
Abstract
Acidic soils occupy a vast area in the world, and the aluminum (Al) in these soils can directly interact with plant cells and tissues to inhibit their growth and reduce yields. The signalgrass Urochloa decumbens cv. Basilisk (syn. Brachiaria decumbens cv. Basilisk), a widely sown tropical forage grass, is recognized for its high productivity under intensive use, vigorous growth, ease of establishment, and good forage value throughout the year, as well as its exceptional adaptation to infertile acid soils. We sequenced the transcriptome from roots of U. decumbens cv. Basilisk under two conditions, with and without Al, using Illumina paired-end sequencing technology and performed de novo assembly of those reads, which yielded 164,920 transcripts. Of these transcripts, 113,918 were assigned a putative protein function through comparisons with different gene set databases. Additionally, 13,375 simple sequence repeat (SSR) markers were identified. Digital gene expression analyses were conducted to identify 6698 differentially expressed genes between treatments, revealing a great differences in the root transcriptional landscape when exposed to aluminum. An extensive annotation of the differentially expressed genes (DEGs), made possible to identify several transcripts with putative functions correlated to aluminum exposure, most belonging to vesicle transportation, cell wall modifications and metal handling ontologies. In this work, abundant, high-quality transcripts were obtained, providing a reference platform for future biotechnological studies and breeding programs for this species and its close relatives.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tropical and subtropical countries account for approximately half the global beef cattle production (Jank et al. 2011). In Brazil 169 million hectares of pastures are used for feeding 208 million cattle (ABIEC 2013). The main grasses that are used as forage belong to species of the Brachiaria genus (syn. Urochloa P. Beauv.).
Although a large amount of genetic information has been deposited in public databases involving Poaceae family (28 genome sequencing projects, and 7,727,397 DNA and RNA sequences) (NCBI 2016a), the majority of the sequences are derived from a few species of high agricultural importance (rice, maize, wheat, and sorghum). In contrast with well-investigated crops, the Panicodae subfamily is of high economical importance (Toledo-Silva et al. 2013), but is genomically poorly explored.
Nevertheless, the production of genomic information is high demanding in terms of time and money; therefore, comparative genomics becomes a convenient method for studying the genetics of different species, providing resources in terms of molecular tools for genetic and genomic studies. As demonstrated by Surget-Groba and Montoya-Burgos (2010) and Liu et al. (2014), transcriptomic studies can be greatly improved with scaffolding steps that use closely genetically related protein sets as a proxy to assemble transcripts.
The species of the genus Urochloa were brought to America from Africa and were widely used in pasture for livestock grazing, especially Brachiaria decumbens and Brachiaria brizantha, which have shown wide adaptation and are extensively used as pasture grasses in this ecosystem (do Valle et al. 1996). Urochloa decumbens (Stapf) R.D. Webster, known as signalgrass, has gained special attention due to its high productivity under intensive use, vigorous growth, ease of establishment, and good forage value throughout the year, as well as its exceptional adaptation to infertile acid soils (Valle et al. 2010), which is mainly due to its high Al tolerance (Arroyave et al. 2013; Ramos et al. 2012; Rao et al. 1993).
Under acidic soil conditions, Al is ionized to trivalent aluminum (Al3+), which is readily available and highly reactive in the soil. Al can interact with a number of extracellular and intracellular structures, inhibiting plant growth and reducing productivity and yield. Al is one of the major limiting factors for growing crops in acidic soils (Delhaize and Ryan 1995).
Given the importance of this abiotic stress, the phytotoxicity of Al3+ has been studied extensively (Foy 1974). The root apex is the first site of Al interaction. There, Al immediately starts to damage cells, inhibiting cell elongation and division. In vegetal cells, Al can modify the cellular wall and membrane structure, harming water and nutrient absorption and compromising plant development (Arroyave et al. 2011).
In Brazil, only one cultivar of U. decumbens has been commercially available since the 1960s, cv. Basilisk, which is apomictic and tetraploid (2n = 4x = 36), with high aluminum tolerance (Oliveira et al. 2015). To develop new cultivars of this species, diploid accessions were artificially tetraploidized using colchicine (Simioni and Valle 2009) in crosses with U. decumbens cv. Basilisk.
Despite the good adaptation to savanna regions of Brazil and the high economic importance of U. decumbens for animal production in many tropical countries, the genomic data on the species are limited; expanding the current knowledge on this species could lead to a more efficient breeding program. Although some genomic resources are available for the genus Urochloa (mainly 2237 EST sequences available) (NCBI 2016b), almost no genetic information is available on molecular markers or gene expression profiles under biological stresses for the genus or this particular species.
SSRs markers are one of the most abundant data correlated to genetic variability obtained by transcriptome sequencing and highly transferable to close related species, typically co-dominant markers are widely used in studies of the Poaceae family given their high abundance and stability (Wang et al. 2015; Santos et al. 2015), and also can be applied to germplasm characterization (Kumari et al. 2013). Although EST (expressed sequence tags) derived SSR markers are less informative, they represent functional sequences and are a cost-effective biotechnological tool for breeders (Salgado et al. 2014; Wang et al. 2015).
Recently, we obtained the transcriptome from roots of U. decumbens cv. Basilisk for study under stressed conditions using aluminum. Extensive information was generated by performing de novo assembly of this transcriptome. In this paper, we describe the analysis of this transcriptome and provide a reference platform for future biotechnological studies and breeding programs for this species and its close relatives.
This study will provide a better understanding of the molecular events underlying Al tolerance and how they vary among plant species and cultivars from same species, considering that these issues are not clearly understood (Ma et al. 2001).
Materials and methods
Plant material
Approximately 600 U. decumbens cv. Basilisk seeds were germinated on seed trays containing sand and vermiculite in a 1:1 proportion. Subsequently, approximately 200 12-day-old seedlings (past germination) were transferred into two hydroponic solutions [(A) 200 CaCl\(_2\), pH 4.2; and (B) 200 CaCl\(_2\) + 200 AlCl\(_3\), pH 4.2, prepared as described by Wenz et al. (2006)] in three experimental replicates with 30 seedlings each. All of the seedlings remained in the solution for 8 h. Then, the root tips were removed and cut approximately one centimeter from the tip, yielding six bundles of root tips that were utilized for RNA extraction.
RNA extraction was performed using Trizol® (Invitrogen, Camarillo, CA, USA), according to the manufacturer’s manual. At the end of the extraction process, the total RNA from the six samples was suspended in 20 \(\upmu\)L of MiliQ sterile water and treated with DNAse (Invitrogen) according to the manufacturer’s manual.
Quantification of the RNA samples was performed on a NanoDrop® 1000 (Thermo, Waltham, MA, USA) at a wavelength of 260 nm. RNA sample quality was evaluated using the A\(_{260\,{\text{nm}}}\)/A\(_{280\,{\text{nm}}}\) wavelength ratio and by direct observation on a 1% agarose gel. After quantification, the samples were suspended in 70% ethanol and sent to the Beijing Genomics Institute (BGI) for de novo transcriptome sequencing.
cDNA library construction and sequencing
Illumina sequencing was performed at the Beijing Genomics Institute (BGI)-Shenzhen, Shenzhen, China (http://www.genomics.cn/index.php), using a HiSeqtm 2000 platform according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Briefly, poly(A) RNA was isolated from 20 \(\upmu\)g of total RNA using Sera-mag Magnetic Oligo (dT) Beads (Illumina). To avoid priming bias when synthesizing the cDNA, the purified mRNA was first fragmented into small pieces. Then, the double-stranded cDNA was synthesized using the SuperScript Double-Stranded cDNA Synthesis kit (Invitrogen) with random hexamer (N6) primers (Illumina). The synthesized cDNA was subjected to end repair and phosphorylation using T4 DNA polymerase, Klenow DNA polymerase and T4 polynucleotide kinase (PNK). The cDNA fragments repaired using these enzymes were 3\({^\prime }\) adenylated using Klenow Exo-(3–5 exo minus, Illumina). Illumina paired-end adapters were ligated to the ends of the 3\({^\prime }\)-adenylated cDNA fragments. To select a template size range for downstream enrichment, the products of the ligation reaction were purified on a 2% TAE agarose gel (Certified Low-Range Ultra Agarose, Bio-Rad). A range of cDNA fragments (200 ± 25 bp) was excised from the gel. Fifteen rounds of polymerase chain reaction (PCR) amplification were performed to enrich the purified cDNA template using PCR Primer PE 1.0 and PE 2.0 (Illumina) with Phusion DNA Polymerase. The cDNA library was constructed with a fragment length range of 200 bp (±25 bp). Finally, after validation on an Agilent Technologies 2100 Bioanalyzer using the Agilent DNA 1000 chip kit, the cDNA library was labeled and sequenced on a PE flow cell using an Illumina Genome Analyzer HiSeq 2000.
The workflow was as follows: template hybridization, isothermal amplification, linearization, blocking, sequencing primer hybridization, and sequencing on the sequencer for Read 1. After the completion of the first read, the templates were regenerated in situ to enable a second 90-bp read from the opposite end of the fragments, i.e., the newly sequenced strands were stripped off, and the complementary strands were bridge-amplified to form clusters. Once the original templates were cleaved and removed, the reverse strands underwent sequencing by synthesis.
Pre-processing, de novo assembly, clustering, and scaffolding
After trimming adaptors, reads with unknown nucleotides longer than 5% and low-quality reads (more than 20% of bases Q \(\le 10\)) were removed. De novo assembly was performed by the BGI using Trinity (Haas et al. 2013). The database was sent to Embrapa for bioinformatic analyses. All reads were deposited in the National Center for Biotechnology Information Short Read Archive (NCBI-SRA) under accession number SRP071168, BioProject number PRJNA314352.
Because several samples were available from the same species, a clustering step was performed by TIGR Gene Indices clustering tools (TGICL) (Pertea et al. 2003) to assemble all the unigenes from different samples to form a single set of non-redundant unigenes. Orientation and CDS (protein coding sequence) predictions were obtained using Blastx (Altschul et al. 1990) against the NCBI non-redundant database (Pruitt et al. 2005). The CDSs from sequences with no hits were predicted using ESTScan (Iseli et al. 1999).
To obtain a comprehensive set of genes, a scaffolding set was assembled using TransPS [Transcriptome Post Scaffold (Liu et al. 2014)] and Setaria italica version 2.1 (Bennetzen et al. 2012) as the reference proteome.
Annotation and screening for plant resistance genes
In an attempt to identify putative functions from the sequences through protein similarity and to identify protein families and metabolic functions, all the sequences were searched against multiple protein databases NCBI-Nr (non redundant database) (Pruitt et al. 2005), Swiss-Prot (European Molecular Biology Laboratory protein database) (Bairoch and Apweiler 2000), KEGG (Kyoto Encyclopedia of Genes and Genomes) (Kanehisa and Goto 2000), and COG (Clusters of Orthologous Groups) (Tatusov et al. 2003) using a Blastx e-value cutoff of \(10^{-5}\). To retrieve Gene Ontology (GO) (Harris et al. 2004) functional annotation, the Blast2GO (Conesa et al. 2005) program was applied to the sequences. The WEGO (Web Gene Ontology Annotation Plot) (Ye et al. 2006) software was used to perform GO annotation analysis and to plot the GO annotations. For a more detailed annotation of the DEGs (Differentially Expressed Genes), the scaffold proxies of those unigenes as well as the unigenes themselves were annotated with agriGO [GO Analysis Toolkit and Database for the Agricultural Community (Du et al. 2010)] and REVIGO (Reduce and Visualization of Gene Ontology) (Supek et al. 2011).
The similarity between the 164,920 de novo transcripts and 104,447 plant resistance genes (R gene) from the Plant Resistance Gene database (PRGdb) (Sanseverino et al. 2010) was analyzed using standalone BLAST tool (Altschul et al. 1990). Only sequences with e-values equal to 0.0 were accepted as result.
Gene expression
By mapping the reads back to the assembled transcriptome, the relative transcript abundance was obtained using the FPKM method (fragments per kb that were allowed to have zero or one gene in each family per million fragments) (Anders and Huber 2010). Whose formula is:
where FPKM denotes the expression of Unigene A, C denotes the number of fragments that uniquely aligned to Unigene A, N denotes the total number of fragments that uniquely aligned to all Unigenes, and L denotes the number of bases in the CDS of Unigene A. By eliminating the influence of different gene length and sequencing level on the calculation of gene expression, the FPKM values can be directly used for comparing the differences in gene expression among samples.
Differentially expressed genes (Audic and Claverie 1997) were identified after a multiple hypothesis test on the relative transcript abundance values, using \(H_0\): a gene has the same expression level in two samples; and H 1: a gene has different expression levels in two samples. Later, a correction test using false discovery rate (FDR) (Yekutieli and Benjamini 2001) combined with the ratio of the FPKMs from control samples was used to determine significant differentially expressed genes. In our analysis, we chose genes with \(\text {FDR}\le 10^{-3}\) and expression \(\log _{2}\) ratios >1.5. To integrate annotation and gene expression levels, MAPMAN (Thimm et al. 2004) was utilized as a visualization tool. Moreover, the top 100 up and down regulated genes with COG annotation were selected and plotted in a M (log ratios) and A (mean average) scale space, where we could relate COG or KEGG annotation codes among the transcripts with the Differential Expression and Protein InteraCTions Visualization Tool (DEPICTViz). DEPICTViz is an interactive JavaScript web tool for visualizing differential gene expression and protein interactions. This tool allows one to visualize genes of interest in an MA graph, including attributes such as COG Id (with category and description), Swiss-Prot, KEGG and KO (KEGG orthology) Ids, in addition to links to protein interactions and gene ontology databases, such as STRING-DB (Search Tool for the Retrieval of Interacting Genes/Proteins database) and COG. If applicable, visualization using the UCSC (University of California, Santa Cruz) Genome Browser is also allowed, which is available at http://git.facom.ufms.br/bioinfo/depictviz.git.
Screening for molecular markers
Searches for SSRs (Simple Sequence Repeats) from the contig dataset were performed using MISA (Microsatellite Identification Tool) (Thiel et al. 2003). The definition of microsatellites was set to 12-fold (1/12), di (2/6), tri (3/5), tetra (4/5), penta (5/5), or hexa (6/4) mononucleotide repeats.
After screening, SSR markers situated more than 150 bp from both ends of the encompassing unigene were retained and used to design primers with Primer3 (Untergasser et al. 2012).
Comparative analysis
Having as input all the protein coding genes from B. decumbens, B. distachyon (GenBank assembly accession—GCA_000005505.1), Leersia perrieri (Department of Energy Joint Genome Institute, DOE-JGI), Oryza sativa (GenBank assembly accession—GCA_000005425.2), Panicum hallii (DOE-JGI), Panicum virgatum (DOE-JGI), S. italica (GenBank assembly accession—GCA_000263155.1), Sorghum bicolor (GenBank assembly accession—GCA_000236745.2) and Zea mays (GenBank assembly accession—GCA_000005005.1) , Orthologsorter (Farias 2013) tool was used to build the whole genome phylogenetic species tree. This package compares essentially all protein coding genes from all species in an all-against-all fashion using OrthoMCL (Li et al. 2003). Among all the families found, only those with exactly one representative gene in each genome were used. The only exception was the outgroup genome, S. italica, which was allowed to have zero or one gene in each family. After selecting those families, Muscle (Edgar 2004) was used to align each of them, and Gblocks (Castresana 2000) was used to remove non-informative sites. All alignments were then concatenated. To build the tree, the final complete alignment was input into RAxML (Stamatakis 2006) with the PROTCATJTT model, rapid bootstrapping and an ML (maximum likelihood) search.
Results
Pre-processing and de novo assembly
All of the 401 million clean reads (66.8 million clean reads per sample on average) generated using the Illumina platform were assembled into 164,920 unigenes, which were defined here as sequences without Ns that could not be extended on either side. The clustering step assembled unigenes from different samples to form a single set of non-redundant unigenes, resulting in 67,123 clusters and 97,797 singletons with lengths ranging from 200 to 18,820 nucleotides (nt) and 63.4% of all sequences with lengths between 500 and 2000+ nt (Table 1).
By retrieving the best BLAST hits from the selected databases, we extracted the CDS information from approximately 62% of the assembled sequences. The BLAST results were also used to train a gene model for ESTScan prediction. CDS prediction by ESTScan successfully resulted in 21,305 CDSs.
Scaffolding and annotation
Using the 116,644 sequences from the Setaria italica protein dataset, the scaffolding step successfully identified 11,634 sequences with one sequence per protein relation. A total of 4979 sequences were assembled into 2230 scaffolds that ranged from 237 to 15,200 nt; the scaffolds were formed from a maximum of 2–8 sequences.
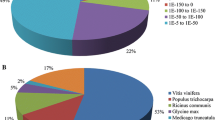
With respect to the identification of putative protein functions, we successfully retrieved functional information from 113,918 sequences (69% of all sequences) against the NCBI-nr, NCBI-nt (NCBI nucleotide database), SwissProt, KEGG and COG databases, with low e-values (76% <1e−05) and high similarity (61% of sequences with >60% identity), mainly with proteins from Sorghum bicolor and Zea mays (Fig. 1).

Annotation statistics. a E-value distribution; b similarity distribution; c species distribution
COG annotation resulted in 55,451 sequences distributed into 121 categories, mainly in “general function prediction only”, “translation, ribosomal structure and biogenesis”, and “function unknown” (Fig. 2). COG annotation of the transcripts provided more insight into the protein function and was used to identify proteins related to broad functions, thus generating a more general sense from the group of transcripts. Although most of the transcript annotations relied on poorly characterized functions, we were able to verify the significant presence of specific pathways related to Al response, toxicity and tolerance, including a mechanism for the modification of the cell wall, plasma membrane transport processes, intracellular capture, and Al3+ DNA binding (Brunner and Sperisen 2013). Specific to this response group, the COG analysis identified 514 transcripts belonging to the “MU” category (“cell wall/membrane/envelope biogenesis; intracellular trafficking, secretion, and vesicular transport”) and 1217 transcripts belonging to the category “V” (defense) annotated as “ABC-type multidrug transport systems”.

Cluster of orthologous groups classification. Overall, 47% of all sequences were successfully annotated into 121 categories mainly under more general terms such as “general function prediction only” and “function unknown”, which accounted for 54% of all annotated transcripts
In an attempt to describe the properties of the identified transcripts and their products, a GO annotation step was performed. We successfully annotated 70,008 sequences into 5370 GO terms. Of these terms, the “cellular component” category under the class “cell” is the most representative, followed by “biological processing”, “single-organism process”, “molecular function”, and “catalytic activity” (Fig. 3).

GO terms histogram. The distribution of GO annotations under “biological process”, “cellular component”, and “molecular function” terms for the 5370 GO entries from 70,008 annotated transcripts
Additionally, to gain more understanding pf our set of transcripts, a KEGG annotation step was performed, and we connected 72,749 sequences to 3290 KO’s and 1065 enzyme commission terms. The most representative hits belonged to glycerophospholipid metabolism (KEGG enzyme 3.1.4.4 and KEGG orthology K01115); in addition, all the main metabolic pathways were identified. A list of biochemical pathways predicted from unigenes by KEGG is provided in electronic supplementary material
Gene expression
From the 164,920 de novo transcripts, we identified 4534 DEGs with \(\text {FDRs}\le 10^{-3}\) between treatments (Fig. 4) and log2-fold-change values \(\ge1.5\) or \(\le-1.5.\) From these highly significant DEGs, 1589 were identified as belonging to 1059 scaffolds with 1726 successfully retrieved GO terms. A gene set enrichment analysis (GSEA) annotated 2240 entries into 131 significant GO terms.

MA plot. Distribution of top 100 annotated genes up and down regulated a in a panoramic view and b zoomed out view. Red points represent genes that were classified as differentially expressed, and orange points represent genes that were expressed in just one of the conditions. X-axis represents the average of the normalized count values, dividing by size factors, taken over all samples, and the y-axis the \(\log _{2}\) fold change. Genes with same COG category are linked by edges
When annotation and gene expression were integrated, our results suggested the increased activity of key cell functions. Transcripts annotated under the “vesicle transport” and “transport” categories were highly up-regulated (Fig. 5). A list of DEGs and the integrated annotation is provided in electronic supplementary material.

Gene set enrichment analysis. a Response to stimulus cluster: the enriched terms from this category are related to processes that occur as a result of the stimulus within or outside the cell or organism, suggesting a change in state or activity due to the presence of aluminum or derived from the chemical response to aluminum toxicity. b Biological regulation cluster: any process that modulates a measurable attribute of any biological process, quality or function. The relationship between the terms points to the negative regulation of cell growth, which is an expected outcome of aluminum exposure. c Cellular component cluster: any constituent part of a cell, the basic structural and functional unit of all organisms. Both internal and external environment terms were enriched. d Legend for arrows and box colors
Screening for molecular markers
A total of 14,932 SSRs (excluding mononucleotide repetitions) were identified in 14,822 transcripts, ranging from 12 to 240 nucleotides in length and possessing four to eleven motif repeats (Table 2). Three-nucleotide motifs were the most common SSRs, accounting for 61% of all SSRs (Fig. 6). The trinucleotides (GCG)*5 and (CGC)*5 were the most frequent.

SSR composition. A bar chart of SSR motif length. Three-nucleotide repetitions accounted for >61% of all SSRs
Additionally, 338 SSRs were found in 292 DEGs. From the scaffolding analysis, the 14,932 markers were placed into 7350 genes. By GSEA analysis, 14,030 transcripts were annotated into 42 significant GO terms. Under the “biological process” category, the most significant terms were “cellular component organization”, “regulation of biological quality”, “transmembrane transport” and “catabolic process”.
Comparative analysis
A complete genome tree generated from the concatenation of orthologous families (see “Materials and methods”) is presented in Fig. 7. Among 48,844 protein families, 561 with exactly one representative gene from each genome were chosen. After aligning each one of these families, all alignments were concatenated, initially resulting in 166,109 columns and then 386 columns after removing non-representative sites. The topology is entirely compatible with that obtained using the online NCBI Taxonomy Browser tree builder, using the same set of species (http://www.ncbi.nlm.nih.gov/Taxonomy/CommonTree/wwwcmt.cgi).

Complete genome tree. Species tree based on the concatenation of alignments of 561 ortholog families. Node labels represent bootstrap support (100 replicates)
Screening for plant resistance gene
A comparison of the 164,920 de novo transcripts and the PRGdb (Sanseverino et al. 2010) yielded 2445 genes with high similarity, 1221 of which were different from each other. The number of sequences similar to the R genes found in U. decumbens may be attributed to the fact that the plant was exposed to Al stress in our study. Pathogen attack is a common stressor for plants. According to Atkinson (2012), conditions such as high salinity and acidity are expected to increase the expression of R genes to protect the plant host from enemy attack and environmental stress, thus maintaining its vital functions.
Under the physiological conditions of this study, U. decumbens expressed 12 domains and multidomains of proteins related to resistance, which were present either alone within a gene or combined with other domains within the same gene. The most expressed domains were serine-threonine kinases (Ser-Thr Kinase), nucleotide binding sites (NBS) and leucine rich repeats (LRR), as indicated in Table 3.
Discussion
Toledo-Silva et al. (2013) sequenced the tropical grass Panicum maximum and presented N50 metric and mean transcript length values (1272 and 942 bp, respectively) similar to ours (Table 1), which were also similar to those of other transcriptomes sequenced using next-generation sequencing from the Poaceae family (Meyer et al. 2012; Xu et al. 2013). Moreover, with respect to reads per library, ploidy levels, and estimated genome size, our results are consistent with other studies that measured differential expression under stress conditions (Li et al. 2015; Xu et al. 2013; Wu et al. 2014).
The difference between the numbers of transcripts obtained by sequencing and sequences actually used for further analysis is due to the adoption of redundancy elimination and sequence complementation steps between and among libraries (Table 1). By systematically comparing different strategies and resources, Duan et al. (2012) optimized the transcriptome assembly of common wheat and found that a scaffolding step significantly reduces redundancy. After optimization, the assembly consisted of four steps: pre-assembly, merging different samples, removal of redundancy, and scaffolding. The resulting assembled transcripts were comparable to Sanger-derived ESTs in terms of both continuity and accuracy.
Sequenced genes identities can be predicted on the basis of the most similar protein deposited and annotated in a protein database (Conesa et al. 2005). The number of annotated sequences in this work for the different databases are summarized in Table 4. Functional annotation provides valuable information, allowing to connect information between gene expression levels and the putative biological role of a transcript, and it can be used to pre-select candidates genes that show linkage to a physiological response for further investigation. With respect to the identification of putative protein functions, we successfully retrieved functional information from 113,918 sequences (69% of all sequences) against NCBI nr, NCBI nt, SwissProt, KEGG (72,749 sequences to 3290 KEEG orthology and 1065 enzyme commission terms), COG (70,008 sequences into 5370 GO terms) databases, with low e-values (76% ≤1e−06) and high similarity (61% of sequences with >60% identity), mainly with proteins from S. bicolor and Z. mays (Fig. 5). The variety of distinct group functions, the number of annotated transcripts and the presence of the main metabolic pathways indicate that this study successfully captured the root transcriptional events in response to early Al exposure.
The Ser-Thr Kinase domain was the most abundant in the U. decumbens sequences. A study of Nicotiana benthamiana demonstrated that genes containing such receptors significantly contribute to resistance against Phytophthora infestans (Chaparro-Garcia et al. 2011). The LRR domain was found in 873 transcribed sequences of U. decumbens, alone or with other domains. Genes with this domain were observed in Oryza sativa L., in which they conferred resistance to the rice blast disease caused by Magnaporthe oryzae (Wu et al. 2015). Alone or with other domains, the NBS domain was found in 763 transcripts from U. decumbens. The majority of the plant R genes have an NBS and LRR site. These domains are highly conserved through generations and have evolved in different species (Pan et al. 2000).
The high-throughput sequencing of RNA (RNA-seq) provides tools that allow researchers to perform gene expression profiling to identify the transcriptional complexity of biological stress responses. As demonstrated by Delhaize and Ryan (1995), the excess Al causes a rapid (minutes to few hours) inhibition of root growth, resulting in a reduced and damaged root system which limits mineral nutrient and water uptake, extending the root tips exposure until nine hours we expect to be able to capture the early molecular events of the exposure to aluminum through the counting and relativization of the sequence data, thus lucidating how the roots of U. decumbens cv. Basilisk first react to Al exposure.
The enrichment of cellular component terms highlights the participation of structural components, such as the cell wall, apoplast (Fig. 5), membrane vesicle. Al binds to the cell wall, particularly to the pectic matrix, and to the apoplastic face of the plasma membrane, which leads to injured cells and the formation of reactive oxygen species (ROS). Increasing physiological, biochemical and molecular evidence suggests that the modification of apoplastic Al binding modulates Al toxicity by reducing interactions between Al and cell wall components, which emphasizes the involvement of cell wall properties in Al resistance (Horst et al. 2010).
The two main mechanisms of Al tolerance in plants include internal detoxification and exclusion from their tissues. The former is related to the ability to capture and enclose Al and form harmless complexes. The strategy involved in the latter is to inhibit aluminum uptake by releasing organic acids that chelate Al (Simões et al. 2012).
Both Al exclusion and internal tolerance mechanisms require the intense participation of plasma membrane transporters (Brunner and Sperisen 2013). Two main transporter families have been reported to play a role in Al tolerance: ABC transporters and MATE-type transporters. MATE transporters are likely responsible for citrate efflux, a strong chelator of Al, and are linked to two major aluminum tolerance quantitative trait loci (QTLs) in maize (Maron et al. 2010). ABC-transporters are involved in various plant systems and largely contribute to the membrane transport of endogenous secondary metabolites in the plant body. Using the NCBI BLAST annotations, we successfully identified 223 DEGs directly related to Al-tolerance mechanisms. Of these transcripts, 38 were annotated as multidrug and toxic compound extrusion (MATE)genes (Yazaki 2006).
COG annotation from DEGs successfully identified 190 transcripts annotated as ABC-type multidrug transport systems. ABC-type transporters are present in vacuolar membranes and have been hypothesized to act in the enclosure of Al, segregating this metal away from sensitive tissues (Huang et al. 2012). Arabidopsis mutants for the ALS1 and ALS3 (Al3+-sensitive) genes, which encode proteins that belong to the ATP-binding cassette (ABC) transporter superfamily, exhibit significantly increased sensitivity to Al and accumulate increased amounts of Al in the cytosol and nucleus. This finding suggests that these gene products are involved in Al transport across the plasma membrane and subcellular sequestration (Larsen et al. 2007). Moreover, the OsALS1 gene, which encodes a half-sized ABC transporter that is a member of the TAP (transporter associated with antigen processing) sub-group, is also related to Al sequestration in O. sativa, mutants of which display significantly increased Al sensitivity but not increased sensitivity toward other metals or low pH (Huang et al. 2012).
By comparing the SSR markers from nine Poaceae species (O. sativa L. ssp. japonica, O. sativa L. ssp. Indica, Z. mays, S. bicolour, Brachypodium distachyon, Foxtail millet, Moso bamboo, Triticum urartu and Aegilops tauschii) Wang et al. (2015) showed that the SSR density of the grass family varies greatly between species. Moreover, the authors found similar numbers of minimum-length SSRs (12 base pairs, bp), a high guanine-cytosine (GC) content (especially in trimers), and hexamers as the most abundant SSR type. In contrast, our results indicated a major percentage of trimers, although also with a high GC content. As demonstrated by Šmarda et al. (2014), Poaceae species exhibit an increased percentage of C/G bases, which facilitates more complex gene regulation and leads to more stable genetic materials.
Conclusions
This is the first large-scale transcriptomic study of the Urochloa genus. The amount of sequencing data generated in this work greatly exceeds the existing resources in public databases and significantly increases the number of reads and annotated transcripts available.
The amount of reads generated, the high number of identified transcripts associated with the diversity of their locations, and the variety of predicted functions demonstrate the success of our approach in capturing the root transcriptome.
The differential gene expression analysis in response to Al stress enabled us to identify genes that are related to Al response in both intracellular and extracellular environments and provided candidates for more detailed assays concerning Al tolerance in U. decumbens cv. Basilisk and future assays concerning elucidate molecular mechanisms of aluminum tolerance. The data presented in this manuscript represent an important contribution for the research community interested in plant stress response at a genomic level.
Additionally, the high number of identified molecular markers provides a valuable source of genetic information that will enable downstream analysis for biotechnological purposes and breeding programs.
Availability of supporting data
All the reads resulting from the sequenced libraries used for de novo transcriptome assembly and downstream analysis are deposited on the National Center for Biotechnology Information Short Read Archive (NCBI-SRA) Number SRP071168, Bioproject Number PRJNA314352.
References
ABIEC (2013) Association of Brazilian beef exporters: ABIEC. Brasilian Beef Profile, 2013. http://www.brazilianbeef.org.br/texto.asp?id=9
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11(10):R106
Arroyave C, Barceló J, Poschenrieder C, Tolrà R (2011) Aluminium-induced changes in root epidermal cell patterning, a distinctive feature of hyperresistance to Al in Brachiaria decumbens. J Inorgan Biochem 105(11):1477–1483
Arroyave C, Tolrà R, Thuy T, Barceló J, Poschenrieder C (2013) Differential aluminum resistance in Brachiaria species. Environ Exp Bot 89:11–18
Atkinson NJ, Urwin PE (2012) The interaction of plant biotic and abiotic stresses: from genes to the field. J Exp Bot 63(10):3523–3543
Audic S, Claverie JM (1997) The significance of digital gene expression profiles. Genome Res 7(10):986–995
Bairoch A, Apweiler R (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28(1):45–48
Bennetzen JL, Schmutz J, Wang H, Percifield R, Hawkins J, Pontaroli AC, Estep M, Feng L, Vaughn JN, Grimwood J, Jenkins J, Barry K, Lindquist E, Hellsten U, Deshpande S, Wang X, Wu X, Mitros T, Triplett J, Yang X, Ye CY, Mauro-Herrera M, Wang L, Li P, Sharma M, Sharma R, Ronald PC, Panaud O, Kellogg EA, Brutnell TP, Doust AN, Tuskan GA, Rokhsar D, Devos KM (2012) Reference genome sequence of the model plant Setaria. Nat Biotechnol 30(6):555–561
Brunner I, Sperisen C (2013) Aluminum exclusion and aluminum tolerance in woody plants. Front Plant Sci 4(June):172
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17(4):540–552
Chaparro-Garcia A, Wilkinson RC, Gimenez-Ibanez S, Findlay K, Coffey MD, Zipfel C, Rathjen JP, Kamoun S, Schornack S (2011) The receptor-like kinase SERK3/BAK1 is required for basal resistance against the late blight pathogen phytophthora infestans in Nicotiana benthamiana. PLoS ONE 6(1):e16, 608
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21(18):3674–3676
Delhaize E, Ryan PR (1995) Aluminum toxicity and tolerance in plants. Plant Physiol 107(2):315–321
do Valle CB, Maass BL, Miles JW, Kumble V (1996) brachiaria: biology, agronomy, and improvement. CIAT Publication, Empresa Brasileira de Pesquisa Agropecuria, Campo Grande. http://isbnplus.org/9789589439579
Du Z, Zhou X, Ling Y, Zhang Z, Su Z (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38(Web Server issue):W64–W70
Duan J, Xia C, Zhao G, Jia J, Kong X (2012) Optimizing de novo common wheat transcriptome assembly using short-read RNA-Seq data. BMC Genom 13:392
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform 5:113
Farias NC (2013) Orthologsorter: inferindo genotipagem e funcionalidade a partir de famlias de protenas ortlogas. PhD thesis, UFMS (Universidade Federal de Mato Grosso do Sul), Campo Grande, Brazil
Foy C (1974) Effects of aluminum on plant growth. In: Carson EW (ed) The plant root and its environment. The University Press of Virginia, Charlottesville, pp 601–641
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, Macmanes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, Leduc RD, Friedman N, Regev A (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protocols 8(8):1494–1512
Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la Cruz N, Tonellato P, Jaiswal P, Seigfried T, White R (2004) The gene ontology (GO) database and informatics resource. Nucleic Acids Res 32(Database issue):D258–D261
Horst WJ, Wang Y, Eticha D (2010) The role of the root apoplast in aluminium-induced inhibition of root elongation and in aluminium resistance of plants: a review. Ann Bot 106(1):185–197
Huang CF, Yamaji N, Chen Z, Ma JF (2012) A tonoplast-localized half-size ABC transporter is required for internal detoxification of aluminum in rice. Plant J 69(5):857–867
Iseli C, Jongeneel CV, Bucher P (1999) EST Scan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. In: Proceedings of international conference on intelligent systems for molecular biology (ISMB), pp 138–148
Jank L, Valle C, Resende R (2011) Breeding tropical forages. Crop Breed Appl Biotechnol 34(spe):11–27
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28(1):27–30
Kumari K, Muthamilarasan M, Misra G, Gupta S, Subramanian A, Parida SK, Chattopadhyay D, Prasad M (2013) Development of eSSR-markers in Setaria italica and their applicability in studying genetic diversity, cross-transferability and comparative mapping in millet and non-millet species. PLoS ONE 8(6):1–15
Larsen PB, Cancel J, Rounds M, Ochoa V (2007) Arabidopsis ALS1 encodes a root tip and stele localized half type ABC transporter required for root growth in an aluminum toxic environment. Planta 225(6):1447–1458
Li L, Stoeckert CJ, Roos DS (2003) Ortho MCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13(9):2178–2189
Li H, Yao W, Fu Y, Li S, Guo Q (2015) De novo assembly and discovery of genes that are involved in drought tolerance in Tibetan Sophora moorcroftiana. PLoS ONE 10(1):e111, 054
Liu M, Adelman ZN, Myles KM, Zhang L (2014) A transcriptome post-scaffolding method for assembling high quality contigs. Comput Biol J 2014:1–4
Ma JF, Ryan PR, Delhaize E (2001) Aluminium tolerance in plants and the complexing role of organic acids. Trends Plant Sci 6(6):273–278
Maron LG, Ma Piñeros, Guimarães CT, Magalhaes JV, Pleiman JK, Mao C, Shaff J, Belicuas SNJ, Kochian LV (2010) Two functionally distinct members of the MATE (multi-drug and toxic compound extrusion) family of transporters potentially underlie two major aluminum tolerance QTLs in maize. Plant J 61(5):728–740
Meyer E, Logan TL, Juenger TE (2012) Transcriptome analysis and gene expression atlas for Panicum hallii var. filipes, a diploid model for biofuel research. Plant J 70(5):879–890
NCBI (2016a) Entrez search. http://www.ncbi.nlm.nih.gov/gquery/?term=poaceae. Accessed 04 Feb 2016
NCBI (2016b) Entrez search. http://www.ncbi.nlm.nih.gov/gquery/?term=Urochloa. Accessed 07 May 2015
Oliveira KMBd, Barrios SCL, Chiari L, Laura VAA, Valle CBd (2015) Evaluation of aluminum resistance in hybrids of Brachiaria decumbens stapf. Crop Breed Appl Biotechnol 15:251–257
Pan Q, Wendel J, Fluhr R (2000) Divergent evolution of plant NBS-LRR resistance gene homologues in dicot and cereal genomes. J Mol Evol 50(3):203–213
Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B, Tsai J, Quackenbush J (2003) TIGR gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19(5):651–652
Pruitt KD, Tatusova T, Maglott DR (2005) NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 33(Database issue):D501–D504
Ramos FT, França MG, Alvim MN, Rossiello RO, Zonta E (2012) Aluminum tolerance measured by root growth and mucilage protection in Urochloa brizantha and Urochloa decumbens. J Plant Interact 7(3):225–229
Rao IM, Zeigler RS, Vera R, Sarkarung S (1993) Selection and breeding for acid-soil tolerance in crops. BioScience 43(7):454–465
Salgado LR, Koop DM, Pinheiro DG, Rivallan R, Le Guen V, Nicolás MF, de Almeida LG, Rocha VR, Magalhães M, Gerber AL, Figueira A, Cascardo JC, de Vasconcelos AR, Silva WA, Coutinho LL, Garcia D (2014) De novo transcriptome analysis of Hevea brasiliensis tissues by RNA-seq and screening for molecular markers. BMC Genom 15(1):236
Sanseverino W, Roma G, De Simone M, Faino L, Melito S, Stupka E, Frusciante L, Ercolano MR (2010) PRGdb: a bioinformatics platform for plant resistance gene analysis. Nucleic Acids Res 38(Database issue):D814–D821
Santos J, Barreto MA, Oliveira FA, Vigna B, Souza AP (2015) Microsatellite markers for Urochloa humidicola (Poaceae) and their transferability to other Urochloa species. BMC Res Notes 8(1):83
Simioni C, Valle VD (2009) Chromosome duplication in brachiaria (a. rich.) stapf allows intraspecific crosses. Crop Breed Appl Biotechnol 9(4):328–333
Simões CC, Melo JO, Magalhaes JV, Guimarães CT (2012) Genetic and molecular mechanisms of aluminum tolerance in plants. Genet Mol Res 11(3):1949–1957
Šmarda P, Bureš P, Horová L, Leitch IJ, Mucina L, Pacini E, Tichý L, Grulich V, Rotreklová O (2014) Ecological and evolutionary significance of genomic GC content diversity in monocots. Proc Natl Acad Sci USA 111(39):E4096–E4102
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22(21):2688–2690
Supek F, Bošnjak M, Škunca N, Šmuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6(7):e21, 800
Surget-Groba Y, Montoya-Burgos JI (2010) Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res 20(10):1432–1440
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA (2003) The COG database: an updated version includes eukaryotes. BMC Bioinform 4:41
Thiel T, Michalek W, Varshney R, Graner A (2003) Exploiting est databases for the development and characterization of gene-derived ssr-markers in barley (hordeum vulgare l.). Theor Appl Genet 106(3):411–422
Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37(6):914–939
Toledo-Silva G, Cardoso-Silva CB, Jank L, Souza AP (2013) De novo transcriptome assembly for the tropical grass Panicum maximum Jacq. PLoS ONE 8(7):e70, 781
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3 - new capabilities and interfaces. Nucleic Acids Res 40(15):e115
Valle CB, Macedo MCM, Euclides VPB, Jank L, Resende R (2010) Gnero Brachiaria. In: Martuscello DMDF, Azevedo J (eds) Plantas forrageiras, 1st edn. Editora UFV, Vicosa, pp 30–77
Wang Y, Yang C, Jin Q, Zhou D, Wang S, Yu Y, Yang L (2015) Genome-wide distribution comparative and composition analysis of the SSRs in Poaceae. BMC Genet 16(1):18
Wenzl P, Arango A, Chaves AL, Buitrago ME, Patiño GM, Miles J, Rao IM (2006) A greenhouse method to screen Brachiariagrass genotypes for aluminum resistance and root vigor. Crop Sci 46(2):968
Wu J, Wang L, Li L, Wang S (2014) De novo assembly of the common bean transcriptome using short reads for the discovery of drought-responsive genes. PLoS ONE 9(10):e109, 262
Wu Y, Xiao N, Yu L, Pan C, Li Y, Zhang X, Liu G, Dai Z, Pan X, Li A (2015) Combination patterns of major R genes determine the level of resistance to the M. oryzae in rice (Oryza sativa L.). PLoS ONE 10(6):e0126, 130
Xu P, Liu Z, Fan X, Gao J, Zhang X, Zhang X, Shen X (2013) De novo transcriptome sequencing and comparative analysis of differentially expressed genes in Gossypium aridum under salt stress. Gene 525(1):26–34
Yazaki K (2006) ABC transporters involved in the transport of plant secondary metabolites. FEBS Lett 580(4):1183–1191
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34(Web Server issue):W293–W297
Yekutieli D, Benjamini Y (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29(4):1165–1188
Acknowledgements
CNPq (National Council for Scientific and Technological Development) Award Number: 031/210, FUNDECT (Foundation for Support to the Development of Education, Science and Technology of the State of Mato Grosso do Sul), Award Number: 0096/2012, FUNDECT (Foundation for Support to the Development of Education, Science and Technology of the State of Mato Grosso do Sul) Award Number: 0048/12.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Salgado, L.R., Lima, R., Santos, B.F.d. et al. De novo RNA sequencing and analysis of the transcriptome of signalgrass (Urochloa decumbens) roots exposed to aluminum. Plant Growth Regul 83, 157–170 (2017). https://doi.org/10.1007/s10725-017-0291-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10725-017-0291-2