
Abstract
This review aims to elucidate the different mechanisms of blood brain barrier (BBB) disruption that may occur due to invasion by different types of bacteria, as well as to show the bacteria–host interactions that assist the bacterial pathogen in invading the brain. For example, platelet-activating factor receptor (PAFR) is responsible for brain invasion during the adhesion of pneumococci to brain endothelial cells, which might lead to brain invasion. Additionally, the major adhesin of the pneumococcal pilus-1, RrgA is able to bind the BBB endothelial receptors: polymeric immunoglobulin receptor (pIgR) and platelet endothelial cell adhesion molecule (PECAM-1), thus leading to invasion of the brain. Moreover, Streptococcus pneumoniae choline binding protein A (CbpA) targets the common carboxy-terminal domain of the laminin receptor (LR) establishing initial contact with brain endothelium that might result in BBB invasion. Furthermore, BBB disruption may occur by S. pneumoniae penetration through increasing in pro-inflammatory markers and endothelial permeability. In contrast, adhesion, invasion, and translocation through or between endothelial cells can be done by S. pneumoniae without any disruption to the vascular endothelium, upon BBB penetration. Internalins (InlA and InlB) of Listeria monocytogenes interact with its cellular receptors E-cadherin and mesenchymal-epithelial transition (MET) to facilitate invading the brain. L. monocytogenes species activate NF-κB in endothelial cells, encouraging the expression of P- and E-selectin, intercellular adhesion molecule 1 (ICAM-1), and Vascular cell adhesion protein 1 (VCAM-1), as well as IL-6 and IL-8 and monocyte chemoattractant protein-1 (MCP-1), all these markers assist in BBB disruption. Bacillus anthracis species interrupt both adherens junctions (AJs) and tight junctions (TJs), leading to BBB disruption. Brain microvascular endothelial cells (BMECs) permeability and BBB disruption are induced via interendothelial junction proteins reduction as well as up-regulation of IL-1α, IL-1β, IL-6, TNF-α, MCP-1, macrophage inflammatory proteins-1 alpha (MIP1α) markers in Staphylococcus aureus species. Streptococcus agalactiae or Group B Streptococcus toxins (GBS) enhance IL-8 and ICAM-1 as well as nitric oxide (NO) production from endothelial cells via the expression of inducible nitric oxide synthase (iNOS) enhancement, resulting in BBB disruption. While Gram-negative bacteria, Haemophilus influenza OmpP2 is able to target the common carboxy-terminal domain of LR to start initial interaction with brain endothelium, then invade the brain. H. influenza type b (HiB), can induce BBB permeability through TJ disruption. LR and PAFR binding sites have been recognized as common routes of CNS entrance by Neisseria meningitidis. N. meningitidis species also initiate binding to BMECs and induces AJs deformation, as well as inducing specific cleavage of the TJ component occludin through the release of host MMP-8. Escherichia coli bind to BMECs through LR, resulting in IL-6 and IL-8 release and iNOS production, as well as resulting in disassembly of TJs between endothelial cells, facilitating BBB disruption. Therefore, obtaining knowledge of BBB disruption by different types of bacterial species will provide a picture of how the bacteria enter the central nervous system (CNS) which might support the discovery of therapeutic strategies for each bacteria to control and manage infection.
Similar content being viewed by others


Introduction
The specialized micro-environment of the central nervous system (CNS) is maintained by the blood–brain barrier (BBB), which enables communication activities with the systemic compartment. The state of brain capillaries and their polarized microvascular endothelial cells are responsible for BBB structure and functional integrity, by possessing tight junctions (TJs) (Kniesel and Wolburg 2000). Occludin, claudin, junctional adhesion molecules (JAMs), and zonula occludens (ZO)-1 are the main elements of intercellular TJ proteins (Hawkins and Davis 2005). Control paracellular passage of substrates across the BBB is the main function of these proteins. However, changes may occur during several CNS pathological events involving bacterial infection.
Neurological disorders such as bacterial meningitis, sepsis, and brain abscess formation are mostly linked to bacterial pathogens (Join-Lambert et al. 2010; Iovino et al. 2013b). Scientifically, the commencement of bacterial meningitis takes place when blood-borne bacteria infiltrate into the BBB, gaining entrance into the CNS. The hallmark events within the pathophysiology of bacterial meningitis are; increased cytokines/chemokine levels in infected patients (Møller et al. 2005), deteriorating endothelium barrier integrity which can be caused by many meningeal pathogens through adherens junction (AJ)/TJ deformation, and BBB disruption (van Sorge and Doran 2012). For example, brain invasion has been occurred during the adhesion of pneumococcal pilus-1, RrgA to brain endothelium receptors such as platelet endothelial cell adhesion molecule-1 (PECAM-1) and polymeric immunoglobulin receptor (pIgR), together with platelet-activating factor receptor (PAFR) (Radin et al. 2005; Iovino et al. 2013a, 2016, 2017). In terms of pneumococcal meningitis, TNF-α, IL-1β, IL-6, and IL-10 increase in order to enhance the immune response for pathogen elimination (Kornelisse et al. 1996; Paul et al. 2003; Østergaard et al. 2004). A previous report has described that BBB integrity can be affected due to microorganisms releasing and expressing cytokines, chemokines, and cell-adhesion molecules, resulting in BBB disruption (Kim 2003). In contrast to the studies mentioned above, it has been reported that Streptococcus pneumoniae invade the endothelial cells without any disruption to the BBB and the vascular endothelium, suggesting an intracellular or paracellular path for BBB translocation (Iovino et al. 2013b). Listeriolysin O (LLO) is the pore-forming toxin of Listeria monocytogenes which facilitates and enhances the expression of the surface adhesion molecules P- and E-selectin, intercellular adhesion molecule 1 (ICAM-1), and Vascular cell adhesion protein 1 (VCAM-1), as well as IL-6 and IL-8 and monocyte chemoattractant protein-1 (MCP-1), allowing neutrophil and monocyte adhesion to the endothelial cells (Kayal et al. 2002), which might encourage BBB disruption. InhA and BsIA Bacillus anthracis invade the endothelial cells by breaking down the TJ protein (ZO-1), and thus contributes to hemorrhaging in the CNS via BBB disruption in vitro and in vivo (Ebrahimi et al. 2009). McLoughlin et al. (2017) demonstrated that BMEC permeability was induced due to Staphylococcus aureus infection via the reduction of vascular endothelial cadherin (VEC), claudin-5, and ZO-1. Recent data have demonstrated that the levels of myeloperoxidase (MPO), cytokine-induced cytokine-induced neutrophil chemoattractant-1 (CINC-1), IL-1β, IL-6, IL-10, and TNF-α were increased after Group B Streptococcus (GBS) infection in the hippocampus (Barichello et al. 2011a). Surprisingly, previous studies showed that GBS crosses the BBB without any evidence of intracellular TJ disruption or detection of micro-organisms between cells (Kim 2008). Also, it has been revealed that B. anthracis degrades endothelial cells accompanied by ZO-1 degradation, leading to bacterial adherence to the endothelium via an S-layer adhesin (BslA) (Ebrahimi et al. 2009).
Moreover, the main portals to let Haemophilus influenzae type b (HiB) enter the CNS are laminin receptor (LR) and PAFR, which subsequently might cause meningitis (Swords et al. 2001). It has been also revealed that BBB permeability might be facilitated by HiB lipopolysaccharide (Patrick et al. 1992) via TJ deformation (Quagliarello et al. 1986; Schubert-Unkmeir et al. 2010) and this damages the brain cells in vivo (Doran et al. 2003). Additionally, the first mechanism of BBB disruption in the presence of Neisseria meningitidis bacterium is that the bacterial adhesins (PilQ, an outer membrane protein involved in secretion of type IV pili in N. meningitidis) targets a common carboxy-terminal domain of LR to establish initial contact with the brain endothelium that might lead to bacterial pathogen invasion into the brain (Orihuela et al. 2009). N. meningitidis prompts the specific cleavage of occludin protein by secretion of host matrix metalloproteinase-8 (MMP-8), subsequently, this might lead to endothelial cell detachment and increased paracellular permeability (Schubert-Unkmeir et al. 2010). Pili type IV of N. meningitidis was shown as the main factor which disturbs functional proteins, resulting in anatomical gaps that were used by the bacteria to penetrate into the CNS (Coureuil et al. 2009, 2010). Similarly, Escherichia coli K1 is able to disrupt BBB integrity by a signaling process that facilitates detachment of β-catenins from cadherins (Sukumaran and Prasadarao 2003). Another study has described that brain microvascular endothelial cells (BMECs) monolayer leakage is increased upon E. coli interaction with Ec-gp96 (a receptor on human BMEC), via OmpA during invasion (Prasadarao 2002), which might lead to a disruption of BBB integrity. Therefore, this review aims to elucidate the different mechanisms of BBB disruption that may occur by different types of bacterial pathogen, as well as to show the bacteria–host interactions that assist the bacterial pathogen in invading the brain. The role of some medically common bacteria on BBB disruption and the mechanisms which are responsible for the interactions between the cerebral host cell and the bacteria contributing to CNS entry are elucidated on. Therefore, obtaining knowledge of BBB disruption by bacterial infection will therefore provide a picture of how bacteria enter the CNS and could develop novel therapeutic strategies to combat these bacteria, particularly those responsible for meningitis.
BBB Structure
Preserving the homeostatic neural micro-environment of the brain is vital for normal neuronal activity and function, which is the role of the BBB. The BBB is a structural and functional barrier that controls the passage of any substances that can be transferred by blood cells into the brain (Abbott et al. 2006). Selective permeability is the main feature of BBB, where some substances are selected upon entering CNS. For examples, lipohphilic molecules may pass the endothelial cells membranes and enter CNS, while, hydrophilic molecules encounter difficulties penetration into the brain (Levin 1980). The brain and neural function should be protected from any agents circulating such as neurotransmitters and xenobiotics by BBB function (Abbott et al. 2006). Additionally, BBB inhibits the entrance of pathogens and severely controls the entry of molecules into the brain parenchyma while promoting the efflux of several molecules (Brito et al. 2014). It is well known that CNS barriers including the BBB are able to provide a stable fluid microenvironment that is essential for neural function and protects the CNS tissue from any damage (Abbott et al. 2010). Thus, BBB is vital for restricting the access some of xenobiotics and metabolites to the CNS via taking out from the brain or blocking their entrance into the brain (De Lange 2004). BMECs that line cerebral microvessels are the main component of the BBB. Pericytes, astrocytes, and a basal membrane are the main periendothelial structures of the BBB (Moura et al. 2017). Pericytes and smooth muscle cells, which are surround and stabilize the endothelium, acting to reduce endothelial apoptosis, whereas astrocyte tasks include the support of CNS tissue. Scientifically, researchers utilize BMECs instead of the peripheral endothelial cells due to the fact that these types of cells have unique features including; many intercellular TJs that retains high transendothelial electrical resistance and hinder paracellular flux; fenestrae albescence and a decreased level of fluid-phase endocytosis; and asymmetrically localized enzymes and carrier-mediated transport systems (Biegel et al. 1995).
Tight Junctions and its Adhesion Molecules in Cerebral Endothelial Cells
The apical end of the basolateral membrane is the main place of TJs, which play a major role in forming endothelial polarity. High transendothelial electrical resistance values of up to 2000 Ω cm2 due to TJs presence are existed, resulting in lesser paracellular permeability, as compared with other peripheral tissues (3–33 Ω cm2) (Crone and Olesen 1982). TJs also aid to separate the apical and basolateral domains of the plasma membranes in epithelial cells, leading to inhibit the dispersion of integral proteins and lipids from one to the other (Tsukita et al. 2001). Occludin and claudins (Furuse et al. 1998), JAMs (Martìn-Padura et al. 1998), and the endothelial cell-selective adhesion molecule (AM) are the essential membrane proteins of the cerebral microvasculate TJs (Nasdala et al. 2002). For example, PECAM-1 plays a main role in endothelial integrity and endothelial–leukocyte interactions (Privratsky and Newman 2014). A former study revealed that adhesion of S. pneumoniae to the BBB endothelium was intermediated by adhesion molecule expression of brain endotheleial cells (Iovino et al. 2014). These four components are linked through cytoplasmic proteins (e.g., ZO-1, -2, -3, cingulin) to the actin cytoskeleton (Wolburg and Lippoldt 2002). ZO-1 is associated with TJ proteins and the actin cytoskeleton, which is essential for the steadiness, organization and signaling of TJ proteins. Any damage to or detachment of this protein from its counterparts may result in barrier permeability enhancement (Mukherjee et al. 2011).
Pathogenesis of Bacteria Correlated with Meningitis
The most common route for bacterial entrance into the meninges is through the hematogenous route and then through the BBB. It has been stated that meningeal pathogens can be disseminated via hematogenous pathogenesis into the cerebrospinal fluid (CSF) (Ferrieri et al. 1980; Huang et al. 1995; Heninger and Collins 2013). Former studies have described bacterial dispersal strategies, whereby bacteria successfully colonize the host respiratory mucosal epithelium, then invade the bloodstream and multiply, cross the BBB, and proliferate in the CSF (Kim 2003), such as in the case of N. meningitidis (Melican and Dumenil 2012). Additionally, adjacent sources such as otitis or sinusitis are another route which allows the bacteria to enter the CNS by CSF inoculation from several experimental models (Heninger and Collins 2013). Several species of bacteria, including S. pneumoniae and N. meningitidis, secrete IgA1 proteases that cleave in the hinge region of IgA (Koedel et al. 2002), resulting in evasion of the immune response. Moreover, the epithelial and endothelial cells of the nasopharynx may be destroyed through bacterial adherence and colonization (Doran et al. 2016). For instance, during pneumococcal colonization, IgA1 protease, which exists in several pathogenic species of Streptococcus, is able to protect the pneumococcus from type-specific antibodies. On the other hand, IgA is the best effector element of the mucosal immune system. Once this component has been cleaved, the host defence is weakened (Janoff et al. 1999), leading to uncontrolled bacterial replication due to local immunodeficiency, that is the most probable cause of BBB penetration and disruption (Koedel et al. 2002).
On the other hand, it has been postulated that complex bacteria–host interaction was the main causative for neuronal injury. Once the bacteria proliferate in the CSF, the permeability of the BBB is enhanced, causing a penetration of inflammatory cells and the release of several pro-inflammatory substances. Consequently, this leads to damage of the neurons, edema, and secondary neuronal damage (Koedel et al. 2002). On the contrary, it has recently been found that BBB dysfunction is related to CNS diseases due to neuro-infalmmation (Weiss et al. 2009). Remarkably, IL-17 is able to disrupt brain endothelium TJs, because of CD4 + IL-17-producing T lymphocytes (Th17) which were recently recognized as the main factor in disease progression (Miller et al. 2007). Moreover, penetration of Th17 lymphocytes across the BBB may occur by discharging chemokines, and acquiring dendritic cells in the infection sites (Kebir et al. 2007). Thus, BBB permeability is linked with leukocyte infiltration in the brain, leading to CNS neuro-inflammation.
The Correlation Between Inflammatory Response and BBB Disruption in Meningitis
Several brain cells such as glial cells, astrocytes, endothelial cells, ependymal cells, and resident macrophages are the main source for cytokines and pro-inflammatory molecules (TNF-α, IL-1β, IL-6, IFN-γ, and chemokines) secretion in response to bacterial reproduction (Moreillon and Majcherczyk 2003). For example, TNF-α is considered to be a pro-inflammatory molecule which acts to enhance the immune response for pathogen elimination (Kronfol 2000). This inflammatory molecule aids to activate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in the brain cells, which controls the expression of several pro-inflammatory mediators (Ichiyama et al. 2002). An earlier report has shown that TNF-α was increased in the first 6 h of pneumococcal meningitis in animal models (Barichello et al. 2010), this might lead to BBB disruption (Rosenberg et al. 1995). Additionally, Dinarello (2005) has reported that neutrophil and monocyte adhesion in endothelial cells is encouraged by IL-1β to eliminate the bacterial pathogen. A previous study revealed that IL-1β was increased in the CSF of patients with bacterial meningitis (Østergaard et al. 2004), as well as in animal models where it was raised in the first 24 h after pneumococcal meningitis induction (Barichello et al. 2010) by increasing the endothelium permeability. Furthermore, monocytes, endothelial cells, and astrocytes also produce IL-6 in response to IL-1β (Parizzi et al. 1991). IL-1β has pro-inflammatory effects, as a potent inducer of acute-phase proteins, fever and leukocytes (Gruol and Nelson 1997) and it also has a second function as an anti-inflammatory cytokine. Indeed, vascular permeability was declined and inflammatory response was increased upon IL-6 deficiency in bacterial meningitis (Paul et al. 2003). In contrast, it has been shown that defence against bacterial pneumonia was compromised in mice which lack IL-6 (van der Poll et al. 1997). B and T lymphocytes, macrophages, monocytes, and brain cells (neurons and microglia) are able to produce IL-10, which is considered as a potent immunosuppressive cytokine (Howard and O’Garra 1992). Elevated levels of this cytokine have been found in the CSF of patients with bacterial meningitis (Kornelisse et al. 1996), resulting in deactivation of macrophages and monocytse, which subsequently inhibits the production of cytokines such as TNF-α and IL-6 and then enhances reactive oxygen species (ROS) formation (Koedel et al. 1996). A lack of IL-10 in mice was linked with higher levels of TNF-α and IL-6 in animal models of pneumococcal meningitis (Zwijnenburg et al. 2003). Therefore, we understand from these previous studies that all these cytokines and pro-inflammatory molecules contribute to an increase in endothelial cell permeability, subsequently disrupting the BBB, particularly in meningitis infected by bacterial pathogens (Rosenberg et al. 1995).
Cell Movement Across the BBB
For any pathological condition in CNS, mononuclear leukocytes, monocytes, and macrophages are recruited, playing complementary roles to those of the resident microglia (Davoust et al. 2008). During BBB inflammation, mononuclear cells and circulating neutrophils are attracted to the site of infection, leading to penetration of the barrier and production of cuffs in the perivascular space around small vessels particularly venules; where immune response can be coordinated by this perivascular space (Konsman et al. 2007). Additionally, cytokines and other agents and mononuclear cells are also recruited upon BBB inflammation to open the TJs between endothelial cells, then it may enter by transcellular and paracellular routes (Konsman et al. 2007). Nevertheless, in normal BBB, the diapedesis process may occur, by which mononuclear cells penetrate through the cytoplasm of the endothelial cells directly, but not via the paracellular route, which involves TJ opening (Wolburg et al. 2005).
Gram-Positive Bacteria and BBB Disruption
Endothelium barrier integrity may be affected by several meningeal pathogens, including Gram-positive bacteria. Several factors may contribute in endothelium barrier integrity including direct toxic effects; interfering specifically with AJs, which are an element of cell–cell junctions between neighboring cells, and TJ formation or by a high expression of inflammatory cytokines, chemokines, and molecules (Fig. 1). For instance, S. pneumoniae and GBS stimulate barrier disruption through secreting a pore-forming toxin in infected BMECs (Nizet et al. 1997; Zysk et al. 2001; Lembo et al. 2010). Based on previous studies, meningitis is initiated by GBS, which is associated with higher toxin production through a process involving IL-8 and ICAM-1 enhancement (Doran et al. 2003). Moreover, as mentioned earlier, the host response to infection can harmfully affect BBB function via a high expression of inflammatory cytokines, chemokines and molecules. Previous studies have shown that a higher level of TNF-α is associated with BBB permeability in infant rats infected with S. pneumoniae (Barichello et al. 2011b). It is reported that BBB invasion has occurred during the pneumococcal adhesion into the brain endothelial cells receptors such as PECAM-1 and pIgR, together with PAFR (Iovino et al. 2016, 2017). Moreover, S. pneumoniae and GBS enhance nitric oxide (NO) production from endothelial cells via expression of inducible nitric oxide synthase (iNOS) enhancement (Leib et al. 1998; Winkler et al. 2001), this might lead to disruption of BBB integrity (Mittal and Prasadarao 2010). Additionally, B. anthracis has the ability to affect both AJs and TJs via secreted non-pore-forming factors including lethal toxin complexes, proteases, and edema (van Sorge and Doran 2012). Similarly, former studies have revealed that immune inhibitor A (InhA), a metalloprotease that exists in specific types of bacteria as a toxin agent, disrupts ZO-1, a TJ component (Ebrahimi et al. 2009, 2011; Mukherjee et al. 2011), indicating BBB disruption. Thus, in the following paragraphs below, specific Gram-positive bacteria related to BBB disruption mechanisms and bacterial-host interactions that facilitate brain invasion are reviewed in details.

Mechanisms of BBB disruption, and bacterial-host interactions upon Gram-positive bacterial infection
Streptococcus pneumoniae
Genus Streptococcus consists of several species, S. pneumoniae is one of them. It is alpha-hemolytic or beta-hemolytic and facultative anaerobic (Kenneth and Ray 2004). S. pneumoniae is identified as the main cause of pneumonia in several humoral immunity studies. Respiratory tract, sinuses,, and nasal cavity are the most colonized parts by S. pneumoniae asymptomatically in healthy subjects. However, individuals with compromised immune systems, especially young children and the elderly, are more susceptible to this bacterium, as the bacteria may become more pathogenic and spread to other sites to cause disease, and can be a cause of neonatal infections (Baucells and Hally et al. 2016). Infants and adults are diagnosed with pneumococcus by 40% and 15% respectively, where human nasopharynx mucosa is considered the main habitat of pneumococcus (Barichello et al. 2012a). Coughing and sneezing are the two main ways where the bacteria is transferred to people. The main S. pneumoniae-related diseases are community-acquired pneumonia and meningitis in children and elderly (van de Beek et al. 2006). Polysaccharide capsule is one of the main component of this bacterium, functioning as a virulence factor for the microorganism. There are approximately more than 90 various serotypes identified within this species, which differ in virulence, prevalence, as well as level of drug resistance. This bacterium can also induce meningitis, which shows signs and symptoms including vomiting, headache, photophobia, stiff neck, fever nausea, and mental status changes including lethargy and confusion (Heninger and Collins 2013). According to cundell et al. who is the first author confirmed that PAFR serves as a ligand for S. pneumoniae that has surface-exposed phosphorylcholine on their surface, where PAFR mediates pneumococcal infection in respiratory cells (Cundell et al. 1995). Former studies have found that several receptors are responsible for brain invasion during the adhesion of pneumococci to brain endothelial cells such as PECAM-1 and pIgR, together with PAFR (Radin et al. 2005; Iovino et al. 2013a, 2016). Similarly, another study has been conducted by Iovino et al. (2017), who stated that pneumococcal pilus-1, RrgA, bound into the two BBB endothelial receptors: (pIgR) and PECAM-1, contributing to brain invasion. In the same study, pneumococcal entry was prevented into the brain and meningitis improvement by using a bacteremia-derived meningitis model and mutant mice, as well as antibodies against those two receptors. Likewise, S. pneumoniae CbpA targets the common carboxy-terminal domain of the LR establishing initial contact with brain endothelium in experimental meningitis models (Orihuela et al. 2009) that might result in BBB penetration. Similarly, CbpA enables pneumococci to adhere and colonize the nasopharyngeal through binding to pIgR (Zhang et al. 2000). Moreover, previous study proved that CbpA is responsible in adherence activity by recruiting a vacuole targeted for transcytosis upon pneumococci, proposing CbpA interacts with the PAFR and serve as a pneumococcal element driving the transcytotic mechanism, consequently, BBB might be invaded and crossed (Ring et al. 1998). Additionally, Neuraminidase A (NanA) which is a surface protein, is expressed by S. pneumoniae, which promoted infiltration of the BBB by inducing chemokine discharge from brain endothelial cells (Uchiyama et al. 2009; Banerjee et al. 2010). Moreover, pneumolysin of pneumococci has able to breach endothelial cells, then enter CNS (Zysk et al. 2001). It is proposed that pneumolysin neutralization might be a potential approach for new therapeutic intervention for pneumococcal diseases treatment (Hirst et al. 2004). Therefore, the disruption or modulation of the interaction between S. pneumoniae bacterial adhesins and LR, PECAM-1, pIgR and PAFR and pneumolysin might encourage a researchers to explore effective therapeutic agent treating pneumococcal bacterial meningitis, as shown in (Fig. 1) (Orihuela et al. 2009; Iovino et al. 2016).
As mentioned before, former study reported that cytokines and pro-inflammatory molecules (TNF-α, IL-1β, IL-6, and IL-10) can be produced in response to bacterial replication in several brain cells (Moreillon and Majcherczyk 2003). In term of pneumococcal meningitis, TNF-α, IL-1β, IL-6, and IL-10 are increased to enhance the immune response for pathogen elimination (Kornelisse et al. 1996; Paul et al. 2003; Østergaard et al. 2004). A previous report has described that BBB integrity can be affected due to micro-organisms releasing and expressing cytokines, chemokines, and cell-adhesion molecules, resulting in a developing BBB disruption (Kim 2003). Thus, the increase of pro-inflammatory markers may have an adverse effect; disrupting the BBB and increasing endothelial permeability (Barichello et al. 2010) for this bacterium. Additionally, it has been revealed that CINC-1 levels were also augmented, related with BBB breakdown between 12 and 24 h in the hippocampus and at 12 and 18 h in the cortex, in wistar rats infected with S. pneumoniae (Barichello et al. 2012b). CINC-1 is a neutrophil chemoattractant and may be associated to events in pneumococcal meningitis pathophysiology, due to its ability to promote leukocyte migration (Barichello et al. 2012b). TNF-α plays an essential role in bacterial meningitis related to brain damage (Sellner et al. 2010). Besides, IL-1β also encourages BBB injury and meningitis in the animal model (Quagliarello et al. 1991); thus, chemokine secretion is normally persuaded by pro-inflammatory cytokines (Baggiolini et al. 1993). In contrast to the studies mentioned above, it has been reported that adhesion, invasion and translocation through or between endothelial cells can be done by S. pneumoniae without any disruption to the vascular endothelium, upon BBB penetration, suggesting that an intracellular or paracellular path is used for BBB translocation (Iovino et al. 2013b). The pneumococcal pathogens cross the BBB by several ways: for example pneumococcal pneumolysin destruct the endothelial cell layers (Marriott et al. 2008). While in another study, BBB can be crosses between the cells via TJs disruption, in S. pneumoniae (Attali et al. 2008). Additionally, BBB might be transversed by transcytosis process, where an intracellular transport route designed to transport molecules and vesicles through cells from the apical to basolateral side (Ring et al. 1998).
Listeria monocytogenes
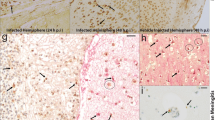
Listeria monocytogenes is facultative intracellular bacterium, which causes invasive diseases in humans and animals, such as CNS infections (Vázquez-Boland et al. 2001). Contaminated food is the main source of L. monocytogenes infection in humans. An epidemiological study of bacterial meningitis in the United States in 1995, showed that L. monocytogenes is approximately tenfold more effective at invading the CNS than other neuroinvasive Gram-positive bacteria, including S. pneumoniae and GBS (Schuchat et al. 1997). Bacterial meningitis are mostly caused by L. monocytogenes in Western Europe and North America (Kyaw et al. 2002). There are increasingly more studies concerning CNS infections caused by L. monocytogenes, which report the mechanisms of how the bacteria enter the CNS and disrupt the BBB. L. monocytogenes is distinctive among neuro-invasive bacteria where in vitro and in vivo data propose that it has the possibility to enter the CNS by several various mechanisms (Drevets et al. 2004). These mechanisms include (1) transportation across the BBB within parasitized leukocytes, (2) direct invasion of endothelial cells by extracellular hematogenous spread, or (3) retrograde (centripetal) migration into the brain within the axons of cranial nerves (Drevets et al. 2004). Internalins (InlA and InlB) are the main pathogenic proteins of L. monocytogenes that interact with its cellular receptors E-cadherin and mesenchymal-epithelial transition (MET), respectively, for intestinal and placental barriers crossing purposes. Therefore, there is one potential explanation stating that L. monocytogenes might cross BBB, due to these receptors are expressed at the surface of choroid plexus epithelial cells, and MET which is also expressed at the brain endothelial level, leading these mechanisms might to occur for blood-CSF and BBB crossing purposes (Gründler et al. 2013). In vitro studies showed that L. monocytogenes can enter and reproduce inside a wide variety of endothelial cells such as the umbilical vein and BMECs (Drevets et al. 2004), and might lead to an increase in the permeability and disruption of the BBB. Similarly, previous analysis has also revealed that the invasion protein InlB of L. monocytogenes mediates invasion of BMECs and human umbilical vein endothelial cells (HUVEC) in vitro (Greiffenberg et al. 1998, 2000; Parida et al. 1998). Additionally, listeriolysin O (LLO) is the pore-forming toxin that allows L. monocytogenes to activate NF-κB cultured endothelial cells. In brain microvessels, LLO facilitates and enhances the expression of the surface adhesion molecules P- and E-selectin, ICAM-1 and Vascular cell adhesion protein 1 (VCAM-1), as well as IL-6 and IL-8 and MCP-1, allowing neutrophil and monocyte adhesion to the endothelial cells (Kayal et al. 2002). These effects may disrupt BBB function and lets L. monocytogenes access the CNS. Nine humans who died of brainstem encephalitis revealed that the inflammatory profiles infiltrate inside nuclei, tracts, and intra-parenchymal parts of cranial nerves in different areas of the oropharynx after a careful analysis of autopsy (Antal et al. 2005). Therefore, it is clear that adherence molecules, as well as inflammatory markers, are the main factors stimulating endothelium permeability, which then leads to BBB disruption.
In terms of therapeutic purposes, as mentioned earlier, it has been suggested that NF-κB activation in endothelial cells plays a main role in L. monocytogenes crossing the BBB (Kayal and Charbit 2006). There are two pathways to activate NF-κB. First, LLO stimulates IκB kinase β-dependent phosphorylation of the physiological NF-κB inhibitor IκBα, followed by proteasomal degradation of the latter, subsequently allowing nuclear translocation of NF-κB (Kayal et al. 2002). InlC directly binds with IKKα, a subunit of the IκB kinase complex critical for the phosphorylation of IκB and NF-κB activation (Gouin et al. 2010). Second, L. monocytogenes has been demonstrated to activate mitogen-activated protein kinase (MAPK) through LLO early in infection (Tang et al. 1994) upon escape from the phagosome (Opitz et al. 2006). The MAPK cascade has been involved both in bacterial entry into endothelial cells (Tang et al. 1998) and in the induction of cytokine expression in vitro (Schmeck et al. 2005) and in vivo (Lehner et al. 2002). As a result, all these markers for NF-κB, MAPK, InlA and InlB activations are responsible for L. monocytogenes spreading into the brain; however, targeting them will facilitate the reduction of the pathogenesis and may be considered as a potential therapeutic agent against meningitis.
Bacillus anthracis
Rod-shaped and spore-forming are the main traits of Bacillus anthracis. The main disease related to B. anthracis infection is anthrax, which has three different clinical forms depending on the major routes of infection: cutaneous, inhalational, and gastrointestinal (Inglesby et al. 2002). All types of anthrax can be fatal if left untreated as it leads to the systemic spreading of this lethal bacteria through lymphatic and hematogenous routes. A previous clinical case report has shown that this type of bacteria disperses to the brain, subsequently resulting in haemorrhagic meningitis (Inglesby et al. 2002). In Sverdlovsk-Russia, 50% of fatal haemorrhagic meningitis infected with B. anthracis was recorded during the epidemic of anthrax inhalation (Inglesby et al. 2002). In another study, anthrax histological analysis was shown that the secreted pathogenic factors such as InhA induces BBB destruction, by making it leaky and permeable (Inglesby et al. 2002; Ramarao and Lereclus 2005). Numerous mechanisms are involved for pathogenicity by this factor (InhA) upon B. anthracis infection, such as escape of bacteria from macrophages, cleavage of antibacterial proteins, control of blood coagulation, and degradation of matrix-associated proteins (Ramarao and Lereclus 2005; Chung et al. 2008, 2009; Kastrup et al. 2008; Guillemet et al. 2010). A previous study has shown that InhA and BsIA invade the endothelial cells by breaking down the TJ protein (ZO-1), thus contributes to hemorrhaging in the CNS via BBB disruption in vitro and in vivo (Ebrahimi et al. 2009). Similarly, purified anthrax lethal toxin-induced human lung microvascular endothelial cells permeability occurred through TJ disruption (Warfel et al. 2005). Additionally, BslA of B. anthracis acts as a global adherence factor for anthrax disease pathogenesis, where the adherence to BMEC was reduced by the BslA-deficient mutant in vitro, related with a decreased risk for development of CNS infection in vivo (Kern and Schneewind 2010). Moreover, anthrax lethal toxin also enhances endothelial barrier dysfunction, leading to vascular leakage following intraperitoneal injection (Gozes et al. 2006). There is one explanation shows how this bacterium penetrate the brain described by van Sorge et al. (2008). van Sorge et al. (2008) reported that B. anthracis infected-BMEC enhanced the neutrophil recruitment response into the infection sites. Nevertheless, the innate defence pathway was down-regulated upon the presence of the toxin-encoding pXO1 plasmid of B. anthracis (van Sorge et al. 2008). This result leads to inhibition of the neutrophil chemotaxis, allowing the dissemination of the bacteria into the CNS (van Sorge et al. 2008).
Therefore, it is clarified that B. anthracis surface-associated adhesin may encourages BMEC binding and BBB infiltration, contributing to the pathogenesis of B. anthracis-caused meningitis. As a result, pXO1, InhA, and BslA may serve as an attractive new target for drug or vaccine development, aiming to inhibit development of the disease upon systemic anthrax infection.
Staphylococcus aureus
Staphylococcus aureus is a facultative anaerobe and round-shaped bacterium which is often found in the nose, on the skin, and in respiratory tract. Although S. aureus is not always pathogenic, skin infections including abscesses (Esen et al. 2010), respiratory infections such as sinusitis (Manarey et al. 2004; Huang and Hung 2006), and food poisoning are common causes of it (Chiang et al. 2008). Virulence factors of pathogenic strains such as potent protein toxins and expression of a cell-surface protein that interacts and inactivates antibodies, may stimulate infections (Bebbington and Yarranton 2008). Although many studies have been conducted on various aspects biologically and epidemiologically, there has yet to be any approved vaccine for S. aureus. Additionally, it is considered the most prevalent opportunistic bacterial pathogen responsible for community- and hospital-acquired infections worldwide. The mortality rate associated with S. aureus-mediated sepsis and meningitis is 36% (Aguilar et al. 2010). The varied range of proteins on its surface, act as virulence factors that assist the bacterium to adhere and invade host cells, including vascular endothelial cells (Foster et al. 2014). McLoughlin et al. (2017) have demonstrated that BMEC permeability was induced due to S. aureus infection via the reduction of VEC, claudin-5 and ZO-1 in a dose-dependent manner. The main mechanism of BBB disruption due to junctional protein disruption is related to pro-inflammatory cytokine signaling, which is associated with ROS generation (Rochfort and Cummins 2015; Rochfort et al. 2016). There is a strong correlation between ZO-1 levels and ROS signaling. It is found that the ZO-1 was disrupted in murine cells upon exposure to hypoxia with reoxygenation (MHR) due to Nicotinamide adenine dinucleotide phosphate (NADPH) activation. Additionally, ZO-1 was also disrupted in murine BMECs treated with oxidized low-density lipoprotein (ox-LDL) (Wang et al. 2012) and Amyloid β Protein Fragment 1–42 (Aβ1–42) (Carrano et al. 2011) approved by NADPH oxidase enhancement. It is shown that high mRNA expression levels of several cytokines: IL-1α, IL-1β, IL-6, TNF-α, MCP-1, and macrophage inflammatory proteins-1 alpha (MIP1α) in S. aureus infected rat brain abscess models, have led to BBB disruption (Kielian and Hickey 2000). In addition, S. aureus infection induced IL-6 production in HUVEC (Park et al. 2007). ROS generation has also been observed in S. aureus infection especially in neutrophils, monocytes, macrophages, and resident bone marrow stem cells (Nandi et al. 2015), resulting in enhancement of the inflammatory response. Furthermore, it has been shown that the expression of adhesin protein (SpA) protein is also able to increase BMEC permeability, accompanied by VEC protein reduction and NF-κB/p65 activation by this pathogenic bacteria (McLoughlin et al. 2017). As a result, barrier integrity maybe disrupted due to S. aureus infection of BMECs through induction of pro-inflammatory cytokines, oxidative stress, and NF-κB activation, in parallel with the reduction of TJ protein expression. For therapeutic purposes, a previous study has demonstrated a role for membrane-anchored lipoteichoic acid (LTA), mediating S. aureus adhesion and cellular invasion into immortalized human BMECs, resulting in BBB penetration (Sheen et al. 2010). A previous report has also shown that IL-17 enchantment of the host defence circuit may offer a basis for innovative therapeutic approaches to treat S. aureus infectious diseases (Cho et al. 2010). Accordingly, to prevent the disease development during this bacterial infection, it is speculated that LTA, IL-17 and SpA may serve as an attractive new target for drug or vaccine development.
Streptococcus agalactiae
Catalase-negative, beta-hemolytic, and facultative anaerobe are the characteristics of Streptococcus agalactiae, or group B Streptococcus (GBS). GBS is a harmless human microbiota colonizing the genitourinary and gastrointestinal tracts of up to 30% of healthy adults, asymptomatically. Nevertheless, severe invasive infection might occur due to GBS (Edwards and Baker 2005). Polysaccharides (exopolysacharide) is the main component of GBS which surrounds the bacterial capsule. Based on the GBS capsular polysaccharide immunologic reactivity, 10 GBS serotypes have been identified (Ia, Ib, II, III, IV, V, VI, VII, VIII, IX) (Rosa-Fraile et al. 2014). It is found that serotype III of GBS has been related to meningitis (Edmond et al. 2012) and other types of diseases (Whidbey et al. 2013, 2015; Leclercq et al. 2016). Meningitis is the most common disease related to GBS in neonates (Brouwer et al. 2010). Although intensive care management and antibiotic treatment are applied, the mortality rate is still 10%, while 25–50% of survivors show neurological sequelae, such as seizures, mental retardation, deafness, blindness, and cerebral palsy (Edwards and Baker 2005). Several virulence factors exist in GBS contributing to the interaction with brain endothelium, such as LTA (Doran et al. 2005), β-hemolysin / cytolysin (β-H/C) (Doran et al. 2003), pili (Maisey et al. 2007; Banerjee et al. 2011a), serine-rich repeat (Srr) proteins (van Sorge et al. 2009; Seo et al. 2012), and hypervirulent GBS adhesion (HvgA) (Tazi et al. 2010). Surprisingly, GBS crosses the BBB without any evidence of intracellular TJ disruption or detection of microorganisms between cells (Kim 2008). Lately, it has been proved that the GBS pilus protein PilA and Srr-1 bind with host extracellular matrix (ECM) components, stimulating BBB disruption and the enhancement of meningitis (Banerjee et al. 2011a; Seo et al. 2012). Specific microorganisms can proliferate within the CSF, resulting in release of bacterial components, which encourage the release of neuro-inflammatory molecules (Yadav et al. 2009). Cytokines and other pro-inflammatory molecules such as TNF-α, IL-1β, and IL-6 can be induced by various brain cells, in response to bacterial stimuli (Moreillon and Majcherczyk 2003), subsequently, other cascade of inflammatory mediators are triggered such as MMP, MPO and ROS. As mentioned earlier, cytokines themselves may contribute to impairment of the BBB and brain damage (Miric et al. 2010). A previous study has measured the levels of cytokine, chemokine, MPO, and oxidative stress in the hippocampus and cortex of neonatal wistar rats in a meningitis model induced by GBS (Barichello et al. 2011a). The data demonstrated that the levels of MPO, cytokine-induced CINC-1, IL-1β, IL-6, IL-10, and TNF-α were increased after GBS infection in the hippocampus (Barichello et al. 2011a). It is speculated that neonatal bacterial infections of the CNS are severe, where complex network of cytokines and chemokines, other inflammatory mediators and oxidants tend to amplify the disease and might lead to BBB destruction (Barichello et al. 2011a). Neutrophilic penetration during acute bacterial meningitis was noticed, where pilus protein (PilA) mediates GBS attachment to the brain endothelium (Banerjee et al. 2011b). In vivo study has shown higher levels of bacterial CNS penetration and BBB permeability are associated with increased neutrophilic infiltrate (Banerjee et al. 2011a). Thus, we may speculate from previous studies that the pro-inflammatory cytokines and chemokines are the main causative agent of BBB integrity in GBS infection.
In terms of therapy strategies, a previous study has demonstrated that Srr-1, which are glycoproteins responsible for adhesion function, mediating the interaction of GBS to fibrinogen, the major protein in human blood. This interaction probably takes place through a DLL-like mechanism, involving the C-terminus of the fibrinogen Aα chain. It is shown that this adhesion marker binds to fibrinogen by DDL as a general mechanism for attachment by Gram-positive organisms to the host cell. The most important point is that this binding of Srr-1 to fibrinogen seems to be vital for the bacterium adherence to brain endothelium and then meningitis development. Additionally, PilA interacts with the extracellular matrix component collagen, which then involves α2β1 integrins on brain endothelium to stimulate bacterial attachment and pro-inflammatory chemokine release. As a result, Srr-1 and PiIA appear to be expressed by most clinical isolates of GBS, therefore this bindings might confirm to be a favorable candidate for innovative therapies targeting bacterial pathogen (Banerjee et al. 2011a; Seo et al. 2012).
Gram-Negative Bacteria and BBB Disruption
In terms of meningeal pathogens, Gram-negative bacteria have also able to target AJ protein and VEC, by using their surface adhesion molecules to exploit brain endothelial cell signaling to enhance paracellular translocation (Coureuil et al. 2009, 2010). Brief mechanisms have been shown in Fig. 2, illustrating the main markers which are responsible for BBB disruption upon Gram-negative bacterial infection. For example, attachment of meningococci (type IV pili) to brain endothelial cells acts to disrupt AJ formation, subsequently opening up the paracellular route for the translocation into the CNS, leading to meningococcal meningitis (Coureuil et al. 2009). In addition, this category of bacteria might stimulate specific cleavage of the TJ component occludin through the release of host MMP-8, causing endothelial cell detachment and developed paracellular permeability (Schubert-Unkmeir et al. 2010). In addition, IL-6 levels were increased significantly in Gram-negative bacteria infection (Abe et al. 2010). Therefore, this group of bacteria may disrupt the BBB via TJ and AJ deformation and also enhance pro-inflammatory parameters in the case of meningitis. Therefore, in the following paragraphs below, specific Gram-negative bacteria related to BBB disruption mechanisms and bacterial-host interactions that facilitate brain invasion are reviewed in detail.

Mechanisms of BBB disruption, and bacterial-host interactions upon Gram-negative bacterial infection
Haemophilus influenzae
Haemophilus influenzae is a Gram-negative, facultative anaerobic, and pathogenic bacterium belonging to the Pasteurellaceae family. Globally, HiB is one of the most common types of this bacterium that causes meningitis in infants and adults. In the 1980s, half of the cases of meningitis which lead to death in infants were due to HiB alone, before the conjugate HiB vaccine was introduced. However, the cases decreased dramatically (78%) following routine vaccination (Peltola 2000). Nevertheless, lack of HiB vaccination programs in developing countries (Peltola 2000), 173,000 cases of meningitis are recorded globally (Watt et al. 2009). Basically, vaginal tracts, skin and upper respiratory tracts of healthy people could be colonized by HiB. In addition, HiB carriage (transient or intermittent) is able to remain asymptomatically (Stephens 1999). The main portals that let HiB enter the CNS are LR and PAFR, subsequently H. influenza pathogens might cause meningitis (Swords et al. 2001). Similarly, H. influenzae OmpP2 has the ability to target the common carboxy-terminal domain of LR to start initial interaction with brain endothelium (Orihuela et al. 2009). According to several studies, it was stated that Haemophilus species pili probably interacted with PAFR on brain endothelial cells (Kolberg et al. 1997; Weiser et al. 1998). Thus, it is suggested that LR and OmpP2 (See Fig. 2) are a potential therapeutic target to block BBB infiltration upon H. influenzae infection (Orihuela et al. 2009).
It has been shown that pili of HiB or fibrils can interact with BMEC (St. Geme and Cutter 1995, St Geme 3rd and, 1996). Additionally, it has also been revealed that BBB permeability might occur by HiB lipopolysaccharide (Patrick et al. 1992) through damaging the brain cells in vivo (Wellmer et al. 2002; Doran et al. 2003). Furthermore, disruption of intercellular TJs was also observed upon H. influenzae infection by alterations of the BBB functionally and morphologically in animal models (Quagliarello et al. 1986; Schubert-Unkmeir et al. 2010). As previously described, neutrophil infiltration also encourages BBB permeability (Banerjee et al. 2011a). Interestingly, HiB meningitis outcomes have been improved after the prevention of leukocyte infiltration into the CNS using anti-CD18 antibody (Tuomanen et al. 1989; Saez-Llorens et al. 1991). Thus, it can be suggested that the TJ deformation that occurs upon H. influenza dissemination is the sole mechanism responsible for BBB disruption.
Neisseria meningitidis
Meningitis and other forms of meningococcal disease such as meningococcemia, a life-threatening sepsis can be caused by Gram-negative bacteria, where Neisseria meningitidis is one of them. Nasopharynx is the main reservoir of this bacterium carried by 10% of adults (Stephens 2007). In addition, skin, upper respiratory, and vaginal tract are the other places for N. meningitidis colonization in healthy individuals. Death in 10% children and youth adults have been estimated due to this type of bacteria. Saliva and respiratory secretions are the main factors for N. meningitidis dissemination during such activities such as chewing, kissing and coughing (Er et al. 2009). Notable, several serotypes which belong to N. meningitidis are limiting the vaccination strategies against it (Gray et al. 2006). Therefore, the possible way to treat meningococcal meningitis infected by N. meningitidis is a vaccine that includes all serotypes, or when there is preserved antigen that exists in all disease isolates. Although, this bacterium is sometimes stable, transient (Stephens 1999) and often remain asymptomatic in the carriage, however, the bacteria may penetrate host cellular barriers to start local infection leading to systemic spreading. The adherence of N. meningitidis to cell host can be occurred by pili and other virulence factors such as surface-exposed Opa and Opc proteins. Similar to S. pneumoniae, pili or fibrils are the main organelles that can be utilized by N. meningitidis to initiate binding to BMECs. The first mechanism of BBB penetration in the presence of this bacterium is that the bacterial adhesins (PilQ), targets a common carboxy-terminal domain of LR to establish initial contact with the brain endothelium that might lead to bacterial pathogen invasion into the brain (Orihuela et al. 2009). The other determinants of host cell binding are existed upon this bacterium infection are complex protein (ACP) as well as the autotransporter meningococcal serine protease A (MspA) (Virji 2009). Thus, all these markers are mentioned above might be considered as the main target for therapeutic purposes in N. meningitidis meningitis.
To show the similarities and differences between three types of bacteria (S. pneumonia, S. agalactiae, and N. meningitidis), Table 1 has illustrated the mechanisms of BBB disruption and bacterial-host interaction sites that enabling brain invasion among these types of bacteria.
The mechanism for BBB disruption is via inflammatory activation of brain endothelial cells by cytokines, which are typically elevated in meningitis patients (Fida et al. 2006; Nagesh Babu et al. 2008), where it can stimulate host receptor expression, resulting in an enhanced bacterial invasion. A former study has revealed that N. meningitidis triggers IL-6 and IL-8 production in BMECs by activating MAPK pathways (Sokolova et al. 2004; Banerjee et al. 2010, 2011a), that most probably resulted in BBB disruption. The other mechanism of BBB disruption in this bacterium is through AJ and TJ deformation. A former study has shown that pili (Type IV) bind to BMECs in meningococci species inducing AJ deformation, by reducing one of its components (VEC), subsequently, the bacterial pathogen penetrates into the CNS, due to the opening up of the paracellular route (Coureuil et al. 2009). Similarly, N. meningitidis has able to disrupt TJ component (occluding) via MMP-8 production, leading to endothelial cell detachment and developed paracellular permeability (Schubert-Unkmeir et al. 2010). Overall, all these studies, it is found that N. meningitidis may disrupt the BBB via accelerated cytokine expression and cause a defect in TJ components resulting in expression of MMP-8, subsequently enhancing paracellular permeability.
Escherichia coli
The lower intestine of warm-blooded organisms is the main place of Escherichia coli. E. coli is facultatively anaerobic and coliform bacterium (Tenaillon et al. 2010). Most of E. coli strains are harmless, which produce vitamin K2, and in the same time prevent any colonization of the intestine with pathogenic bacteria, as these strains are considered part of the normal flora (Hudault et al. 2001). However, some serotypes of E. coli are intermittently responsible for product recalls due to food contamination, and can cause severe food poisoning in their hosts (Vogt and Dippold 2005). An correlation between high-level bacteremia and enhancement of meningitis was observed in E. coli, in experimental models of hematogeneous meningitis (Bell et al. 1985). It is stated that Pili (or fimbriae) or the fibrils (Danne and Dramsi 2012), is responsible for initiating binding to BMECs in E. coli K1 (Teng et al. 2005). According to Khan et al. (2002), cytotoxic necrotizing factor-1 (CNF-1) activates Ras homolog gene family, member A (RhoA), which leads to E. coli K1 invasion of BMECs in vitro and increased penetration of BBB in vivo. The LR is known as the main internalization receptor for E. coli K1 to BMECs (Kim et al. 2005), that might facilitates brain invasion. Additionally, another way for penetration to CNS is through E. coli OmpA binds to C4-binding protein (C4 bp), a classical complement pathway regulator to block the complement cascade reaction, thus avoids bacteriolysis and recognition by immune cells (Prasadarao et al. 2002). Therefore, these mechanisms highlight that the pharmacological inhibition of the host receptor and host cell signaling molecules, which contributes to E. coli invasion of BMEC, is a good indicator to prevent meningitis occurred by E. coli. Table 2 illustrates the mechanisms of BBB disruption and the bacterial-host interaction sites that enabling brain invasion among Gram-positive and -negative bacteria.
As mentioned above, once the host responds to infection, inflammatory cytokine and chemokine molecules are elevated by the host, resulting in BBB dysfunction and disease. Previous studies have demonstrated that E. coli binds to BMECs, resulting in IL-6 and IL-8 release in vitro (Zhou et al. 2012). Furthermore, through the predominant cell-surface antigen OmpA, E. coli K1 increases NO production from brain endothelial cells by inducing expression of iNOS, subsequently weakening BBB integrity and stimulating bacterial invasion (Mittal and Prasadarao 2010). A higher level of NO has been detected in animal models of bacterial meningitis and also in human patients with the meningeal disease (Murawska-Cialowicz et al. 2000). There is a growing indication that NO is an important modulator of cerebral vascular permeability (Mayhan 2000), confirming that this marker induces vascular permeability, which then leads to BBB disruption. Another study has described that BMEC monolayers leakage are increased upon E. coli interaction with Ec-gp96 (a receptor on human BMEC), via OmpA during invasion (Prasadarao 2002), which might lead to an disruption of BBB integrity. Another mechanism of BBB disruption by E. coli is the separation of VEC from other molecules of the TJs in endothelial cells (Sukumaran and Prasadarao 2003), that most propably lead to BBB disruption.
Conclusion
It can be concluded from this review that each bacteria has a different mechanism for BBB disruption, as well as a different mechanism for antigen-host cell interaction to invade the brain.
For brain invasion status and therapeutic purposes, several mechanisms have been described as below:
-
S. pneumoniae CbpA targets a common carboxy-terminal domain of LR to establish initial contact with brain endothelium (Orihuela et al. 2009), that might assist to invade the brain. Additionally, other receptors were also found to be responsible for brain invasion during adhesion of pneumococci to brain endothelial cells including PECAM-1 and pIgR, together with PAFR (Radin et al. 2005; Iovino et al. 2013a, 2016).
-
InlA and InlB of L. monocytogenes interact with its cellular receptors E-cadherin and MET, respectively, (Gründler et al. 2013), that might facilitates BBB invasion.
-
InhA and BslA may encourage BMEC binding and BBB infiltration in vivo upon B. anthracis infection, (Ebrahimi et al. 2009). Additionally, pXO1 B. anthracis inhibits the innate defence pathway, resulting in neutrophil chemotaxis prevention, allowing the dissemination of the bacteria into the CNS (van Sorge et al. 2008).
-
LTA, mediating S. aureus adhesion and cellular invasion into immortalized human BMECs, resulting in BBB penetration (Sheen et al. 2010). LTA, IL-17 and SpA are being considered as a good indicators for S. aureus treatment (Cho et al. 2010; Sheen et al. 2010; McLoughlin et al. 2017).
-
PilA of GBS interacts with the extracellular matrix component to stimulate bacterial attachment on brain endothelium (Banerjee et al. 2011a; Seo et al. 2012), which might enable brain invasion.
-
In terms of H. influenza OmpP2, LR and PAFR, are the main portals to let the pathogen enter the CNS and BBB invasion (Swords et al. 2001; Orihuela et al. 2009).
-
The bacterial adhesins of N. meningitidis (PilQ) targets a common carboxy-terminal domain of LR to establish initial contact with brain endothelium (Orihuela et al. 2009), which encourages BBB invasion.
-
E. coli K1 encouraged BMECs invasion and increased penetration of BBB through CNF-1 activates Ras homolog gene family, member A (RhoA) (Khan et al. 2002). Additionally, another way for penetration of E. coli to CNS through OmpA binds to C4 bp (Prasadarao et al. 2002).
Thus, all these interactions mentioned above may prove to be promising mechanisms for novel therapies targeting bacterial virulence, where they might stimulate protection against bacterial meningitis and may provide a therapeutic target for the prevention and treatment of the disease.
The following points are the mechanisms which are responsible for BBB disruption in Gram-negative and -positive bacteria:
-
S. pneumoniae species induced meningitis and BBB disruption through TNF-α, IL-1β, IL-6 and IL-10, CINC-1 production (Kornelisse et al. 1996; Paul et al. 2003; Østergaard et al. 2004; Barichello et al. 2012b). In contrast, S. pneumoniae lead to adhesion, invasion and translocation through or between endothelial cells without any disruption to the vascular endothelium, upon BBB penetration, suggesting that an intracellular or paracellular path is used for BBB translocation (Iovino et al. 2013b).
-
L. monocytogenes encouraged the expression of P- and E-selectin, ICAM-1 and VCAM-1, as well as IL-6 and IL-8 and MCP-1 (Kayal et al. 2002), resulting in endothelium permeability and then BBB disruption.
-
Down regulation of the TJ protein (ZO-1) levels by B. anthracis contributing to BBB disruption (Warfel et al. 2005; Ebrahimi et al. 2009).
-
BMEC permeability and BBB disruption were induced by S. aureus infection via VEC, claudin-5 and ZO-1 reduction and induction of pro-inflammatory cytokines (IL-1α, IL-1β, IL-6, TNF-α, MCP-1, MIP1α), oxidative stress and NF-κB activation (Kielian and Hickey 2000; McLoughlin et al. 2017).
-
Levels of cytokine, chemokine, MPO and oxidative stress in a meningitis-induced model were increased during GBS infection (Barichello et al. 2011a), which might encourage to BBB dysfunction.
-
H. influenza bacteria is able to disrupt the BBB due to TJ deformation (Quagliarello et al. 1986; Schubert-Unkmeir et al. 2010).
-
N. meningitidis induced AJs deformation, leading to the opening up of the paracellular route for N. meningitidis translocation into the CNS. N. meningitidis also triggered IL-6 and IL-8 production in BMECs that most probably results in BBB disruption (Sokolova et al. 2004; Banerjee et al. 2010, 2011a). N. meningitidis also induced specific cleavage of the TJ component occluding, leading to endothelial cell detachment and increased paracellular permeability (Coureuil et al. 2009; Schubert-Unkmeir et al. 2010).
-
E. coli infection encouraged IL-6 and IL-8, and iNOS production (Zhou et al. 2012), that facilitates BBB disruption. The disassembly of TJs between endothelial cells was induced by E.coli due to the separation of VEC from other TJ molecules (Sukumaran and Prasadarao 2003).
Overall, all these studies have found that each bacteria has their own way to disrupt the BBB and enter into the CNS, which has assisted the researchers in obtaining more knowledge about bacteria pathogenicity, especially in meningitis.
References
Abbott NJ, Rönnbäck L, Hansson E (2006) Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 7:41–53. https://doi.org/10.1038/nrn1824
Abbott NJ, Patabendige AAK, Dolman DEM et al (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37:13–25. https://doi.org/10.1016/j.nbd.2009.07.030
Abe R, Oda S, Sadahiro T et al (2010) Gram-negative bacteremia induces greater magnitude of inflammatory response than Gram-positive bacteremia. Crit Care 14:R27. https://doi.org/10.1186/cc8898
Aguilar J, Urday-Cornejo V, Donabedian S et al (2010) Staphylococcus aureus meningitis: case series and literature review. Medicine 89:117–125. https://doi.org/10.1097/MD.0b013e3181d5453d
Antal E-A, Løberg E-M, Dietrichs E, Maehlen J (2005) Neuropathological findings in 9 cases of listeria monocytogenes brain stem encephalitis. Brain Pathol 15:187–191
Attali C, Durmort C, Vernet T, Di Guilmi AM (2008) The interaction of Streptococcus pneumoniae with plasmin mediates transmigration across endothelial and epithelial monolayers by intercellular junction cleavage. Infect Immun 76:5350–5356. https://doi.org/10.1128/IAI.00184-08
Baggiolini M, Dewald B, Moser B (1993) lnterleukin-8 and related chemotactic cytokines—CXC and CC chemokines. Adv Immunol 55:97–179. https://doi.org/10.1016/S0065-2776(08)60509-X
Banerjee A, van Sorge NM, Sheen TR et al (2010) Activation of brain endothelium by pneumococcal neuraminidase NanA promotes bacterial internalization. Cell Microbiol 12:1576–1588. https://doi.org/10.1111/j.1462-5822.2010.01490.x
Banerjee A, Kim BJ, Carmona EM et al (2011a) Bacterial Pili exploit integrin machinery to promote immune activation and efficient blood-brain barrier penetration. Nat Commun 2:462. https://doi.org/10.1038/ncomms1474
Banerjee A, Kim BJ, Carmona EM et al (2011b) Bacterial Pili exploit integrin machinery to promote immune activation and efficient blood-brain barrier penetration. Nat Commun. https://doi.org/10.1038/ncomms1474
Barichello T, dos Santos I, Savi GD et al (2010) TNF-α, IL-1β, IL-6, and cinc-1 levels in rat brain after meningitis induced by Streptococcus pneumoniae. J Neuroimmunol 221:42–45. https://doi.org/10.1016/j.jneuroim.2010.02.009
Barichello T, Lemos JC, Generoso JS et al (2011a) Oxidative stress, cytokine/chemokine and disruption of blood-brain barrier in neonate rats after meningitis by streptococcus agalactiae. Neurochem Res 36:1922–1930. https://doi.org/10.1007/s11064-011-0514-2
Barichello T, Pereira JS, Savi GD et al (2011b) A kinetic study of the cytokine/chemokines levels and disruption of blood–brain barrier in infant rats after pneumococcal meningitis. J Neuroimmunol 233:12–17. https://doi.org/10.1016/j.jneuroim.2010.10.035
Barichello T, Generoso JS, Collodel A et al (2012a) Pathophysiology of acute meningitis caused by Streptococcus pneumoniae and adjunctive therapy approaches. Arq Neuropsiquiatr 70:366–372. https://doi.org/10.1590/S0004-282X2012000500011
Barichello T, Generoso JS, Silvestre C et al (2012b) Circulating concentrations, cerebral output of the CINC-1 and blood-brain barrier disruption in Wistar rats after pneumococcal meningitis induction. Eur J Clin Microbiol Infect Dis 31:2005–2009. https://doi.org/10.1007/s10096-011-1533-2
Baucells JB, Hally MM et al (2016) Probiotic associations in the prevention of necrotising enterocolitis and the reduction of late-onset sepsis and neonatal mortality in preterm infants under 1500 g. A systematic review. An Pediatr (Barc)JFigueras Aloy 85:247–255. https://doi.org/10.1016/j.anpede.2015.07.021
Bebbington C, Yarranton G (2008) Antibodies for the treatment of bacterial infections: current experience and future prospects. Curr Opin Biotechnol 19:613–619
Bell LM, Alpert G, Campos JM, Plotkin SA (1985) Routine quantitative blood cultures in children with Haemophilus influenzae or Streptococcus pneumoniae bacteremia. Pediatrics 76:901–904
Biegel D, Spencer DD, Pachter JS (1995) Isolation and culture of human brain microvessel endothelial cells for the study of blood-brain barrier properties in vitro. Brain Res 692:183–189. https://doi.org/10.1016/0006-8993(95)00511-N
Brito MA, Palmela I, Cardoso FL et al (2014) Blood–brain barrier and bilirubin: clinical aspects and experimental data. Arch Med Res 45:660–676
Brouwer MC, Tunkel AR, Van De Beek D (2010) Epidemiology, diagnosis, and antimicrobial treatment of acute bacterial meningitis. Clin Microbiol Rev 23:467–492
Carrano A, Hoozemans JJM, van der Vies SM et al (2011) Amyloid beta induces oxidative stress-mediated blood–brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal 15:1167–1178. https://doi.org/10.1089/ars.2011.3895
Chiang Y-C, Liao W-W, Fan C-M et al (2008) PCR detection of Staphylococcal enterotoxins (SEs) N, O, P, Q, R, U, and survey of SE types in Staphylococcus aureus isolates from food-poisoning cases in Taiwan. Int J Food Microbiol 121:66–73. https://doi.org/10.1016/j.ijfoodmicro.2007.10.005
Cho JS, Pietras EM, Garcia NC et al (2010) IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest 120:1762–1773. https://doi.org/10.1172/JCI40891
Chung MC, Popova TG, Jorgensen SC et al (2008) Degradation of circulating von Willebrand factor and its regulator ADAMTS13 implicates secreted Bacillus anthracis metalloproteases in anthrax consumptive coagulopathy. J Biol Chem 283:9531–9542. https://doi.org/10.1074/jbc.M705871200
Chung MC, Jorgensen SC, Popova TG et al (2009) Activation of plasminogen activator inhibitor implicates protease InhA in the acute-phase response to Bacillus anthracis infection. J Med Microbiol 58:737–744. https://doi.org/10.1099/jmm.0.007427-0
Coureuil M, Mikaty G, Miller F et al (2009) Meningococcal type IV pili recruit the polarity complex to cross the brain endothelium. Science 325:83–87. https://doi.org/10.1126/science.1173196
Coureuil M, Lécuyer H, Scott MGH et al (2010) Meningococcus hijacks a β2-adrenoceptor/β-arrestin pathway to cross brain microvasculature endothelium. Cell 143:1149–1160. https://doi.org/10.1016/j.cell.2010.11.035
Crone C, Olesen SP (1982) Electrical resistance of brain microvascular endothelium. Brain Res 241:49–55. https://doi.org/10.1016/0006-8993(82)91227-6
Cundell DR, Gerard NP, Gerard C et al (1995) Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature 377:435–438. https://doi.org/10.1038/377435a0
Danne C, Dramsi S (2012) Pili of Gram-positive bacteria: roles in host colonization. Res Microbiol 163:645–658. https://doi.org/10.1016/j.resmic.2012.10.012
Davoust N, Vuaillat C, Androdias G, Nataf S (2008) From bone marrow to microglia: barriers and avenues. Trends Immunol 29:227–234
De Lange ECM (2004) Potential role of ABC transporters as a detoxification system at the blood-CSF barrier. Adv Drug Deliv Rev 56:1793–1809
Dinarello CA (2005) Interleukin-1. Crit Care Med 33:S460–S462
Doran KS, Liu GY, Nizet V (2003) Group B streptococcal β-hemolysin/cytolysin activates neutrophil signaling pathways in brain endothelium and contributes to development of meningitis. J Clin Invest 112:736–744. https://doi.org/10.1172/JCI200317335
Doran KS, Engelson EJ, Khosravi A et al (2005) Blood-brain barrier invasion by group B. Streptococcus depends upon proper cell-surface anchoring of lipoteichoic acid. J Clin Invest 115:2499–2507. https://doi.org/10.1172/JCI23829
Doran KS, Fulde M, Gratz N et al (2016) Host–pathogen interactions in bacterial meningitis. Acta Neuropathol 131:185–209
Drevets DA, Leenen PJM, Greenfield RA (2004) Invasion of the central nervous system by intracellular bacteria. Clin Microbiol Rev 17:323–347
Ebrahimi CM, Kern JW, Sheen TR et al (2009) Penetration of the blood–brain barrier by Bacillus anthracis requires the pXO1-encoded BslA protein. J Bacteriol 191:7165–7173. https://doi.org/10.1128/JB.00903-09
Ebrahimi CM, Sheen TR, Renken CW et al (2011) Contribution of lethal toxin and edema toxin to the pathogenesis of anthrax meningitis. Infect Immun 79:2510–2518. https://doi.org/10.1128/IAI.00006-11
Edmond KM, Kortsalioudaki C, Scott S et al (2012) Group B streptococcal disease in infants aged younger than 3 months: systematic review and meta-analysis. Lancet 379:547–556. https://doi.org/10.1016/S0140-6736(11)61651-6
Edwards MS, Baker CJ (2005) Group B streptococcal infections in elderly adults. Clin Infect Dis an Off Publ Infect Dis Soc Am 41:839–847. https://doi.org/10.1086/432804
Er TK, Cheng BH, Ginés MÁR (2009) Importance of cerebrospinal fluid analysis in stat laboratory. Am J Emerg Med. https://doi.org/10.1016/j.ajem.2008.10.022
Esen N, Wagoner G, Philips N (2010) Evaluation of capsular and acapsular strains of S. aureus in an experimental brain abscess model. J Neuroimmunol 218:83–93. https://doi.org/10.1016/j.jneuroim.2009.10.006
Ferrieri P, Burke B, Nelson J (1980) Production of bacteremia and meningitis in infant rats with group B streptococcal serotypes. Infect Immun 27:1023–1032
Fida NM, Al-Mughales J, Farouq M (2006) Interleukin-1alpha, interleukin-6 and tumor necrosis factor-alpha levels in children with sepsis and meningitis. Pediatr Int 48:118–124. https://doi.org/10.1111/j.1442-200X.2006.02152.x
Foster TJ, Geoghegan JA, Ganesh VK, Höök M (2014) Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. NatRevMicrobiol 12:49–62. https://doi.org/10.1038/nrmicro3161
Furuse M, Fujita K, Hiiragi T et al (1998) Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 141:1539–1550. https://doi.org/10.1083/jcb.141.7.1539
Gouin E, Adib-Conquy M, Balestrino D et al (2010) The Listeria monocytogenes InlC protein interferes with innate immune responses by targeting the I{kappa}B kinase subunit IKK{alpha}. Proc Natl Acad Sci USA 107:17333–17338. https://doi.org/10.1073/pnas.1007765107
Gozes Y, Moayeri M, Wiggins JF, Leppla SH (2006) Anthrax lethal toxin induces ketotifen-sensitive intradermal vascular leakage in certain inbred mice. Infect Immun 74:1266–1272. https://doi.org/10.1128/IAI.74.2.1266-1272.2006
Gray SJ, Trotter CL, Ramsay ME et al (2006) Epidemiology of meningococcal disease in England and Wales 1993/94 to 2003/04: contribution and experiences of the meningococcal reference unit. J Med Microbiol 55:887–896. https://doi.org/10.1099/jmm.0.46288-0
Greiffenberg L, Goebel W, Kim KS et al (1998) Interaction of Listeria monocytogenes with human brain microvascular endothelial cells: InlB-dependent invasion, long-term intracellular growth, and spread from macrophages to endothelial cells. Infect Immun 66:5260–5267
Greiffenberg L, Goebel W, Kim KS et al (2000) Interaction of Listeria monocytogenes with human brain microvascular endothelial cells: an electron microscopic study. Infect Immun 68:3275–3279. https://doi.org/10.1128/IAI.68.6.3275-3279.2000
Gründler T, Quednau N, Stump C et al (2013) The surface proteins InlA and InlB are interdependently required for polar basolateral invasion by Listeria monocytogenes in a human model of the blood-cerebrospinal fluid barrier. Microbes Infect 15:291–301. https://doi.org/10.1016/j.micinf.2012.12.005
Gruol DL, Nelson TE (1997) Physiological and pathological roles of interleukin-6 in the central nervous system. Mol Neurobiol 15:307–339. https://doi.org/10.1007/BF02740665
Guillemet E, Cadot C, Tran SL et al (2010) The InhA metalloproteases of Bacillus cereus contribute concomitantly to virulence. J Bacteriol 192:286–294. https://doi.org/10.1128/JB.00264-09
Hawkins BT, Davis TP (2005) The blood–brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57:173–185. https://doi.org/10.1124/pr.57.2.4
Heninger M, Collins KA (2013) Acute bacterial meningitis with coincident methamphetamine use: a case report and review of the literature. J Forensic Sci 58:1088–1091. https://doi.org/10.1111/1556-4029.12176
Hirst RA, Kadioglu A, O’Callaghan C, Andrew PW (2004) The role of pneumolysin in pneumococcal pneumonia and meningitis. Clin Exp Immunol 138:195–201
Howard M, O’Garra A (1992) Biological properties of interleukin 10. Immunol Today 13:198–200. https://doi.org/10.1016/0167-5699(92)90153-X
Huang W-H, Hung P-K (2006) Methicillin-resistant Staphylococcus aureus infections in acute rhinosinusitis. Laryngoscope 116:288–291. https://doi.org/10.1097/01.mlg.0000197316.36698.c4
Huang SH, Wass C, Fu Q et al (1995) Escherichia coli invasion of brain microvascular endothelial cells in vitro and in vivo: molecular cloning and characterization of invasion gene ibe10. Infect Immun 63:4470–4475
Hudault S, Guignot J, Servin AL (2001) Escherichia coli strains colonising the gastrointestinal tract protect germfree mice against Salmonella typhimurium infection. Gut 49:47–55. https://doi.org/10.1136/gut.49.1.47
Ichiyama T, Isumi H, Yoshitomi T et al (2002) NF-kappaB activation in cerebrospinal fluid cells from patients with meningitis. Neurol Res 24:709–712. https://doi.org/10.1179/016164102101200627
Inglesby TV, O’Toole T, Henderson D et al (2002) Anthrax as a biological weapon, 2002. JAMA 287:2236. https://doi.org/10.1001/jama.287.17.2236
Iovino F, Brouwer MC, van de Beek D et al (2013a) Signalling or binding: the role of the platelet-activating factor receptor in invasive pneumococcal disease. Cell Microbiol 15:870–881
Iovino F, Orihuela CJ, Moorlag HE et al (2013b) Interactions between blood-borne Streptococcus pneumoniae and the blood–brain barrier preceding meningitis. PLoS ONE. https://doi.org/10.1371/journal.pone.0068408
Iovino F, Molema G, Bijlsma JJE (2014) Platelet endothelial cell adhesion molecule-1, a putative receptor for the adhesion of Streptococcus pneumoniae to the vascular endothelium of the blood-brain barrier. Infect Immun 82:3555–3566. https://doi.org/10.1128/IAI.00046-14
Iovino F, Seinen J, Henriques-Normark B, van Dijl JM (2016) How does Streptococcus pneumoniae invade the brain? Trends Microbiol 24:307–315. https://doi.org/10.1016/j.tim.2015.12.012
Iovino F, Engelen-Lee J-Y, Brouwer M et al (2017) pIgR and PECAM-1 bind to pneumococcal adhesins RrgA and PspC mediating bacterial brain invasion. J Exp Med 214:1619–1630. https://doi.org/10.1084/jem.20161668
Janoff EN, Fasching C, Orenstein JM et al (1999) Killing of Streptococcus pneumoniae by capsular polysaccharide-specific polymeric IgA, complement, and phagocytes. J Clin Invest 104:1139–1147. https://doi.org/10.1172/JCI6310
Join-Lambert O, Morand PC, Carbonnelle E et al (2010) Mechanisms of meningeal invasion by a bacterial extracellular pathogen, the example of Neisseria meningitidis. Prog Neurobiol 91:130–139
Kastrup CJ, Boedicker JQ, Pomerantsev AP et al (2008) Spatial localization of bacteria controls coagulation of human blood by “quorum acting”. Nat Chem Biol 4:742–750. https://doi.org/10.1038/nchembio.124
Kayal S, Charbit A (2006) Listeriolysin O: a key protein of Listeria monocytogenes with multiple functions. FEMS Microbiol Rev 30:514–529
Kayal S, Lilienbaum A, Join-Lambert O et al (2002) Listeriolysin O secreted by Listeria monocytogenes induces NF-kappaB signalling by activating the IkappaB kinase complex. Mol Microbiol 44:1407–1419
Kebir H, Kreymborg K, Ifergan I et al (2007) Human TH17 lymphocytes promote blood–brain barrier disruption and central nervous system inflammation. Nat Med 13:1173–1175. https://doi.org/10.1038/nm1651
Kenneth JR, Ray CG (2004) Sherris Medical Microbiology
Kern J, Schneewind O (2010) BslA, the S-layer adhesin of B. anthracis, is a virulence factor for anthrax pathogenesis. Mol Microbiol 75:324–332. https://doi.org/10.1111/j.1365-2958.2009.06958.x
Khan NA, Wang Y, Kim KJ et al (2002) Cytotoxic necrotizing factor-1 contributes to Escherichia coli K1 invasion of the central nervous system. J Biol Chem 277:15607–15612. https://doi.org/10.1074/jbc.M112224200
Kielian T, Hickey WF (2000) Proinflammatory cytokine, chemokine, and cellular adhesion molecule expression during the acute phase of experimental brain abscess development. Am J Pathol 157:647–658. https://doi.org/10.1016/S0002-9440(10)64575-0
Kim KS (2003) Pathogenesis of bacterial meningitis: from bacteraemia to neuronal injury. Nat Rev Neurosci 4:376. https://doi.org/10.1038/nrn1103
Kim KS (2008) Mechanisms of microbial traversal of the blood–brain barrier. Nat Rev Microbiol 6:625–634. https://doi.org/10.1038/nrmicro1952
Kim KJ, Chung JW, Kim KS (2005) 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J Biol Chem 280:1360–1368. https://doi.org/10.1074/jbc.M410176200
Kniesel U, Wolburg H (2000) Tight junctions of the blood-brain barrier. Cell Mol Neurobiol 20:57–76. https://doi.org/10.1023/A:1006995910836
Koedel U, Bernatowicz A, Frei K et al (1996) Systemically (but not intrathecally) administered IL-10 attenuates pathophysiologic alterations in experimental pneumococcal meningitis. J Immunol 157:5185–5191
Koedel U, Scheld WM, Pfister HW (2002) Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis 2:721–736
Kolberg J, Høiby EA, Jantzen E (1997) Detection of the phosphorylcholine epitope in streptococci, Haemophilus and pathogenic Neisseriae by immunoblotting. Microb Pathog 22:321–329. https://doi.org/10.1006/mpat.1996.0114
Konsman JP, Drukarch B, Van Dam A-M (2007) (Peri)vascular production and action of pro-inflammatory cytokines in brain pathology. Clin Sci 112:1–25. https://doi.org/10.1042/CS20060043
Kornelisse RF, Savelkoul HF, Mulder PH et al (1996) Interleukin-10 and soluble tumor necrosis factor receptors in cerebrospinal fluid of children with bacterial meningitis. J Infect Dis 173:1498–1502
Kronfol Z (2000) Cytokines and the brain: implications for clinical psychiatry. Am J Psychiatry 157:683–694. https://doi.org/10.1176/appi.ajp.157.5.683
Kyaw MH, Christie P, Jones IG, Campbell H (2002) The changing epidemiology of bacterial meningitis and invasive non-meningitic bacterial disease in Scotland during the period 1983–1999. Scand J Infect Dis 34:289–298. https://doi.org/10.1080/00365540110080403
Leclercq SY, Sullivan MJ, Ipe DS et al (2016) Pathogenesis of Streptococcus urinary tract infection depends on bacterial strain and β-hemolysin/cytolysin that mediates cytotoxicity, cytokine synthesis, inflammation and virulence. Sci Rep. https://doi.org/10.1038/srep29000
Lehner MD, Schwoebel F, Kotlyarov A et al (2002) Mitogen-activated protein kinase-activated protein kinase 2-deficient mice show increased susceptibility to Listeria monocytogenes infection. J Immunol 168:4667–4673
Leib SL, Kim YS, Black SM et al (1998) Inducible nitric oxide synthase and the effect of aminoguanidine in experimental neonatal meningitis. J Infect Dis 177:692–700
Lembo A, Gurney MA, Burnside K et al (2010) Regulation of CovR expression in Group B Streptococcus impacts blood-brain barrier penetration. Mol Microbiol 77:431–443. https://doi.org/10.1111/j.1365-2958.2010.07215.x
Levin VA (1980) Relationship of octanol/water partition coefficient and molecular weight to rat brain capillary permeability. J Med Chem 23:682–684. https://doi.org/10.1021/jm00180a022
Maisey HC, Hensler M, Nizet V, Doran KS (2007) Group B streptococcal pilus proteins contribute to adherence to and invasion of brain microvascular endothelial cells. In: Journal of Bacteriology. pp 1464–1467
Manarey CRA, Anand VK, Huang C (2004) Incidence of methicillin-resistant staphylococcus aureus causing chronic rhinosinusitis. Laryngoscope 114:939–941
Marriott H, Mitchell T, Dockrell D (2008) Pneumolysin: a double-edged sword during the host–pathogen interaction. Curr Mol Med 8:497–509. https://doi.org/10.2174/156652408785747924
Martìn-Padura I, Lostaglio S, Schneemann M et al (1998) Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol 142:117–127. https://doi.org/10.1083/jcb.142.1.117
Mayhan WG (2000) Nitric oxide donor-induced increase in permeability of the blood-brain barrier. Brain Res 866:101–108. https://doi.org/10.1016/S0006-8993(00)02254-X
McLoughlin A, Rochfort KD, McDonnell CJ et al (2017) Staphylococcus aureus-mediated blood–brain barrier injury: an in vitro human brain microvascular endothelial cell model. Cell Microbiol. https://doi.org/10.1111/cmi.12664
Melican K, Dumenil G (2012) Vascular colonization by Neisseria meningitidis. Curr Opin Microbiol 15:50–56
Miller SD, McMahon EJ, Schreiner B, Bailey SL (2007) Antigen presentation in the CNS by myeloid dendritic cells drives progression of relapsing experimental autoimmune encephalomyelitis. Ann NY Acad Sci 1103:179–191
Miric D, Katanic R, Kisic B et al (2010) Oxidative stress and myeloperoxidase activity during bacterial meningitis: effects of febrile episodes and the BBB permeability. Clin Biochem 43:246–252. https://doi.org/10.1016/j.clinbiochem.2009.09.023
Mittal R, Prasadarao NV (2010) Nitric oxide/cGMP signalling induces Escherichia coli K1 receptor expression and modulates the permeability in human brain endothelial cell monolayers during invasion. Cell Microbiol 12:67–83. https://doi.org/10.1111/j.1462-5822.2009.01379.x
Møller K, Tofteng F, Qvist T et al (2005) Cerebral output of cytokines in patients with pneumococcal meningitis. Crit Care Med 33:979–983. https://doi.org/10.1097/01.CCM.0000162494.84354.9D
Moreillon P, Majcherczyk PA (2003) Proinflammatory activity of cell-wall constituents from Gram-positive bacteria. Scand J Infect Dis 35:632–641. https://doi.org/10.1080/00365540310016259
Moura RP, Almeida A, Sarmento B (2017) The role of non-endothelial cells on the penetration of nanoparticles through the blood–brain barrier. Prog Neurobiol 159:39–49. https://doi.org/10.1016/j.pneurobio.2017.09.001
Mukherjee DV, Tonry JH, Kim KS et al (2011) Bacillus anthracis protease InhA increases blood–brain barrier permeability and contributes to cerebral hemorrhages. PLoS ONE. https://doi.org/10.1371/journal.pone.0017921
Murawska-Cialowicz E, Szychowska Z, Tr busiewicz B (2000) Nitric oxide production during bacterial and viral meningitis in children. Int J Clin Lab Res 30:127–131
Nagesh Babu G, Kumar A, Kalita J, Misra UK (2008) Proinflammatory cytokine levels in the serum and cerebrospinal fluid of tuberculous meningitis patients. Neurosci Lett 436:48–51. https://doi.org/10.1016/j.neulet.2008.02.060
Nandi A, Dey S, Biswas J et al (2015) Differential induction of inflammatory cytokines and reactive oxygen species in murine peritoneal macrophages and resident fresh bone marrow cells by acute Staphylococcus aureus infection: contribution of toll-like receptor 2 (TLR2). Inflammation 38:224–244. https://doi.org/10.1007/s10753-014-0026-8
Nasdala I, Wolburg-Buchholz K, Wolburg H et al (2002) A transmembrane tight junction protein selectively expressed on endothelial cells and platelets. J Biol Chem 277:16294–16303. https://doi.org/10.1074/jbc.M111999200
Nizet V, Kim KS, Stins M et al (1997) Invasion of brain microvascular endothelial cells by group B streptococci. Infect Immun 65:5074–5081
Opitz B, Püschel A, Beermann W et al (2006) Listeria monocytogenes activated p38 MAPK and induced IL-8 secretion in a nucleotide-binding oligomerization domain 1-dependent manner in endothelial cells. J Immunol 176:484–490. https://doi.org/10.4049/jimmunol.176.1.484
Orihuela CJ, Mahdavi J, Thornton J et al (2009) Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J Clin Invest 119:1638–1646. https://doi.org/10.1172/JCI36759
Østergaard C, Brandt C, Konradsen HB, Samuelsson S (2004) Differences in survival, brain damage, and cerebrospinal fluid cytokine kinetics due to meningitis caused by 3 different Streptococcus pneumoniae serotypes: evaluation in humans and in 2 experimental models. J Infect Dis 190:1212–1220. https://doi.org/10.1086/423852
Parida SK, Domann E, Ronde M et al (1998) Internalin B is essential for adhesion and mediates the invasion of Listera monocytognes into human endothelial cells. Mol Microbiol 28:81–93. https://doi.org/10.1046/j.1365-2958.1998.00776.x
Parizzi F, Garlaschi L, Assael BM et al (1991) Interleukin 6 activity in infants and children with bacterial meningitis. the collaborative study on meningitis. Pediatr Infect Dis J 10:117–121
Park WB, Kim S-H, Cho JH et al (2007) Effect of salicylic acid on invasion of human vascular endothelial cells by Staphylococcus aureus. FEMS Immunol Med Microbiol 49:56–61
Patrick D, Betts J, Frey EA et al (1992) Haemophilus influenzae lipopolysaccharide disrupts confluent monolayers of bovine brain endothelial cells via a serum-dependent cytotoxic pathway. J Infect Dis 165:865–872. https://doi.org/10.1093/infdis/165.5.865
Paul R, Koedel U, Winkler F et al (2003) Lack of IL-6 augments inflammatory response but decreases vascular permeability in bacterial meningitis. Brain 126:1873–1882. https://doi.org/10.1093/brain/awg171
Peltola H (2000) Worldwide Haemophilus influenzae type b disease at the beginning of the 21st century: global analysis of the disease burden 25 years after the use of the polysaccharide vaccine and a decade after the advent of conjugates. Clin Microbiol Rev 13:302–317
Prasadarao NV (2002) Identification of Escherichia coli outer membrane protein A receptor on human brain microvascular endothelial cells. InfectImmun 70:4556–4563. https://doi.org/10.1128/IAI.70.8.4556
Prasadarao NV, Blom AM, Villoutreix BO, Linsangan LC (2002) A novel interaction of outer membrane protein A with C4b binding protein mediates serum resistance of Escherichia coli K1. J Immunol 169:6352–6360. https://doi.org/10.4049/jimmunol.169.11.6352
Privratsky JR, Newman PJ (2014) PECAM-1: regulator of endothelial junctional integrity. Cell Tissue Res 355:607–619
Quagliarello VJ, Long WJ, Scheld WM (1986) Morphologic alterations of the blood–brain barrier with experimental meningitis in the rat. Temporal sequence and role of encapsulation. J Clin Invest 77:1084–1095. https://doi.org/10.1172/JCI112407
Quagliarello VJ, Wispelwey B, Long WJ, Scheld WM (1991) Recombinant human interleukin-1 induces meningitis and blood-brain barrier injury in the rat: characterization and comparison with tumor necrosis factor. J Clin Invest 87:1360–1366. https://doi.org/10.1172/JCI115140
Radin JN, Orihuela CJ, Murti G et al (2005) β-arrestin 1 participates in platelet-activating factor receptor-mediated endocytosis of Streptococcus pneumoniae. Infect Immun 73:7827–7835. https://doi.org/10.1128/IAI.73.12.7827-7835.2005
Ramarao N, Lereclus D (2005) The InhA 1 metalloprotease allows spores of the B. cereus group to escape macrophages. Cell Microbiol 7:1357–1364. https://doi.org/10.1111/j.1462-5822.2005.00562.x
Ring A, Weiser JN, Tuomanen EI (1998) Pneumococcal trafficking across the blood–brain barrier molecular analysis of a novel bidirectional pathway. J Clin Invest 102:347–360. https://doi.org/10.1172/JCI2406
Rochfort KD, Cummins PM (2015) Cytokine-mediated dysregulation of zonula occludens-1 properties in human brain microvascular endothelium. Microvasc Res 100:48–53. https://doi.org/10.1016/j.mvr.2015.04.010
Rochfort KD, Collins LE, McLoughlin A, Cummins PM (2016) Tumour necrosis factor-α-mediated disruption of cerebrovascular endothelial barrier integrity in vitro involves the production of proinflammatory interleukin-6. J Neurochem 136:564–572. https://doi.org/10.1111/jnc.13408
Rosa-Fraile M, Dramsi S, Spellerberg B (2014) Group B streptococcal haemolysin and pigment, a tale of twins. FEMS Microbiol Rev 38:932–946. https://doi.org/10.1111/1574-6976.12071
Rosenberg GA, Estrada EY, Dencoff JE, Stetler-Stevenson WG (1995) Tumor necrosis factor-α-induced gelatinase B causes delayed opening of the blood–brain barrier: an expanded therapeutic window. Brain Res 703:151–155. https://doi.org/10.1016/0006-8993(95)01089-0
Saez-Llorens X, Jafari HS, Severien C et al (1991) Enhanced attenuation of meningeal inflammation and brain edema by concomitant administration of anti-CD18 monoclonal antibodies and dexamethasone in experimental Haemophilus meningitis. J Clin Invest 88:2003–2011. https://doi.org/10.1172/JCI115527
Schmeck B, Beermann W, van Laak V et al (2005) Intracellular bacteria differentially regulated endothelial cytokine release by MAPK-dependent histone modification. J Immunol 175:2843–2850
Schubert-Unkmeir A, Konrad C, Slanina H et al (2010) Neisseria meningitidis induces brain microvascular endothelial cell detachment from the matrix and cleavage of occludin: a role for MMP-8. PLoS Pathog 6:e1000874. https://doi.org/10.1371/journal.ppat.1000874
Schuchat a, Robinson K, Wenger JD et al (1997) Bacterial meningitis in the United States in 1995. Active surveillance team. N Engl J Med 337:970–976. https://doi.org/10.1056/NEJM199710023371404
Sellner J, Täuber MG, Leib SL (2010) Pathogenesis and pathophysiology of bacterial CNS infections. Handb Clin Neurol 96:1–16. https://doi.org/10.1016/S0072-9752(09)96001-8
Seo HS, Mu R, Kim BJ et al (2012) Binding of glycoprotein Srr1 of Streptococcus agalactiae to fibrinogen promotes attachment to brain endothelium and the development of meningitis. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1002947
Sheen TR, Ebrahimi CM, Hiemstra IH et al (2010) Penetration of the blood–brain barrier by Staphylococcus aureus: contribution of membrane-anchored lipoteichoic acid. J Mol Med 88:633–639. https://doi.org/10.1007/s00109-010-0630-5
Sokolova O, Heppel N, Jägerhuber R et al (2004) Interaction of Neisseria meningitidis with human brain microvascular endothelial cells: Role of MAP- and tyrosine kinases in invasion and inflammatory cytokine release. Cell Microbiol 6:1153–1166. https://doi.org/10.1111/j.1462-5822.2004.00422.x
St Geme 3rd JW, Cutter D (1996) Influence of pili, fibrils, and capsule on in vitro adherence by Haemophilus influenzae type b. Mol Microbiol 21:21–31
St. Geme JW, Cutter D (1995) Evidence that surface fibrils expressed by Haemophilus influenzae type b promote attachment to human epithelial cells. Mol Microbiol 15:77–85
Stephens DS (1999) Uncloaking the meningococcus: dynamics of carriage and disease. Lancet 353:941
Stephens DS (2007) Conquering the meningococcus. FEMS Microbiol Rev 31:3–14
Sukumaran SK, Prasadarao NV (2003) Escherichia coli K1 invasion increases human brain microvascular endothelial cell monolayer permeability by disassembling vascular-endothelial cadherins at tight junctions. J Infect Dis 188:1295–1309. https://doi.org/10.1086/379042
Swords WE, Ketterer MR, Shao J et al (2001) Binding of the non-typeable Haemophilus influenzae lipooligosaccharide to the PAF receptor initiates host cell signalling. Cell Microbiol 3:525–536. https://doi.org/10.1046/j.1462-5822.2001.00132.x
Tang P, Rosenshine I, Finlay BB (1994) Listeria monocytogenes, an invasive bacterium, stimulates MAP kinase upon attachment to epithelial cells. Mol Biol Cell 5:455–464
Tang P, Sutherland CL, Gold MR, Finlay BB (1998) Listeria monocytogenes invasion of epithelial cells requires the MEK-1/ERK-2 mitogen-activated protein kinase pathway. Infect Immun 66:1106–1112
Tazi A, Disson O, Bellais S et al (2010) The surface protein HvgA mediates group B streptococcus hypervirulence and meningeal tropism in neonates. J Exp Med 207:2313–2322. https://doi.org/10.1084/jem.20092594
Tenaillon O, Skurnik D, Picard B, Denamur E (2010) The population genetics of commensal Escherichia coli. NatRevMicrobiol 8:207–217. https://doi.org/10.1038/nrmicro2298
Teng CH, Cai M, Shin S et al (2005) Escherichia coli K1 RS218 interacts with human brain microvascular endothelial cells via type 1 fimbria bacteria in the fimbriated state. Infect Immun 73:2923–2931. https://doi.org/10.1128/IAI.73.5.2923-2931.2005
Tsukita S, Furuse M, Itoh M (2001) Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol 2:285–293. https://doi.org/10.1038/35067088
Tuomanen EI, Saukkonen K, Sande S et al (1989) Reduction of inflammation, tissue damage, and mortality in bacterial meningitis in rabbits treated with monoclonal antibodies against adhesion-promoting receptors of leukocytes. J Exp Med 170:959–969
Uchiyama S, Carlin AF, Khosravi A et al (2009) The surface-anchored NanA protein promotes pneumococcal brain endothelial cell invasion. J Exp Med 206:1845–1852. https://doi.org/10.1084/jem.20090386
van Sorge NM, Doran KS (2012) Defense at the border: the blood–brain barrier versus bacterial foreigners. Future Microbiol 7:383–394. https://doi.org/10.2217/fmb.12.1
van Sorge NM, Ebrahimi CM, McGillivray SM et al (2008) Anthrax toxins inhibit neutrophil signaling pathways in brain endothelium and contribute to the pathogenesis of meningitis. PLoS ONE. https://doi.org/10.1371/journal.pone.0002964
van Sorge NM, Quach D, Gurney M et al (2009) The group B streptococcal serine-rich repeat 1 glycoprotein mediates penetration of the blood–brain barrier. J Infect Dis 199:1479–1487. https://doi.org/10.1086/598217
van de Beek D, de Gans J, Tunkel AR, Wijdicks EFM (2006) Community-acquired Bacterial meningitis in adults. N Engl J Med 354:44–53. https://doi.org/10.1093/qjmed/hcl131
van der Poll T, Keogh CV, Guirao X et al (1997) Interleukin-6 gene-deficient mice show impaired defense against Pneumococcal pneumonia. J Infect Dis 176:439–444. https://doi.org/10.1086/514062
Vázquez-Boland JA, Kuhn M, Berche P et al (2001) Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev 14:584–640
Virji M (2009) Pathogenic neisseriae: surface modulation, pathogenesis and infection control. Nat Rev Microbiol 7:274–286
Vogt RL, Dippold L (2005) Escherichia coli O157:H7 outbreak associated with consumption of ground beef, June–July 2002. Public Health Rep 120:174–178. https://doi.org/10.1177/003335490512000211
Wang J, Sun L, Si YF, Li BM (2012) Overexpression of actin-depolymerizing factor blocks oxidized low-density lipoprotein-induced mouse brain microvascular endothelial cell barrier dysfunction. Mol Cell Biochem 371:1–8. https://doi.org/10.1007/s11010-012-1415-7
Warfel JM, Steele AD, D’Agnillo F (2005) Anthrax lethal toxin induces endothelial barrier dysfunction. Am J Pathol 166:1871–1881. https://doi.org/10.1016/S0002-9440(10)62496-0
Watt JP, Wolfson LJ, O’Brien KL et al (2009) Burden of disease caused by Haemophilus influenzae type b in children younger than 5 years: global estimates. Lancet 374:903–911. https://doi.org/10.1016/S0140-6736(09)61203-4
Weiser JN, Pan N, McGowan KL et al (1998) Phosphorylcholine on the lipopolysaccharide of Haemophilus influenzae contributes to persistence in the respiratory tract and sensitivity to serum killing mediated by C-reactive protein. J Exp Med 187:631–640. https://doi.org/10.1084/jem.187.4.631
Weiss N, Miller F, Cazaubon S, Couraud PO (2009) The blood–brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta 1788:842–857
Wellmer A, Zysk G, Gerber J et al (2002) Decreased virulence of a pneumolysin-deficient strain of Streptococcus pneumoniae in murine meningitis. Infect Immun 70:6504–6508. https://doi.org/10.1128/IAI.70.11.6504-6508.2002
Whidbey C, Harrell MI, Burnside K et al (2013) A hemolytic pigment of Group B Streptococcus allows bacterial penetration of human placenta. J Exp Med 210:1265–1281. https://doi.org/10.1084/jem.20122753
Whidbey C, Vornhagen J, Gendrin C et al (2015) A streptococcal lipid toxin induces membrane permeabilization and pyroptosis leading to fetal injury. EMBO Mol Med 7:488–505. https://doi.org/10.15252/emmm.201404883
Winkler F, Koedel U, Kastenbauer S, Pfister H-W (2001) Differential expression of nitric oxide synthases in bacterial meningitis: role of the inducible isoform for blood–brain barrier breakdown. J Infect Dis 183:1749–1759. https://doi.org/10.1086/320730
Wolburg H, Lippoldt A (2002) Tight junctions of the blood–brain barrier: development, composition and regulation. Vascul Pharmacol 38:323–337
Wolburg H, Wolburg-Buchholz K, Engelhardt B (2005) Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol 109:181–190. https://doi.org/10.1007/s00401-004-0928-x
Yadav A, Malik GK, Trivedi R et al (2009) Correlation of CSF neuroinflammatory molecules with leptomeningeal cortical subcortical white matter fractional anisotropy in neonatal meningitis. Magn Reson Imaging 27:214–221. https://doi.org/10.1016/j.mri.2008.06.010
Zhang JR, Mostov KE, Lamm ME et al (2000) The polymeric immunoglobulin receptor translocates pneumococci across human nasopharyngeal epithelial cells. Cell 102:827–837. https://doi.org/10.1016/S0092-8674(00)00071-4
Zhou Y, Tao J, Yu H et al (2012) Hcp family proteins secreted via the type VI secretion system coordinately regulate Escherichia coli K1 interaction with human brain microvascular endothelial cells. Infect Immun 80:1243–1251. https://doi.org/10.1128/IAI.05994-11
Zwijnenburg PJG, Van der Poll T, Florquin S et al (2003) Interleukin-10 negatively regulates local cytokine and chemokine production but does not influence antibacterial host defense during murine Pneumococcal meningitis. Infect Immun 71:2276–2279. https://doi.org/10.1128/IAI.71.4.2276-2279.2003
Zysk G, Schneider-Wald BK, Hwang JH et al (2001) Pneumolysin is the main inducer of cytotoxicity to brain microvascular endothelial cells caused by Streptococcus pneumoniae. Infect Immun 69:845–852. https://doi.org/10.1128/IAI.69.2.845-852.2001
Acknowledgements
This study was supported by the fundamental research grant scheme, (Grant No. 5524924), and long term research grant scheme (Grant No. 5526406), Ministry of education Malaysia.
Author information
Authors and Affiliations
Contributions
MMJ designed the manuscript, searched the literatures and wrote the review. MNMD revised the review.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Al-Obaidi, M.M.J., Desa, M.N.M. Mechanisms of Blood Brain Barrier Disruption by Different Types of Bacteria, and Bacterial–Host Interactions Facilitate the Bacterial Pathogen Invading the Brain. Cell Mol Neurobiol 38, 1349–1368 (2018). https://doi.org/10.1007/s10571-018-0609-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-018-0609-2