
Abstract
A collection of 46 pea (Pisum sativum L.) accessions, mostly from Europe, were analysed for genetic diversity using the GenoPea 13.2 K SNP Array chip. Of these accessions were 24 nomal-leaved and 22 semi-leafless. Principal components analysis (PCA) separated the peas into two groups characterized by the two different leaf types, although some genotypes were exceptions and appeared in the opposite group. Cluster analysis confirmed the two groups. A dendrogram showed larger genetic distances between genotypes in the normal-leafed group compared to semi-leafless genotypes. Both PCA and cluster analysis show that the two leave types are genetically divergent. So normal-leaved peas are an interesting genetic resource, even if the breeding goal is to develop semi-leafless varieties.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Peas (Pisum sativum L.) first appeared in early Neolithic farming villages of the Near East (7000 to 6000 B.C.). They were closely associated with wheat and barley production (Zohary and Hopf 1973). Peas were then quickly distributed across Southwest Asia, the Mediterranean basin and temperate Europe, Ethiopia, and northwestern India (Weiss and Zohary 2011). Peas are adapted to a wide range of climates and altitudes and, as a result, the diversity of the pea germplasm is very large (Burstin et al. 2015).
Today, peas are grown in many countries across Europe, North America, and Asia (Smýkal et al. 2012). Nevertheless, as for many other crops, in present plant breeding only a small part of the diversity is used. Especially, genetic diversity available in wild Pisum species has been poorly exploited (Jing et al. 2010; Ellis 2011; Smýkal et al. 2011).
Pea is a diploid (2n = 2x = 14) species of the Fabaceae and has a genome size of about 4,500 Mb (Jain et al. 2014). Since the first genetic map, already published in 1948 (Lamprecht 1948), knowledge of the pea genome has expanded consistently along with the development of molecular markers including RAPDs, SSRs, SRAPs and SNPs (Pandey et al. 2021). SNP markers are abundant and amenable to high-throughput genotyping (Tayeh et al. 2015b; Jha et al. 2017; Desgroux et al. 2018; Gali et al. 2018; Aznar-Fernández et al. 2020).
Thanks to the development of molecular markers in recent decades, various genetic diversity studies have been performed in pea. In an early study of Baranger et al. (2004), 121 protein-based and PCR-based markers (RAPD, ISSR, STS, SSR, isozymes, storage proteins) were used to investigate the genetic relationships and access the genetic diversity of 148 pea accessions mostly from Europe, and from China and USA. Three types of molecular markers RAPD, ISSR, SSR were used to estimate the genetic relation among 65 pea varieties (Tar’an et al. 2005). Smýkal et al. (2008) screened materials originating from Europe, America and Asia by using SSR and RBIP markers. Zong et al. (2009) used 21 informative microsatellite loci to assess and compare the genetic diversity among pea genotypes from within and outside China. Kwon et al. (2012) analysed genetic diversity and population structure of 285 accessions in a pea collection of Western Regional Plant Introduction Station, United States Department of Agriculture (USDA) by using RAPD, SCAR, and SSR markers. Burstin et al. (2015) concluded that despite the limited number of SNP markers used in their study, these markers proved to be most efficient in describing the genetic structure or the pea collection compared to SSR and RBIP markers. Benefiting from next-generation sequencing technology, a large number of gene-based SNP markers have been developed for pea genetic improvement. The first SNP array, GenoPea 13.2 K SNP, was developed from 12,802 transcript-derived SNPs following the re-sequencing of 16 diverse pea accessions (Tayeh et al. 2015a).
In pea there are four basic leaf types: the wild type or normal-leafed with leaflets and tendrils, tendril-less (homozygous recessive mutation for the Tendrilless gene), afila or semi-leafless without leaflets (homozygous recessive mutation of the Afila gene), and afila-tendril-less (the combination of both mutations) (Mikić et al. 2011). Today, semi-leafless cultivars are mainly used in the main production areas of pea such as Western Canada, European Union, Russia, and Australia (Mikić et al. 2011; French 2016), as this leaf type shows very much reduced lodging and can easier be harvested. However, the development of new cultivars by exploiting only semi-leafless peas could narrow down the genetic diversity.
The production of pea is facing new challenges such as resistance to biotic and abiotic stresses and higher seed quality. Therefore, it is necessary to exploit a wide range of genetic resources including normal-leafed accessions. A comprehensive comparison of the two leaf types with a larger number of accessions using SNPs markers has not yet been carried out to our knowledge. The main aim of this study was to assess the genetic diversity of 46 pea cultivars of different leaf type, and to evaluate if there is a relationship between leaf type and genetic distance.
Materials and methods
Plant materials
Germplasm consisted of 46 pea genotypes from various gene banks, seed companies and research institutes released from before 1935 to 2016. It consists of 24 normal-leafed and 22 semi-leafless accessions. The majority are European spring cultivars for grain usage, but some winter types, green fodder cultivars and exotic accessions from Asia and Africa are included (Table 1). Information on agronomic traits can be found in Tran et al. (2022).
Molecular markers
The material was genotyped with the GenoPea 13.2 K SNP Array chip (Tayeh et al. 2015a) by the company Trait Genetics, Gatersleben, Germany. The company received seeds. DNA extraction and SNP analyses were performed according to the standard protocols of the company.
Filtering
Loci with minor allele frequency (MAF) lower than 0.05 were removed. Markers with missing data were arranged as minor frequency alleles. Finally, 11,069 high quality markers were used for the analysis.
Data analysis
Principal component analysis was calculated using adegenet (Jombart et al. 2021), poppr (Kamvar et al. 2014), factoextra (Kassambara and Mundt 2020), adegraphic (Dray et al. 2018), ape (Paradis et al. 2021) packages for R software (3.6.2 version).
A K-means clustering approach (Hartigan and Wong 1979) was used to determine the most likely number of groups. Genetic distances were computed according to Nei’s Genetic Distance (Nei 1972), and a dendrogram with bootstrap values was created by using ape and poppr packages for R software. The number/cycle of bootstrap was set to 1,000. Ward’s minimum variance method, in which dissimilarities are squared before clustering (Ward.D2) (Murtagh and Legendre 2011) was used as the algorithm of clustering analysis.
The R syntaxes for data analysis are presented in the Supplementary Information.
Results
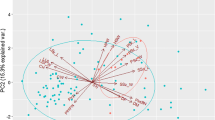
Principal component (PC) analysis for 46 pea genotypes illustrates that the first two PCs (PC1, PC2) explained about 20.5% of the total variation, with the first component explaining 14.2% and the second component explaining 6.3% (Fig. 1).

Principal component analysis of 46 pea genotypes by SNP markers, percentage of explained variance by each PC
The first principal component (Fig. 2) separated normal-leafed and semi-leafless genotypes. The normal-leafed cultivars Natura, Protecta, Bohatyr and Grana are present in the semi-leafless group. For the second principal component, the two leaf types are not distinct. Both groups of leaf types have some genotypes (AF447, AF448, GR409, GR 293EW, Pandora of normal-leafed, and Specter, James, Myster of semi-leafless) which were located far from the position of their main groups. All these are winter cultivars, except AF447 aqnd AF448.

The first two principal components of a principal component analysis of 46 pea genotypes by SNP markers (round shatpe, red colour: normal-leafed; triangle shape, cyan color: semi-leafless)
For the 46 pea genotypes, two main clusters were revealed by the dendrogram using Nei’s genetic distance (Fig. 3). This number of clusters is in correspondence with the result of the K-mean analysis (Supplementary Information, Figure S1). The assignment of the genotypes to the groups essentially agrees with the allocation made by principal component analysis (Supplemenatary Information, Table S1). The upper cluster represents normal-leafed genotypes while the lower cluster mainly contains semi-leafless genotypes. The upper normal-leafed group contains four semi-leafless genotypes (Radley, KA-L11, KA-258, Specter), while the lower semi-leafless group contains four normal-leafed accessions (Grana, Natura, Bohatyr, Protecta). The mean Nei’s genetic distance between the two groups is about 0.7. The sub-clusters in each group were determined with quite low bootstrap values (38.6% for both groups of leaf type). The bootstrap values of the following sub-clusters in the semi-leafless group are generally much higher than in the normal-leafed group and the distance between semi-leafless genotypes is in most cases clearly smaller that between normal-leafed genotypes.

Dendrogram with bootstrap values for 46 pea genotypes. The X axis presents Nei genetic distances. Red labels represent normal-leafed genotypes, and cyan labels represent semi-leafless genotypes
Discussion
Our hypothesis was that normal-leafed and semi-leafless breeding materials would form two distinct groups with a clear genetic distinction, even though the phenotypic difference in leaf type is due to only one gene (Mikić et al. 2011). If confirmed, this hypothesis implies that older normal-leafed cultivars may therefore be an interesting genetic resource for breeding new semi-leafless cultivars.
For our study we used 46 genotypes. These are mainly European summer peas. but some winter peas and some accessions from Afghanistan and Ethiopia were also included. This is a good basis for assessing the relationship between leaf type and genetic diversity. With a much larger number of genotypes, it would have been possible to study marker-trait associations as well. However, this was not our goal, as such studies are available in large numbers (for review see Sharma et al. 2020).
The allocation of 46 pea genotypes in principal component analysis was generally in agreement with the results of clustering by the K-mean method and the dendrogram based on Nei’s genetic distance. The dendrogram shows two very clearly separated clusters, one made up of mainly semi-leafless genotypes, the other one made up of mainly normal-leafed genotypes. The genetic distance between the two leaf types obviously reflects the breeding history of the cultivars, since in current breeding programs semi-leafless cultivars are normally developed from crosses between other semi-leafless cultivars. This is clearly demonstrated by the cluster of present European semi-leafless cultivars from different breeding companies with rather limited diversity (Fig. 3: Salamanca, Cheyenne, Astronaute, Santana, Casablanca, Alvesta, Navarro, Cleopatra and Camilla). Baranger et al. (2004) and Tar’an et al. (2005) also observed that the genetic diversity within recently released pea cultivars in Western Europe is low, especially in the spring-sown feed pea breeding pool. The normal-leafed materials have a larger diversity including the cultivars from Russia, Afghanistan and Ethiopia.
Our results are in agreement with other studies: Baranger et al. (2004) used 121 protein- and PCR-based markers (RAPD, SSR, STS, Isozymes) to analyse the genetic relationships in a collection of 148 Pisum genotypes. Ward hierarchical classification based on Jaccard’s distances revealed that almost all normal-leafed and semi-leafless genotypes were distributed in different groups, however some of them were also in the same sub-groups. Some winter peas were grouped within spring sown cultivars, although there is generally a genetic distance between winter and spring sown genotypes.
Tar’an et al. (2005) used ISSR, RAPD, and SSR markers for genotyping 65 pea cultivars from North America, Europe, and Australia and 11 wild Pisum accessions. The results of PCA and the cluster analysis were generally consistent, in which the majority of the varieties that were bred in Europe and many Canadian varieties were separated from those that were developed in Australia and the USA, but clustered together into one large group far away from wild-type accessions. Leaf types were generally not clearly separated, but a small group of cultivars released before 1991 contained mainly normal-leafed genotypes. Jain et al. (2014) used 42 molecular markers (SSR and EST-derived markers) with 108 alleles to assess genetic diversity and population structure among 96 pea cultivars. The results showed that the effect of leaf type or geographical origin was faintly visible, and the dendrogram illustrated that normal-leafed cultivars were allocated scattered among semi-leafless genotypes in all three main clusters. The first cluster was dominated by the cultivars that had similar geographical origin and plant characteristics, while the second and the third cluster were a mixture of varieties with yellow and green seed coat of different geographical origin. That genotypes from different geographic origins are grouped together was explained by the exchange of germplasm among different countries, suggesting that these varieties may share common parents.
Our results show a few interesting exceptions to the otherwise clear clustering into semi-leafless and normal-leafed cultivars. The cluster of semi-leafless cultivars (lower part of the dendrogram in Fig. 3) contains four normal-leafed cultivars. They all come from Czech breeding programs, in which the two leaf types apparently are not as strongly separated from one another as in breeding programs in other countries. In the normal-leafed cluster (upper part in Fig. 3) there is a group of winter cultivars (GR409, GR293EW, Pandora) also containing the semi-leafless winter cultivar Specter, because in breeding of winter pea there was obviously not such a strong distinction between the two leave types.
Simioniuc et al. (2002) used 20 RAPD primers with 175 polymorphic bands and 11 AFLP primer pairs with 462 polymorphic bands for genotyping 21 pea cultivars of which were 16 semi-leafless and 5 normal-leafed. The UPGMA cluster analysis based on genetic similarity revealed two main groups, in which two normal-leafed genotypes are forming the first group, while the second one included all semi-leafless genotypes plus the three remaining normal-leafed genotypes.The normal-leafed cultivar Bohatyr and the semi-leafless cultivar Eiffel are clustered closely together, as is also the case in our results (Fig. 3). In this case the explanation is obvious: Bohatyr is one of the parents of Eiffel (Simioniuc et al. 2002). The other exceptions to the clear separation of the two leaf types can probably be explained in a similar way.
Ahmad et al. (2015) observed that cluster analysis classified pea genotypes into groups that were somewhat consistent with their geographical origins with some exceptions, that may have been introduced from other regions by germplasm exchange programs. Jing et al. (2010) observed a clear separation between wild and cultivated accessions, but the subgroups of cultivated peas did not coincide well with the country of origin. Rana et al. (2017), when analyzing a worldwide collection of 151 accessions, found three separate groups, that were not in close agreement with different geographic origins.
In our material, three accessions originate from Afghanistan and two from Ethiopia (Table 1). Two accessions from Afghanistan (AF447 and AF448) have extreme values for the second PC in the PCA (Fig. 2). In the dendrogram, however, the five accessions from Afghanistan and Ethiopia form one normal-leafed cluster together with four other accessions from Europe (Fig. 3). The “exotic” accessions probably were introduced from Europe in the past, especially the genotypes from Ethiopia.
The low diversity of European pea varieties was mentioned by Baranger et al. (2004) and Tar’an et al. (2005), suggesting that it may encourage breeders to include exotic material in their breeding programs. Our results showed a larger genetic distance between genotypes in the normal-leafed group compared to semi-leafless genotypes. Probably all semi-leafless cultivars carry the same original afila mutation. We suggest, that a first step to increase genetic diversity should be to include normal-leafed pea genotypes as an interesting genetic resource, even when breeding semi-leafless cultivars.
Data availability
The data are available from the corresponding author on reasonable request.
References
Ahmad S, Kaur S, Lamb-Palmer ND, Lefsrud M, Singh J (2015) Genetic diversity and population structure of Pisum sativum accessions for marker-trait association of lipid content. Crop J 3:238–245. https://doi.org/10.1016/j.cj.2015.03.005
Annicchiarico P, Filippi L (2007) A field pea ideotype for organic systems of northern Italy. J Crop Improv 20:193–203. https://doi.org/10.1300/J411v20n01_11
Aznar-Fernández T, Barilli E, Cobos MJ, Kilian A, Carling J, Rubiales D (2020) Identification of quantitative trait loci (QTL) controlling resistance to pea weevil (Bruchus pisorum) in a high-density integrated DArTseq SNP-based genetic map of pea. Sci Rep 10:33. https://doi.org/10.1038/s41598-019-56987-7
Baranger A, Aubert G, Arnau G, Lainé AL, Deniot G, Potier J, Weinachter C, Lejeune-Hénaut I, Lallemand J, Burstin J (2004) Genetic diversity within Pisum sativum using protein- and PCR-based markers. Theor Appl Genet 108:1309–1321. https://doi.org/10.1007/s00122-003-1540-5
Burstin J, Salloignon P, Chabert-Martinello M et al (2015) Genetic diversity and trait genomic prediction in a pea diversity panel. BMC Genomics 16:105. https://doi.org/10.1186/s12864-015-1266-1
CPVO (2021) CPVO variety finder. https://Vf.Plantvarieties.Eu/Varieties, Accessed on 28 January 2021.
Desgroux A, Baudais VN, Aubert V, Le Roy G, De Larambergue H, Miteul H, Bourion V (2018) Comparative genome-wide-association mapping identifies common loci controlling root system architecture and resistance to Aphanomyces euteiches in pea. Front Plant Sci 8:2195. https://doi.org/10.3389/fpls.2017.02195
Dray S, Siber- Chicot A, Thioulouse J, Julien-Laferrière A (2018) Package “adegraphics” (pp. 1–159). https://cran.r-project.org/web/packages/adegraphics/adegraphics.pdf
Ellis THN (2011) Pisum. In: Kole C (ed) Wild Crop Relatives: Genomic and Breeding Resources. Springer-Verlag, Berlin-Heidelberg, Germany, pp 237–248. https://doi.org/10.1007/978-3-642-14387-8
European commission (2017) Plant variety database. http://ec.europa.eu/food/plant/plant_propagation_material/plant_variety_catalogues_databases/search/public/index.cfm, accessed on 9th October 2017. (in German).
Federal Plant Variety Offic (1997) Beschreibende Sortenliste 1997. Federal Plant Variety Office, Hannover, Germany
Federal Plant Variety Office (2017) Sorteninformationen. http://www.bundessortenamt.de/internet30/index.php?id=21, accessed on 13rd July 2017. (in German),
French RJ (2016) Field pea: Agronomy. In: Encyclopedia of Food Grains 2nd edn. Elsevier Inc. Vol 4–4, pp. 240–250. https://doi.org/10.1016/B978-0-12-394437-5.00196-0
Genbankinformationssytem (GBIS) IPK (2017). Leibniz-Institut für Pflanzengenetik und Kulturpflanzenforschung. https://Gbis.Ipkgatersleben.de/GBIS_I/Home.Jsf;Jsessionid=1SYK5anjxgCUqJJv9y0X3Il4vw_t05M1tSr7VMbJdoLc6iCAgV58!-1446071765!1507716344291, accessed on 09 October 2017.
Hartigan JA, Wong MA (1979) Algorithm AS 136: A K-Means clustering algorithm. J Royal Stat Soc Series C 28:100–108
Jain S, Kumar A, Mamidi S, McPhee K (2014) Genetic diversity and population structure among pea (Pisum sativum L.) cultivars as revealed by Simple Sequence Repeat and Novel Genic Markers. Mol Biotechnol 56:925–938. https://doi.org/10.1007/s12033-014-9772-y
Jha AB, Gali KK, Tar’an B. & Warkentin T. D. (2017) Fine mapping of QTLs for ascochyta blight resistance in pea using heterogeneous inbred families. Front Plant Sci. https://doi.org/10.3389/fpls.2017.00765
Jing R, Vershinin A, Grzebyta J, Shaw P, Smýkal P, Marshall D, Flavell AJ (2010) The genetic diversity and evolution of field pea (Pisum) studied by high throughput retrotransposon based insertion polymorphism (RBIP) marker analysis. BMC Evolut Biol 10(1):1–20
Jombart T, Kamvar ZN, Collins C et al (2021) Package “Adegenet” , pp 1–180. https://github.com/thibautjombart/adegenet
Kamvar ZN, Tabima JF, Everhart SE et al (2014) Package “POPPR” Version 2.9.0. In: Peer J 2014: 1–134). https://doi.org/10.7717/peerj.281
Kassambara A, Mundt F (2020) Package “factoextra”, pp.1–84. https://cran.r-project.org/web/packages/factoextra/factoextra.pdf
Kwon SJ, Brown AF, Hu J, McGee R, Watt C, Kisha T, Timmerman-Vaughan G, Grusak M, McPhee KE, Coyne CJ (2012) Genetic diversity, population structure and genome-wide marker-trait association analysis emphasizing seed nutrients of the USDA pea (Pisum sativum L.) core collection. Genes Genomics 34:305–320. https://doi.org/10.1007/s13258-011-0213-z
Lamprecht H (1948) The variation of linkage and the course of crossing over. Agri Hortic Genet 6:10–48. https://doi.org/10.1371/journal.pcbi.1002462
Mikić A, Mihailović V, Ćupina B, Kosev V, Warkentin T, Mcphee K, Ambrose M, Hofer J, Ellis N (2011) Genetic background and agronomic value of leaf types in pea (Pisum sativum). Ratarstvo Povrtarstvo 48:275–284. https://doi.org/10.5937/ratpov1102275m
Murtagh F, Legendre P (2011) Ward’s Hierarchical Clustering Method: Clustering Criterion and Agglomerative Algorithm, 1–21. https://doi.org/10.1007/s00357-014-9161-z
Nei M (1972) Genetic distance between populations. Amer Naturalist 106:283–292
Pandey AK, Rubiales D, Wang Y, Fang P, Sun T, Liu N, Xu P (2021) Omics resources and omics-enabled approaches for achieving high productivity and improved quality in pea (Pisum sativum L.). Theor Appl Genet 134:755–776. https://doi.org/10.1007/s00122-020-03751-5
Paradis E, Blomberg S, Bolker B et al (2021) Package “ape” version 5.5, pp 1–294. https://cran.r-project.org/web/packages/ape/ape.pdf
Rana JC, Rana M, Sharma V, Nag A, Chahota RK, Sharma TR (2017) Genetic diversity and structure of pea (Pisum sativum L.) germplasm based on morphological and SSR markers. Plant Mol Biol Rep 35:118–129. https://doi.org/10.1007/s11105-016-1006-y
Sharma A, Sekhon BS, Kumar R, Sharma S, Mahajan R (2020) Marker-assisted selection in pea breeding. In: Gosal S, Wani S (eds) Accelerated plant breeding, vol 2. Springer, Cham, pp 137–154. https://doi.org/10.1007/978-3-030-47298-6_6
Simioniuc D, Uptmoor R, Friedt W, Ordon F (2002) Genetic diversity and relationships among pea cultivars revealed by RAPDs and AFLPs. Plant Breed 121:429–435. https://doi.org/10.1046/j.1439-0523.2002.733320.x
Smýkal P, Hýbl M, Corander J, Jarkovský J, Flavell AJ, Griga M (2008) Genetic diversity and population structure of pea (Pisum sativum L.) varieties derived from combined retrotransposon, microsatellite and morphological marker analysis. Theor Appl Genet 117:413–424. https://doi.org/10.1007/s00122-008-0785-4
Smýkal P, Kenicer G, Flavell AJ et al (2011) Phylogeny, phylogeography and genetic diversity of the Pisum genus. Plant Genetic Resour 9:4–18. https://doi.org/10.1017/S147926211000033X
Smýkal P, Aubert G, Burstin J et al (2012) Pea (Pisum sativum L.) in the genomic era. Agronomy 2:74–115. https://doi.org/10.3390/agronomy2020074
Tar’an B, Warkentin T, Somers DJ, Miranda D, Vandenberg A, Blade S, Bing D (2004) Identification of quantitative trait loci for grain yield, seed protein concentration and maturity in field pea (Pisum sativum L.). Euphytica 136(3):297–306. https://doi.org/10.1186/s12870-018-1368-4
Tar’an B, Zhang C, Warkentin T, Tullu A, Vandenberg A (2005) Genetic diversity among varieties and wild species accessions of pea (Pisum sativum L.) based on molecular markers, and morphological and physiological characters. Genome 48(2):257–272. https://doi.org/10.1139/G04-114
Tayeh N, Aluome C, Falque M et al (2015a) Development of two major resources for pea genomics: the GenoPea 13.2K SNP Array and a high-density, high-resolution consensus genetic map. Plant J 84:1257–1273. https://doi.org/10.1111/tpj.13070
Tayeh N, Klein A, Le Paslier MC, Jacquin F, Houtin H, Rond C, Burstin J (2015) Genomic prediction in pea: Effect of marker density and training population size and composition on prediction accuracy. Front Plant Sci 6:941. https://doi.org/10.3389/fpls.2015.00941
Tran CT, Becker HC, Horneburg B (2022) Agronomic performance of normal-leafed and semi-leafless peas (Pisum sativum L) genotypes. Crop Sci 62:1430-1442 https://doi.org/10.1002/csc2.20746
Weiss E, Zohary D (2011) The neolithic Southwest Asian founder crops, their biology and archaeobotany. Curr Anthropol 4:S237–S254. https://doi.org/10.1086/658367
Zohary D, Hopf M (1973) Domestication of pulses in the Old World. Science 182:887–894
Zong X, Redden RJ, Liu Q et al (2009) Analysis of a diverse global Pisum sp. collection and comparison to a Chinese local P. sativum collection with microsatellite markers. Theor Appl Genet 118:193–204. https://doi.org/10.1007/s00122-008-0887-z
Acknowledgements
For supplying pea genotypes, we thank Getreidezüchtung Peter Kunz, Paolo Annicchiarico of CREA-FLC, Selgen, Naturland, the IPK Gatersleben Genebank, KWS, and Norddeutsche Pflanzenzucht Hans-Georg Lembke KG.
Funding
Open Access funding enabled and organized by Projekt DEAL. The authors wish to thank the Software AG Foundation for financial support for this study. CTT received a scholarship from the government of Vietnam and the DAAD, a scholarship from Professor-Werner-Schulze Foundation, and a Finishing Grant from the Göttingen Graduate School Forest and Agricultural Sciences.
Author information
Authors and Affiliations
Contributions
CTT, HCB, and BH contributed to the study conception and design. CTT performed the data analysis and wrote the first draft of the manuscript. BH, HCB, and TMB acquired funds and supervised the analyses. All authors commented on previous versions of the manuscript and read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We are dedicating this paper to Wolfgang Ecke on the occasion of his retirement. For more than 25 years he supported countless students and scientists in our division in the analysis of molecular markers.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tran, C.T., Beissinger, T.M., Becker, H.C. et al. Genetic diversity of pea (Pisum sativum L.) genotypes differing in leaf type using SNP markers. Genet Resour Crop Evol 70, 1085–1095 (2023). https://doi.org/10.1007/s10722-022-01487-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-022-01487-3