
Abstract
Cardiovascular disease (CVD) has long been the leading cause of death worldwide, and myocardial infarction (MI) accounts for the greatest proportion of CVD. Recent research has revealed that inflammation plays a major role in the pathogenesis of CVD and other manifestations of atherosclerosis. Overwhelming evidence supports the view that macrophages, as the basic cell component of the innate immune system, play a pivotal role in atherosclerosis initiation and progression. Limited but indispensable resident macrophages have been detected in the healthy heart; however, the number of cardiac macrophages significantly increases during cardiac injury. In the early period of initial cardiac damage (e.g., MI), numerous classically activated macrophages (M1) originating from the bone marrow and spleen are rapidly recruited to damaged sites, where they are responsible for cardiac remodeling. After the inflammatory stage, the macrophages shift toward an alternatively activated phenotype (M2) that promotes cardiac repair. In addition, extensive studies have shown the therapeutic potential of macrophages as targets, especially for emerging nanoparticle-mediated drug delivery systems. In the present review, we focused on the role of macrophages in the development and progression of MI, factors regulating macrophage activation and function, and the therapeutic potential of macrophages in MI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Despite advances in prevention, diagnosis, and treatment, cardiovascular disease (CVD) remains the primary cause of death worldwide, and myocardial infarction (MI) makes the greatest contribution to CVD [1]. Furthermore, the World Health Organization has predicted that annual deaths from CVD will increase from 18.1 million in 2010 to 24.2 million in 2030 globally [2]. CVDs, including hypertension, atherosclerosis, ischemic heart disease like MI, and ischemic stroke [3], result in significant death and disability [4]. Considering the poor prognosis associated with CVD, new therapeutic strategies are needed to facilitate cardiac repair following MI.
Previous studies have demonstrated that vertebrate zebrafish is capable of complete cardiac regeneration [5,6,7], while some regenerative capacity of mammals can be maintained for just a few days [8, 9]. Macrophages are basic cell components of the innate immune system that infiltrate into injured myocardium during neonatal heart regeneration [8]. Accumulating evidence has revealed that inflammation plays a major role in the pathogenesis of coronary artery disease and atherosclerosis [10,11,12] and is necessary for correct and timely repair [13, 14]. After MI, circulating blood monocytes rapidly infiltrate into the infarcted area and differentiate into the appropriate macrophages [15]. Based on surface markers and functions, macrophages are divided into two major subtypes: classically activated macrophages (M1), which are related to inflammatory response, and alternatively activated macrophages (M2), which are associated with regeneration and injury repair. In the inflammatory phase of MI, M1 macrophages activated by tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ), and lipopolysaccharide (LPS) are the leading subtypes that initially respond to the removal of dead cells and cellular and extracellular matrix (ECM) debris [16, 17]. In the proliferative phase of MI, M2 macrophages gradually predominate to facilitate the repair and regeneration of damaged cardiac tissues [18, 19]. Therefore, the correct and timely regulation of macrophage polarization is a promising therapeutic target for the treatment of MI. In addition, stem cell transplantation and nanoparticle-mediated drug delivery systems have made extensive breakthroughs. Together, macrophages modulated by all kinds of therapeutic strategies, particularly the nanoparticle-mediated drug delivery system, have become the promising therapeutic target in the field of cardiac repair.
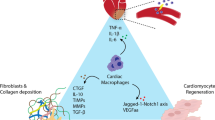
In the present review, we discuss recent findings on the association of macrophages with the development of post-MI. More specifically, we focus on the phenotypes and functions of macrophages in a steady state and during MI, as well as possible mechanisms underlying macrophage polarization in the heart. Importantly, we discuss potential therapeutic strategies to improve injury control and functional recovery by modulating macrophage polarization, which involves self-assembly/engineered extracellular vesicles (EVs), nanomedicine, and stem cells (Fig. 1).

Macrophages function in homeostasis and in/post-myocardial infarction. The three overlapping stages are involved in the repair response after obstruction of the blood flow: infarction, inflammatory, and proliferative. In the infarction phase, damaged cardiomyocytes, active DAMPs, and neutrophils are recruited to the infarcted site and release many inflammatory mediators, which are indispensable for the subsequent inflammatory stage. In the inflammatory stage, proinflammatory subset M1 macrophages and NK cells secrete inflammatory cytokines, such as IL-1β, IL-6, iNOS, TNF-α, and IFN-γ, which promote clearance of dead cells and cellular and ECM debris. Last, the proliferative stage involves cell proliferation, collagen formation, and tissue repair that mainly contribute to anti-inflammatory subset M2 macrophages that secrete IL-10, TGF-β, and Arg-1. Modulation (i.e., self-assembly/engineered extracellular vesicles including exosomes[exo], nanoparticles, stem cells) of macrophages may repair damaged myocardium by promoting angiogenesis and reducing hypertrophy, fibrosis, and cell apoptosis.
ONTOGENY OF CARDIAC MACROPHAGES
Macrophages are the first immune cells that develop during the development of an organism. They play a crucial role in immunity (homeostasis and inflammation) and also regulate organ development and function [20]. Macrophages primarily originate from circulating blood monocytes [21]. Monocytes pertain to the population of mononuclear leukocytes derived from hematopoietic stem cells in fetal liver, adult bone marrow, and splenic reservoir under the stimulation of some cytokines, such as M-CSF, GM-CSF, interleukin-1β (IL-1β), and IL-3 [22, 23], and are released in the bloodstream. Monocytes can be divided into two classifications after being mobilized into the peripheral circulation [24]: (1) Ly6Cl° CCR2− CX3CR1hi patrolling monocytes (CD14l°CD16+ in humans), which are responsible for surveying the vascular lumen and for clearing dead cells and cellular and ECM debris, and (2) Ly6Chi CCR2+ CX3CR1l° inflammatory monocytes (CD14hiCD16− in humans), which produce proinflammatory cytokines, such as IL-1, IL-10, and TNF-α. Over the past 40 years, it has been accepted that all forms of macrophages, including resident macrophages, originate from monocytes; however, this has been challenged in the past few years [25]. Recent research has shown that tissue-resident macrophages represent a different population of cells that are derived from diverse lineages [21, 26, 27]. Studies using mouse models have revealed that many tissue-resident macrophages in the kidney, lung, skin, brain, liver, and heart originate from an embryonic lineage and are maintained throughout life free of monocyte recruitment [28,29,30,31,32]. In recent years, advances in gene fate-mapping techniques have revealed the two distinct populations of tissue-resident macrophages originating from the prenatal yolk sac and fetal liver [28, 29]. It is now clear that the majority of cardiac resident macrophages originate from the yolk sac [30, 31, 33], and these cardiac resident macrophages are divided into two populations that coexist at homeostasis: MHC-IIl°CCR2− and MHC-IIhiCCR2− cells. The others are known as circulating monocyte-derived MHC-IIhiCCR2+ cardiac resident macrophages that are responsible for inflammation initiation (Table 1).
MACROPHAGES: INFLAMMATION, ACTIVATION, AND FUNCTION
Macrophages, as the basic cell component of the innate immune system, are involved in all stages of atherosclerosis. Macrophages exhibit extensive functional plasticity that is dependent on activation (in vitro) or microenvironmental milieu (in vivo). Macrophages not only possess the essential functions of phagocytic killing of pathogens and antigen presentation activating an adaptive immune response but also maintain tissue homeostasis by eliminating dead cells, cellular debris, and ECM debris and by promoting adaptive remodeling of the ECM [34]. Macrophages acquire a proinflammatory or an anti-inflammatory subtype under proper environmental stimuli [34]. Therefore, macrophages have been divided into M1 and M2 [35, 36] based on the type of in vitro stimulation, surface molecule expression pattern, secretory profile, and function. Although this distinction has deficiencies in adequately including the entire macrophage biological complexity, it provides a common scheme to classify macrophage function. In addition, the switch of macrophage polarization is tightly regulated by signaling networks at the transcriptional and translational levels (Figs. 2 and 3).

Signaling pathways regulating M1 macrophage polarization. Naïve macrophage is induced into M1 macrophage by LPS, IFN-γ, and IFN-α/β through specific receptors, such as TLR4, IFNγR, and IFNAR. And the related signaling pathways such as STAT1, NF-κB, and IRF3 have an important role in the process, which results in the secretion of proinflammatory cytokines, such as IL-1β, TNF-α, and IL-6.

Signaling pathways regulating M2 macrophage polarization. Naïve macrophage is induced into M2 macrophage by IL-4, IL-13, IL-10, and IL-21 through interacting with specific receptors, such as IL-4Rα, IL-13Rα, IL-10R, and il-21R. And the related signaling pathways such as STAT6, STAT3, HIF-2α, KLF-4, PPARγ, and IRF4 are activated in the process, which promotes the secretion of anti-inflammatory cytokines, such as TGF-β and IL-10.
M1 macrophages are characterized by increased microbicidal or tumoricidal capacity and secrete high amounts of proinflammatory cytokines and mediators. Classically activated M1 macrophages are triggered by IFN-γ and LPS and are characterized by upregulated biomarkers, such as IL-1β, IL-6, IL-12, and IL-23; inducible nitric oxide synthase (iNOS); TNF-α; chemokine (C-C motif) ligand 2 (CCL2), CCL15, and CCL20; C-X-C motif chemokine 9 (CXCL9), CXCL10, and CXCL11; and CD80 and CD86 [37,38,39]. M1 macrophages detect and recognize damage-associated molecular patterns (DAMPs) present in the debris of necrotic cells and pathogen-associated molecular patterns (PAMPs) such as LPS and chitin through toll-like receptors (TLRs) located on the surface of macrophages to promote inflammation response. During an innate immune response, TLR agonists engage the MyD88-dependent pathway, including IRAK4, TRAF6, and IKKβ, ultimately leading to the activation of NF-κB to induce M1 polarization [40]. In addition, a TLR ligand can induce the transcription of TNF through an MyD88-dependent manner, further cooperating with IFN-γ in an autocrine manner to activate these macrophages [34]. IFNs mediate the activation of IRF/STAT signaling pathways via the JAK/STAT signaling pathway, favoring M1 polarization [41, 42]. IFN-γ, which is produced by natural killer (NK) cells responding to stress and infections, can promote macrophages to produce proinflammatory cytokineswhich in turn increase the killing ability of NK cells [43]. Typically, research has shown that mice and humans who lack IFN-γ expression are more susceptible to protozoal and certain kinds of bacterial or viral infections [44]. Furthermore, activated M1 macrophages by LPS via an exogenous TLR ligand can clear the parasite completely [34]. Finally, M1-secreted proinflammatory cytokines play an important role in host defense but also can cause extensive damage to the host.
M2 macrophages are characterized as having wound healing and proliferative properties. M2 macrophages, which are activated by the stimulation of IL-4, IL-13, or IL-21, are involved in wound repair, homeostasis, and tumor metastasis and tumor promotion. They also secrete anti-inflammatory cytokines [45,46,47]. M2 macrophages are characterized by decreased expression levels of biomarkers, such as TGF-β, CD163, CD206, chil3 chitinase-like 3 (known as Ym-1), resistin-like-α (known as Fizz1), arginase 1 (Arg-1), and IL-10 to promote cell proliferation, collagen formation, and tissue repair [47]. Aberrant activation of M2 macrophages is associated with tissue fibrosis. Accumulating evidence has indicated that macrophages lacking expression of IL-4 receptor (IL-4R) are incapable of promoting wound healing. In terms of mechanism, M2 macrophages can be triggered by IL-4/L-13 and IL-10/IL-21, depending on the activation of the IRF/STAT signaling pathway via STAT6 and STAT3 [48,49,50]. IRF4, PPARγ, Krüppel-like factor 4 (KLF-4), and HIF-2α also mediate the induction of the M2 phenotype [40]. M2 macrophages are further divided into different phenotypes denoted by the stimulus and effector function. IL-4 and IL-13 can induce M2a polarization, whereas Fc-γ receptors and TLR stimulation can trigger M2b macrophages, and GC, IL-10, or TGF-β ligands are responsible for M2c activation [37]. In function, both M2a and M2c can enhance the adaptive immune response, whereas M2b plays a key role in suppressing and regulating inflammation and immunity [51].
MACROPHAGES IN CARDIAC HOMEOSTASIS
Cardiac resident macrophages, which account for 6% to 8% of the noncardiomyocyte population according to the data from healthy adult mouse heart, are indispensable for maintaining the cardiac homeostasis and neonatal heart regeneration [52]. The long-held perspective is that the majority of cardiac resident macrophages originate from peripheral blood monocytes and present an M2 polarization profile [53]. Recently, numerous researchers have demonstrated that cardiac resident macrophages consist of a heterogeneous population that includes resident macrophages derived from the yolk sac: MHC-IIl°CCR2− and MHC-IIhiCCR2− cells, macrophages derived from fetal monocytes and macrophages derived from adult monocytes [15, 33, 54, 55]. The populations seed the heart at specific developmental stages so that they can maintain cardiac homeostasis.
As noted, yolk sac–derived CCR2− macrophages play a major role in the coronary development and cardiac repair. Deletion of CCR2− macrophages in the embryonic period could cause abnormal remodeling, diminished LV systolic function, larger LV chamber dimensions, and increased akinetic myocardium [56]. Circulating monocyte-derived CCR2+ cardiac resident macrophages are abundant in proinflammatory genes involved in inflammation initiation and exhibit an M1 polarization profile to maintain cardiac homeostasis. Interestingly, more substantial contributions of circulating monocytes have been observed in the aging heart, indicating that circulating blood monocytes may differentiate into CCR2− macrophages [57, 58], which seemingly coincide with the decreased self-renewal ability of yolk sac–derived resident CCR2− macrophages with age, as demonstrated by Molawi et al. [58].
MACROPHAGES IN THE PROCESS OF ATHEROSCLEROSIS AND THROMBUS FORMATION
Atherosclerosis is considered to be a chronic inflammatory disease. Perturbation of lipid metabolism and local inflammation are the two major causes in the pathogenesis of atherosclerosis, including cells such as platelets, which play an initiating role in the development of atherosclerosis and the atherosclerotic plaque [59], monocytes and endothelial cells, connective-tissue elements, lipids, and debris. First, low-density lipoproteins (LDLs) accumulate in the intima, activating endothelial cells to initiate atherosclerosis formation [60]. This accumulation of lipoproteins is located in the subendothelial space and is modified by reactive oxygen species and enzymatic cleavage to be involved in the inflammatory process [61]. LDL, especially oxidized LDL (ox-LDL), and activated platelets are responsible for recruiting circulating monocytes into the endothelial space where they are further differentiated into macrophages that engulf ox-LDL and LDL to form foam cells, which finally produces proinflammatory cytokines (e.g., TNF-α, IL-1, and IL-6) and exacerbate local inflammation [62]. In addition, Lindemann et al. reported that progenitor cells adhere to lipid-laden platelets and turn into macrophages that internalize the lipid-rich platelets and develop into foam cells [63]. Next, macrophages, mast cells, and T cells infiltrate the atherosclerotic plaques, exhibit signs of activation, and secrete inflammatory cytokines responding to proinflammatory cytokines [10]. The apoptosis of foam cells in the lipid core is a key reason for atherosclerosis progression. Next, these apoptotic foam cells are removed primarily by macrophages, a process known as efferocytosis. Further inflammation, necrosis, and thrombosis originating from the gradual accumulation of apoptotic debris in the lipid core have occurred when the balance in which efferocytosis internalizes all dead cells is disturbed [64].
THE ROLE OF MACROPHAGES DURING AND POST-MYOCARDIAL INFARCTION
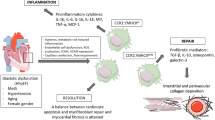
MI is defined as pathologically myocardial cell death resulting from prolonged myocardial ischemia. MI is the leading cause of mortality and disability worldwide [1] and results in a major socioeconomic burden. Such events result from the imbalance between myocardial oxygen supply and demand. Although multiple other mechanisms have been reported to contribute to MI, coronary thrombosis promoted by rupture of the atheroma plaque accounts for most of the cases of MI [65]. Cardiomyocyte necrosis resulting from coronary artery ischemia triggers both a systemic inflammatory response and a local reaction to recruit circulating monocytes into the infarcted site. The post-MI repair response includes three sequential stages: inflammation, tissue replacement, and healing or maturation. In the early inflammatory stage, infiltrated neutrophils and recruited circulating monocytes and cardiac resident macrophages contribute to clear the dead cells and matrix debris. Next, these inflammatory responses are gradually replaced by proliferative monocytes and macrophages, which results in angiogenesis and myofibroblast differentiation. Finally, in the healing phase, fibroblasts, immune cells, and microvasculature form a mature scar [66]. In the following phases, we analyze the role of macrophages in the inflammatory and healing stages after MI.
M1 Macrophages in Myocardial Infarction
MI can lead to necrosis of cardiac myocytes within a few minutes. To maintain tissue integrity and function, inflammatory cells, including neutrophils and macrophages, are activated. After MI, neutrophils are the first immune cells to occur in the infarcted area in large numbers and are responsible for clearing cellular debris and further recruiting leucocytes such as Ly-6Chi monocytes and macrophages [67, 68]. Monocytes and macrophages are the two major cell populations infiltrating the damaged site. Depletion of monocytes and macrophages may result in a thromboembolic event [69]. The two sequential monocyte and macrophage phases demonstrate a significant difference in healing after MI: first, bone marrow– and spleen-derived Ly-6Chi monocytes are recruited to the infarcted site via MCP-1. Its receptor, CCR2 [70], and corresponding M1 macrophages predominate in the infarcted region during days 2 to 5 post-MI. During the inflammatory phase of MI, significant proinflammatory mediators, such as TNF-α, IL-1β, and proteases, originating from activated Ly-6Chi/M1, are released in the damaged site, which contributes to the clearance of dead cells and debris in the infarcted region through the activated M1 macrophages. The process of phagocytosis is indispensable for proper initiation of the wound repair after MI [71]. However, prolonged inflammatory response to such compounds can result in extensive damage to infarcted myocardium.
M2 Macrophages in Myocardial Infarction
MI results in necrosis of cardiac myocytes within a few minutes, contributing to chamber dilatation, contractile dysfunction, and eventually heart failure. The regenerative capacity of mammals persists for only a short period [8, 9]. Therefore, the correct and timely repair after MI is necessary to maintain the constructive and functional integrity of the heart. Macrophages can promote the infarcted repair as regulators and effectors. In addition, different effects on fibrosis and scarring versus regeneration as a result of the depletion of macrophages at different stages post-injury in a model of liver fibrosis have been demonstrated [72].
From days 4 to 14 post-MI, Ly-6Chi monocytes and M1 macrophages are replaced by the Ly-6Clow monocytes and M2 macrophages. The proliferative phase after MI origin from the macrophages shifts from inflammatory (M1) to reparative phenotypes (M2). M2 macrophages gradually dominate in the infarcted sites. Then, M2 macrophages establish an anti-inflammatory environment by downregulating inflammatory cytokines and upregulating anti-inflammatory cytokines, such as IL-10, VEGF, and TGF-β [73]. TGF-β and IL-10 can trigger myofibroblasts to produce collagen, and VEGF promotes cell proliferation and blood vessel development. The lack of the Trib1 gene in the myocardial-infarcted mouse model presented selective deletion of M2 macrophages, which resulted from an impaired ability to form M2 macrophages in the spleen, liver, and adipose tissue. Compared with a control group, diminished reparative function after MI, similar to frequent cardiac rupture, was observed. In addition, IL-4 has been demonstrated to improve the post-MI prognosis of wild-type mice with an increased number of M2 macrophages. Together, M2 macrophages play a vital role in facilitating myocardial wound healing in adult murine heart [74]. Moreover, in the GRLysMCre mice, Galuppo et al. reported that the glucocorticoid receptor in macrophages critically determines post-MI repair by regulating myofibroblast differentiation in the infarct microenvironment during the early phase of wound healing [75].
THE ROLE OF MACROPHAGES IN MYOCARDIAL ISCHEMIA/REPERFUSION INJURY
As noted, the most timely and effective treatment involves amelioration of myocardial ischemia and restriction of the size of MI. Ischemia/reperfusion (I/R) such as percutaneous coronary intervention and intravenous thrombolysis has so far been the principal or only strategy for MI treatment, thereby promptly restoring blood supply [76]. However, deteriorated ischemic damages and further swelling of the infarct size will be accompanied with sudden reperfusion, which results in secondary cascade damages, known as myocardial I/R injury [77]. I/R injury may trigger all kinds of pathological changes, including local acute inflammatory reactions, metabolic disorders, and cell apoptosis or necrosis, even resulting in cardiac dysfunction. Macrophages, a major type of inflammatory cells, have a crucial effect on myocardial ischemic injury with reperfusion [78] and have multiple roles because of their specific phenotypes and the stage of disease.
M1 Macrophages in Myocardial Ischemia/Reperfusion Injury
Though M1 macrophages are believed to damage the heart in the early period of reperfusion by releasing reactive oxygen species, inflammatory mediators, and proteases [79, 80], some researchers have found that the process of phagocytosis performed by the M1 macrophages is essential for further repair. Fan et al. reported that M1 macrophages polarized by Dectin-1 expressed largely on cardiac macrophages aggravate myocardial I/R injury [81]. In contrast, a previous study has reported that soluble receptor for advanced glycation end products can improve heart function in mice after I/R by promoting infiltration and differentiation of macrophages into M1 and IFN-γ production [82]. M1 macrophages may involve cardioprotection primarily in the period of ischemia while damaging the heart in the following stages by releasing inflammatory cytokines and recruiting the inflammatory cells. In clinical practice, therapeutic strategies are applied mainly to recover blood flow in a timely manner and ameliorate myocardial I/R injury. Thus, methods limiting M1 while promoting M2 polarization of macrophages in myocardial I/R injury have been researched extensively and represent a unique therapeutic strategy to suppress inflammatory responses and ameliorate myocardial I/R injury.
M2 Macrophages in Myocardial Ischemia/Reperfusion Injury
M2 macrophages, polarized by Th2 cytokines and characterized by the production of high levels of anti-inflammatory cytokines and pro-fibrogenic factors, exhibit anti-inflammatory and tissue repair properties. Accumulated evidence has demonstrated that M2 macrophages play an important role in alleviating myocardial I/R injury. A previous study reported that alternatively activated M2 macrophages by Chemerin15 protect against myocardial I/R injury in mice by significantly suppressing proinflammatory cytokines and markedly increasing the level of anti-inflammatory cytokine IL-10 [83]. In recent years, M2b macrophages, as one subtype of M2 macrophages and regulatory cells, have drawn considerable attention for the treatment of myocardial I/R injury. In vivo experiments have shown that transplantation of M2b macrophages into the myocardium that had been subjected to I/R injury improved cardiac function and reduced the cardiac fibrosis and myocardial remodeling caused by I/R injury [84]. Yue et al. reported that M2b macrophages modulate inflammatory immune responses without participating in wound healing and enhance protective effects on myocardial remodeling after myocardial I/R injury [85].
TARGETING THE THERAPEUTIC ROLE OF MACROPHAGES POST-MI
Exogenous Cardioprotection by Modulation of Macrophage Polarization
It has been reported that stem cells and stromal cells could be utilized for the treatment of myocardial injury by modulating macrophage polarization. Cardiosphere-derived cells (CDCs), essentially cardiac stromal cells (CSCs), represent a promising stem cell source for repairing damaged heart tissue [86,87,88]. These cells can regulate macrophage activation, leading to the promotion of a phenotypic switch from M1 to M2 [89, 90]. In a CDC-treated MI model, functional and structural benefits, such as decreased infarcted area, improved cardiac function, and enhanced angiogenesis after MI, have been observed by modulating M1/M2 macrophage polarization and neutrophil recruitment [89,90,91]. Human embryonic stem cell–derived cardiovascular progenitor cells (hESC-CVPCs) also are known to be attractive cell sources for cardiac repair. hESC-CVPCs, which modulate cardiac macrophages toward an M2 phenotype, play an important role in ameliorating worsening heart function and reducing scar formation through a paracrine effect–activated STAT6 [92]. In addition, many studies have demonstrated that mesenchymal stem cells (MSCs) play a key role in post-MI repair by reversing cardiac dysfunction and enhancing angiogenesis, which may result from regulating the M1/M2 balance [93, 94]. MSCs have been reported to regulate a macrophage subtype toward an M2-like status in vitro and in vivo [95].
EVs Mediate Cardioprotection in MI
EVs have been demonstrated to play a crucial role in cell-cell communication during different pathological and physiological processes [96, 97]. EVs have received increasing attention as cell-free therapeutics for regenerative medicine because of the structure of their lipid bilayer and cargos, such as miRNA, protein, and lipids [98,99,100,101] (Table 2). As noted earlier in this review, CDCs and MSCs can promote M2 polarization in cases in which the EVs may play, at least partly, a major role.
MSCs have long become a promising therapeutic strategy for ischemic heart disease, although the mechanism remains elusive. Several recent studies have implicated that MSC-exo could polarize macrophage to create an anti-inflammatory environment under myocardial I/R injury or MI [102, 103]. MSC-exo, which transfers miR-182 and miR-101a into macrophage, has been demonstrated to reduce the number of M1 macrophages, polarize macrophages into M2 phenotype, and reduce infarct size and inflammatory response under myocardial I/R injury [102, 104].
CDC or CDC-derived EVs polarize the macrophage into a special phenotype that is highly phagocytic and anti-inflammatory to display cardioprotection [105, 106]. De Couto et al. further revealed that CDC-derived EVs enhance macrophage efferocytosis and cardioprotection response via EV transfer of miR-26a to modulate the expression of MerTK and C1qa [107]. Moreover, miR-181b–enriched exosomes secreted from CDCs play a critical role in modulating macrophage polarization in vitro and confer cardioprotection in vivo by minimizing infarct size, decreasing the total number of CD68+ macrophages, and inducing macrophages to develop into a distinct subtype [105]. Another study has suggested that CDC-derived EVs can shift M1 macrophage into the pro-angiogenic subtype [108]. Furthermore, the short non-coding RNA, Y RNA fragment (the Y RNAs consist of 83–112 nucleotides known as one poorly understood class of non-coding RNA), is particularly plentiful in EVs and was first discovered in complex with ribonucleoproteins in the serum of patients with lupus. The characteristic stem-loop secondary structure and high sequence conservation between the upper and lower stem have been found in the four human Y RNAs. Highly enriched CDC-derived EVs also have a crucial cardioprotection response by promoting IL-10 gene expression and secretion in macrophages [109].
Nguyen et al. reported that atherogenic macrophage-secreted EVs can inhibit macrophage migration via transferring miRNAs, especially the miR-146a, which downregulates target genes IGF2BP1 and HuR in recipient cells, thereby promoting the progression of atherosclerosis [110]. Moreover, miR-155–containing exosomes derived from activated macrophages are adverse to post-MI repair. A recent study has shown that exosomes inhibit cardiac fibroblast proliferation and promote inflammation by downregulating Son of Sevenless 1 expression and decreasing suppressor of cytokine signaling 1 expression, while in vivo experiments revealed a lower incidence of cardiac rupture and improved cardiac function in the miR-155–deficient mice compared with controls [111]. In addition, M2 macrophages secreting miR-148a–enriched exosomes can reduce the size of the infarct and improve cardiac function following MI [112]. Furthermore, Wu et al. obtained molecularly engineered M2 macrophage exosomes, which were further electroporated with a US Food and Drug Administration (FDA)–approved hexyl 5-aminolevulinate hydrochloride (HAL). These HAL-containing M2 exosomes exhibited anti-inflammatory capabilities and ultimately alleviated atherosclerosis because of the anti-inflammatory effects of the M2 exosomes and the encapsulated HAL affect. The exosomes were involved in endogenous biosynthesis and metabolism of heme to produce carbon monoxide and bilirubin, which have known anti-inflammatory capabilities [113].
Although EVs have been demonstrated to be an attractive therapeutic approach to ischemic heart disease, low retention and short-lived therapeutic effects remain significant challenges. To reduce off-target delivery, engineered exosomes and prior blocking of endocytosis of exosomes by macrophages have been utilized to enhance the delivery efficiency of exosomes to specific cells, offering therapeutic benefit [114,115,116]. Thus, these have improved delivery efficiency, including platelet nanovesicles [117], monocyte mimic–modified EVs [118], EVs incorporated in alginate hydrogel [119], overexpressed targeting sequences or those modified by DMPE-PEG-streptavidin (DPS), and biotin-conjugated antibody or peptide.
Cardioprotection Using a Nanoparticle-Mediated Drug Delivery System
To improve the safety and efficiency of therapeutic agents, specially designed nanocarriers, including liposomes, polymeric nanoparticles, and complexes, have been widely applied, particularly in the cardiovascular and oncology fields. These nanocarriers containing active substances, such as siRNAs or statins, effectively polarize macrophages into the anti-inflammatory phenotype or mediate gene expression in macrophages (Table 3). Increasing evidence suggests that these changes in macrophages promote cardiac repair in infarcted animal models.
Liposomes
Liposomes are microscopic phospholipid bubbles, which include one or more biocompatible lipid bilayers with an aqueous core inside, ranging from a few hundreds to thousands of nanometers in size [120,121,122,123]. Liposomes have been used mostly in basic and clinical medicine since the first FDA-approved liposomal drug Doxil® was used and have exhibited advantageous performance in facilitating encapsulation of a broad variety of pharmaceuticals, depending on the intrinsic amphiphilicity of their lipid bilayer shell(s) [124]. Moreover, compared with the conventional (classical) liposomes, immunoliposomes, which are target liposomes with different surface-targeted ligands, including antibodies and peptides, can target the special cells and reduce the recognition of MPS to eliminate the off-target effects. Tamar et al. demonstrated that systemic administration of phosphatidylserine (PS)-presenting liposomes significantly increases the number of CD206+ macrophages and the level of anti-inflammatory cytokines, such as TGF-β and IL-10, and downregulates the expression of proinflammatory markers at the same time, which promotes angiogenesis and halts ventricular dilatation and adverse remodeling in a rat model of acute MI [125]. Moreover, Ruvinov et al. showed a similar therapeutic effect in a rat model of acute MI by activating macrophages into an anti-inflammatory state [126].
Polymeric Nanoparticles and Complexes
Polymeric nanoparticles (NPs) mainly consist of biodegradable and biocompatible polymers such as natural polymers (e.g., albumin), polylactide, and poly(D,L-lactide co-glycolic acid) (PLGA) and are heterogeneous in size, often ranging from a few tens to thousands of nanometers in diameter [127]. Because of both the hydrophilic and hydrophobic character of polymeric NPs, they have been used in all kinds of pharmaceuticals. Polymeric NPs are promising candidates for drug delivery and have received public attention since the albumin-paclitaxel complex (Abraxane) was approved for IV treatment. This review describes the excellent properties of polymeric NPs: first, these prevent cargo degradation; second, they decrease phagocytosis by MPS; third, they break the absorption barrier formed by biological membranes; and last, they provide a method of sustained drug release. Even more encouraging is that PLGA-based NPs can escape the endolysosomal compartment and release the encapsulated payload in the cytoplasm following cell internalization into the cell by endocytosis [128].
Monocytes/macrophages have been known as targets that are modulated by polymeric NP– and complex-based carriers in the field of cardiac repair and have caught special attention in the past few years. Recent studies have demonstrated that systemic administration of siRNA against messenger (mRNA) loaded into NPs imparts beneficial outcomes in cardiac repair [115, 116]. Acid-sensitive polyketal PK3 particle–encapsuled Nox2-specific siRNA has been reported to contribute to improvement in cardiac function by silencing the Nox2 gene in cardiac macrophages [129]. Leuschner et al. evaluated the therapeutic effect of an optimized lipid NP–loaded siCCR2 on cardiac repair. siCCR2-NPs accumulate in splenic phagocytic cells when administered systemically in mice [130]. Treatment with siCCR2-NPs resulted in a marked reduction in the number of inflammatory monocytes and M1 macrophages along with significant attenuation of MI progression.
Furthermore, NPs containing pioglitazone have been demonstrated to modulate monocyte/macrophage subtype [131, 132]. Pioglitazone, a peroxisome proliferator–activated receptor-γ (PPARγ) agonist, shows a marked impact on monocyte and macrophage polarization, transferring them into the anti-inflammatory subtype [133]. Matoba et al. performed a randomized placebo-controlled study in a mouse model of plaque rupture and suggested that PLGA NPs containing pioglitazone enhance the proportion of M2 macrophages [131]. In a similar approach, pioglitazone NPs administered intravenously to an atherosclerosis ApoE−/− mouse model showed a significant reduction in the number of fibrous caps by decreasing proinflammatory monocytes along with a moderate increase in anti-inflammatory phenotypes [132].
Moreover, the timely endocytosis of dead cells by macrophages can trigger the anti-inflammatory response and transform M1 macrophages into M2 macrophages. Given these findings, a theranostic nanosystem with mimicking apoptosis was established and showed a remarkable ability in resolving inflammation and promoting cardiac repair [134].
CONCLUDING REMARKS
Macrophages play an indispensable role in the mammalian heart and respond to both the post-MI regeneration and repair by mediating inflammation and immunity. M1 and M2 macrophages are indispensable in repairing the myocardium and in retaining functional architecture. Different populations of macrophages respond to a special stage after MI. Several studies have focused on the mechanism of macrophage polarization that is related to the cardioprotective potential. With advancement in therapeutic approaches, especially the burgeoning NP-mediated drug delivery system, modulating macrophage activation to guide cardiac repair and regeneration is now a promising therapeutic strategy. These strategies are also available to other CVDs associated with macrophages, such as atherosclerosis and myocarditis.
As this review highlights, the roles of macrophages in terms of cardiac repair and regeneration are complex. To better understand the mechanisms of macrophages during the various stages of acute and chronic myocardial disease, further research is warranted. Moreover, although specially designed nanocarriers have been applied to improve the efficiency of therapeutic agents and a major breakthrough has been reached, many challenges remain, including the off-target delivery and security issues, which require additional investigation.
Abbreviations
- Arg-1:
-
arginase 1
- CCL:
-
chemokine (C-C motif) ligand
- CDCs:
-
cardiosphere-derived cells
- CSCs:
-
cardiac stromal cells
- CVD:
-
cardiovascular disease
- CXCL:
-
C-X-C motif chemokine
- DAMPs:
-
damage-associated molecular patterns
- ECM:
-
extracellular matrix
- EVs:
-
extracellular vesicles
- GM-CSF:
-
granulocyte macrophage colony-stimulating factor
- hESC-CVPCs:
-
human embryonic stem cell–derived cardiovascular progenitor cells
- HIF:
-
hypoxia-inducible factor
- IFN:
-
interferon
- IL:
-
interleukin
- IKKβ :
-
IκB kinase β
- iNOS:
-
inducible nitric oxide synthase
- I/R:
-
ischemia/reperfusion
- IRAK4:
-
interleukin-1 receptor–associated kinase 4
- IRF:
-
interferon regulatory factor
- JAK:
-
Janus kinase
- KLF-4:
-
Krüppel-like factor 4
- LDL:
-
low-density lipoprotein
- LPS:
-
lipopolysaccharide
- M-CSF:
-
macrophage colony-stimulating factor
- MI:
-
myocardial infarction
- MPS:
-
mononuclear phagocyte system
- MSCs:
-
mesenchymal stromal cells
- Mrc1:
-
mannose receptor (CD206)
- MyD88:
-
myeloid differentiation primary response gene 88
- NPs:
-
nanoparticles
- PLGA:
-
poly(lactide co-glycolic acid)
- PPARγ :
-
peroxisome proliferator–activated receptor-γ
- SOCS3:
-
suppressor of cytokine signaling 3
- STAT:
-
signal transducers and activators of transcription
- TGF-β :
-
transforming growth factor
- TLR4:
-
toll-like receptor 4
- TNF-α :
-
tumor necrosis factor-α
- TRAF:
-
TNF receptor–associated factor
- TRIF:
-
TIR domain–containing adapter-inducing interferon
References
Sadek, Hesham, and Eric N. Olson. 2020. Toward the goal of human heart regeneration. Cell Stem Cell 26: 7–16.
Kura, Branislav, Barbara Szeiffova Bacova, Barbora Kalocayova, Matus Sykora, and Jan Slezak. 2020. Oxidative stress-responsive microRNAs in heart injury. International Journal of Molecular Sciences 21 (1): 358.
Roth, Gregory A., Catherine Johnson, Amanuel Abajobir, Foad Abd-Allah, Semaw Ferede Abera, Gebre Abyu, Muktar Ahmed, et al. 2017. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. Journals of the American College of Cardiology 70: 1–25.
Braunwald, Eugene. 2015. The war against heart failure: the Lancet lecture. Lancet 385 (9970): 812–824.
Poss, Kenneth D., Lindsay G. Wilson, and Mark T. Keating. 2002. Heart regeneration in zebrafish. Science 298: 2188–2190.
Kikuchi, Kazu, Jennifer E. Holdway, Andreas A. Werdich, Ryan M. Anderson, Yi Fang, Gregory F. Egnaczyk, Todd Evans, et al. 2010. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 464: 601–605.
Jopling, Chris, Eduard Sleep, Marina Raya, Mercè Martí, Angel Raya, and Juan Carlos Izpisúa Belmonte. 2010. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 464: 606–609.
Porrello, Enzo R., Ahmed I. Mahmoud, Emma Simpson, Joseph A. Hill, James A. Richardson, Eric N. Olson, and Hesham A. Sadek. 2011. Transient regenerative potential of the neonatal mouse heart. Science 331: 1078–1080.
Porrello, Enzo R., Ahmed I. Mahmoud, Emma Simpson, Brett A. Johnson, David Grinsfelder, Diana Canseco, Pradeep P. Mammen, et al. 2013. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proceedings of The National Academy of Sciences of The United States of America 110 (1): 187–192.
Hansson, Göran K. 2005. Inflammation, atherosclerosis, and coronary artery disease. New England Journal of Medicine 352: 1685–1695.
Ross, Russell. 1999. Atherosclerosis — an inflammatory disease. New England Journal of Medicine 340: 115–126.
Wayne Alexander, R. 1994. Inflammation and coronary artery disease. New England Journal of Medicine 331: 468–469.
Redd, Michael J., Lisa Cooper, Will Wood, Brian Stramer, and Paul Martin. 2004. Wound healing and inflammation: embryos reveal the way to perfect repair. Philosophical Transactions of The Royal Society B-Biological Sciences 359: 777–784.
Eming, Sabine A., Thomas A. Wynn, and Paul Martin. 2017. Inflammation and metabolism in tissue repair and regeneration. Science 356: 1026–1030.
Heidt, Timo, Gabriel Courties, Partha Dutta, Hendrik B. Sager, Matt Sebas, Yoshiko Iwamoto, Yuan Sun, Nicolas da Silva, Peter Panizzi, Anja M. van der Laan, Filip K. Swirski, Ralph Weissleder, and Matthias Nahrendorf. 2014. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circulation Research 115: 284–295.
van der Laan, Anja M., Ellis N. ter Horst, Ronak Delewi, Mark P.V. Begieneman, Paul A.J. Krijnen, Alexander Hirsch, Mehrdad Lavaei, M. Nahrendorf, A.J. Horrevoets, H.W.M. Niessen, and J.J. Piek. 2014. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. European Heart Journal 35: 376–385.
Frangogiannis, Nikolaos G. The inflammatory response in myocardial injury, repair, and remodelling. Nature Reviews Cardiology 11 (5): 255–265.
Kanisicak, Onur, Hadi Khalil, Malina J. Ivey, Jason Karch, Bryan D. Maliken, Robert N. Correll, and Matthew J. Brody, et al. 2016. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nature Communications 7: 12260.
Serini, Guido, Marie-Luce Bochaton-Piallat, Patricia Ropraz, Antoine Geinoz, Laura Borsi, Luciano Zardi, and Giulio Gabbiani. 1998. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. The Journal of Cell Biology 142 (3): 873–881.
Frodermann, Vanessa, and Matthias Nahrendorf. 2018. Macrophages and cardiovascular health. Physiological Reviews 98: 2523–2569.
van Furth, Ralph, and Zanvil A. Cohn. 1968. The origin and kinetics of mononuclear phagocytes. Journal of Experimental Medicine 128 (3): 415–435.
Robbins, Clinton S., Aleksey Chudnovskiy, Philipp J. Rauch, Jose-Luiz Figueiredo, Yoshiko Iwamoto, Rostic Gorbatov, Martin Etzrodt, Georg F. Weber, et al. 2012. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation 125 (2): 364–374.
Swirski, Filip K., Matthias Nahrendorf, Martin Etzrodt, Moritz Wildgruber, Virna Cortez-Retamozo, Peter Panizzi, Jose-Luiz Figueiredo, et al. 2009. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325 (5940): 612–616.
Auffray, Cedric, Darin Fogg, Meriem Garfa, Gaelle Elain, Olivier Join-Lambert, Samer Kayal, Sabine Sarnacki, A. Cumano, G. Lauvau, and F. Geissmann. 2007. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317 (5838): 666–670.
Bajpai, Geetika, Caralin Schneider, Nicole Wong, Andrea Bredemeyer, Maarten Hulsmans, Matthias Nahrendorf, Slava Epelman, Daniel Kreisel, Yongjian Liu, Akinobu Itoh, Thirupura S. Shankar, Craig H. Selzman, Stavros G. Drakos, and Kory J. Lavine. 2018. The human heart contains distinct macrophage subsets with divergent origins and functions. Nature Medicine 24 (8): 1234–1245.
Volkman, A., N.C. Chang, P.H. Strausbauch, and P.S. Morahan. 1983. Differential effects of chronic monocyte depletion on macrophage populations. Laboratory Investigation 49: 291–298.
Sawyer, R.T., P.H. Strausbauch, and A. Volkman. 1982. Resident macrophage proliferation in mice depleted of blood monocytes by strontium-89. Laboratory Investigation 46 (2): 165–170.
Ginhoux, Florent, Melanie Greter, Marylene Leboeuf, Sayan Nandi, Peter See, Solen Gokhan, and Mark F Mehler, et al. 2010. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330 (6005): 841–845.
Schulz, Christian, Elisa Gomez Perdiguero, Laurent Chorro, Heather Szabo-Rogers, Nicolas Cagnard, Katrin Kierdorf, Marco Prinz, Bishan Wu, Sten Eirik W. Jacobsen, Jeffrey W. Pollard, Jon Frampton, Karen J. Liu, and Frederic Geissmann. 2012. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336 (6077): 86–90.
Yona, Simon, Ki-Wook Kim, Yochai Wolf, Alexander Mildner, Diana Varol, Michal Breker, Dalit Strauss-Ayali, Sergey Viukov, Martin Guilliams, Alexander Misharin, David A. Hume, Harris Perlman, Bernard Malissen, Elazar Zelzer, and Steffen Jung. 2013. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38 (1): 79–91.
Hashimoto, Daigo, Andrew Chow, Clara Noizat, Pearline Teo, Mary Beth Beasley, Marylene Leboeuf, and Christian D Becker, et al. 2013. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38 (4): 792–804.
Guilliams, Martin, Ismé De Kleer, Sandrine Henri, Sijranke Post, Leen Vanhoutte, Sofie De Prijck, Kim Deswarte, et al. 2013. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. Journal of Experimental Medicine 210 (10): 1977–1992.
Epelman, Slava, Kory J. Lavine, Anna E. Beaudin, Dorothy K. Sojka, Javier A. Carrero, Boris Calderon, Thaddeus Brija, et al. 2014. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40 (1): 91–104.
Mosser, David M., and Justin P. Edwards. 2008. Exploring the full spectrum of macrophage activation. Nature Reviews Immunology 8 (12): 958–969.
Gordon, Siamon. 2003. Alternative activation of macrophages. Nature Reviews Immunology 3 (1): 23–35.
Gordon, Siamon, and Philip R. Taylor. 2005. Monocyte and macrophage heterogeneity. Nature Reviews Immunology 5: 953–964.
Martinez, Fernando Oneissi, Antonio Sica, Alberto Mantovani, and Massimo Locati. 2008. Macrophage activation and polarization. Frontiers in Bioscience 13: 453–461.
Martinez, Fernando O., Siamon Gordon, Massimo Locati, and Alberto Mantovani. 2006. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. Journal of Immunology 177 (10): 7303–7311.
Martinez, Fernando O., and Siamon Gordon. 2014. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Reports 6: 13.
Cheng, Yuanyuan, and Jianhui Rong. 2018. Macrophage polarization as a therapeutic target in myocardial infarction. Current Drug Targets 19 (6): 651–662.
Krausgruber, Thomas, Katrina Blazek, Tim Smallie, Saba Alzabin, Helen Lockstone, Natasha Sahgal, Tracy Hussell, Marc Feldmann, and Irina A. Udalova. 2011. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nature Immunology 12: 231–238.
Toshchakov, Vladimir, Bryan W. Jones, Pin-Yu Perera, Karen Thomas, M. Joshua Cody, Shuling Zhang, Bryan R.G. Williams, et al. 2002. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nature Immunology 3: 392–398.
Dale, David C., Laurence Boxer, and W. Conrad Liles. 2008. The phagocytes: neutrophils and monocytes. Blood 112 (4): 935–945.
Filipe-Santos, Orchidée, Jacinta Bustamante, Ariane Chapgier, Guillaume Vogt, Ludovic de Beaucoudrey, Jacqueline Feinberg, Emmanuelle Jouanguy, Stéphanie Boisson-Dupuis, Claire Fieschi, Capucine Picard, and Jean-Laurent Casanova. 2006. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Seminars in Immunology 18 (6): 347–361.
Murray, Peter J., Judith E. Allen, Subhra K. Biswas, Edward A. Fisher, Derek W. Gilroy, Sergij Goerdt, Siamon Gordon, et al. 2014. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41 (1): 14–20.
Gordon, Siamon, and Fernando O. Martinez. 2010. Alternative activation of macrophages: mechanism and functions. Immunity 32: 593–604.
Rőszer, Tamás. 2015. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators of Inflammations 2015: 816460.
Lang, Roland, Divyen Patel, John J. Morris, Robert L. Rutschman, and Peter J. Murray. 2002. Shaping gene expression in activated and resting primary macrophages by IL-10. Journal of Immunology 169: 2253–2263.
Nelms, Keats, Achsah D. Keegan, Jose Zamorano, John J. Ryan, and William E. Paul. 1999. The IL-4 receptor: signaling mechanisms and biologic functions. Annual Review of Immunology 17: 701–738.
O’Farrell, Anne-Marie, Ying Liu, Kevin W. Moore, and Alice L.-F. Mui. 1998. IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanisms: evidence for Stat3-dependent and -independent pathways. EMBO Journal 17: 1006–1018.
Navegantes, Kely Campos, Rafaelli de Souza Gomes, Priscilla Aparecida Tártari Pereira, Paula Giselle Czaikoski, Carolina Heitmann Mares Azevedo, and Marta Chagas Monteiro. 2017. Immune modulation of some autoimmune diseases: the critical role of macrophages and neutrophils in the innate and adaptive immunity. Journal of Translational Medicine 15 (1): 36.
Aurora, Arin B., Enzo R. Porrello, Wei Tan, Ahmed I. Mahmoud, Joseph A. Hill, Rhonda Bassel-Duby, Hesham A. Sadek, et al. 2014. Macrophages are required for neonatal heart regeneration. Journal of Clinical Investigation 124 (3): 1382–1392.
Pinto, Alexander R., Rosa Paolicelli, Ekaterina Salimova, Janko Gospocic, Esfir Slonimsky, Daniel Bilbao-Cortes, James W. Godwin, et al. 2012. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One 7 (5): e36814.
Perdiguero, Elisa Gomez, Kay Klapproth, Christian Schulz, Katrin Busch, Emanuele Azzoni, Lucile Crozet, Hannah Garner, et al. 2015. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518 (7540): 547–551.
Epelman, Slava, Kory J. Lavine, and Gwendalyn J. Randolph. 2014. Origin and functions of tissue macrophages. Immunity 41: 21–35.
Bajpai, Geetika, Andrea Bredemeyer, Wenjun Li, Konstantin Zaitsev, and Andrew L Koenig, Inessa Lokshina, Jayaram Mohan, et al. 2019. Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circulation Research 124 (2): 263–278.
Lavine, Kory J., Slava Epelman, Keita Uchida, Kassandra J. Weber, Colin G. Nichols, Joel D. Schilling, David M. Ornitz, et al. 2014. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proceedings of The National Academy of Sciences of The United States of America 111 (45): 16029–16034.
Molawi, Kaaweh, Yochai Wolf, Prashanth K. Kandalla, Jeremy Favret, Nora Hagemeyer, Kathrin Frenzel, Alexander R. Pinto, et al. 2014. Progressive replacement of embryo-derived cardiac macrophages with age. Journal of Experimental Medicine 211: 2151–2158.
Weyrich, Andrew S., Mark R. Elstad, Rodger P. McEver, Thomas M. McIntyre, Kevin L. Moore, James H. Morrissey, Stephen M. Prescott, and Guy A. Zimmerman. 1996. Activated platelets signal chemokine synthesis by human monocytes. Journal of Clinical Investigation 97 (6): 1525–1534.
Moore, Kathryn J., and Ira Tabas. 2011. Macrophages in the pathogenesis of atherosclerosis. Cell 145: 341–355.
Witztum, Joseph L., and Daniel Steinberg. 2001. The oxidative modification hypothesis of atherosclerosis: does it hold for humans? Trends in Cardiovascular Medicine 11: 93–102.
Remmerie, Anneleen, and Charlotte L. Scott. 2018. Macrophages and lipid metabolism. Cellular Immunology 330: 27–42.
Lindemann, Stephan, Björn Krämer, Karin Daub, Konstantinos Stellos, and Meinrad Gawaz. 2007. Molecular pathways used by platelets to initiate and accelerate atherogenesis. Current Opinion Lipidology 18 (5): 566–573.
Tabas, Ira. 2005. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arteriosclerosis, Thrombosis, and Vascular Biology 25 (11): 2255–2264.
De Filippis, Andrew P., Andrew R. Chapman, Nicholas L. Mills, James A. de Lemos, Armin Arbab-Zadeh, L. Kristin Newby, and David A. Morrow. 2019. Assessment and treatment of patients with type 2 myocardial infarction and acute nonischemic myocardial injury. Circulation 140 (20): 1661–1678.
Forte, Elvira, Milena Bastos Furtado, and Nadia Rosenthal. 2018. The interstitium in cardiac repair: role of the immune-stromal cell interplay. Nature Reviews Cardiology 15 (10): 601–616.
Döring, Yvonne, Maik Drechsler, Oliver Soehnlein, and Christian Weber. 2015. Neutrophils in atherosclerosis: from mice to man. Arteriosclerosis, Thrombosis, and Vascular Biology 35: 288–295.
Soehnlein, Oliver, Alma Zernecke, Einar E. Eriksson, Antonio Gigliotti Rothfuchs, Christine T. Pham, Heiko Herwald, Kiril Bidzhekov, et al. 2008. Neutrophil secretion products pave the way for inflammatory monocytes. Blood 112: 1461–1471.
Frantz, Stefan, Ulrich Hofmann, Daniela Fraccarollo, Andreas Schäfer, Stefanie Kranepuhl, Ina Hagedorn, Bernhard Nieswandt, Matthias Nahrendorf, Helga Wagner, Barbara Bayer, Christina Pachel, Michael P. Schön, Susanne Kneitz, Tobias Bobinger, Frank Weidemann, Georg Ertl, and Johann Bauersachs. 2013. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB Journal 27 (3): 871–881.
Dewald, Oliver, Pawel Zymek, Kim Winkelmann, Anna Koerting, Guofeng Ren, Tareq Abou-Khamis, Lloyd H. Michael, et al. 2005. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circulation Research 96 (8): 881–889.
Wan, Elaine, Xin Yi Yeap, Shirley Dehn, Rachael Terry, Margaret Novak, Shuang Zhang, Shinichi Iwata, Xiaoqiang Han, Shunichi Homma, Konstantinos Drosatos, Jon Lomasney, David M. Engman, Stephen D. Miller, Douglas E. Vaughan, John P. Morrow, Raj Kishore, and Edward B. Thorp. 2013. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circulation Research 113 (8): 1004–1012.
Duffield, Jeremy S., Stuart J. Forbes, Christothea M. Constandinou, Spike Clay, Marina Partolina, Srilatha Vuthoori, Shengji Wu, Richard Lang, and John P. Iredale. 2005. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. Journal of Clinical Investigation 115: 56–65.
Fadok, Valerie A., Donna L. Bratton, Anatole Konowal, Peter W. Freed, Jay Y. Westcott, and Peter M. Henson. 1998. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. Journal of Clinical Investigation 101 (4): 890–898.
Shiraishi, Manabu, Yasunori Shintani, Yusuke Shintani, Hidekazu Ishida, Rie Saba, Atsushi Yamaguchi, Hideo Adachi, Kenta Yashiro, and Ken Suzuki. 2016. Alternatively activated macrophages determine repair of the infarcted adult murine heart. Journal of Clinical Investigation 126 (6): 2151–2166.
Galuppo, Paolo, Sabine Vettorazzi, Julian Hövelmann, Claus-Jürgen Scholz, Jan Peter Tuckermann, Johann Bauersachs, and Daniela Fraccarollo. 2017. The glucocorticoid receptor in monocyte-derived macrophages is critical for cardiac infarct repair and remodeling. FASEB Journal 31 (11): 5122–5132.
Vincent, Anne, Aurélie Covinhes, Christian Barrère, Laura Gallot, Soulit Thoumala, Christophe Piot, Catherine Heurteaux, Michel Lazdunski, Joël Nargeot, and Stéphanie Barrère-Lemaire. 2017. Acute and long-term cardioprotective effects of the traditional Chinese medicine MLC901 against myocardial ischemia-reperfusion injury in mice. Scientific Reports 7 (1): 14701.
Pælestik, Kim B., Nichlas R. Jespersen, Rebekka V. Jensen, Jacob Johnsen, Hans Erik Bøtker, and Steen B. Kristiansen. 2017. Effects of hypoglycemia on myocardial susceptibility to ischemia-reperfusion injury and preconditioning in hearts from rats with and without type 2 diabetes. Cardiovascular Diabetology 16 (1): 148.
Vagnozzi, Ronald J., Marjorie Maillet, Michelle A. Sargent, Hadi Khalil, Anne Katrine Z. Johansen, Jennifer A. Schwanekamp, Allen J. York, et al. 2020. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 577 (7790): 405–409.
Steffens, Sabine, Fabrizio Montecucco, and François Mach. 2009. The inflammatory response as a target to reduce myocardial ischaemia and reperfusion injury. Thrombosis And Haemostasis 102 (2): 240–247.
Formigli, Lucia, Lidia Ibba Manneschi, Chiara Nediani, Elena Marcelli, Geri Fratini, Sandra Zecchi Orlandini, and Avio M. Perna. 2001. Are macrophages involved in early myocardial reperfusion injury? Annals of Thoracic Surgery 71 (5): 1596–1602.
Fan, Qin, Rong Tao, Hang Zhang, Hongyang Xie, Lin Lu, Ting Wang, Su Min, et al. 2019. Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation 139 (5): 663–678.
Zhang, Xiuling, Xianxian Cao, Mengqiu Dang, Hongxia Wang, Buxing Chen, Du Fenghe, Huihua Li, Xiangjun Zeng, and Caixia Guo. 2019. Soluble receptor for advanced glycation end-products enhanced the production of IFN-γ through the NF-κB pathway in macrophages recruited by ischemia/reperfusion. International Journal of Molecular Medicine 43 (6): 2507–2515.
Chang, Chao, Qingwei Ji, Bangwei Wu, Kunwu Yu, Qiutang Zeng, Shuanli Xin, Jixiang Liu, and Yujie Zhou. 2015. Chemerin15-ameliorated cardiac ischemia-reperfusion injury is associated with the induction of alternatively activated macrophages. Mediators of Inflammation 2015: 563951.
Yue, Yuan, Suiqing Huang, Lexun Wang, Zixuan Wu, Mengya Liang, Huayang Li, Linhua Lv, Wei Li, and Wu. Zhongkai. 2020. M2b macrophages regulate cardiac fibroblast activation and alleviate cardiac fibrosis after reperfusion injury. Circulation Journal 84 (4): 626–635.
Yue, Yuan, Suiqing Huang, Huayang Li, Wei Li, Jian Hou, Li Luo, Quan Liu, Cuiping Wang, Song Yang, Linhua Lv, Jinghua Shao, and Zhongkai Wu. 2020. M2b macrophages protect against myocardial remodeling after ischemia/reperfusion injury by regulating kinase activation of platelet-derived growth factor receptor of cardiac fibroblast. Annals of Translational Medicine 8 (21): 1409.
Smith, Rachel Ruckdeschel, Lucio Barile, Hee Cheol Cho, Michelle K. Leppo, Joshua M. Hare, Elisa Messina, Alessandro Giacomello, M. Roselle Abraham, and Eduardo Marbán. 2007. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 115: 896–908.
Cheng, Ke, Konstantinos Malliaras, Rachel Ruckdeschel Smith, Deliang Shen, Baiming Sun, Agnieszka Blusztajn, Yucai Xie, Ahmed Ibrahim, Mohammad Amin Aminzadeh, Weixin Liu, Tao-Sheng Li, Michele A. de Robertis, Linda Marbán, Lawrence S.C. Czer, Alfredo Trento, and Eduardo Marbán. 2014. Human cardiosphere-derived cells from advanced heart failure patients exhibit augmented functional potency in myocardial repair. Jacc-Heart Failure 2: 49–61.
Tang, Junnan, Jinqiang Wang, Ke Huang, Yanqi Ye, Su Teng, Li Qiao, Michael Taylor Hensley, et al. 2018. Cardiac cell-integrated microneedle patch for treating myocardial infarction. Science Advances. Science Advance 4 (11): eaat9365.
Hasan, Al Shaimaa, Lan Luo, Chen Yan, Tian-Xia Zhang, Yoshishige Urata, Shinji Goto, Safwat A. Mangoura, Mahmoud H. Abdel-Raheem, Shouhua Zhang, and Tao-Sheng Li. 2016. Cardiosphere-derived cells facilitate heart repair by modulating M1/M2 macrophage polarization and neutrophil recruitment. PLoS One 11 (10): e0165255.
Kanazawa, Hideaki, Eleni Tseliou, James F. Dawkins, Geoffrey De Couto, Romain Gallet, Konstantinos Malliaras, Kristine Yee, et al. 2016. Durable benefits of cellular postconditioning: long-term effects of allogeneic cardiosphere-derived cells infused after reperfusion in pigs with acute myocardial infarction. Journal of the American Heart Association 5 (2): e002796.
Makkar, Raj R., Rachel R. Smith, Ke Cheng, Konstantinos Malliaras, Louise Ej Thomson, Daniel Berman, Lawrence Sc Czer, et al. 2012. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet 379 (9819): 895–904.
Wang, Jinxi, Meilan Liu, Qiang Wu, Qiang Li, Ling Gao, Yun Jiang, Boxiong Deng, Wei Huang, Wei Bi, Zhongyan Chen, Y. Eugene Chin, Christian Paul, Yigang Wang, and Huang-Tian Yang. 2019. Human embryonic stem cell-derived cardiovascular progenitors repair infarcted hearts through modulation of macrophages via activation of signal transducer and activator of transcription 6. Antioxidants & Redox Signaling 31 (5): 369–386.
Cho, Dong-Im, Mi Ra Kim, Hye-yun Jeong, Hae Chang Jeong, Myung Ho Jeong, Sung Ho Yoon, Yong Sook Kim, and Youngkeun Ahn. 2014. Mesenchymal stem cells reciprocally regulate the M1/M2 balance in mouse bone marrow-derived macrophages. Experimental And Molecular Medicine 46 (1): e70.
Dayan, Victor, Gustavo Yannarelli, Filio Billia, Paola Filomeno, Xing-Hua Wang, John E. Davies, and Armand Keatingl. 2011. Mesenchymal stromal cells mediate a switch to alternatively activated monocytes/macrophages after acute myocardial infarction. Basic Research In Cardiology 106 (6): 1299–1310.
Ben-Mordechai, Tamar, Radka Holbova, Natalie Landa-Rouben, Tamar Harel-Adar, Micha S. Feinberg, Ihab Abd Elrahman, Galia Blum, et al. 2013. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. Journal of the American College of Cardiology 62 (20): 1890–1901.
Maas, Sybren L.N., Xandra O. Breakefield, and Alissa M. Weaver. 2017. Extracellular vesicles: unique intercellular delivery vehicles. Trends in Cell Biology 27 (3): 172–188.
Qiao, Li, Shiqi Hu, Suyun Liu, Hui Zhang, Hong Ma, Ke Huang, Zhenhua Li, et al. 2019. MicroRNA-21-5p dysregulation in exosomes derived from heart failure patients impairs regenerative potential. Ca-A Cancer Journal For Clinicians 129 (6): 2237–2250.
Mol, Emma A., Zhiyong Lei, Marieke T. Roefs, Maarten H. Bakker, Marie-José Goumans, Pieter A. Doevendans, Patricia Y.W. Dankers, et al. 2019. Injectable supramolecular ureidopyrimidinone hydrogels provide sustained release of extracellular vesicle therapeutics. Advanced Healthcare Materialsr 8 (20): e1900847.
Kalluri, Raghu, and Valerie S. LeBleu. 2020. The biology, function, and biomedical applications of exosomes. Science 367 (6478): eaau6977.
Zhang, Jianchao, Xiaolin Cui, Jiacheng Guo, Chang Cao, Zenglei Zhang, Bo Wang, Li Zhang, D. Shen, K. Lim, T. Woodfield, J. Tang, and J. Zhang. 2020. Small but significant: insights and new perspectives of exosomes in cardiovascular disease. Journal of Cellular And Molecular Medicine 24 (15): 8291–8303.
Tang, Jun-Nan, Jhon Cores, Ke Huang, Xiao-Lin Cui, Lan Luo, Jin-Ying Zhang, Tao-Sheng Li, Li Qian, and Ke Cheng. 2018. Concise review: is cardiac cell therapy dead? Embarrassing trial outcomes and new directions for the future. Stem Cells Translational Medicine 7 (4): 354–359.
Zhao, Jinxuan, Xueling Li, Jiaxin Hu, Fu Chen, Shuaihua Qiao, Xuan Sun, Ling Gao, Jun Xie, and Biao Xu. 2019. Mesenchymal stromal cell-derived exosomes attenuate myocardial ischaemia-reperfusion injury through miR-182-regulated macrophage polarization. Cardiovascular Research 115 (7): 1205–1216.
Xu, Ruqin, Fangcheng Zhang, Renjie Chai, Wenyi Zhou, Ming Hu, Bin Liu, Xuke Chen, Mingke Liu, Qiong Xu, Ningning Liu, and Shiming Liu. 2019. Exosomes derived from pro-inflammatory bone marrow-derived mesenchymal stem cells reduce inflammation and myocardial injury via mediating macrophage polarization. Journal of Cellular And Molecular Medicine 23 (11): 7617–7631.
Wang, Jinli, Christine J. Lee, Michael B. Deci, Natalie Jasiewicz, Anjali Verma, John M. Canty, and Juliane Nguyen. 2020. MiR-101a loaded extracellular nanovesicles as bioactive carriers for cardiac repair. Nanomedicine 27: 102201.
de Couto, Geoffrey, Romain Gallet, Linda Cambier, Ervin Jaghatspanyan, Nupur Makkar, James Frederick Dawkins, Benjamin P. Berman, et al. Exosomal microRNA transfer into macrophages mediates cellular postconditioning. Circulation 136: 200–214.
de Couto, Geoffrey, Weixin Liu, Eleni Tseliou, Baiming Sun, Nupur Makkar, Hideaki Kanazawa, Moshe Arditi, and Eduardo Marbán. 2015. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. Journal of Clinical Investigation 125: 3147–3162.
de Couto, Geoffrey, Ervin Jaghatspanyan, Matthew DeBerge, Weixin Liu, Kristin Luther, Yizhou Wang, Jie Tang, Edward B. Thorp, and Eduardo Marbán. 2019. Mechanism of enhanced MerTK-dependent macrophage efferocytosis by extracellular vesicles. Arteriosclerosis Thrombosis And Vascular Biology 39 (10): 2082–2096.
Mentkowski, Kyle I., Asma Mursleen, Jonathan D. Snitzer, Lindsey M. Euscher, and Jennifer K. Lang. 2020. CDC-derived extracellular vesicles reprogram inflammatory macrophages to an arginase 1-dependent proangiogenic phenotype. American Journal of Physiology-Heart And Circulatory Physiology 318 (6): H1447–H1460.
Cambier, Linda, Geoffrey de Couto, Ahmed Ibrahim, and Antonio K Echavez, Jackelyn Valle, Weixin Liu, Michelle Kreke, et al. 2017. Y RNA fragment in extracellular vesicles confers cardioprotection via modulation of IL-10 expression and secretion. Embo Molecular Medicine 9 (3): 337–352.
Nguyen, My-Anh, Denuja Karunakaran, Michèle Geoffrion, and Henry S Cheng, Kristofferson Tandoc, Ljubica Perisic Matic, Ulf Hedin, et al. 2018. Extracellular vesicles secreted by atherogenic macrophages transfer microRNA to inhibit cell migration. Arteriosclerosis Thrombosis And Vascular Biology 38 (1): 49–63.
Wang, Chunxiao, Congcong Zhang, Luxin Liu, A. Xi, Boya Chen, Yulin Li, and Du. Jie. 2017. Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Molecular Therapy-Nucleic Acids 25 (1): 192–204.
Dai, Yuxiang, Wang Shen, Shufu Chang, Daoyuan Ren, Shalaimaiti Shali, Chenguang Li, Hongbo Yang, et al. 2020. M2 macrophage-derived exosomes carry microRNA-148a to alleviate myocardial ischemia/reperfusion injury via inhibiting TXNIP and the TLR4/NF-κB/NLRP3 inflammasome signaling pathway. Journal of Molecular And Cellular Cardiology 142: 65–79.
Wu, Guanghao, Jinfeng Zhang, Qianru Zhao, Wanru Zhuang, Jingjing Ding, Chi Zhang, Haijun Gao, Dai-Wen Pang, Kanyi Pu, and Hai-Yan Xie. 2020. Molecularly engineered macrophage-derived exosomes with inflammation tropism and intrinsic heme biosynthesis for atherosclerosis treatment. Angewandte Chemie International Edition 59 (10): 4068–4074.
Pironti, Gianluigi, Ryan T. Strachan, Dennis Abraham, Samuel Mon-Wei Yu, Minyong Chen, Wei Chen, Kenji Hanada, et al. 2015. Circulating exosomes induced by cardiac pressure overload contain functional angiotensin II type 1 receptors. Circulation 131 (24): 2120–2130.
Antes, Travis J., Ryan C. Middleton, Kristin M. Luther, Takeshi Ijichi, Kiel A. Peck, Weixin Jane Liu, Jackie Valle, et al. 2018. Targeting extracellular vesicles to injured tissue using membrane cloaking and surface display. Journal of Nanobiotechnology 16 (1): 61.
Wan, Zhuo, Lianbi Zhao, Fan Lu, Xiaotong Gao, Yan Dong, Yingxin Zhao, Mengying Wei, Guodong Yang, Changyang Xing, and Li Liu. 2020. Mononuclear phagocyte system blockade improves therapeutic exosome delivery to the myocardium. Theranostics 10 (1): 218–230.
Tang, Junnan, Su Teng, Ke Huang, Phuong-Uyen Dinh, Zegen Wang, Adam Vandergriff, Michael T. Hensley, et al. 2018. Targeted repair of heart injury by stem cells fused with platelet nanovesicles. Nature Biomedical Engineering 2: 17–26.
Zhang, Ning, Yanan Song, Zheyong Huang, Jing Chen, Haipeng Tan, Hongbo Yang, Mengkang Fan, Qiyu Li, Qiaozi Wang, Jinfeng Gao, Zhiqing Pang, Juying Qian, and Junbo Ge. 2020. Monocyte mimics improve mesenchymal stem cell-derived extracellular vesicle homing in a mouse MI/RI model. Biomaterials 255: 120168.
Lv, Kaiqi, Qingju Li, Ling Zhang, Yingchao Wang, Zhiwei Zhong, Jing Zhao, Xiaoxiao Lin, Jingyi Wang, Keyang Zhu, Changchen Xiao, Changle Ke, Shuhan Zhong, Xianpeng Wu, Jinghai Chen, Hong Yu, Wei Zhu, Xiang Li, Ben Wang, Ruikang Tang, Jian'an Wang, Jinyu Huang, and Xinyang Hu. 2019. Incorporation of small extracellular vesicles in sodium alginate hydrogel as a novel therapeutic strategy for myocardial infarction. Theranostics 9 (24): 7403–7416.
Chen, Wenjie, Ewa M. Goldys, and Wei Deng. 2020. Light-induced liposomes for cancer therapeutics. Progress In Lipid Research 79: 101052.
Bulbake, Upendra, Sindhu Doppalapudi, Nagavendra Kommineni, and Wahid Khan. 2017. Liposomal formulations in clinical use: an updated review. Pharmaceutics 9 (2): 12.
Wang, Yuwei, and David W. Grainger. 2019. Lyophilized liposome-based parenteral drug development: reviewing complex product design strategies and current regulatory environments. Advanced Drug Delivery Reviews 151-152: 56–71.
Torchilin, Vladimir P. 2005. Recent advances with liposomes as pharmaceutical carriers. Nature Reviews Drug Discovery 4 (2): 145–160.
Gabizon, Alberto, Hilary Shmeeda, and Yechezkel Barenholz. 2003. Pharmacokinetics of pegylated liposomal doxorubicin: review of animal and human studies. Clinical Pharmacokinetics 42 (5): 419–436.
Harel-Adar, Tamar, Tamar Ben Mordechai, Yoram Amsalem, Micha S. Feinberg, Jonathan Leor, and Smadar Cohen. 2011. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proceedings of The National Academy of Sciences of The United States of America 108 (5): 1827–1832.
Ruvinov, Emil, Tamar Harel-Adar, and Smadar Cohen. 2011. Bioengineering the infarcted heart by applying bio-inspired materials. Journal of Cardiovascular Translational Research 4 (5): 559–574.
Aizik, Gil, Etty Grad, and Gershon Golomb. 2018. Monocyte-mediated drug delivery systems for the treatment of cardiovascular diseases. Drug Delivery And Translational Research 8 (4): 868–882.
Elazar, Victoria, Hassan Adwan, Tobias Bäuerle, Keren Rohekar, Gershon Golomb, and Martin R. Berger. 2010. Sustained delivery and efficacy of polymeric nanoparticles containing osteopontin and bone sialoprotein antisenses in rats with breast cancer bone metastasis. International Journal of Cancer 126 (7): 1749–1760.
Somasuntharam, Inthirai, Archana V. Boopathy, Raffay S. Khan, Mario D. Martinez, Milton E. Brown, Niren Murthy, and Michael E. Davis. 2013. Delivery of Nox2-NADPH oxidase siRNA with polyketal nanoparticles for improving cardiac function following myocardial infarction. Biomaterials 34 (31): 7790–7798.
Leuschner, Florian, Partha Dutta, Rostic Gorbatov, Tatiana I. Novobrantseva, Jessica S. Donahoe, Gabriel Courties, Kang Mi Lee, et al. 2011. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nature Biotechnology 29 (11): 1005–1010.
Matoba, Tetsuya, Jun-Ichiro Koga, Kaku Nakano, Kensuke Egashira, and Hiroyuki Tsutsui. 2017. Nanoparticle-mediated drug delivery system for atherosclerotic cardiovascular disease. Journal of Cardiology 70 (3): 206–211.
Nakashiro, Soichi, Tetsuya Matoba, Ryuta Umezu, Jun-Ichiro Koga, Masaki Tokutome, Shunsuke Katsuki, Kaku Nakano, Kenji Sunagawa, and Kensuke Egashira. 2016. Pioglitazone-incorporated nanoparticles prevent plaque destabilization and rupture by regulating monocyte/macrophage differentiation in ApoE(−/−) mice. Arteriosclerosis Thrombosis And Vascular Biology 36 (3): 491–500.
Duan, Sheng Zhong, Michael G. Usher, and Richard M. Mortensen. 2008. Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circulation Research 102 (3): 283–294.
Chen, Jingli, Jun Yang, Ruiyuan Liu, Chenmeng Qiao, Zhiguo Lu, Yuanjie Shi, Zhanming Fan, Zhenzhong Zhang, and Xin Zhang. 2017. Dual-targeting theranostic system with mimicking apoptosis to promote myocardial infarction repair via modulation of macrophages. Theranostics 7 (17): 4149–4167.
Availability of Data and Materials
Not applicable.
Funding
This study was funded by the National Natural Science Foundation of China (No. 81800267, 81870328), Henan Medical Science and Technology Joint Building Program (No. 2018020002), Henan Thousand Talents Program (No. ZYQR201912131), Henan Province Youth Talent Promoting Project (No. 2020HYTP051), and Excellent Youth Science Foundation of Henan Province (No. 202300410362).
Author information
Authors and Affiliations
Contributions
(I) Conception and design: all authors. (II) Administrative support: none. (III) Provision of study materials or patients: none. (IV) Collection and assembly of data: none. (V) Data analysis and interpretation: none. (VI) Manuscript writing: all authors. (VII) Final approval of manuscript: all authors.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Written informed consent for publication was obtained from all participants.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Z., Tang, J., Cui, X. et al. New Insights and Novel Therapeutic Potentials for Macrophages in Myocardial Infarction. Inflammation 44, 1696–1712 (2021). https://doi.org/10.1007/s10753-021-01467-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-021-01467-2