
Abstract
The thermal gravimetric analysis (TG) is a common method for the examination of the carbonation progress of cement-based materials. Unfortunately, the thermal properties of some components complicate the evaluation of TG results. Various hydrate phases, such as ettringite (AFt), C–S–H and AFm, decompose almost simultaneously in the temperature range up to 200 °C. Additionally, physically bound water is released in the same temperature range. In the temperature range between 450 °C and 600 °C, the decomposition of calcium hydroxide and amorphous or weakly bound carbonates takes place simultaneously. Carbonates, like calcite, from limestone powder or other additives may be already contained in the noncarbonated sample material. For this research, an attempt was made to minimise the influence of these effects. Therefore, differential curves from DTG results of noncarbonated areas and areas with various states of carbonation of the same sample material were calculated and evaluated. Concretes based on three different types of cement were produced and stored under accelerated carbonation conditions (1% CO2 in air). The required sample material was obtained by cutting slices from various depths of previously CO2-treated specimen and subsequent grinding. During the sample preparation, a special attention was paid that no additional carbonation processes took place. As reference method for the determination of the carbonation depth, the sprayed application of phenolphthalein solution was carried out. Microscopic analysis was examined to confirm the assumptions made previously. Furthermore, the observed effect of encapsulation of calcium hydroxide by carbonates caused by the accelerated carbonation conditions was examined more closely.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The different thermal behaviour of the components in cementitious materials can be used for the analysis and characterisation of the material composition. Moreover, these properties allow to monitor temporal changes in the composition of the material. The thermogravimetric analysis (TG) is a widely used method for the examination of cementitious material. This method allows to determine the different hydrates and carbonates. Additionally, supplementary cementitious materials (SCMs) such as fly ash and blast furnace slags can be examined. The measurement principle is based on the mass loss determination during heating sample material under defined environmental conditions. Thereby, material-specific reactions such as dihydroxylation, decarbonization, decomposition or changes in the crystalline structure can be determined. Additionally, the absorption and emission of heat from the sample material are recognisable by heat flow measurements. In general, the measurement systems can be divided into thermal gravimetric (TG) analysis systems for mass loss detection and differential calorimeter systems (differential scanning calorimetry (DSC) and differential thermal analysis (DTA)) for detection of the heat flow related to the reaction temperatures. The principles of these methods are described in reference books [1,2,3]. Quite often, combinations of TG and DTA or rather DSC are used for material analysis. By evaluation of the specific peak position and peak width, it is possible to distinguish the different compounds or rather the decomposition products. Most frequently, the release of water (H2O) and carbon dioxide (CO2) is examined by TG analysis. The decomposition temperature ranges of the different hydrates and carbonates are well known. By stoichiometric calculations, the amount of the hydrates and carbonates can be determined.
The amount of calcium hydroxide can be calculated (1), using the mass loss between 400 °C and 500 °C and the molar masses of calcium hydroxide \(\left( {M_{{{\text{Ca}}\left( {\text{OH}} \right)_{2} }} = ~74.092~g/mol} \right)~\) and water \(\left( {M_{{{\text{H}}_{2} {\text{O}}}} = 18.015~ {\text{g}}\,{\text{mol}}^{-1}} \right)\). In their reference book, Lothenbach et al. [4] published further molar masses of hydrate and carbonate phases. From these data, the amount of C–S–H and ettringite can also be calculated. Nevertheless, the precondition for this evaluation is a clear differentiability of the hydrate phases by TG analysis, especially in the temperature range up to 200 °C. Therefore, the quantification of TG results is often limited to calcium hydroxide and calcite.
Calcium carbonate decomposes in the temperature range between 500 °C and 900 °C to calcium oxide and carbon dioxide. By using the mass loss in this temperature range and the molar masses of calcium carbonate \(\left( {{\text{M}}_{{\text{CaCO}}_{3}} = ~100.0869~{\text{g}}\,{\text{mol}}^{-1} } \right)~\) and carbon dioxide \(\left( {M_{\text{CO}_{2} } = 44.01~{\text{g}}\,{\text{mol}}^{-1} } \right)\), the amount of calcium carbonate can be calculated (2). A further subdivision of the different carbonate modifications (amorphic carbonates, aragonite and vaterite) in the temperature range from 500 °C to 700 °C is in principal possible but difficult because of quite similar decomposition temperature ranges. In contrast, calcite can be determined more easily because of the high decomposition temperature above 700 °C. An extensive collection of material specific data related to different reaction temperatures can be found in the publication of Collier et al. [5]. Moreover, Blaine [6] examined calcium sulphate hydrates, Borges et al. [7] calcium hydroxide (CH) and calcium silicate hydrate (C–S–H), Chen et al. [8] C–S–H and Dweck et al. [9] ettringite phases. Additionally, different types of cement paste, mortars and concretes with various supplementary materials were also examined [10,11,12,13,14,15,16]. An excellent overview about thermal gravimetric analysis provides chapter 5 of the reference book “A Practical Guide to Microstructural Analysis of Cementitious Materials”, published by Scrivener, Snellings and Lothenbach [4].
Already the sample preparation for thermal analysis of cementitious materials provides a major challenge. Particularly, the hydrate phases are very sensitive to treatment by CO2 and temperature. Since 0.04 percentage of CO2 is contained in ambient air (recent global CO2) [17], this is already problematic. For this reason, the sample preparation should be carried out under inert gas atmosphere or rather in organic solvents. Furthermore, different drying methods for the sample material are available. The common methods are the exchange of physically bound water by organic solvents, thermal treatment (evaporation of H2O due to heating), storing in vacuum atmosphere (decreasing the boiling point and accelerated evaporation of H2O) and freeze-drying (sublimation of the arising ice crystals). However, the pre-treatment can also affect the phases contained in cementitious materials [4].
The thermogravimetric analysis is a widely used method for examination of carbonates in case of cementitious materials. During the carbonation process, the hydrate phases, such as CH (4), C–S–H (5) and ettringite (6), react with CO2 or more precisely with carbonic acid (3) [18, 19].
Thereby, the hydrates become transformed into carbonates such as aragonite, vaterite and calcite. The types of the resulting carbonates and the reaction velocity are strongly dependent on the composition of the hardened cement paste and the environmental conditions. A more detailed description of the processes taking place during carbonation can be found in [20,21,22,23,24,25,26,27,28]. However, precise statements for the quantities of carbonates based on the released CO2 prove difficult and may lead to large variations in the results. Carbonates can already arise during cement production or storage due to CO2 and humidity in the ambient air. Furthermore, CaCO3 is already included in cements containing limestone powder or rather aggregates, even without natural or accelerated carbonation. According to DIN EN 197-1 [29], up to 5% of minor additional constituents are permitted. In case of Portland limestone cements and CEM II-based systems, up to 35% limestone can be contained [29]. Carbonates based on limestone decompose in the temperature range of 760–1000 °C. Calcite arisen by carbonation of hydrate phases already decomposes in a range of 550 °C to 760 °C [30]. Additionally, weakly bound or rather amorphous carbonates occur. During carbonation of C–S–H, mainly vaterite and amorphous and weakly bound carbonates appear (approximately 480–700 °C) [31,32,33]. By long-term CO2 treatment, these carbonates become more stable and were also transformed into calcite. Thus, the reaction temperature increases. Mainly, calcite arises during carbonation of calcium hydroxide (700–800 °C). The evaluation of thermogravimetric measurements from samples material containing hydrate phases and carbonates same time proves difficult, especially in the temperature range of 450–600 °C. Within this range, dehydration of portlandite and decomposition of carbonates take place simultaneously. As both effects are linked to mass loss and endothermic reaction, the proper separation by TG or DTA/DSC is excluded. Villain et al. [30] tried to pass by this problem by combination of TG, chemical analysis and gamma densitometry. Chang et al. [34] correlated results from TG, X-ray diffraction analysis (XRD) and Fourier transform infrared spectroscopy (FTIR). In all cases, the effort to perform qualitative or rather quantitative analysis is considerably high.
Within this research, the effect of encapsulation of calcium hydroxide by carbonates based on accelerated carbonation conditions was examined more closely. It is assumed that the higher amount of CO2 during accelerated carbonation leads to encapsulation of the unreacted Ca(OH)2 particles by CaCO3. The permeability of this “passivation” layer is very low. For this reason, the diffusion of CO2 is slowed down. This affects the carbonation rate of Ca(OH)2 [31, 35,36,37]. For this research, an approach was made to receive clear results only by using TG measurements. Samples from different concretes based on three types of cement were exposed to accelerated carbonation. Based on these results, differential curves from carbonated and noncarbonated areas were calculated and changes in the phase composition determined and quantified. Additionally, the sprayed application of phenolphthalein solution, optical microscopic investigations and studies with scanning electron microscopy (SEM) were performed to confirm the previously made assumptions.
Materials and Methods
Three different types of cements (Table 1) were used for preparing concrete specimens with sample size 40 × 40 × 160 mm according to DIN EN 1766 [38]. For the sample preparation, a water/cement ratio (w/c) of 0.7 and the maximum aggregate grain size of 8 mm were used. Then, a notation for the sample designation is used that contains the type of cement (CEM I, CEM II and CEM III) used by a Roman numeral and appended the water/cement ratio used for the mixture. Accordingly, a sample with the designation II_70 was produced from CEM II with a water/cement ratio of 0.7. After demoulding, the specimens were cured for 28 days under water and stored for another 14 days in 20 °C and 65% relative humidity (RH) environment.
The CO2 treatment was carried out in a climate chamber with 1% CO2 in ambient air, 21 °C and 60% RH.
After 56 days, the samples were removed from the climate chamber and fractured as shown in Fig. 1. The specimen part “B” (Fig. 1) was embedded in fluorescent resin, sliced and polished for microscopic examinations. After each preparation step, the specimens were stored in a desiccator (20% RH) in argon atmosphere. For optical characterisation, the stereomicroscope Olympus SZX16 with 500-fold magnification was used. By using the motorised linear positioning table MT mot 50 × 50 (Märzhäuser Wetzlar), the sample surface was scanned. Changes in the microstructure were additionally analysed by a scanning electron microscope (Zeiss EVO MA10). For this purpose, the sample size had to be reduced to 20 × 20 mm, due to the physical dimension of the sample holder. The sample segment was chosen to capture carbonated and noncarbonated areas simultaneously.

Sample preparation for the performed examinations: A: initial carbonation depth; B: optical microscopic analysis and scanning electron microscopy (SEM); C: TG, chemical analysis, D: phenolphthalein test
The specimen part “C” (Fig. 1) was prepared for TG analysis. For these examinations, the samples were cut into slices of 2.0 mm thickness. Subsequently, they were ground by hand with mortar and pestle. To prevent additional carbonation effects, all preparation steps were performed in isopropanol. By using isopropanol for the sample preparation, the interaction with the hydrate phases of the sample material was much weaker compared to methanol or acetone [4]. Furthermore, the storage time in the organic solvent was limited to prevent additional effects. At the end, the liquid phase was separated by vacuum filtration and the solid content was dried in a desiccator in argon atmosphere at room temperature. For thermal analyses, an STA 449C system from Netzsch was used. The sample material was heated from 35 °C to 1000 °C, with a heating rate of 10 °C min−1 and 80 mL min−1 nitrogen flow. For each measurement, 35 mg of the sample material was filled in an Al2O3 crucible and placed in the TG system. Due to the technical equipment and the smallest available crucible with a volume of 300 µl, it was necessary to use this large sample quantity. The used quantity of 35 mg was required to completely enclose the thermocouple with sample material. Only in this way, it was possible to measure the actual sample temperature and not the temperature of the environment. The furnace was evacuated and filled with nitrogen to eliminate oxygen and to prevent any oxidation process during the measurement. For exact analysis of the TG curves and classification of reactions, several hydrate and carbonate phases were previously examined one by one. Therefore, calcium hydroxide (Ca(OH)2), calcium sulphate dihydrate (CaSO4·2H2O) and calcium carbonate (CaCO3) as ground powder were used. Furthermore, calcium silicate hydrate (C–S–H) with a calcium/silica ratio of 1.75 was synthesised and carbonated subsequently. The synthesis was carried out in accordance with the method proposed by Merlin et al. [39]. Additionally, pure calcium hydroxide was also carbonated in the climate chamber at 1% CO2 in ambient air, 21 °C and 60% RH.
The fractured surface of the specimen part “D” (Fig. 1) was processed by sprayed application of phenolphthalein solution. The test was carried out according to DIN EN 13,295:2004-08 [40] in conjunction with DIN EN 14,630:2007-01 [41]. The fresh fractured surfaces were treated with solution of 1% phenolphthalein dissolved in ethanol. The carbonation depth was determined after 60 min by measuring the distances between the boundary of colour change and sample edge for each side of the sample. The average of the values represented the carbonation depth for each sample. The phenolphthalein test was used as reference method for preliminary examinations of the test specimens. The results of more extensive indicator tests on the specimens used here can be taken from reference [36].
For the clarification of the encapsulation phenomena, Ca(OH)2 was stored under accelerated carbonation conditions at 1% CO2 in air and 60% RH. After 1d, 2d, 3d, 6d, 9d, 12d and 28d of treatment, the sample material was examined by TG. Since CaCO3 has a 11.8% higher volume compared to Ca(OH)2 [21], the transformation process should be also evident by an increase in the particle size distribution. For this purpose, the laser granulometric system HELOS from Sympatec, with an optical range of 0.1–875 µm, was used. The sample material was dispersed with ultrasonic in isopropyl alcohol solution. In order to exclude further effects caused by the storing conditions and dispersion properties (hydration shell or hygroscopic properties), Ca(OH)2 was also stored in a desiccator under argon atmosphere. To achieve the necessary relative humidity of approximately 60% of RH, saturated salt solution of magnesium nitrate (Mg(NO3)2) was added to the desiccator.
Results and discussion
TG
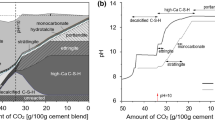
To distinguish the different hydrate and carbonate phases, the first derivation of the TG curves (i.e. DTG) was calculated. Figure 2a, b and c represents the results from different types of concrete (specimens I_70 (a), II_70 (b) and III_70 (c)).

Results of thermal gravimetric analysis. a, b and c: Progress of carbonation by depth depending on DTG results and d, e and f: differential curves from DTG results
The exact assignment of the different curves is shown in Fig. 1. Thereby, the number of curves (1–5) corresponds to the number of the appropriate sample slices (1–5). Curve (5) represents the core area of the examined specimens and is not noticeably influenced by carbonation yet. Accordingly, each curve represents another state of carbonation. Coincidentally, the carbonation depth can be determined. For all three examined types of concrete, the calcium hydroxide peak at 450 °C temperature disappears between curve (2) and curve (3). Regarding the total depth, each slice has 2 mm thickness plus two times 1 mm for the saw thickness. In sum, the relevant structural changes based on carbonation became evident down to the region of approximately 6 mm. The strong decrease of Ca(OH)2 and the associated reduction of hydroxide ions inevitably lead to the lowering of the pH value in this area. However, the transformation processes due to carbonation were already taking place at this point in deeper areas of the examined specimens.
Additionally, the carbonation behaviour of the three investigated concretes was evaluated by the DTG results. In case of CEM I- and CEM II-based systems, mainly calcium hydroxide (450 °C) [5] was formed (Fig. 2a, b). In contrast, a lower amount of calcium hydroxide arose in case of CEM III-based system. Also, AFm phases (approximately 140 °C) [5, 42] and C–S–H and ettringite (AFt) (approximately 120 °C) [5, 42] were verifiable in all three types of concrete (Fig. 2a, c).
Unfortunately, physically bound water, and for example limestone powder, makes it difficult to analyse the hydration and carbonation behaviour accurately. For this research, the differential curves of DTG results from carbonated and noncarbonated areas were calculated. The main goal of this approach was to eliminate all artefacts which are not related to the accelerated carbonation. This method provides a signal which occurs negative for decomposing components and positive for emerging components of the hardened cement paste based on carbonation. The DTG curves from the core area (Fig. 2a, c, curve (5)) were used as reference. By calculating the differential curve of the reference and the respective DTG curves (Fig. 2a–c, curves (1)–(4)), the desired results are achieved (Fig. 2d–f). It must be considered that the initial material composition of the whole specimen was assumed as homogeneous before CO2 treatment. Thus, the initial differences of the composition between core area (reference) and affected areas (carbonated) were estimated as low compared to the changes based on carbonation. Curve (IV) of samples II_70 and III_70 (Fig. 2e, f) shows an increase in the temperature range up to 200 °C. This behaviour is not related to hydration or carbonation effects. The examined sample material contained a considerable amount of aggregates. These aggregates were unevenly distributed to the individual samples. Consequently, the share of hydrates fluctuated. Effects based on this variation are dominating when carbonation has only a minor impact. Consequently, a faulty signal was obtained.
A comparison of the results with the previous examined main phases of hydrates and carbonates allows an appropriate interpretation of the differential curves based on the DTG results (Fig. 3).

The differential curves from the core area (noncarbonated) and surface area (fully carbonated) of the samples I_70 (CEM I), II_70 (CEM II) and III_70 (CEM III) combined with the determined thermal reactions based on the comparative studies (grey value distribution)
The release of water took place up to 500 °C, which corresponds to the results in the literature [5, 13, 36, 37]. By the synthesis of C–S–H according to Merlin et al. [39], a gel-like substance was obtained. Subsequently, it was dried and stored in a desiccator in argon atmosphere at room temperature. Based on these results, a mass loss in the range of 80 °C to 150 °C can be attributed to water release of C–S–H. This agrees with results from the literature [5, 42]. Lothenbach et al. [4] specify the decomposition range of C–S–H up to 600 °C. This could not be confirmed during these preliminary examinations. In case of carbonated C–S–H, the release of CO2 took place between 450 °C and 700 °C. During carbonation of Ca(OH)2, carbonates with higher calcination temperatures (700–800 °C) arose (Fig. 3). Additionally, it became visible that the Ca(OH)2 was not fully transformed into CaCO3 (Fig. 2a, b and Fig. 3). It is assumed that the higher amount of CO2 during accelerated carbonation led to encapsulation of the unreacted Ca(OH)2 particles by CaCO3. In case of carbonated Ca(OH)2, it was also recognisable that in the temperature range between 450 °C and 550 °C, H2O and CO2 were released same time [23]. The examination of pure CaCO3 (marble powder), shown in Fig. 3 (CaCO3), led to an even higher reaction temperature compared to carbonated Ca(OH)2. Synthesis of ettringite (AFt) and AFm phases could not be carried out for this research. Therefore, results from the literature were used for evaluation [4, 5, 9].
In the noncarbonated area (Fig. 2a, curve (5)) of the CEM I-based concrete, C–S–H, ettringite (AFt) and AFm phases (100–180 °C) and portlandite (450 °C) were contained. Simultaneously, only small quantities of carbonates were apparent (> 500 °C). The calculated differential curves showed that the carbonation started simultaneously on C–S–H, ettringite (AFt), AFm phases and Ca(OH)2. Thereby, mainly calcite (approximately 750 °C) and a small amount of poorly crystallised carbonates (< 700 °C) arose. At the time, when a larger quantity of C–S–H, ettringite (AFt) and AFm phases got transformed, particularly amorphic or poorly crystallised carbonates arose (Fig. 2d, curve II).
In the CEM II-based concrete, calcite (based on limestone) was already contained in the noncarbonated area (Fig. 2b, curve 5). The first clear signs for changes based on carbonation were apparent on the decreased amount of Ca(OH)2 (Fig. 2e, curves (IV)–(III)). Thereby, calcite and poorly crystallised carbonates arose (Fig. 2e, curve III). Therefore, it cannot be excluded that C–S–H, ettringite (AFt) or AFm phases already started to convert. It is also possible that Ca(OH)2 got transformed into calcite via an intermediate stage of poorly crystallised carbonates. Here too, the connection between disappearing C–S–H, ettringite (AFt) and AFm phases and the increasing amount of poorly crystallised carbonates (Fig. 2e, curve (II)) was evident.
In the CEM III-based concrete, calcite (temperature range 690 °C to 780 °C) was already contained in the noncarbonated area (Fig. 2f, curve (5)). The Ca(OH)2 transformation into calcite was not evident at this time. However, the calculated mass loss of CO2 from TG analysis was higher compared to the results determined by the loss of ignition (Table 1) of the pure cement. As shown in Fig. 2f (curve (IV)), the amount of AFm phases, C–S–H and ettringite (AFt) was already decreased at this early stage. Consequently, it can be assumed that the contained calcite was attributed to an advanced carbonation process of AFm phases, C–S–H and ettringite (AFt). Compared to the CEM I- and CEM II-based concretes, a higher amount of C–S–H (100–150 °C) was available and got transformed into poorly crystallised carbonates (450–680 °C). Simultaneously, the Ca(OH)2 content and the arisen calcite were considerably low compared to the CEM I- and CEM II-based systems. It was found that the carbonation of the CEM III-based sample started on C–S–H and ettringite (AFt). Accordingly, the results published by Castellote et al. [43] were confirmed by the results of thermogravimetric analysis. At the time, when the amount of Ca(OH)2 has started to decrease, the calcite peak increases (Fig. 6f, curve (II)). It can be assumed that the quick transformation of Ca(OH)2 is attributed to the open pore structure caused by disappearing C–S–H and ettringite (AFt) and the resulting carbonation shrinkage. Consequently, a larger amount of CO2 can penetrate the matrix.
The results indicate that Ca(OH)2 was transformed directly or very quickly via an intermediate carbonation state into calcite (> 700 °C). The resulting carbonates in the range between 450 °C and approximately 700 °C were particularly based on transformation of C–S–H, ettringite (AFt) and AFm. From the literature, it can be assumed that beside amorphic- and poorly crystallised carbonates also vaterite and aragonite emerged. These carbonates arise especially during carbonation of C–S–H due to appropriate calcium-to-silica ratio [13]. Furthermore, it is assumed that in case of the CEM III-based concrete, the formation of aragonite was increased by the higher quantity of magnesium compared to the investigated CEM I and CEM II systems [36, 37, 39]. Both modifications are metastable and become transformed into calcite in the later stage of carbonation. In all three cases, it was evident that the carbonates change to more stable carbonate modifications with increasing carbonation time.
By using numerical integration methods to recalculate the mass loss from the differential curves, also quantitative analysis can be performed. Based on the curve slope, the mass loss can be calculated by using the Simpson’s rule (7).
For the application of the Simpson’s rule, the area between starting temperature \(\left( {\vartheta _{\rm{start}} } \right)\) and final temperature \(\left( {\vartheta _{\rm{end}} } \right)~\) becomes subdivided into an even number (N) of strips with the same width (h). In this case, the value of h corresponds with the sampling rate. The number of the measured values is stated by N. The function \(f\left( \vartheta \right)\) corresponds to the distance between baseline and the respective differential curve values.
For this approach, the calculated area between differential curve and zero line corresponds with the mass loss. It should be noted that the signal shape for carbonates is positive in the case of the calculated differential curves. However, the decomposition of carbonates is associated with mass loss. Accordingly, all positive differential curve values must be reversed (negative algebraic sign). It is also important that the determined mass loss is only related to the transformation caused by the carbonation. Consequently, the results do not correspond to the actual amount of hydrates and carbonates in the sample material. The considerable differences between the determined TG curves and corrected curves are shown in Fig. 4a–f. Moreover, the individual mass loss steps of the different hydrate and carbonate phases became more apparent.

Corrected TG curves, created by numerical integration from the differential curves. a CEM I—hydrates (< 200 °C), b CEM I—carbonates, c CEM II—hydrates (< 200 °C), d CEM II—carbonates, e CEM III—hydrates (< 200 °C), (f) CEM III—carbonates
The DTG curves of the samples I_70 and II_70 indicate portlandite even in the apparently fully carbonated areas. Accordingly, the Ca(OH)2 was not fully transformed into CaCO3. In contrast, this effect was not detected in case of sample III_70. To exclude leaching effects during the underwater storage, the total amount of calcium oxide was determined. Therefore, untreated (noncarbonated) sample material was ground and examined by ICP-OES. Subsequently, the results were compared with the results of the used pure cements. However, the comparison showed no considerable differences.
The additionally performed experiments for the encapsulation effect of calcium hydroxide by carbonates showed a significant correlation between emerging carbonates and shifting of the particle size distribution. For this purpose, pure calcium hydroxide was exposed to CO2 and subsequently examined. The results of the simultaneously performed experiments under argon atmosphere yielded no considerable shift of the particle size distribution (Fig. 5). The results of the parameter d50 are slightly scattered but unaffected by the storage time. The parameter d50 means that 50% of all measured particles are smaller compared to this value. The thermogravimetric analysis showed no significant changes in the composition of the sample material. Consequently, it can be assumed that a hydration shell or hygroscopic properties had no influence on the results. The examinations of the sample material treated by accelerated carbonation exhibit a significant shift of the particle size distribution after 1d. In the period between 12 and 28 days, an additional shift of the particle size distribution to a higher d50 value was determined.

Changes of the particle size distributions in conjunction with an increase of the CaCO3 content based on carbonation effects. Determined by laser granulometry. Rectangular symbol: stored in atmosphere with 1% CO2. Triangular symbol: stored in desiccator in argon atmosphere. Treatment time: 1d to 28d
Considerable changes in the TG results (increasing quantity of calcite) were only apparent up to three days of treatment (Fig. 5). Subsequently, only minor changes in the results were recognisable. It can be assumed that calcite crystals already arose during the first 24 h of treatment. The size of these crystals already led to a significant shift of the particle size distribution. These crystals had not yet formed a high dense shell, and CO2 was able to penetrate. Consequently, the carbonation process was further in progress. After three days, the tightness of the shell reached a high level and the transformation from calcium hydroxide to calcite was stopped. Up to 12 days, the particle size was constant. The considerable increase of the d50 value in the period up to 28 days was not attributed to additional transformation of calcium hydroxide. An exact explanation for this additional shift in the particle size distribution was not possible by the methods used here.
Microstructural examinations
For the microscopic examinations, the samples were embedded in fluorescent resin and subsequently ground and polished. A correlation between depth of resin penetration and carbonation depth was determined during optical microscopic examinations (Fig. 6). The intensity of the UV light source was reduced for the examination of the CEM III-based sample. This was necessary because of a considerable outshine of the edge region. Consequently, a direct comparison based on the intensity was excluded.

Imbedded sample material in fluorescent resin, captured by optical microscopy (ultraviolet light source) and samples treated by sprayed application of phenolphthalein. a–d I_70 (CEM I, w/b 0.7), e–h III_70 (CEM III, w/b 0.7)
Nevertheless, from Fig. 6c it is apparent that the noncarbonated area fluoresces more intense than the carbonated edge region of the sample I_70. Due to the progress of carbonation, pores and cracks were filled with carbonates. This is caused by an 11.8% higher volume of calcium carbonate compared to calcium hydroxide [21]. Accordingly, the tightness of the hardened cement pastes was increased [31, 32]. By this reason, a lower amount of fluorescent resin penetrated to the edge region and it appeared darker. The reverse effect was observed on the CEM III-based sample. The core area of sample III_70 (Fig. 6g) which was noncarbonated appeared darker. Accordingly, a higher tightness of the noncarbonated hardened cement past can be assumed. For the clarification of this effect, the microstructure of the hardened cement paste was examined by SEM.
In both cases, an SEM overview scan at lower magnification showed a clear separation between noncarbonated and carbonated areas (Fig. 7).

Overview image of the transition zone between noncarbonated and carbonated areas of CEM I-based (a) and CEM III-based (b) systems generated by SEM
The transition zone was in a scale of a few micrometres. Accordingly, there was only narrow range where recognisable microstructural changes took place. Additionally, the crack width before and after treatment with CO2 was determined by means of SEM images. The evaluation of the shrinkage cracks showed decreasing crack widths due to carbonation. This is regardless of the used type of cement (Fig. 8a, b).

Distribution curves of the crack width before and after carbonation. a I_70 (CEM I, w/b 0.7), b III_70 (CEM III, w/b 0.7)
The results correspond to the expectations from the literature [44]. The maximum of the crack width distribution curve from the noncarbonated sample III_70 was in the region of 0.9 to 1.1 µm. After carbonation, the maximum was shifted to 0.1 to 0.3 µm. The distribution curve maximum of sample I_70 decreased only slightly. Under both noncarbonated and carbonated condition, the maximum was in the region of 0.3 µm to 0.5 µm. Nevertheless, changes in the microstructure based on carbonation were clearly apparent.
In Fig. 9, b, the hardened cement past (noncarbonated (a) and carbonated (b)) of sample I_70 is shown. As expected, the structure became more compact by the arisen CaCO3. In case of sample III_70 (Fig. 9c, d), a clear statement of the microstructural behaviour was not possible. Indeed, the emerging CaCO3 led to sealing of shrinkage cracks and air voids. Consequently, the crack and pore structure of sample III_70 was not responsible for the better penetration of fluorescent resin. By the hand of this SEM image, it was not possible to determine whether the structure of the hardened cement paste becomes rougher at all due to carbonation shrinkage.

Hardened cement paste of (a, b) CEM I-based concrete (w/b 0.7) and (c, d) CEM III-based concrete (w/b 0.7) before and after carbonation generated by SEM analysis
Joint consideration of TG results and microscopic examinations
Compared to sample I_70, a considerable smaller quantity of Ca(OH)2 was determined by TG analysis for sample III_70. Accordingly, the quantity of arisen carbonates was also lower (Table 2). The SEM analysis of the sample III_70 yielded a decrease in the shrinkage crack width due to carbonation. In contrary, the microscopic examinations showed a correlation between carbonation depth and resin penetration depth. As a result, it can be assumed that the arisen carbonates were sufficient to refill the shrinkage cracks. Simultaneously, the open pore structure based on the carbonation shrinkage probably caused the correlation of carbonation depth and the penetration of the resin. When larger quantities of C–S–H were affected by carbonation, the quantity of remaining calcium was too low to form sufficient calcium carbonate crystals and refill the open pore structure. It was also conceivable that a two-step process takes place during carbonation. First, C–S–H, ettringite (AFt) and AFm were decomposed and aragonite and vaterite arose. By the presence of H2O, vaterite got solved [33, 45, 46]. Subsequently, it got recrystallized as calcite. Whether the higher compactness of calcite compared to vaterite and aragonite or additional migration effects of the arisen carbonates played a role during these processes could not be clarified here.
Conclusions and outlook
For this research, an approach was made to describe the progress of carbonation by using thermal gravimetric analysis. Concretes based on CEM I, CEM II and CEM III were treated by 1% CO2 in air atmosphere and examined subsequently. For all admixtures, a water–binder ratio of 0.7 was chosen. The evaluation of TG results proves difficult because of overlapping reactions which are not related to carbonation effects. Mass losses based on physically bound water overlap the signals induced by the different hydrate phases. Additionally, already available carbonates such as limestone powder generate signals which are also not related to carbonation effects. During evaluation of TG results, it must be noted that signals may vary slightly based on device-specific fluctuations. For this reason, preliminary TG investigations of different components from hydrated and carbonated concretes were carried out. By the means of these results, the evaluation of the examined types of concretes was implemented. During these preliminary investigations, an effect was examined which was mentioned before but not described that precisely. It has been shown that accelerated carbonation (1% CO2 in air atmosphere) of calcium hydroxide leads to encapsulation of the hydrated particle by CaCO3 covering. The progress of the calcium hydroxide transformation was clearly evident by TG analysis during the first 72 h of CO2 treatment. The additionally examined particle size distribution showed an increase in the PSD within the first 24 h. Subsequently, the particle size remained on a constant level. Based on these results, a two-stage process is conceivable. During the first 24 h, individual carbonate crystals arose, and the particle size increased noticeable. Thereby, the arisen crystals do not form a dense shell of carbonates. Accordingly, the carbonation process was further in progress. During the next 48 h, more crystals arose, and the covering became dense. Simultaneously, the crystals achieved a certain size and the hydrate–carbonate particles stopped growing for now. After 72 h, the dense of the shell reached a certain value and the carbonation was stopped. After a period of 12 days, the particle size increased again without any additional arose of carbonates. A clarification for the additional growing of the particle size was not possible with the methods used for this research. Finally, the encapsulation effect is based on an oversupply of CO2. Following, the accelerated carbonation already with 1% CO2 or higher in air atmosphere leads to effects which are not observable during natural carbonation.
The former described encapsulation effect of calcium hydroxide was also detected in case of the CEM I- and CEM II-based systems. Additionally, it was ascertained that microstructural changes have already taken place in areas which are supposed as not carbonated by the indicator. Especially in case of CEM I-based system, these changes became apparent. Analysis by optical microscopy and SEM of the hardened cement paste showed also microstructural changes. During carbonation of CEM I- and CEM II-based systems, the structure became denser because of the arisen carbonates. The microscopic examinations on the CEM III-based system with UV light on samples imbedded in fluorescents resin suggested a lower density in the already carbonated area. Primary leaching effects based on the underwater storage of the specimens were excluded by quantification of the chemical composition (ICP-OES). Therefore, all changes fed back to carbonation. It can be assumed that the amount of calcium was not sufficient to create adequate quantities of calcium carbonate and refill the open pore structure. Nevertheless, by means of SEM it became apparent that the shrinkage cracks were filled by carbonates. Another possible cause might be that mainly aragonite and vaterite arose during carbonation of C–S–H, ettringite (AFt) and AFm phases. These two CaCO3 modifications are metastable. By the presence of H2O, they got solved and became transformed into calcite at a later stage. Nevertheless, it was currently difficult to estimate whether additional transport effects of the solved carbonates in the pore solution increase the open pore structure finally.
References
Hemminger WF, Cammenga HK. Methoden der thermischen Analyse. Berlin Heidelberg New York: Springer; 1989.
Haines PJ. Principles of Thermal Analysis and Calorimetry. Cambridge: Royal Society of Chemistry; 2002.
Höhne G, Hemminger WF, Flammersheim H-J. Differential Scanning Calorimetry. Berlin Heidelberg: Springer; 2003.
Scrivener K, Snellings R, Lothenbach B. A practical guide to microstructural analysis of cementitious materials. Boca Raton: CRC Press Taylor & Francis Group; 2016.
Collier NC. Transition and decomposition temperatures of cement phases – a collection of thermal analysis data. Ceram-Silikaty. 2016;60–4:338–43.
Blaine R. Determination of calcium sulfate hydrates in building materials using thermal analysis. Am Lab. 1995;27–14:24–8.
Borges PHR, Costa JO, Milestone NB, Lynsdale CJ, Streatfield RE. Carbonation of CH and C-S-H in composite cement pastes containing high amounts of BFS. Cem Concr Res. 2010;40:284–92.
Chen W, Xu W, Li Y. Microstructural investigations of carbonation of calcium silicate hydrate in hydrated cement paste. Adv Mat Res. 2011;261–263:601–5.
Dweck J, Ferreira da Silva PF, Büchler PM, Cartledge FK. Study by thermogravimetry of the evaluation of ettringite phase during type II Portland cement hydration. J Therm Anal Calorim. 2002;69:179–86.
Dweck J, da Cunha ALC, Pinto CA, Gonçalves JP. Thermogravimetry on calcined mass basis – hydrated cement phases and pozzolanic activity quantitative analysis. J Therm Anal Calorim. 2009;97:85–9.
Gruyaert E, Robeyst N, De Belie N. Study of hydration of Portland cement blended with blast-furnace slag by calorimetry and thermogravimetry. J Therm Anal Calorim. 2010;102:941–51.
Pavlík Z, Trník A, Kulovaná T, Scheinherrová L, Rahhal V, Irassar E, Černý R. DSC and TG analysis of a blended binder based on waste ceramic powder and Portland cement. Int J Thermophys. 2016;37:32.
Torréns-Martín D, Fernández-Carrasco L, Blanco-Varela MT. Thermal analysis of blended cements. J Therm Anal Calorim. 2015;121:1197–204.
Petkova V, Stoyanov V, Pelovski Y. TG–DTG–DTA in studying white selfcompacting cement mortars. J Therm Anal Calorim. 2012;109:797–806.
Rodriguez ET, Garbev K, Merz D, Black L, Richardson IG. Thermal stability of C-S-H phases and applicability of Richardson and Groves’ and Richardson C-(A)-S-H(I) models to synthetic C-S-H. Cem Concr Res. 2017;93:45–56.
Trník A, Scheinherrová L, Medve I, Černý R. Simultaneous DSC and TG analysis of high performance concrete containing natural zeolite as a supplementary cementitious material. J Therm Anal Calorim. 2015;121:67–73.
NOAA (National Oceanic and Atmospheric Administration), Earth System Research Laboratory, Trends in Atmospheric Carbon Dioxide. https://www.esrl.noaa.gov/gmd/ccgg/trends/. Accessed 03 Aug 2020
Reschke T, Gräf H. Influence of the alkali content of cement on the carbonate of mortar and concrete, Betontechnische Berichte. Nr.27, Verein Deutscher Zementwerke e.V. VDZ, Forschungsinstitut der Zementindustrie. 1998;145–153
Urbonas L, Heinz D. Einfluss der beschleunigten Carbonatisierung mit überkritischem CO2 auf die Eigenschaften von Zementstein, Wissenschaftlicher Kurzbericht Nr. 24, Centrum Baustoffe und Materialprüfung TU München. (2011) Available online: https://www.cbm.bgu.tum.de/fileadmin/w00bdr/www/pdf/Kurzberichte/KB_24.pdf .Accessed 12 July 2018
Stark J, Wicht B. Zement und Kalk: Der Baustoff als Werkstoff. Basel-Boston-Berlin: Springer; 2000.
Leemann A, Moro F. Carbonation of concrete: the role of CO2 concentration, relative humidity and CO2 buffer capacity. Mater Struct. 2017;50:30.
Leemann A, Nygaard P, Kaufmann J, Loser R. Relation between carbonation resistance, mix design and exposure of mortar and concrete. Cem Concr Compos. 2015;62:33–43.
Morandeau A, Thiéry M, Dangla P. Investigation of the carbonation mechanism of CH and C-S-H in terms of kinetics, microstructure changes and moisture properties. Cem Concr Res. 2014;56:153–70.
Fernández Bertos M, Simons SJR, Hills CD, Carey PJ. A review of accelerated carbonation technology in the treatment of cement-based materials and sequestration of CO2. J Hazard Mater. 2004;B112:193–205.
Ekolu SO. A review on effects of curing, sheltering, and CO2 concentration upon natural carbonation of concrete. Constr Build Mater. 2016;127:306–20.
Šavija B, Lukovic M. Carbonation of cement paste: understanding, challenges, and opportunities. Constr Build Mater. 2016;117:285–301.
Castellote M, Fernandez L, Andrade C, Alonso C. Chemical changes and phase analysis of OPC pastes carbonated at different CO2 concentrations. Mater Struct. 2009;42:515–25.
Cui H, Tang W, Liu W, Dong Z, Xing F. Experimental study on effects of CO2 concentrations on concrete carbonation and diffusion mechanisms. Constr Build Mater. 2015;93:522–7.
DIN EN 197–1 Cement Part 1: Composition, specifications and conformity criteria for common cements. 2011
Villain G, Thiery M, Platret G. Measurement methods of carbonation profiles in concrete: thermogravimetry, chemical analysis and gammadensimetry. Cem Concr Res. 2007;37:1182–92.
Groves GW, Brough A, Richardson IG, Dobsont CM. Progressive changes in the structure of hardened C3S cement pastes due to carbonation. J Am Ceram Soc. 1991;74–11:2891–6.
Neves Junior A, Dias Toledo Filho R, Fairbairn EDMR, Dweck J. A study of the carbonation profile of cement pastes by thermogravimetry and its effect on the compressive strength. J Therm Anal Calorim. 2014;116:69–76.
Branch JL, Kossona DS, Garrabrants AC, He PJ. The impact of carbonation on the microstructure and solubility of major constituents in microconcrete materials with varying alkalinities due to fly ash replacement of ordinary Portland cement. Cem Concr Res. 2016;89:297–309.
Chang C-F, Chen J-W. The experimental investigation of concrete carbonation depth. Cem Concr Res. 2006;36:1760–7.
Vogler N, Lindemann M, Drabetzki P, Kühne H-C. Alternative pH-indicators for determination of carbonation depth on cement-based concretes. Cem Concr Compos. 2020;109:103565.
Galan I, Glasser FP, Baza D, Andrade C. Assessment of the protective effect of carbonation on portlandite crystals. Cem Concr Res. 2015;74:68–77.
Hidalgo A, Domingo C, Garcia C, Petit S, Andrade C, Alonso C. Microstructural changes induced in Portland cement-based materials due to natural and supercritical carbonation. J Mater Sci. 2008;43:3101–11.
DIN EN 1766 Products and systems for the protection and repair of concrete structures. Test methods-Reference concretes for testing. 2017
Merlin F, Lombois H, Joly S, Lequeux N, Halary J-L, v. Damme H. . Cement-polymer and clay-polymer nano- and meso-composites: spotting the difference. J Mater Chem. 2002;12:3308–15.
DIN EN 13295 Products and systems for the protection and repair of concrete structures. Test methods-Determination of resistance to carbonation. 2004
DIN EN 14630 Products and systems for the protection and repair of concrete structures. Test methods-Determination of carbonation depth in hardened concrete by the phenolphthalein method. 2007
Taylor HF. Cement Chemistry. 2nd ed. London UK: Thomas Telford Publishing; 1997.
Castellote M, Andrade C, Turrillas X, Campo J, Cuello GJ. Accelerated carbonation of cement pastes in situ monitored by neutron diffraction. Cem Concr Res. 2008;38:1365–73.
Auroy M, Poyet S, Le Bescop P, Torrenti J-M, Charpentier T, Moskura M, Bourbon X. Comparison between natural and accelerated carbonation (3% CO2): Impact on mineralogy, microstructure, water retention and cracking. Cem Concr Res. 2018;109:64–80.
Plummer LN, Busenberg E. The solubilities of calcite, aragonite and vaterite in CO2-H2O solutions between 0 and 90°C, and an evaluation of the aqueous model for the system CaC03-CO2-H20. Geochim Cosmochim Acta. 1982;46:1011–40.
De Visscher A, Vanderdeelen J. Estimation of the solubility constant of calcite, aragonite, and vaterite at 25°c based on primary data using the Pitzer ion interaction approach. Monatsh Chem. 2003;134:769–75.
Acknowledgements
The authors would like to thank all colleagues from the Federal Institute for Materials Research and Testing (BAM) for support and input while executing this research.
Funding
Open Access funding enabled and organized by Projekt DEAL. This research did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
All authors were involved in the conception and implementation of the manuscript. The material preparation, the execution of experiments as well as data collection and analysis were carried out by Nico Vogler, Philipp Drabetzki and Mathias Lindemann. In his function as head of department, Hans-Carsten Kühne supported this research by providing the infrastructure and by contributing scientific content. The first draft of the manuscript was written by Nico Vogler and all authors commented on earlier versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vogler, N., Drabetzki, P., Lindemann, M. et al. Description of the concrete carbonation process with adjusted depth-resolved thermogravimetric analysis. J Therm Anal Calorim 147, 6167–6180 (2022). https://doi.org/10.1007/s10973-021-10966-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-021-10966-1