
Abstract
The polymer structure impact on the adsorption layer conformation formed on the chromium (III) oxide surface was investigated. As adsorbates, three macromolecular compounds were tested: poly(l-aspartic acid) homopolymer and two block copolymers containing the poly(amino acid) segments as well as the poly(ethylene glycol) ones in the chain structure. Due to the ionic nature of the poly(amino acid), all measurements were carried out as a function of solution pH value. The most probable polymer chain binding mechanism was determined on the basis of the values obtained from the adsorption, potentiometric titration, turbidimetry and thermogravimetry experiments. The acquired results indicate that the solution pH has a great influence on the adsorbed macromolecule conformation. Additionally, in the case of the block copolymers, the individual structural unit affinity for the solid particles can be modified by the pH changes. Therefore, a significant difference in the mass decrement measured for the systems at pH 3 and 10 can be explained by the contribution of electrostatic forces and hydrogen bond formation to the polymer adsorption mechanism. Structure of the adsorbed polymer layer determines also the suspension stability. The solid surface charge neutralization by the macromolecules bound to the Cr2O3 surface at pH 3 impairs the sample stability, whereas the presence of the extended polymer chains in the basic solution leads to the durability improvement.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polymers adsorption at the solid–liquid interface is a very sophisticated process depending on various factors, but at the same time this phenomenon finds numerous applications in many industrial branches as well as in human activities [1–4]. However, it is a complex process depending on numerous factors related to the macromolecular compound structure, nature of adsorbent surface as well as the conditions of the entire process [5–8].
Chromium (III) oxide (Cr2O3) is a dark green, amorphous powder, forming hexagonal crystals on heating that are insoluble in water or acids. It is broadly used in many industry branches. Cr2O3 finds application in the metallurgical processing of ferrochromium and other metallurgical products to impart corrosion resistance, chiefly stainless steel. It is one of the most frequently used dyes in the industry. Chromium (III) oxide is applied for coloring a concrete, as a stable green pigment for toys, plastics and leather. The aqueous suspension of Cr2O3 is very popular substance in the production of banknotes and green ink. It is also used in paint and ceramic industries, a heterogeneous catalyst as well as a coating for providing mechanical and thermal protection. Due to the light and chemical resistance, chromium (III) oxide is required for glass, ceramics and polymers. Moreover, Cr2O3 can be used as an ingredient in cosmetics (production of eye shadows), dye in bronzers, lipsticks and cheek rose. It is possible due to the fact that this solid is chemically stable under a wide range of solution pH conditions [9–12].
The presence of the polymers in aqueous medium has a significant influence on the colloidal system stability. As a result of macromolecular compound adsorption, the polymer chains assembled on the solid surface modify the interactions present in the system. As a consequence, two opposite effects can be observed. First of all, the presence of polymer can increase the particle suspension durability (on account of steric or electrosteric stabilization mechanism). In the second case, the macromolecules adsorbed at the solid–liquid interface lead to a complete system destabilization (bridging flocculation, depletion interactions or charge neutralization) [13, 14]. Therefore, in terms of the applications involving preparation of stable aqueous suspensions (as in case of coloring a concrete, leather, production of banknotes and magnetic composites containing Cr2O3 particles), the addition of suitable polymer is necessary to obtain a stable system [15–20].
Searching for new stabilizers, the suspension-destabilizing agents, or the surface modifiers has led to the increased interest in copolymers—the compounds consisting of two (or more) types of structural units connected via a covalent bond. All blocks present in the copolymer macromolecules can exhibit a different spatial structure and chain length as well as the opposite ionic character, polarity or affinity for the solid surface. Hence, these substances show considerably different properties compared with those of proper homopolymers which are their building blocks [21]. In relation to the applied solution pH conditions, the ionic blocks may achieve a variety of conformations at the solid–liquid interface depending on the electrostatic forces occurring in the studied sample. Usually under appropriate conditions, one of the polymer structural units undergoes a strong adsorption (an anchor), whereas the other is extended into the bulk solution (a buoy) [22–26].
Polyamino acids are a group of synthetic polymers whose backbone is made up of individual amino acid monomers linked by a stable peptide bond. Besides the synthetic origin, the prepared polymers exhibit a number of important properties relevant to medicine, pharmacy and environmental protection. First of all, the macromolecules are characterized by complete biodegradability and biodegradability. Secondly, these compounds are characterized by an excellent solubility in water and resistance to pH changes. As a result, in the future, they can replace polymeric substances currently used which are not decomposed in the environment (or they are slightly biodegradable) [27–29].
Since interactions between various metal oxides and polymers containing only one type of the functional groups were repeatedly tested, the copolymer adsorption mechanism on the solid surface should be precisely investigated. In the presented paper, three different polymers were used as adsorbates: the poly(l-aspartic acid) homopolymer (ASP) and two block copolymers containing ASP and poly(ethylene glycol) (PEG) segments in the chain structure. It is worth noting that poly(aspartic acid) is anionic, water-soluble compound. Because of the unique properties such as dispersing activity and biodegradability, ASP can be used as a component of laundry detergents, dishwashing as well as water treatment chemical [28, 30]. Both the anionic polyamino acid and the nonionic PEG play an important role applied as a dispersing agent not only in the industrial processes but also in biomedicine to obtain stable systems used in MRI probes [27, 28, 31–33].
The aim of the paper was to investigate the polymer structure impact on the adsorption layer conformation. Application of the different types of measurements (adsorption, potentiometric titration, turbidimetry, thermogravimetry) allows to determine the homopolymer and copolymer binding mechanism at the Cr2O3–polymer solution interface. Moreover, the conclusions resulting from the comparison of dependencies obtained from different techniques can be very useful due to the potential application of both the solid and the polyamino acid homopolymer (or the PEGylated copolymers).
Experimental
Materials
BET surface area and particle size of adsorbent
Chromium (III) oxide (Cr2O3) (POCh Gliwice, Poland) was used as an adsorbent. The Cr2O3 specific surface area determined by the BET method was found to be 7.12 m2 g−1 (Micromeritics ASAP 2405 analyzer). The point of charge (pHpzc) value was equal to 7.6 [34]. The average Cr2O3 particles size (dynamic light scattering, Zetasizer 3000, Malvern) was 265 nm [35]. The particle diameter is calculated on the basis of the Brownian motions. The detector (at the angle of 90°) analyzes the scattered light fluctuations intensity. The dependency between the size and the particle velocity is given by the Stokes–Einstein equation:
r, the radius of the spherical particle/nm; k, Boltzmann’s constant; T, the absolute temperature/K; η, the dynamic viscosity/kg m−1s−1; D, the diffusion coefficient/m2 s−1.
Structure of applied homopolymer and block copolymers
As macromolecular compounds, three classes of polymers were applied (all from Alamanda Polymers, USA). One of them is simple polyamino acid: poly(l-aspartic acid sodium salt)—ASP. The second group consists of the two block copolymers:
-
poly(l-aspartic acid sodium salt)-block-poly(ethylene glycol), designated as AP,
-
poly(l-aspartic acid sodium salt)-block-poly(ethylene glycol)-block-poly(l-aspartic acid sodium salt), described as APA.
The average molecular weights of the applied polymers were 6800 Da for ASP, 26,800 Da for AP diblock copolymer, and 29,000 Da for APA triblock copolymer. The analyzed polymer structure is shown in Fig. 1. The polydispersity index (PDI) which is a measure of the molecular weight distribution in the polymer is in the range of 1.02–1.1. Therefore, polymers exhibit a uniform chain length. The polydispersity index is measured by gel permeation chromatography (GPC) in DMF with 0.1 M LiBr at 60 °C; a calibration curve constructed from narrow polydispersity PEG standards is used. The average molecular weight is provided by proton NMR spectroscopy using the amino acid repeating unit to the incorporated initiator peak integration ratio. The calculated dissociation constant value (pKa) for the carboxyl groups belonging to the poly(l-aspartic acid) segments was 3.73 [36].

Structure of the studied polymers: ASP—poly(l-aspartic acid) homopolymer, AP—poly(l-aspartic acid)-block-poly(ethylene glycol) copolymer, APA—poly(l-aspartic acid)-block-poly(ethylene glycol)-block-poly(l-aspartic acid) triblock copolymer
Methods
Thermogravimetric measurements
Thermal analysis was carried out on a STA 449 Jupiter F1, Netzsch (Germany) under the following operational conditions: heating rate of 10 °C min−1, a dynamic atmosphere of synthetic air (50 mL min−1), temperature range of 30–930 °C, sample mass ~70 mg, sensor thermocouple type S TG–DTA. As a reference, empty platinum crucible was used. The gaseous products emitted during decomposition of materials were analyzed by FTIR spectrometer Brucker (Germany) and by QMS 403C Aeölos (Germany) coupling on-line to STA instrument. The QMS data were gathered in the range from 10 to 300 amu. The FTIR spectra were recorded in the spectral range of 600–4000 cm−1 with 16 scans per spectrum at a resolution of 4 cm−1.
Surface tension measurements
In order to investigate the copolymer aggregation ability (especially the micelle-like aggregate formation), the surface tension of the polymers solution was measured using the pendant drop method (CAM Theta goniometer; KSV Instruments, Finland). This analysis is based on fitting the full equations for drop profiles for pendant drops (surface tension) derived from the Young–Laplace equation. The values obtained for both compounds throughout the concentration range (10–200 mg L−1) were constant and practically equal to those of doubly distilled water (73 mN m−1) indicating a lack of aggregation.
Adsorption tests
Adsorption measurements were conducted by the static method. The polymer concentration after the adsorption process (Γ/mg m−2) was estimated by using the UV–Vis spectrometer (Cary 100, Varian) at a wavelength of 210 nm. This wavelength was chosen after the UV–Vis absorption tests. The peptide bonds began to absorb at 280 nm and were most sensitive in the range of 210–230 nm. However, in that region, a severe interference from the absorption of OH− ions was observed. Thus, 210 nm was picked and the absorption of “blank” samples without polymers at the same pH was subtracted. A linear calibration curve was obtained from the standard solution, and it was used to determine the unknown polymer concentrations in the supernatants [28]. In order to eliminate the interferences originating from the hydroxyl ions, the calibration curves were made as a function of the solution pH. A suitable amount of Cr2O3 was added into the Erlenmeyer flasks containing 10 cm3 of polymer solution with fixed concentrations. After adjusting the appropriate pH values, the samples were shaken in water bath (OLS 200, Grant) for 24 h in order to obtain the polymer adsorption equilibrium. Then, the sediments were centrifuged twice (MPW-223e, Centrifuge) and 5 cm3 of the polymer solution was taken for the spectrophotometric analysis. Each measurement was repeated twice, and the uncertainty in the analysis was up to 3 %.
Surface charge density
The surface charge density of chromium (III) oxide was calculated from the difference in the volume of base added to the suspension in order to obtain the desired pH value
where ∆V, the difference in the volume of base added to the suspension to obtain the desired pH of the solution; c, the molar concentration of base/mol L−1; F, the Faraday constant (9.648 × 104 C mol−1); m, the mass of the metal oxide/g; S, the specific surface area of applied metal oxide/m2 g−1.
The potentiometric titrations of Cr2O3 with and without the analyzed polymers were performed in the thermostated Teflon vessel. 1.5 g of solid was added to a thermostated Teflon vessel containing 50 cm3 of supporting electrolyte solution or polymer solution with a fixed concentration. The suspensions were titrated with the NaOH solution with a concentration of 0.1 mol dm−3. A thermostated Teflon vessel with a stirrer, an automatic burette (Dosimat 765, Methrom), glass and calomel electrodes (Beckman Instruments), and the pH meter PHM 240 (Radiometer) were the parts of the measurement set. The process was controlled by a computer. The surface charge density (σ 0) was calculated using the “Titr_v3” program written by W. Janusz. The tests were performed twice, and the measurement uncertainty was at the level of 2 %.
Stability of Cr2O3 in the absence and presence of analyzed polymers
Stability measurements were conducted using a turbidimeter Turbiscan Lab Expert connected to the cooling module TLab Cooler. This device possesses an electroluminescence diode which emits a collimated light beam (λ = 880 nm), passing through the studied suspension and two synchronized detectors. The transmission detector records the light passing through the sample at the angle of 0˚ in relation to the incident light direction. The second one is the backscattering detector, which registers the light scattered at the angle of 135°. The obtained results are presented in the form of transmissions and backscattering curves as a function of time. The analysis of the turbidimetric data allows for the assessment of process dynamics occurring in the sample during the measurement. Moreover, due to the specialized computer software connected with Turbiscan, it was possible to calculate the TSI parameter (Turbiscan Stability Index) that is very useful in the evaluation of colloidal system stability [34, 35].
The Turbiscan Stability Index (TSI) coefficient is calculated from the following formula:
where x i denotes the average backscattering for each minute of measurement, x BS is the average value of x i, and n is the number of scans. The particle diameter (d) was determined using the general law of sedimentation, which is Stokes’ law extended to the concentrated dispersions [37]:
where V, the particle migration velocity/µm min−1; ρ c, the continuous phase density/kg m−3; ρ p, the particle density/kg m−3; d, the particle mean diameter/µm; v, the continuous phase viscosity/kg m−1s−1; φ, the volume of the dispersed solid fraction/%.
Results and discussion
Influence of the solution pH and the macromolecule structure on the polymer adsorption on the Cr2O3 surface
In the case of the polymers containing the functional groups capable of electrolytic dissociation process, the solution pH is the key factor determining the adsorption mechanism. Changes involving the hydrogen ion concentration modification have influence on both the solid surface charge and the polymer coil conformation. Moreover, the copolymer block affinity for the metal oxide surface can be easily affected by fixing the proper pH value.
As one can see in Fig. 2, both the solid surface structure and the analyzed polymer macromolecule conformation strongly depend on the solution pH. At the acidic pH conditions, the Cr2O3 surface is positively charged whereas the carboxyl groups belonging to the ASP chains remain undissociated. With the hydroxyl ion concentration growth, a larger number of the negative charges appear on the metal oxide surface. Simultaneously, the ASP segment dissociation degree increase leads to the formation of the more extended spatial conformation of the polymer chains. With regard to the nonionic poly(ethylene glycol) (PEG) fragment, the solution pH adjustments do not affect the polymer coil structure, but they can contribute to the changes of the macromolecule affinity for the solid particles. This is related to the PEG segment hydrogen bond formation ability; such connections can be created only between the positively charged surface groups or the amphoteric ones. Therefore, the adsorbent and adsorbate property changes described above have significantly influence on the polymer adsorption mechanism.

Changes of the solid surface charge density (in the background electrolyte) and the ASP segment dissociation degree as a function of the solution pH
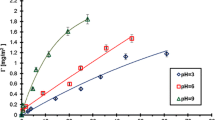
The adsorbed amounts of the tested macromolecular compounds (both homo- and copolymers) are placed in Table 1. At pH 3, the Cr2O3 particle surface is positively charged (σ 0 = 11.23 μC cm−2). Under these conditions, the ASP segments can either interact electrostatically with Cr2O3 or form hydrogen bridges (just as PEG). As one can see, the adsorption of all of the compounds gradually increases with the growth of the polymer initial concentration. The maximum of the polymeric chains linked to the adsorbent surface is reached in the case of the AP diblock copolymer, whereas the ASP homopolymer exhibits the lowest adsorption. The differences in the quantities of the polymer macromolecules bound at the metal oxide–aqueous solution interface can be explained by the analyzed substance structure. The AP copolymer chains consist of a long nonionic block and the relatively short polyamino acid ones. Under the acidic pH conditions, both copolymer structural units can undergo the adsorption on the Cr2O3 surface. The low ASP segment dissociation degree favors the more closely packed adsorption layer. Moreover, the presence of long PEG tail contributes to the screening of charges originating from the adjacent adsorbed ASP coils which contribute to further adsorption amount increase (related to the homopolymer). This effect is considerably less visible regarding the APA triblock copolymer. In this case, the PEG fragment is too short (in comparison with the ASP units length) in order to improve the proper screening of charges. A higher APA adsorption in relation to the homopolymer containing only the poly(aspartic acid) monomers comes from a larger number of the carboxylic groups capable of interacting with the solid particle surface.
At pH 7.6, the number of the positively and negatively charged groups present on the Cr2O3 particle surface is nearly equal (pHpzc—point of zero charge). The solution pH growth leads to reduction of the adsorption of all studied polymers by half in relation to the values obtained at pH 3 (except for APA—in this case, the decrease is significantly larger). The reasons for such a behavior are the electrostatic repulsion forces acting between the ASP segments in the macromolecules and the solid surface as well as between the adjacent adsorbed ASP chains. The presence of the strongly extended macromolecules on the metal oxide surface results that the lower number of polymer chains can be bound to the Cr2O3 surface. As regards the block copolymers, the PEG fragment has less capabilities of hydrogen bond formation due to a smaller content of the positively charged surface groups. The adsorption maximum is reached for the AP diblock copolymer, similar to pH 3. The APA adsorbed amount reduction is associated with the presence of the two large ASP units which can block the surface active sites making them inaccessible for the successive macromolecules.
The solution pH increase to 10 results in the further polymer adsorption amount reduction. Under these conditions, the PEG structural unit loses the affinity for the Cr2O3 surface. At pH 10, the negatively charged surface groups predominate at the Cr2O3–aqueous solution interface. Under these conditions, the repulsion forces between the ether groups belonging to the PEG chains and the solid surface significantly impede the nonionic polymer binding. As a consequence, the external part of copolymer adsorption layer consists of the nonionic structural units directed toward the bulk solution. In the case of the APA triblock copolymer, the loop formed by the PEG segments located between the two spatially extended ASP blocks provides another steric hindrance precluding the adsorption of polymer chains.
Solid particle surface charge density changes in the presence of the poly(l-aspartic acid) and the block copolymers
The potentiometric data obtained for the Cr2O3 particle surface charge density (σ 0) changes in the absence and presence of the analyzed homo- and copolymers as a function of the solution pH are shown in Figs. 3 and 4. The analysis of the results at lower polymer concentration (C p = 10 mg L−1; Fig. 3) reveals that the curves obtained for the Cr2O3 particles in the background electrolyte and the Cr2O3/ASP homopolymer system overlap. The addition of the other two compounds causes the surface charge density reduction up to pH 10. At the initial pH values, the highest σ 0 drop is observed for the solid particles covered with numerous adsorbed AP copolymer macromolecules. A considerable surface charge decrease in the presence of APA triblock copolymer (despite lower adsorption) can be explained by a larger number of the dissociated carboxyl groups in the polymer chains. Another conclusion which may be drawn is that the lower the adsorption is, the closer and closer to the results obtained for the Cr2O3/NaCl system the potentiometric curves are (Fig. 3).

The Cr2O3 particle surface charge density in the absence and presence of analyzed polymers at the concentration of 10 mg L−1. ASP—poly(l-aspartic acid) homopolymer, AP—poly(l-aspartic acid)-block-poly(ethylene glycol) copolymer, APA—poly(l-aspartic acid)-block-poly(ethylene glycol)-block-poly(l-aspartic acid) triblock copolymer

The Cr2O3 particle surface charge density in the absence and presence of analyzed polymers at the concentration of 100 mg L−1. ASP—poly(l-aspartic acid) homopolymer, AP—poly(l-aspartic acid)-block-poly(ethylene glycol) copolymer, APA—poly(l-aspartic acid)-block-poly(ethylene glycol)-block-poly(l-aspartic acid) triblock copolymer
For the systems containing the studied polymers at a concentration of 100 mg L−1, the noticeable surface charge density drop is observed (compared with the solid suspensions with a lower polymer content). A greater number of the macromolecules adsorbed on the solid surface is responsible for the significant σ 0 value decrease. In addition, the change of curves order can be explained by the impact of the macromolecule structure on the polymer adsorption layer conformation. In the samples containing a lower concentration, the polymer chains adopt more parallel conformation, while at higher concentration the adsorption layer is rich in the loops and tails directed toward the bulk solution. Increase in the number of polymer macromolecules linked to the Cr2O3 surface results in higher amount of the functional groups able to interact with the adsorbent active sites.
Despite the fact that the highest adsorption is reached for the samples containing the AP copolymer, the lowest surface charge values are measured in the presence of both the ASP homopolymer and the APA triblock copolymer. The reason for such a behavior is a larger number of the negatively charged functional groups located in the mentioned macromolecular compound chains in relation to the AP copolymer. In the case of the AP diblock copolymer, a slight changes in the surface charge density values (compared with the lower concentration) is related to the adsorption mechanism. Due to the presence of the long PEG chain in the macromolecule structure, AP can be bonded to the Cr2O3 surface on account of the hydrogen bond formation between the nonionic block and the adsorbent surface active sites (especially in the acidic solution). The tests carried out previously indicate that the presence of PEG presence in the by surface by-surface layer of the metal oxide does not change the surface charge density [1]. Therefore, the inconsiderable drop of the surface charge density values can be associated with the marked contribution of the PEG block in the copolymer adsorption process (Fig. 5). Amount of the polymer charged functional groups occurring near the adsorbent particle surface plays a crucial role in the suspension stabilization or destabilization process.

Conformation of the polymer chains adsorbed on the Cr2O3 surface at pH 3 and 10. Symbol of the negative charges belonging to ASP block was omitted
The polymer adsorption impact on the aqueous Cr2O3 suspension stability
In order to prepare a comprehensive analysis of the examined systems, the TSI parameter values in the absence and presence of the ionic polyamino acid and their copolymers were calculated. The analysis of the data collected in Fig. 6 indicates that in the absence of the studied polymers, the Cr2O3 suspensions are stable only at pH 3, and under other pH conditions, the solid samples exhibit a low durability. At pH 3, the Cr2O3 particles contain mainly positively charged surface groups contributing to the electrostatic sample stabilization. As one can see from the TSI analysis, at pH 7.6, the Cr2O3 suspensions without the polymer exhibit the lowest stability (TSI = 62.91). Under these conditions, the overall surface charge is equal to zero which means that there is the same number of positively and negatively charged groups. The attraction forces between them are responsible for the stability reduction. A lower TSI parameter value was reached at pH 10 (TSI = 49.82). It can be driven by electrostatic repulsion between the negative surface groups.

The TSI parameter values obtained for the in the absence and presence of tested polymers
The analysis of the turbidimetric data (Fig. 6; Table 2) exhibited that the lowest durability of the sample containing the ASP homopolymer at pH 3 (in comparison with the other studied polymers) is related to the absence of PEG fragment in the macromolecule structure. The nonionic structural unit performs two functions. First of all, it ensures the screening of charges originating from the ASP blocks. Secondly, under these conditions, the PEG segments can undergo adsorption process at the Cr2O3 surface providing to the more densely packed polymer adsorption layer formation. As a result, the sample aggregation is partially impeded by the impact of steric effects. In the presence of ASP homopolymer, the solid particle charge is more neutralized due to the electrostatic interactions with the dissociated carboxyl groups leading to the formation of flocs characterized by a small diameter. In the case of the AP or APA block copolymer, much more larger aggregates are the results of the PEG ability of the formation polymer bridges between different solid particles.
At higher pH values (7.6 or 10), the Cr2O3 suspension stability improvement in the presence of all tested polymers is observed (in comparison with pH 3 and the solid in the background electrolyte under the same conditions). Another conclusion that can be drawn on the basis of data analysis in Fig. 6 is that the polymer chain structure has a considerable impact on the suspension stability. At pHpzc, the sample containing the ASP homopolymer exhibits the highest durability among other analyzed compounds, whereas the samples with the APA triblock copolymer are unstable. In the basic environment, this tendency is maintained. Such a behavior can be explained by the two effects. First of all, the adsorption of all tested compounds decreases with the solution pH growth. Therefore, the solid surface is not fully covered. Secondly, with the pH increase, the PEG block present in the AP and APA copolymers chain loses the affinity for the Cr2O3 surface. The nonionic tails directed toward the bulk solution can form the hydrogen bonds leading to the suspension destabilization. Such a behavior is not observed for the ASP homopolymer. In this case, the extended negatively charged chains impede the sample aggregation. The proposed mechanism is confirmed by the average floc size and sedimentation velocity analysis (Table 2). The smallest values at both pH are noted for ASP, whereas the large aggregates are formed in the presence of the copolymers. Slight differences between AP and APA follow from the higher adsorption of the diblock copolymer causing decrease in the solid active sites available for the polymer macromolecules.
Thermogravimetric analysis of the poly(l-aspartic acid) homopolymer and copolymers impact on the Cr2O3 particles
The interactions between various polymers and the solid particles using the thermogravimetric methods are subject of numerous research [38–50]. Figures 7–10 present the TG, DTA and DTG curves for the chromium (III) oxide samples, both unmodified and modified with the adsorption layer formed by the analyzed homo- and copolymers. Changes in the solid particle thermal stability in the presence and absence of the macromolecular compounds were measured at two extreme pH values in order to investigate the polymer influence on the Cr2O3 properties.

TG and DTA curves for the chromium (III) oxide systems modified by poly(l-aspartic acid) or the PEG copolymers adsorbed at pH 3. ASP—poly(l-aspartic acid) homopolymer, AP—poly(l-aspartic acid)-block-poly(ethylene glycol) copolymer, APA—poly(l-aspartic acid)-block-poly(ethylene glycol)-block-poly(l-aspartic acid) triblock copolymer
Analyzing the DTG curve obtained for Cr2O3 without the adsorbed polymer one can distinguish two regions [51–53]. The first region ranges from 30 to 360 °C with the minimum at 96.9 °C, and the mass loss of 0.16 % corresponds to the endothermic desorption of physically (the BET measurements indicate that the solid is nonporous and possess only numerous spaces between crystallites). In the second stage (360–930 °C, minimum at 432.6 °C), the mass loss of 0.21 % corresponds to dehydroxylation of the solid particles. The addition of the polymers containing the poly(l-aspartic acid) segments significantly changes the course of thermogravimetric curves. First of all, in the initial temperature range (up to 150 °C), the endothermic process of the physically adsorbed water desorption is observed. Above 150 °C, curved-down peaks indicate the exothermic decomposition of polymeric substances adsorbed on the Cr2O3 surface. The shift of the peak maximum (DTG curves) involves the changes in the type of interactions between the polymer macromolecules and the solid particles. It is worth noting that the course of thermogravimetric curves course as well as the peak position depends on the solution pH value which is associated with the polymer adsorption mechanism.
Under the acidic pH conditions (Figs. 7, 8), at the temperature up to 200 °C, the polymer adsorption on the solid surface is responsible for the lower mass losses compared with the Cr2O3 sample. This is related to the replacement of water molecules by the polymer chain bound to the adsorbent surface. The DTA peaks obtained between 250 and 420 °C depend on the macromolecule structure. The analysis of the dependencies shows that the system containing the APA triblock copolymer is characterized by the greatest mass decrement, whereas the fewest polymer chains undergo decomposition in the case of the ASP homopolymer addition. Simultaneously, the adsorption amount of this macromolecular compound at pH 3 is the lowest. This indicates that the ASP macromolecules linked to the Cr2O3 particles demonstrate the most flat conformation (Fig. 5). As a result, the polymer functional groups are able to interact with numerous solid surface active groups giving a less space accessible to the successive macromolecules present in the solution. Such a polymer chain conformation is responsible for the low ASP adsorption, but it ensures stronger binding to the metal oxide surface. The AP and APA copolymers contain in their structure a larger number of the functional groups able to interact with the solid. This contributes to the formation of more densely packed polymer adsorption layer in which the number of “train” segments (the macromolecule fragments bound directly to the surface) is smaller than in the ASP case. As a result, the copolymer can be more readily removed from the solid particles. The shift of the decomposition peak maximum toward a higher temperature is related to the carboxyl group number interacting with the surface active groups in the electrostatic manner. The opposite situation takes place for the dehydroxylation peak of Cr2O3 surface groups (420–480 °C). Under these conditions, the polymer segment binding density plays a key factor.

DTG curves for the chromium (III) oxide systems modified by poly(l-aspartic acid) or the PEG copolymers adsorbed at pH 3. ASP—poly(l-aspartic acid) homopolymer, AP—poly(l-aspartic acid)-block-poly(ethylene glycol) copolymer, APA—poly(l-aspartic acid)-block-poly(ethylene glycol)-block-poly(l-aspartic acid) triblock copolymer
The analysis of the thermogravimetric curves measured for the samples at pH 10 (Figs. 9, 10) indicates that the samples mass decrement are larger in comparison with those at pH 3. This confirms the polymer adsorption mechanism which assumes that the hydrogen bond formation is a driving force for macromolecule binding. The obtained values are in good agreement with the adsorbed amounts of the analyzed polymeric compounds. In the polymer decomposition temperature range (150–400 °C), the mass increment increases in the following order: APA triblock copolymer, ASP homopolymer, AP diblock copolymer. This phenomenon can be explained on the basis of the macromolecule conformation. In the basic environment, the carboxyl groups belonging to the poly(l-aspartic acid) chains are totally dissociated. As a result, the polymer macromolecules adsorbed on the Cr2O3 surface due to the van der Waals interactions adopt an extended conformation. Additionally, under these conditions, the PEG unit present in the block copolymers cannot form hydrogen bridges. Hence, the AP macromolecules containing a long nonionic chain can be readily removed from the solid surface (two intensive peaks on DTA curve with minimum at 179.4 and 296 °C). The lowest mass loss measured for the sample with APA is related to the low polymer adsorption.

TG and DTA curves for the chromium (III) oxide systems modified by poly(l-aspartic acid) or the PEG copolymers adsorbed at pH 10. ASP—poly(l-aspartic acid) homopolymer, AP—poly(l-aspartic acid)-block-poly(ethylene glycol) copolymer, APA—poly(l-aspartic acid)-block-poly(ethylene glycol)-block-poly(l-aspartic acid) triblock copolymer

DTG curves for the chromium (III) oxide systems modified by poly(l-aspartic acid) or the PEG copolymers adsorbed at pH 10. ASP—poly(l-aspartic acid) homopolymer, AP—poly(l-aspartic acid)-block-poly(ethylene glycol) copolymer, APA—poly(l-aspartic acid)-block-poly(ethylene glycol)-block-poly(l-aspartic acid) triblock copolymer
Interesting observations can be done comparing the DTA data obtained for samples prepared in acidic and basic solutions. As one can see, at pH 3 (Fig. 7), only one peak on DTA curve for each polymeric substance is detected. Moreover, the signals are shifted toward the higher temperatures in the following order: AP, ASP and APA (the curves obtained for the last two substance are nearly overlapping). On the basis of these results, the interactions between the particular suspension constituents can be drawn. As it was mentioned before, the AP diblock copolymer forms a densely packed polymer layer in which the adjacent macromolecules can interact on account of the hydrogen bond formation. Such connections can be easily destroyed. The greater the number of the polymer segments linked with the solid surface, the higher the temperature required to remove the macromolecules is.
At pH 10, two peaks observed for AP copolymer on DTA curves (Fig. 9) can be related to the two-step process of the polymer chain removal. Such a behavior can be connected with the amount of the adsorbed polymer macromolecules and the strength of the polymer–solid surface interactions. As one can see, at pH 10 those two peaks are observed for AP, one signal (with the minimum shifted toward higher temperatures) is detected for ASP homopolymer. In the case of the APA triblock copolymer, there are no significant changes in the curve course. In addition, a higher quantity of the adsorbed polymer chains is noted for the AP, while the lowest value is reached in the system containing APA. Therefore, the explanation of this fact is the peaks is related to the polymer chain removal phenomena from the Cr2O3 particles. First signal detected for the AP copolymer can be associated with the breaking of the hydrogen bonds formed between the fewer PEG segments adsorbed on the solid surface or the destruction of the hydrogen bridges between the adjacent blocks. The second one (with the maximum at 300 °C) corresponds to the ASP block removal. Since these fragments are strongly linked to the adsorbent compared with the nonionic PEG ones (and the number of the adsorbed polyamino acid segments is also greater), the higher temperature is required to destroy the polymer–surface active group interactions. Such a behavior is not observed at pH 3. The main reason is fact that the adjacent adsorbed macromolecules in the polymer layer formed in the acidic solution are connected by the numerous hydrogen bonds. As the pH value increases, the number of the hydrogen bridges is considerably reduced. As a result, the adsorbed layer is less densely packed with a higher distance between the successive chains. In the case of the ASP homopolymer, temperature necessary to the polymer decomposition increases which can be related to the ASP chain conformation. Under these conditions, the anionic polyamino acid macromolecules adopt more flat arrangement with respect to the AP diblock copolymer. A lack of the considerable effect in the probe containing APA is a consequence of the low polymer adsorption. The spatially extended copolymer macromolecules are easily removed from the Cr2O3 surface. These results are in a good agreement with the data obtained from the mass loss analysis.
MS profile of CO2 and H2O released in the decomposition stage versus temperature obtained for the analyzed systems at pH 3 and 10 are presented in Figs. 11 and 12. As one can see, under acidic conditions in 250–420 °C range the highest amount of carbon dioxide (m/z = 44) originates from the APA containing sample while the lowest value is emitted in the case of the AP copolymer. The reason for such a behavior is a lower content of carboxyl groups which undergo decomposition with the CO2 evolution. Considering water (m/z = 18) originating from oxidation of hydrogen atoms located in the polymeric chain, the largest number of the solvent molecules was adsorbed to the Cr2O3 surface in the case of the ASP homopolymer on account of low polymer adsorption level. A more densely packed adsorption film formed in the presence of the block copolymers effectively impedes the water molecule sorption.

MS profile of CO2 versus temperature for the Cr2O3 samples containing the poly(l-aspartic acid) or the PEG copolymers adsorbed at pH 3 and 10

MS profile of H2O versus temperature for the Cr2O3 samples containing the poly(l-aspartic acid) or the PEG copolymers adsorbed at pH 3 and 10
Different situation is observed analyzing the MS profiles of CO2 and H2O emitted in the decomposition stage versus temperature obtained for the analyzed systems at pH 10 (Figs. 11, 12). In the case of both released gases, the highest emission is observed for the particles covered by the AP diblock copolymer. This is associated with a larger number of the polymer functional groups undergoing the oxidation process on account of high adsorption.
Conclusions
Comparison of the adsorption levels obtained for the analyzed polymers exhibits that the solution pH and the chain structure have a significant influence on the polymer adsorption layer. The adsorption maximum for all tested macromolecular compounds is reached at pH 3, whereas the minimum was observed in the basic solution. Considering the chain structure impact on the polymer adsorption, at pH 3 the presence of the nonionic PEG block increases the bounded macromolecules amount in comparison with the homopolymer solution due to the PEG ability to screening of the negative charges from ASP fragments. The solution pH growth contributes to the significant polymer adsorption drop. At pH 10, the extended negatively charged ASP chains contribute to blocking of the adsorbent active sites. Additionally, a lack of the PEG affinity for surface impedes the polymer binding.
Conformation of the adsorbed polymer macromolecules influences the aqueous suspension stability. In relation to the solid sample at pH 3, the addition of the ASP homopolymer leads to the considerable suspension destabilization. This is related to the charge neutralization by the numerous ASP functional groups located near the Cr2O3 surface. Such destabilization effect is less visible for the samples containing the block copolymers on account of the more densely packed adsorption layer formation. In the basic environment, the addition of polymers leads to the suspension stability improvement (in relation to Cr2O3 under the same conditions). Despite the low adsorption, the highest suspension stability is reached in the presence of the ASP homopolymer. The fully dissociated adsorbed polymer chains prevent the particles merging into larger aggregates. In the case of the AP and APA block copolymers, the presence of nonionic PEG fragment contributes to the formation of a larger number of the hydrogen bonds and polymer bridges between the adjacent solid particles. As a consequence, such suspensions are less stable. The proposed binding mechanism was totally confirmed by the thermogravimetric measurements.
References
Chibowski S, Patkowski J, Opala-Mazur E. Adsorption of commercial, filtrated and fractionated polyethylene oxide onto hematite. Mater Chem Phys. 2005;92:519–25.
Grzadka E, Chibowski S. Adsorption and electrokinetic properties of the system: carboxymethylcellulose/manganese oxide/surfactant. Cellulose. 2012;19:23–36.
Grzadka E. Influence of surfactants on the structure of the adsorption layer in the system: Carboxymethylcellulose/alumina. Mater Chem Phys. 2011;126:488–93.
Wiśniewska M, Chibowski S, Urban T. Effect of the type of polymer functional groups on the structure of its film formed on the alumina surface—suspension stability. React Funct Polym. 2012;72:791–8.
Chibowski S. Effect of functional groups of polyacrylamide and polyacrylic acid on their adsorption onto TiO2 surface. J Colloid Interface Sci. 1990;140:444–9.
Vermöhlen K, Lewandowski H, Narres HD, Schwuger MJ. Adsorption of polyelectrolytes onto oxides—the influence of ionic strength, molar mass, and Ca2+ ions. Colloids Surf. 2000;163:45–53.
Steitz R, Leiner V, Siebrecht R, Klitzing R. Influence of the ionic strength on the structure of polyelectrolyte films at the solid/liquid interface. Colloids Surf. 2000;163:63–70.
Wiśniewska M, Chibowski S. Influences of temperature and purity of polyacrylic acid on its adsorption and surface structures at ZrO2–polymer solution interface. Adsorpt Sci Technol. 2005;23:655–67.
Bernardi MIB, Crispim SCL, Maciel AP, Souza AG, Conceicao MM, Leite ER, Longo E. Synthesis and characterization of Al2O3/Cr2O3-based pigments. J Therm Anal Calorim. 2004;75:475–80.
Palimi MJ, Rostami M, Mahdavian M, Ramezanzadeh B. Studying the effects of surface modification of Cr2O3 nanoparticles by 3-aminopropyltrimethoxysilane (APTMS) on its corrosion inhibitive performance. J Sol-Gel Sci Technol. 2015;73:141–53.
Ostolska I, Wiśniewska M. Investigation of the colloidal Cr2O3 removal possibilities from aqueous solution using the ionic polyamino acid block copolymers. J Hazard Mater. 2015;290:69–77.
Makhlouf SA, Bakra ZH, Al-Attara H, Moustafa MS. Structural, morphological and electrical properties of Cr2O3 nanoparticles. J Mater Sci Eng B. 2013;178:337–43.
Semenov AN. Theory of colloid stabilization in semidilute polymer solutions. Macromolecules. 2008;41:2243–9.
Wiśniewska M. A review of temperature influence on adsorption mechanism and conformation of water soluble polymers on the solid surface. J Dispers Sci Technol. 2011;32:1605–23.
Anger G, Halstenberg J, Hochgeschwender K, Scherhag C, Korallus U, Knopf H, Schmidt P, Ohlinger M. Chromium compounds. In: Ullmann’s encyclopedia of industrial chemistry. Weinheim: Wiley-VCH; 2005.
Carrion D, Coleman-Nally D, Graves G. Cosmetic compositions comprising nanoparticles and processes for using the same. Patent no.: US20030064086 A1; 2002.
Singh KK, Senapati KK, Borgohain C, Sarma KC. Newly developed Fe3O4–Cr2O3 magnetic nanocomposite for photocatalytic decomposition of 4-chlorophenol in water. J Environ Sci. 2015. doi:10.1016/j.jes.2015.01.035.
Wei G, Qu J, Yu Z, Li Y, Guo Q, Qi T. Mineralizer effects on the synthesis of amorphous chromium hydroxide and chromium oxide green pigment using hydrothermal reduction method. Dyes Pigments. 2015;113:487–95.
Brock T, Groteklaes M, Mischke P. European coating handbook. Hanover: Vincentz Verla; 2000.
Kumar K, Chandramohan R, Kalyanaraman D. Effect of heat treatment on cobalt and nickel electroplated surfaces with Cr2O3 dispersions. Appl Surf Sci. 2004;227:383–6.
Hamley I. Block copolymers in solution: fundamentals and applications. New York: Wiley; 2005.
Sarkar B, Venugopal V, Tsianou M, Alexandridis P. Adsorption of Pluronic block copolymers on silica nanoparticles. Colloids Surf A. 2013;422:155–64.
Kumagai M, Imai Y, Nakamura T, Yamasaki Y, Sekino M, Ueno S, Hanaoka K, Kikuchi K, Nagano T, Kaneko E, Shimokado K, Kataoka K. Iron hydroxide nanoparticles coated with poly(ethylene glycol)-poly(aspartic acid) block copolymer as novel magnetic resonance contrast agents for in vivo cancer imaging. Colloids Surf B. 2007;56:174–81.
Louguet S, Kumar AC, Sigaud G, Duguet E, Lecommandoux S, Schatz C. A physico-chemical investigation of poly(ethylene oxide)-block-poly(l-lysine) copolymer adsorption onto silica nanoparticles. J Colloid Interface Sci. 2011;359:413–22.
Popelka Š, Machová L, Rypáček F. Adsorption of poly(ethylene oxide)-block-polylactide copolymers on polylactide as studied by ATR-FTIR spectroscopy. J Colloid Interface Sci. 2007;308:291–9.
de Laat AWM, Schoo HFM. Novel poly(vinyl ether) block copolymers: adsorption from aqueous solutions on α-Fe2O3(hematite) and the mechanism of colloidal stabilization. J Colloid Interface Sci. 1997;191:416–23.
Collins IR. Surface electrical properties of barium sulfate modified by adsorption of poly α, β aspartic acid. J Colloid Interface Sci. 1999;212:535–44.
Wang X, Burtrand IL, Mann L. Dispersion of barium titanate with polyaspartic acid in aqueous media. Colloids Surf A. 2002;202:71–80.
Obst M, Steinbüchel A. Microbial degradation of poly(amino acid)s. Biomacromolecules. 2004;5:1166–76.
Kołodyńska D, Hubicki Z, Gęca M. Application of a new-generation complexing agent in removal of heavy metal ions from aqueous solutions. Ind Eng Chem Res. 2008;47:3192–9.
Yoon D-H, Lee BI. Effects on aqueous barium titanate tape properties of passivation of barium ion leaching by using dispersants. J Eur Ceram Soc. 2004;24:3747–52.
Chaussemier M, Pourmohtasham E, Gelus D, Pécoul N, Perrot H, Lédion J, Cheap-Charpentier H, Horner O. State of art of natural inhibitors of calcium carbonate scaling. A review article. Desalination. 2015;356:47–55.
Yang H-M, Park CW, Ahn T, Jung B, Seo B-K, Park J-H, Kim J-D. A direct surface modification of iron oxide nanoparticles with various poly(amino acid)s for use as magnetic resonance probes. J Colloid Interface Sci. 2013;391:158–67.
Wiśniewska M, Szewczuk-Karpisz K. Removal possibilities of colloidal chromium (III) oxide from water using polyacrylic acid. Environ Sci Pollut Res. 2012;20:3657–69.
Ostolska I, Wiśniewska M. Application of the zeta potential measurements to explanation of colloidal Cr2O3 stability mechanism in the presence of the ionic polyamino acids. Colloid Polym Sci. 2014;292:2453–64.
Ostolska I, Wiśniewska M. Comparison of the influence of polyaspartic acid and polylysine functional groups on the adsorption at the Cr2O3—aqueous polymer solution interface. Appl Surf Sci. 2014;311:734–9.
Snabre P, Mills P. Settling of a suspension of hard spheres. Europhys Lett. 1994;25:651–6.
Hong S-G, Huang S-C. Effect of modified silica on the crystallization and degradation of poly(3-hydroxybutyrate). J Therm Anal Calorim. 2015;119:1693–702.
Basilio de Souza JP, Laredo dos Reis JM. Influence of Al2O3 and CuO nanoparticles on the thermal properties of polyester- and epoxy-based nanocomposites. J Therm Anal Calorim. 2015;119:1739–46.
Musuc AM, Dumitru R, Stan A, Munteanu C, Birjega R, Carp O. Synthesis, characterization and thermoreactivity of some methylcellulose–zinc composites. J Therm Anal Calorim. 2015;120:85–94.
Nielsen BV, Nevell TG, Barbu E, Rees GD, Tsibouklis J. A thermogravimetric method for assessing the substantivity of polymer films on dentally relevant substrates. J Therm Anal Calorim. 2010;102:121–6.
Censi R, Gigliobianco MR, Malaj L, Di Martino P. Effect of poly(vinylpyrrolidone) or sodium alginate on the stability of the amorphous form of nimesulide. J Therm Anal Calorim. 2016;123:2415–25.
Pielichowska K. The influence of polyoxymethylene molar mass on the oxidative thermal degradation of its nanocomposites with hydroxyapatite. J Therm Anal Calorim. 2016. doi:10.1007/s10973-015-5196-6.
Jurca B, Tirsoaga A, Ianculescu A, Carp O. Influence of the synthesis parameters on the thermal behavior of some ZnO–starch composites. J Therm Anal Calorim. 2014;115:495–501.
Jurca B, Tirsoaga A, Ianculescu A, Carp O. Influence of the synthesis parameters on the thermal behavior of some ZnO–starch composites. J Therm Anal Calorim. 2013;112:885–91.
Mallakpour S, Dinari M. The effects of reactive organoclay on the thermal, mechanical, and microstructural properties of polymer/layered silicate nanocomposites based on chiral poly(amide-imide)s. J Therm Anal Calorim. 2013;114:329–37.
Unnikrishnan L, Mohanty S, Nayak SK. Evaluation of flammability and shear performance of layered silicate—reinforced styrenic polymer. J Therm Anal Calorim. 2016. doi:10.1007/s10973-016-5331-z.
Wiśniewska M, Szewczuk-Karpisz K, Sternik D. Adsorption and thermal properties of the bovine serum albumin–silicon dioxide system. J Therm Anal Calorim. 2015;120:1355–64.
Natkański P, Kuśtrowski P, Białas A, Surnam J. Effect of Fe3+ ions present in the structure of poly(acrylic acid)/montmorillonite composites on their thermal decomposition. J Therm Anal Calorim. 2013;113:335–42.
Grochowicz M, Gawdzik B, Jaćkowska M, Buszewski B. Investigation of the thermal behavior of new silica-polymer anion exchangers. J Therm Anal Calorim. 2013;112:885–91.
Sternik D, Staszczuk P, Sobieszek J, Płanda-Czyż M, Wasak S. Influence of albumin adsorption on physico-chemical properties of alumina surfaces. J Therm Anal Calorim. 2006;86:77–83.
Wiśniewska M, Chibowski S, Urban T, Sternik D. Investigation of the alumina properties with adsorbed polyvinyl alcohol. J Therm Anal Calorim. 2011;103:329–37.
Wiśniewska M, Chibowski S, Urban T. Investigations of flocculation possibilities of the water alumina suspension in the presence of nonionic polymer. J Ind Eng Chem. 2013;19:263–71.
Acknowledgements
The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/under REA Grant Agreement No. PIRSES-GA-2013-612484.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wiśniewska, M., Ostolska, I. & Sternik, D. Impact of adsorption of poly(aspartic acid) and its copolymers with polyethylene glycol on thermal characteristic of Cr2O3 . J Therm Anal Calorim 125, 1171–1184 (2016). https://doi.org/10.1007/s10973-016-5478-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5478-7