Middendorp S, Boggetto N, Alcaide-Loridan C, Deshayes F (2011) Junctional adhesion molecules are required for melanoma cell lines transendothelial migration in vitro. Pigment Cell Melanoma Res 24:504–511. https://doi.org/10.1111/j.1755-148X.2011.00856.x
Akimoto T, Takasawa A, Murata M, Kojima Y, Takasawa K, Nojima M, Aoyama T, Hiratsuka Y, Ono Y, Tanaka S, Osanai M, Hasegawa T, Saito T, Sawada N (2016) Analysis of the expression and localization of tight junction transmembrane proteins, claudin-1, -4, -7, occludin and JAM-A, in human cervical adenocarcinoma. Histol Histopathol 31:921–931. https://doi.org/10.14670/HH-11-729
Aoyama T, Takasawa A, Murata M, Osanai M, Takano K, Hasagawa T, Sawada N (2018) Immunoreactivity patterns of tight junction proteins are useful for differential diagnosis of human salivary gland tumors. Med Mol Morphol 52:23–35. https://doi.org/10.1007/s00795-018-0199-6
Caykara B, Alsaadoni H, Pence HH, Pence S, Yilmaz Aydogan H, Tastekin D (2019) Investigation of JAM-A (rs790056) and LFA-1 (rs8058823) gene variants in Turkish colorectal cancer patients. Turk J Gastroenterol 30:872–876. https://doi.org/10.5152/tjg.2019.19141
Fong D, Spizzo G, Mitterer M, Seeber A, Steurer M, Gastl G, Brosch I, Moser P (2012) Low expression of junctional adhesion molecule A is associated with metastasis and poor survival in pancreatic cancer. Ann Surg Oncol 19:4330–4336. https://doi.org/10.1245/s10434-012-2381-8
Gutwein P, Schramme A, Voss B, Abdel-Bakky MS, Doberstein K, Ludwig A, Altevogt P, Hansmann ML, Moch H, Kristiansen G, Pfeilschifter J (2009) Downregulation of junctional adhesion molecule-A is involved in the progression of clear cell renal cell carcinoma. Biochem Biophys Res Commun 380:387–391. https://doi.org/10.1016/j.bbrc.2009.01.100
Hamazaki Y, Itoh M, Sasaki H, Furuse M, Tsukita S (2002) Multi-PDZ domain protein 1 (MUPP1) is concentrated at tight junctions through its possible interaction with claudin-1 and junctional adhesion molecule. J Biol Chem 277:455–461. https://doi.org/10.1074/jbc.M109005200
Huang JY, Xu YY, Sun Z, Wang ZN, Zhu Z, Song YX, Luo Y, Zhang X, Xu HM (2014) Low junctional adhesion molecule A expression correlates with poor prognosis in gastric cancer. J Surg Res 192:494–502. https://doi.org/10.1016/j.jss.2014.06.025
Ikeo K, Oshima T, Shan J, Matsui H, Tomita T, Fukui H, Watari J, Miwa H (2015) Junctional adhesion molecule-A promotes proliferation and inhibits apoptosis of gastric cancer. Hepatogastroenterology 62:540–545
Boljevic I, Malisic E, Milovic-Kovacevic M, Jovanic I, Bukumiric Z, Jankovic R (2019) High expression of junctional adhesion molecule-A is associated with poor survival in patients with epithelial ovarian cancer. Int J Biol Markers 34:262–268. https://doi.org/10.1177/1724600819850178
Jiang X, Dai B, Feng L (2019) miR-543 promoted the cell proliferation and invasion of nasopharyngeal carcinoma by targeting the JAM-A. Hum Cell 32:477–486. https://doi.org/10.1007/s13577-019-00274-0
Koshiba H, Hosokawa K, Kubo A, Tokumitsu N, Watanabe A, Honjo H (2009) Junctional adhesion molecule A [corrected] expression in human endometrial carcinoma. Int J Gynecol Cancer 19:208–213. https://doi.org/10.1111/IGC.0b013e31819bc6e9
Kurose M, Kakuki T, Takano K, Kondo A, Obata K, Nomura K, Miyata R, Kaneko Y, Konno T, Kohno T, Kojima T, Himi T (2016) Junctional adhesion molecule-A in head and neck squamous cell carcinoma. Adv Otorhinolaryngol 77:92–97. https://doi.org/10.1159/000441881
Lathia JD, Li M, Sinyuk M, Alvarado AG, Flavahan WA, Stoltz K, Rosager AM, Hale J, Hitomi M, Gallagher J, Wu Q, Martin J, Vidal JG, Nakano I, Dahlrot RH, Hansen S, McLendon RE, Sloan AE, Bao S, Hjelmeland AB, Carson CT, Naik UP, Kristensen B, Rich JN (2014) High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell Rep 6:117–129. https://doi.org/10.1016/j.celrep.2013.11.043
Magara K, Takasawa A, Osanai M, Ota M, Tagami Y, Ono Y, Takasawa K, Murata M, Hirohashi Y, Miyajima M, Yamada G, Hasegawa T, Sawada N (2017) Elevated expression of JAM-A promotes neoplastic properties of lung adenocarcinoma. Cancer Sci 108:2306–2314. https://doi.org/10.1111/cas.13385
Orlandella FM, Mariniello RM, Iervolino PLC, Auletta L, De Stefano AE, Ugolini C, Greco A, Mirabelli P, Pane K, Franzese M, Denaro M, Basolo F, Salvatore G (2019) Junctional adhesion molecule-A is down-regulated in anaplastic thyroid carcinomas and reduces cancer cell aggressiveness by modulating p53 and GSK3 alpha/beta pathways. Mol Carcinog 58:1181–1193. https://doi.org/10.1002/mc.23001
Rosager AM, Sorensen MD, Dahlrot RH, Boldt HB, Hansen S, Lathia JD, Kristensen BW (2017) Expression and prognostic value of JAM-A in gliomas. J Neurooncol 135:107–117. https://doi.org/10.1007/s11060-017-2555-0
Solimando AG, Brandl A, Mattenheimer K, Graf C, Ritz M, Ruckdeschel A, Stuhmer T, Mokhtari Z, Rudelius M, Dotterweich J, Bittrich M, Desantis V, Ebert R, Trerotoli P, Frassanito MA, Rosenwald A, Vacca A, Einsele H, Jakob F, Beilhack A (2018) JAM-A as a prognostic factor and new therapeutic target in multiple myeloma. Leukemia 32:736–743. https://doi.org/10.1038/leu.2017.287
Tian Y, Tian Y, Tu Y, Zhang G, Zeng X, Lin J, Ai M, Mao Z, Zheng R, Yuan Y (2020) microRNA-124 inhibits stem-like properties and enhances radiosensitivity in nasopharyngeal carcinoma cells via direct repression of expression of JAMA. J Cell Mol Med 00:1–12. https://doi.org/10.1111/jcmm.15177
Turaga SM, Silver DJ, Bayik D, Paouri E, Peng S, Lauko A, Alban TJ, Borjini N, Stanko S, Naik U, Keri RA, Connor JR, Barnholtz-Sloan JS, Rubin JB, Berens M, Davalos D, Lathia JD (2020) JAM-A functions as a female microglial tumor suppressor in glioblastoma. Neuro Oncol. https://doi.org/10.1093/neuonc/noaa148
Upadhaya P, Barhoi D, Giri A, Bhattacharjee A, Giri S (2019) Joint detection of claudin-1 and junctional adhesion molecule-A as a therapeutic target in oral epithelial dysplasia and oral squamous cell carcinoma. J Cell Biochem 120:18117–18127. https://doi.org/10.1002/jcb.29115
Xu PP, Sun YF, Fang Y, Song Q, Yan ZX, Chen Y, Jiang XF, Fei XC, Zhao Y, Leboeuf C, Li B, Wang CF, Janin A, Wang L, Zhao WL (2017) JAM-A overexpression is related to disease progression in diffuse large B-cell lymphoma and downregulated by lenalidomide. Sci Rep 7:7433. https://doi.org/10.1038/s41598-017-07964-5
Zhang M, Luo W, Huang B, Liu Z, Sun L, Zhang Q, Qiu X, Xu K, Wang E (2013) Overexpression of JAM-A in non-small cell lung cancer correlates with tumor progression. PLoS ONE 8:e79173. https://doi.org/10.1371/journal.pone.0079173
Zhao C, Wang A, Lu F, Chen H, Fu P, Zhao X, Chen H (2017) Overexpression of junctional adhesion molecule-A and EphB2 predicts poor survival in lung adenocarcinoma patients. Tumour Biol 39:1010428317691000. https://doi.org/10.1177/1010428317691000
Murakami M, Giampietro C, Giannotta M, Corada M, Torselli I, Orsenigo F, Cocito A, d’Ario G, Mazzarol G, Confalonieri S, Di Fiore PP, Dejana E (2011) Abrogation of junctional adhesion molecule-A expression induces cell apoptosis and reduces breast cancer progression. PLoS ONE 6:e21242. https://doi.org/10.1371/journal.pone.0021242
McSherry EA, McGee SF, Jirstrom K, Doyle EM, Brennan DJ, Landberg G, Dervan PA, Hopkins AM, Gallagher WM (2009) JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int J Cancer 125:1343–1351. https://doi.org/10.1002/ijc.24498
McSherry EA, Brennan K, Hudson L, Hill AD, Hopkins AM (2011) Breast cancer cell migration is regulated through junctional adhesion molecule-A-mediated activation of Rap1 GTPase. Breast Cancer Res 13:R31. https://doi.org/10.1186/bcr2853
Brennan K, McSherry EA, Hudson L, Kay EW, Hill AD, Young LS, Hopkins AM (2013) Junctional adhesion molecule-A is co-expressed with HER2 in breast tumors and acts as a novel regulator of HER2 protein degradation and signaling. Oncogene 32:2799–2804. https://doi.org/10.1038/onc.2012.276
Naik MU, Naik TU, Suckow AT, Duncan MK, Naik UP (2008) Attenuation of junctional adhesion molecule-A is a contributing factor for breast cancer cell invasion. Cancer Res 68:2194–2203. https://doi.org/10.1158/0008-5472.CAN-07-3057
Bednarek R, Selmi A, Wojkowska D, Karolczak K, Popielarski M, Stasiak M, Salifu MO, Babinska A, Swiatkowska M (2020) Functional inhibition of F11 receptor (F11R/junctional adhesion molecule-A/JAM-A) activity by a F11R-derived peptide in breast cancer and its microenvironment. Breast Cancer Res Treat 179:325–335. https://doi.org/10.1007/s10549-019-05471-x
Goetsch L, Haeuw JF, Beau-Larvor C, Gonzalez A, Zanna L, Malissard M, Lepecquet AM, Robert A, Bailly C, Broussas M, Corvaia N (2013) A novel role for junctional adhesion molecule-A in tumor proliferation: modulation by an anti-JAM-A monoclonal antibody. Int J Cancer 132:1463–1474. https://doi.org/10.1002/ijc.27772
Götte M, Mohr C, Koo CY, Stock C, Vaske AK, Viola M, Ibrahim SA, Peddibhotla S, Teng YHF, Low JY, Ebnet K, Kiesel L, Yip GW (2010) miR-145-dependent targeting of junctional adhesion molecule A and modulation of fascin expression are associated with reduced breast cancer cell motility and invasiveness. Oncogene 29:6569–6580. https://doi.org/10.1038/onc.2010.386
Leech AO, Vellanki SH, Rutherford EJ, Keogh A, Jahns H, Hudson L, O’Donovan N, Sabri S, Abdulkarim B, Sheehan KM, Kay EW, Young LS, Hill ADK, Smith YE, Hopkins AM (2018) Cleavage of the extracellular domain of junctional adhesion molecule-A is associated with resistance to anti-HER2 therapies in breast cancer settings. Breast Cancer Res 20:140. https://doi.org/10.1186/s13058-018-1064-1
Wang Y, Lui WY (2012) Transforming growth factor-beta1 attenuates junctional adhesion molecule-A and contributes to breast cancer cell invasion. Eur J Cancer 48:3475–3487. https://doi.org/10.1016/j.ejca.2012.04.016
Yao ES, Zhang H, Chen YY, Lee B, Chew K, Moore D, Park C (2007) Increased beta1 integrin is associated with decreased survival in invasive breast cancer. Cancer Res 67:659–664. https://doi.org/10.1158/0008-5472.CAN-06-2768
Cruz RGB, Madden SF, Richards CE, Vellanki SH, Jahns H, Hudson L, Fay J, O’Farrell N, Sheehan K, Jirstrom K, Brennan K, Hopkins AM (2021) Human epidermal growth factor receptor-3 expression is regulated at transcriptional level in breast cancer settings by junctional adhesion molecule-A via a pathway involving beta-catenin and FOXA1. Cancers (Basel). https://doi.org/10.3390/cancers13040871
Kummer D, Ebnet K (2018) Junctional adhesion molecules (JAMs): the JAM-integrin connection. Cells 7:25. https://doi.org/10.3390/cells7040025
Bazzoni G (2003) The JAM family of junctional adhesion molecules. Curr Opin Cell Biol 15:525–530. https://doi.org/10.1016/s0955-0674(03)00104-2
Babinska A, Clement CC, Swiatkowska M, Szymanski J, Shon A, Ehrlich YH, Kornecki E, Salifu MO (2014) Development of new antiatherosclerotic and antithrombotic drugs utilizing F11 receptor (F11R/JAM-A) peptides. Biopolymers 102:322–334. https://doi.org/10.1002/bip.22503
Kostrewa D, Brockhaus M, D’Arcy A, Dale GE, Nelboeck P, Schmid G, Mueller F, Bazzoni G, Dejana E, Bartfai T, Winkler FK, Hennig M (2001) X-ray structure of junctional adhesion molecule: structural basis for homophilic adhesion via a novel dimerization motif. Eur Mol Biol Organ 20:4391–4398
Monteiro AC, Luissint AC, Sumagin R, Lai C, Vielmuth F, Wolf MF, Laur O, Reiss K, Spindler V, Stehle T, Dermody TS, Nusrat A, Parkos CA (2014) Trans-dimerization of JAM-A regulates Rap2 and is mediated by a domain that is distinct from the cis-dimerization interface. Mol Biol Cell 25:1574–1585. https://doi.org/10.1091/mbc.E14-01-0018
Wojcikiewicz EP, Koenen RR, Fraemohs L, Minkiewicz J, Azad H, Weber C, Moy VT (2009) LFA-1 binding destabilizes the JAM-A homophilic interaction during leukocyte transmigration. Biophys J 96:285–293. https://doi.org/10.1529/biophysj.108.135491
Scott DW, Tolbert CE, Graham DM, Wittchen E, Bear JE, Burridge K (2015) N-glycosylation controls the function of junctional adhesion molecule-A. Mol Biol Cell 26:3205–3214. https://doi.org/10.1091/mbc.E14-12-1604
Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley lC (1997) Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 275:73–77
Ebnet K, Schulz CU, Meyer zu Brickwedde M-K, Pendl GG, Vestweber D (2000) Junctional adhesion molecule (JAM) interacts with the PDZ domain containing proteins AF-6 and ZO-1. J Biol Chem 275:27979–88. https://doi.org/10.1074/jbc.M002363200
Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, Dejana E (2000) Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem 275:20520–20526. https://doi.org/10.1074/jbc.M905251199
Nomme J, Fanning AS, Caffrey M, Lye MF, Anderson JM, Lavie A (2011) The Src homology 3 domain is required for junctional adhesion molecule binding to the third PDZ domain of the scaffolding protein ZO-1. J Biol Chem 286:43352–43360. https://doi.org/10.1074/jbc.M111.304089
Monteiro AC, Sumagin R, Rankin CR, Leoni G, Mina MJ, Reiter DM, Stehle T, Dermody TS, Schaefer SA, Hall RA, Nusrat A, Parkos CA (2013) JAM-A associates with ZO-2, afadin, and PDZ-GEF1 to activate Rap2c and regulate epithelial barrier function. Mol Biol Cell 24:2849–2860. https://doi.org/10.1091/mbc.E13-06-0298
Klaus Ebnet K, Suzuki A, Horikoshi Y, Hirose T, Meyer zu Brickwedde M-K, Ohno S, Vestweber D (2001) The cell polarity protein ASIP/PAR-3 directly associates with junctional adhesion molecule (JAM). EMBO J 20:3738–3748
Itoh M, Sasaki H, Furuse M, Ozaki H, Kita T, Tsukita S (2001) Junctional adhesion molecule (JAM) binds to PAR-3: a possible mechanism for the recruitment of PAR-3 to tight junctions. J Cell Biol 154:491–497. https://doi.org/10.1083/jcb.200103047
Reymond N, Garrido-Urbani S, Borg JP, Dubreuil P, Lopez M (2005) PICK-1: a scaffold protein that interacts with Nectins and JAMs at cell junctions. FEBS Lett 579:2243–2249. https://doi.org/10.1016/j.febslet.2005.03.010
Martinez-Estrada OM, Villa A, Breviario F, Orsenigo F, Dejana E, Bazzoni G (2001) Association of junctional adhesion molecule with calcium/calmodulin-dependent serine protein kinase (CASK/LIN-2) in human epithelial caco-2 cells. J Biol Chem 276:9291–9296. https://doi.org/10.1074/jbc.M006991200
Aravindan RG, Fomin VP, Naik UP, Modelski MJ, Naik MU, Galileo DS, Duncan RL, Martin-Deleon PA (2012) CASK interacts with PMCA4b and JAM-A on the mouse sperm flagellum to regulate Ca2+ homeostasis and motility. J Cell Physiol 227:3138–3150. https://doi.org/10.1002/jcp.24000
Peddibhotla SS, Brinkmann BF, Kummer D, Tuncay H, Nakayama M, Adams RH, Gerke V, Ebnet K (2013) Tetraspanin CD9 links junctional adhesion molecule-A to alphavbeta3 integrin to mediate basic fibroblast growth factor-specific angiogenic signaling. Mol Biol Cell 24:933–944. https://doi.org/10.1091/mbc.E12-06-0481
Heibeck TH, Ding SJ, Opresko LK, Zhao R, Schepmoes AA, Yang F, Tolmachev AV, Monroe ME, Camp DG 2nd, Smith RD, Wiley HS, Qian WJ (2009) An extensive survey of tyrosine phosphorylation revealing new sites in human mammary epithelial cells. J Proteome Res 8:3852–3861. https://doi.org/10.1021/pr900044c
Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP (2008) A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci USA 105:10762–10767. https://doi.org/10.1073/pnas.0805139105
Zahedi RP, Lewandrowski U, Wiesner J, Wortelkamp S, Moebius J, Schütz C, Walter U, Gambaryan S, Sickmann A (2008) Phosphoproteome of resting human platelets. J Proteome Res 7:526–534
Gauci S, Helbig AO, Slijper M, Krijgsveld J, Heck AJ, Mohammed S (2009) Lys-N and trypsin cover complementary parts of the phosphoproteome in a refined SCX-based approach. Anal Chem 81:4493–4501. https://doi.org/10.1021/ac9004309
Iden S, Misselwitz S, Peddibhotla SS, Tuncay H, Rehder D, Gerke V, Robenek H, Suzuki A, Ebnet K (2012) aPKC phosphorylates JAM-A at Ser285 to promote cell contact maturation and tight junction formation. J Cell Biol 196:623–639. https://doi.org/10.1083/jcb.201104143
Ozaki H, Ishii K, Arai H, Horiuchi H, Kawamoto T, Suzuki H, Kita T (2000) Junctional adhesion molecule (JAM) is phosphorylated by protein kinase C upon platelet activation. Biochem Biophys Res Commun 276:873–878. https://doi.org/10.1006/bbrc.2000.3574
Naik MU, Naik UP (2006) Junctional adhesion molecule-A-induced endothelial cell migration on vitronectin is integrin alpha v beta 3 specific. J Cell Sci 119:490–499. https://doi.org/10.1242/jcs.02771
Ohno S (2001) Intercellular junctions and cellular polarity: the PAR–aPKC complex, a conserved core cassette playing fundamental roles in cell polarity. Curr Opin Cell Biol 13:641–648
Martin TA, Mansel RE, Jiang WG (2010) Loss of occludin leads to the progression of human breast cancer. Int J Mol Med 26:723–734. https://doi.org/10.3892/ijmm_00000519
Kominsky SL, Argani P, Korz D, Evron E, Raman V, Garrett E, Rein A, Sauter G, Kallioniemi OP, Sukumar S (2003) Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene 22:2021–2033. https://doi.org/10.1038/sj.onc.1206199
Suren D, Yildirim M, Kaya V, Alikanoglu AS, Bulbuller N, Yildiz M, Sezer C (2014) Loss of tight junction proteins (Claudin 1, 4, and 7) correlates with aggressive behavior in colorectal carcinoma. Med Sci Monit 20:1255–1262. https://doi.org/10.12659/MSM.890598
Shimada H, Abe S, Kohno T, Satohisa S, Konno T, Takahashi S, Hatakeyama T, Arimoto C, Kakuki T, Kaneko Y, Takano KI, Saito T, Kojima T (2017) Loss of tricellular tight junction protein LSR promotes cell invasion and migration via upregulation of TEAD1/AREG in human endometrial cancer. Sci Rep 7:37049. https://doi.org/10.1038/srep37049
Lee SK, Moon J, Park SW, Song SY, Chung JB, Kang JK (2005) Loss of the tight junction protein claudin 4 correlates with histological growth-pattern and differentiation in advanced gastric adenocarcinoma. Oncol Rep 13:193–199
Li N, Zhang Y, Naylor MJ, Schatzmann F, Maurer F, Wintermantel T, Schuetz G, Mueller U, Streuli CH, Hynes NE (2005) Beta1 integrins regulate mammary gland proliferation and maintain the integrity of mammary alveoli. EMBO J 24:1942–1953. https://doi.org/10.1038/sj.emboj.7600674
Huck L, Pontier SM, Zuo DM, Muller WJ (2010) beta1-integrin is dispensable for the induction of ErbB2 mammary tumors but plays a critical role in the metastatic phase of tumor progression. Proc Natl Acad Sci USA 107:15559–15564. https://doi.org/10.1073/pnas.1003034107
Cao M, Nie W, Li J, Zhang Y, Yan X, Guan X, Chen X, Zen K, Zhang CY, Jiang X, Hou D (2014) MicroRNA-495 induces breast cancer cell migration by targeting JAM-A. Protein Cell 5:862–872. https://doi.org/10.1007/s13238-014-0088-2
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. https://doi.org/10.3322/caac.21660
Kelly KR, Espitia CM, Zhao W, Wendlandt E, Tricot G, Zhan F, Carew JS, Nawrocki ST (2015) Junctional adhesion molecule-A is overexpressed in advanced multiple myeloma and determines response to oncolytic reovirus. Oncotarget 6:41275–41289
Solimando AG, Da Via MC, Leone P, Borrelli P, Croci GA, Tabares P, Brandl A, Di Lernia G, Bianchi FP, Tafuri S, Steinbrunn T, Balduini A, Melaccio A, De Summa S, Argentiero A, Rauert-Wunderlich H, Frassanito MA, Ditonno P, Henke E, Klapper W, Ria R, Terragna C, Rasche L, Rosenwald A, Kortum KM, Cavo M, Ribatti D, Racanelli V, Einsele H, Vacca A, Beilhack A (2020) Halting the vicious cycle within the multiple myeloma ecosystem: blocking JAM-A on bone marrow endothelial cells restores the angiogenic homeostasis and suppresses tumor progression. Haematologica 105:1–19. https://doi.org/10.3324/haematol.2019.239913
Nava P, Capaldo CT, Koch S, Kolegraff K, Rankin CR, Farkas AE, Feasel ME, Li L, Addis C, Parkos CA, Nusrat A (2011) JAM-A regulates epithelial proliferation through Akt/beta-catenin signalling. EMBO Rep 12:314–320. https://doi.org/10.1038/embor.2011.16
Kirschner N, Poetzl C, von den Driesch P, Wladykowski E, Moll I, Behne MJ, Brandner JM (2009) Alteration of tight junction proteins is an early event in psoriasis: putative involvement of proinflammatory cytokines. Am J Pathol 175:1095–1106. https://doi.org/10.2353/ajpath.2009.080973
Cera MR, Del Prete A, Vecchi A, Corada M, Martin-Padura I, Motoike T, Tonetti P, Bazzoni G, Vermi W, Gentili F, Bernasconi S, Sato TN, Mantovani A, Dejana E (2004) Increased DC trafficking to lymph nodes and contact hypersensitivity in junctional adhesion molecule-A–deficient mice. J Clin Investig 114:729–738. https://doi.org/10.1172/jci21231
Luissint AC, Williams HC, Kim W, Flemming S, Azcutia V, Hilgarth RS, Leary MNO, Denning TL, Nusrat A, Parkos CA (2019) Macrophage-dependent neutrophil recruitment is impaired under conditions of increased intestinal permeability in JAM-A-deficient mice. Mucosal Immunol 12:668–678. https://doi.org/10.1038/s41385-019-0143-7
Bonilha CS, Benson RA, Scales HE, Brewer JM, Garside P (2021) Junctional adhesion molecule-A on dendritic cells regulates Th1 differentiation. Immunol Lett 235:32–40. https://doi.org/10.1016/j.imlet.2021.05.001
Murakami M, Francavilla C, Torselli I, Corada M, Maddaluno L, Sica A, Matteoli G, Iliev ID, Mantovani A, Rescigno M, Cavallaro U, Dejana E (2010) Inactivation of junctional adhesion molecule-A enhances antitumoral immune response by promoting dendritic cell and T lymphocyte infiltration. Cancer Res 70:1759–1765. https://doi.org/10.1158/0008-5472.CAN-09-1703
Bonilha CS, Benson RA, Brewer JM, Garside P (2020) Targeting opposing immunological roles of the junctional adhesion molecule-A in autoimmunity and cancer. Front Immunol 11:602094. https://doi.org/10.3389/fimmu.2020.602094
Pong WW, Walker J, Wylie T, Magrini V, Luo J, Emnett RJ, Choi J, Cooper ML, Griffith M, Griffith OL, Rubin JB, Fuller GN, Piwnica-Worms D, Feng X, Hambardzumyan D, DiPersio JF, Mardis ER, Gutmann DH (2013) F11R is a novel monocyte prognostic biomarker for malignant glioma. PLoS ONE 8:e77571. https://doi.org/10.1371/journal.pone.0077571
Alvarado AG, Turaga SM, Sathyan P, Mulkearns-Hubert EE, Otvos B, Silver DJ, Hale JS, Flavahan WA, Zinn PO, Sinyuk M, Li M, Guda MR, Velpula KK, Tsung AJ, Nakano I, Vogelbaum MA, Majumder S, Rich JN, Lathia JD (2016) Coordination of self-renewal in glioblastoma by integration of adhesion and microRNA signaling. Neuro Oncol 18:656–666. https://doi.org/10.1093/neuonc/nov196
Boman BM, Wicha MS (2008) Cancer stem cells: a step toward the cure. J Clin Oncol 26:2795–2799. https://doi.org/10.1200/JCO.2008.17.7436
Thiagarajan PS, Hitomi M, Hale JS, Alvarado AG, Otvos B, Sinyuk M, Stoltz K, Wiechert A, Mulkearns-Hubert E, Jarrar AM, Zheng Q, Thomas D, Egelhoff TT, Rich JN, Liu H, Lathia JD, Reizes O (2015) Development of a fluorescent reporter system to delineate cancer stem cells in triple-negative breast cancer. Stem Cells 33:2114–2125. https://doi.org/10.1002/stem.2021
Acknowledgements
This work was supported by the Medical University of Lodz, Poland.
Author information
Authors and Affiliations
Contributions
Conception and design: all authors. Literature search: KCP. Analysis and interpretation of data: KCP. Manuscript writing: all authors. Approval of final article: all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Czubak-Prowizor, K., Babinska, A. & Swiatkowska, M. The F11 Receptor (F11R)/Junctional Adhesion Molecule-A (JAM-A) (F11R/JAM-A) in cancer progression. Mol Cell Biochem 477, 79–98 (2022). https://doi.org/10.1007/s11010-021-04259-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-021-04259-2