
Abstract
Cellulose acetate is widely used in films, filters, textiles, lacquer and cosmetic products. Herein we demonstrate the production of cellulose esters under homogeneous conditions using 1,5-diazabicyclo[4.3.0]non-5-ene acetate ([DBNH][OAc]) as solvent. The reagents have been chosen such that the system is recyclable, i.e. by-products are low boiling and easy to remove. It is demonstrated that cellulose acetate can be synthesized with different degree of substitution (DS) values, and that some commonly used acylation regents, like vinyl carboxylates react well without additional base catalyst. Low to high DS values are possible with good recovery of high purity ionic liquid (IL). A linear correlation method of two separate methods, IR and 31P NMR, is proposed to reliably assess the DS of the products. The recyclability of the solvent is demonstrated by acetylating cellulose with isopropenyl acetate to high degree and regeneration into water. After regeneration of cellulose acetate from the IL with addition of water, the residual water was entrained using n-butanol to minimize hydrolysis of [DBNH][OAc], to allow for high recovery and high purity of the ionic liquid. Thus, an overall scheme for batch cellulose acetylation and recovery of [DBNH][OAc] from aqueous solutions is proposed.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cellulose acetate is one of the first synthetic industrial polymers, as the process for cellulose acetate production was patented already in 1894 (Cross et al. 1894). The industrial production of cellulose acetate is based on heterogeneous modification of cellulose using acetic anhydride (Ac2O) with sulfuric acid catalysis (Klemm et al. 1998), which affords cellulose triacetate (DS 3). Afterwards the product is hydrolyzed to desirable degree of substitution (DS). Extensive literature concerning the acetylation of cellulose has been published and more recently there are many articles concerning acetylation in ionic liquids, where cellulose acetate with different DS values and uniform distribution of substituents can be obtained directly (Barthel and Heinze 2006; Jogunola et al. 2016; Wu et al. 2004; Yang et al. 2014).
Ionic liquids (ILs) have gained considerable attention during recent years due to their unique solvation and physiochemical properties (Gericke et al. 2012). The chemical properties of ILs vary according to the composition. For instance, the ability to dissolve cellulose is often dependent on the basicity of the IL (Fukaya et al. 2006, 2008; King et al. 2012; Parviainen et al. 2013), but there are exceptions, e.g., tetraalkylphosphonium salts (Hauru et al. 2012; Holding et al. 2014), which are basic but require a dipolar aprotic co-solvent to dissolve cellulose. In previous literature, the use of imidazolium based ILs, with alkyl substituents (ethyl, methyl, and allyl) and chloride, bromide or acetate anions, typifies most publications on cellulose dissolving ILs (Heinze et al. 2008; Swatloski et al. 2002). In regard to developing commercial processes, the potential environmental impacts of ILs are also of concern. Several recent articles have also assessed the toxicity of different biomass dissolving ionic liquids. Those with shorter anion and cation alkyl chain length are considered to be of lower toxicity (Ruokonen et al. 2016; Witos et al. 2017). This also corresponds with the better capability to dissolve cellulose as cellulose dissolution capability typically decreases as cation or anion volume gets too large (Isik et al. 2014).
Recyclability of ionic liquids is an important and quite often overlooked aspect. One recycling approach for ILs is based on the use of more labile organic superbase-carboxylic acid conjugates. Above processing temperatures these conjugates dissociate to the neutral starting components, which allows for distillability of the IL. One of these that is of current interest is [DBNH][OAc], which can be recycled and purified by distillation to either pure IL or to its starting compounds (Parviainen et al. 2013, 2015). [DBNH][OAc] is a robust solvent for cellulose and pulp, recycled paper and even cotton, e.g., used cotton clothes. Further, high quality cellulosic textile fibers can be produced from cellulose [DBNH][OAc] solutions by spinning thin filaments into an antisolvent, as in IONCELL-F process (Michud et al. 2016; Sixta et al. 2015). Therefore, [DBNH][OAc] is also of interest as a reaction medium, as the same solvent and process could be used to obtain chemically modified cellulosic fibers or materials. The current Lyocell process, using NMMO, is limited due to the undesired side reactions (Rosenau et al. 2001).
The acetylation of cellulose has been studied in different ionic liquids with several esterification reagents. In the first report on the topic of acetylation in IL the reaction was carried out with Ac2O using 1-allyl-3-methyl-imidazolium chloride ([Amim]Cl) as solvent (Wu et al. 2004). Acetic acid is a side product, which can be difficult to remove from the IL solution and may also lead to partial hydrolysis (degradation) of the cellulose polymer, if some water is present. Barthel and Heinze (2006) and Hinner et al. (2016) have studied esterification of cellulose with longer fatty acid chlorides in ionic liquids, e.g., [Bmim]Cl and [Emim]Cl. Isopropenyl acetate (IpeAc) has been used in 1-ethyl-3-methylimidazolium acetate ([Emim][OAc]) to form cellulose acetates (Kakuchi et al. 2015). Notably, the side-product of the acetylation reaction with IpeAc is acetone which is easy to evaporate from the synthesis mixture and does not usually introduce side reactions. The same group have just recently published an article, where [Emim][OAc] and other imidazolium based ionic liquids were used in DMSO mixtures for assessment of the mechanistic effects of ILs (Kakuchi et al. 2017). Vinyl esters can also be used in cellulose esterification reactions, as an acylation reagent, but have been used in “traditional” solvents with catalysts or activators (Chen et al. 2016; Çetin et al. 2009; Heinze et al. 2003; Jebrane et al. 2011; Jebrane and Sebe 2007).
Binary systems consisting of IL and an organic co-solvent have also been used to produce cellulose esters. Typical co-solvents include dimethyl sulfoxide (DMSO) (Kakuchi et al. 2017; Xu et al. 2013) or dimethylacetamide (DMA) (Rinaldi 2011), but acetonitrile (ACN) (Nawaz et al. 2014) and acetone (Jogunola et al. 2016) have also been used. However, having an additional co-solvent may actually complicate the workup and recovery of all components. The use of acidic ILs like 1-vinyl-3-(3-sulfopropyl)imidazolium hydrogensulfate ([VSim]HSO4) has been suggested to increase the efficiency of the acetylation reaction in Ac2O, but the effect on product molecular weight was not reported (Tian et al. 2014) and anhydrides are already highly activated species.
Besides cellulose, acetylation of other biomass components has been of interest. As an example, efficient acetylation of hemicelluloses with Ac2O was achieved in [DBNH][OAc], as a slight excess of the base (DBN) can be used to scavenge the released acid thus avoiding possible hydrolytic reactions (Stepan et al. 2013).
In our studies, we have been concentrating on the organic superbase-derived ILs, as these are efficient solvents for cellulose and can be efficiently recycled. One of these is [DBNH][OAc], which is not only an efficient solvent for cellulose, but high-quality textile fibers can be produced upon regeneration of cellulose solutions into water (IONCELL-F process) (Michud et al. 2016; Parviainen et al. 2013, 2015; Sixta et al. 2015). Therefore, it would be highly beneficial to control the modification to cellulose esters before regeneration to fibers (for textile and non-wovens) or films. In the current work, we describe the reaction conditions for homogeneous esterification of pre-hydrolysis kraft (PHK) pulp, manufactured from Eucalyptus urograndis and microcrystalline cellulose (MCC) in [DBNH][OAc]. The recyclability of the IL after reaction, with mass balances and purity of recovered ionic liquid is also demonstrated in preliminary fashion.
Experimental
Materials
The starting materials were eucalyptus (Eucalyptus urograndis) PHK pulp with DPW 2300, DPn 313 (Bahia Specialty Cellulose, Brazil) and Avicel® PH-101 MCC with DPW 416 and DPn 112 from Sigma Aldrich. Before reactions PHK pulp was ground with a Bamix (bar blender) or Wiley mill, dried under vacuum and stored in a desiccator. All the reagents were used without further purification. 1.5-Diazabicyclo[4.3.0]non-5-ene (DBN) was obtained from Fluorochem and glacial acetic acid (AcOH) from Fisher Chemical. The following additional reagents were purchased from Sigma Aldrich: acetic anhydride (Ac2O), propionic anhydride, vinyl propionate (VinPr), isopropenyl acetate (IpeAc), 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (2-Cl-TMDP), pyridine (py), 4-(dimethylamino)pyridine, deuterated chloroform (CDCl3) and dimethyl sulfoxide-d6 (DMSO-d6), N,N-dimethylacetamide (DMA) and anhydrous lithium chloride (LiCl). Vinyl acetate (VinAc) was obtained from Supelco, Chromium (III) acetylacetonate (Cr(acac)3) from Fluka, endo-N-hydroxy-5-norborenene-2,3-dicarboximide (e-HNDI) from abcr GmbH and ethanol (96 and 99%) from Altia.
The ionic liquid [DBNH][OAc] was prepared from glacial acetic acid and DBN according to Parviainen et al. (2013) with slight modifications: The reaction was performed in a two necked flask with a reflux condenser, under argon atmosphere and stirred magnetically. The superbase DBN (168.5 g, 1.3 mol) was measured to the flask and one equivalent of acetic acid (81.5 g, 1.3 mol) was weighed to a dropping funnel. The reaction media was heated to 60 °C and acetic acid addition was made at a moderate pace (2 h addition time). A pale yellow liquid was formed and the mixture was left to settle at 70 °C for 1 h before cooling. The purity of the liquid was verified with 1H NMR in DMSO-d6.
Typical dissolution and esterification procedure
[DBNH][OAc] was melted (70 °C) and weighed (10 g) to the reaction vessel. Cellulose powder (0.5 g) was weighed and added to the solution. The mixture was stirred under argon atmosphere at 70 °C for 0.5–1 h, until cellulose had dissolved and the reaction medium was a clear viscous solution. The reaction temperature was 60–70 °C, slightly depending on the reactants boiling point and (presumed) reactivity. The acylation reagent IpeAc, VinAc, VinPr or Ac2O was added in one portion through a septum. The amount of acylation reagent depended on the desired obtainable DS being 0.25, 1, 3 or 9 eq’s per AGU. (see supporting information) When acetylation was performed with Ac2O, 1.01 eq of DBN was added prior to the acetylating reagent to scavenge the released acetic acid. The reactions were quenched with addition of 30 ml of ethanol (96%).
The precipitated reaction products were collected by suction filtration using Whatman 1 filter paper. The product was washed with ethanol in a Soxhlet apparatus overnight. The washed sample was filtered and pre-dried in a Büchner funnel, rinsed once with acetone to enhance the drying, and dried under vacuum overnight. The ethanol solution containing [DBNH][OAc] was collected and evaporated using a rotary evaporator. Final traces of the ethanol were removed under high vacuum on a Schlenk line.
Demonstration of IL recycling from aqueous solutions
For IL recycling demonstration from aqueous solutions, the reaction was stopped with addition of ethanol and the reaction product washed with distilled water. The liquid phases were combined. The removal of water and the purification of the IL was carried out by entraining using a rotary evaporator, with repeated additions of n-butanol. Finally, the fractional distillation of the recovered IL was performed in a Kugelrohr distillation apparatus, where two different temperatures (140 and 160 °C) were used to collect different fractions at 0.7 mbar pressure.
Analytical methods
Attenuated total reflectance infrared spectroscopy (ATR-IR)
FTIR spectra were collected (400–4000 cm−1) from solid samples with a Bruker alpha-P FTIR spectrometer with a diamond ATR. The ATR-IR -spectra were baseline corrected and normalized to the cellulose C–O–C backbone signal (1010 cm−1) intensity.
Gel permeation chromatography (GPC)
Molar mass determination was performed using Agilent Infinity 1260 LC-system including degasser, pump, auto sampler, and refractive index detector (RID) from 1200-series. The gel permeation columns were Agilent PLgel guard column (50*7.5 mm) with PLgel MIXED 10 µm (300*7.5 mm) column connected in series. The mobile phase 0.5% LiCl/DMA was used at a flow rate of 1 ml/min. The GPC system was calibrated with Agilent pullulan polysaccharide standard kit, which consists of 10 standards in the range of 180–708,000 Da. The Agilent Open Lab CDS ChemStation Edition (Rev. C.01.03) with Agilent GPC-Addon data analysis software (Rev. B.01.02) was used to calculate the molar mass distributions.
The sample preparation for GPC analysis has been made according to (Timpa 1991) with some modifications. The blank samples (17.5 mg) and low DS -samples from reactions were weighed in small glass vials, 1.25 ml of DMA was pipetted and the vial was stirred and heated at 130 °C for 2 h. The swollen gel was cooled to 90 °C and LiCl was added to the system (7 wt% LiCl in 1.25 ml of DMA). The mixture was let to cool down to r.t. and stirred overnight to obtain a clear solution. The solution was diluted with DMA so that the concentration of LiCl was the same as in the eluent. In the case of samples with higher DS the sample was diluted in the eluent and heated at 60 °C for 2 h. All samples and eluent were filtered through Millipore® HPLC –membrane filters (HVLP 0.45 µm).
NMR spectroscopy
Quantitative 31P NMR spectra were measured using a Varian Unity INOVA 600 spectrometer (600 MHz proton frequency) equipped with a 5-mm direct detection broadband probe-head at 27 °C. Quantitative 31P NMR were collected with 512 transients using 90° flip angle, 84,000 Hz spectral width, 1 s acquisition time and 10 s relaxation delay. The DS values were determined using a method described previously (King et al. 2010). When the presumed DS of the product was low, e.g., one or less, the NMR samples were prepared as described in King et al. (2010), using DMA/LiCl (3 wt%) (solubilized like the GPC samples) instead of [Amim]Cl.
A known amount (between 10 and 25 mg) of the cellulose acetate was weighed in a small 10 ml glass vial. Pyridine (150 µl), CDCl3 (1 ml), 2-Cl-TMDP (200 µl), e-HNDI (125 µl of 1.6:1 v/v in pyridine: CDCl3) and Cr(acac)3 (1 ml of 0.08 M solution in CDCl3) were added in sequence with vortexing after every solvent or reactant addition. The analysis was performed immediately after the derivatization. If the sample was solubilized in DMA/LiCl, no CDCl3 addition was made after the pyridine addition.
The calculation of the results have been made according to King et al. (2010), in which two different formulas were used for complete DS determination. First the amount of substituted hydroxyls (OHs) are calculated (1). In the formula (1) ISmol is the molarity of the used internal standard in mmol/l, the volume of the used internal standard in µl is ISvol. IR stands for the relation of the integrals (Ialkoxy/IIS) obtained from signals and their integrals in the spectrum; IS is seen in the spectrum as e-HNDI-TMDP at 152 ppm. The cellulose free hydroxyls form alkoxy-TMDP and are seen in the spectrum at 143.5–150 ppm (Fig. 3). In addition, in the formula (1) is the mass of the sample in g (Ws) and a constant of 1,000,000, for unit compensation. The actual DS is calculated in the second formula (2) in which, the maximum DS (DSmax) is used to calculate the real DS with the OHs, molecular weight of the substituent (MWs) and free hydroxyl per weight unit of cellulose (OHc = 3/162.14 g/mol = 0.019 mol/g). The maximum DS depends on the amount of used acylation reagent.
1H-13C HSQC spectra were measured with a Varian Unity INOVA 600 (600 MHz proton frequency) using an indirect detection 5 mm probe-head. For these, approximately 20 mg of sample was dissolved in 1 ml of DMSO-d6 and the spectra were recorded at 60 °C due to the sample’s viscosity. The amount of 100 t1 acquisitions with 32 scans were collected with 10.16 s repetition time.
1H NMR spectra for IL purity determination were recorded either with a Varian Unity INOVA 500 spectrometer (500 MHz proton frequency) equipped with a 5-mm triple resonance (1H, 13C, 15N) z-gradient probe-head at 27 °C or with Varian Mercury Plus 300 spectrometer (300 MHz proton frequency) at 27 °C. 1H NMR were collected with 16-64 transients using 45° flip angle, 5000 Hz spectral width, 1 s acquisition time and 1 s relaxation delay.
Results and discussion
Acylation of cellulose samples in [DBNH][OAc] was carried out with acetic anhydride (Ac2O), isopropenyl acetate (IpeAc), vinyl acetate (VinAc) or vinyl propionate (VinPr). When conducting these reactions, a few things need to be considered: the boiling point of the reagent, side products and their properties.
Acetylation with Ac2O produces acetic acid as the side product. Thus, in these reactions, a slight excess of DBN was used to scavenge released acid and to minimize the hydrolysis of cellulose. Notably, this only produces new [DBNH][OAc] to the system and thus avoids contamination of the ionic liquid. For a sustainable process this would require conversion of a portion of [DBNH][OAc] back to DBN. This is possible through distillation of the recovered ionic liquid (King et al. 2011). Recent results have indicated that DBN actually has a higher vapor pressure than acetic acid in the mixture, allowing for partial separation of the components (Ahmad et al. 2016). Thus, the motivation for this method is sound.
Acylation with isopropenyl carboxylates produce acetone as by-product (Scheme 1). In contrast to Kakuchi et al. (2015, 2017), in [DBNH][OAc] no additional base or activation was required to obtain excellent reactivity. As the IL is derived from the conjugation of a superbase (DBN) and acetic acid, traces of superbase (from equilibration) likely contribute to catalyzing the reaction. Reaction with vinyl carboxylates produces acetaldehyde as by-product. Both acetone and acetaldehyde are easy to remove from the reaction mixture and are relatively non-toxic, but flammable. In prior literature, the reactions made with VinAc were either catalyzed with a base; K2CO3, Na2CO3 (Çetin et al. 2009; Jebrane and Sebe 2007; Jebrane et al. 2011), or cellulose was activated with NaOH (Chen et al. 2016).

Acylation of cellulose in [DBNH][OAc]. The forming side products are acetone for IpeAc (a) and acetaldehyde for VinAc and VinPr (b). With Ac2O, an excess DBN is used (c), and thus the released acetic acid is bound to the base, and [DBNH][OAc] is formed. R = CH3CO (Ac), R′ = methyl or ethyl and R″ = Ac or CH3CH2CO (Pr)
In general, the progress of acylation reactions of cellulose is easy to follow by IR spectroscopy, from the appearance of the ester carbonyl signal at ~ 1740 cm−1 associated with the decrease of the intensity of the hydroxyl signal band at ~ 3400 cm−1 (Fig. 1). The cellulose C–O–C signal intensity around 1100 cm−1 stays unchanged during the esterification reaction. Therefore, it is a good reference point to assess intensity or volume changes of carbonyl signals. Even though ATR-IR is not an inherently quantitative technique (the signal intensities depend of several issues, including IR light penetration depth into the sample), the normalized intensity of the carbonyl signal was shown to give good correlation with the DS values, compared to the 31P NMR analyses (see below).

Baseline corrected and normalized ATR-IR spectra of cellulose acetates from the reactions of eucalyptus PHK pulp with IpeAc. The product with the lowest DS value has the strongest hydroxyl (OH) signal at 3400 cm−1and weakest carbonyl (C=O) signal around 1740 cm−1. With the high DS sample (DS = 2.97), lowest 9 eq. line, the hydroxyl signal is essentially absent
The relation of integrals from the ATR-IR spectra show a linear correlation with the DS values determined by 31P NMR and the correlation between these methods is excellent (Fig. 2). The results show that ATR-IR is a rapid but somewhat crude method to access the DS of acetylated celluloses and 31P NMR can be used to obtain external calibration for the ATR-IR. Further, ATR-IR spectroscopy is a sensitive method to track impurities, e.g., identifying possible residual IL in the regenerated product. In the case of [DBNH][OAc], the possible remaining IL could be identified from signals at 1300 (COO), 1370 (CH3), 1580 (COO−) and 1670 (C=O) cm−1.

Linear correlations of the 31P NMR and ATR-IR results. Reactions from: Ac2O (orange), IpeAc (yellow), VinAc (grey) and VinPr (blue). The linear correlation of all data points is represented as a black line. (Color figure online)
More accurate DS values for the reaction products were assessed using 31P NMR (King et al. 2010). An example of a 31P NMR spectrum for the reaction made with 9 eq. of Ac2O is shown in Fig. 3. The spectrum is referenced to the 4,4,5,5-tetramethyl-1,3,2-dioxaphospholane anhydride (TMDP-anhydride) at 132.2 ppm. The signal at 175 ppm is the unreacted (excess) phosphorylating reagent, which needs to be present in order to verify that the amount of reactant has been sufficient for complete phosphitylation. The DS values were calculated from 31P NMR spectra according King et al. (2010) and are listed in Table 1. The reactions were repeated, and also conducted under different conditions to assess the effect of time/temperature, or possible experimental errors. The results of the duplicated reactions were similar to the original ones (< 5% variation in yield and DS). The DS values were determined to be between 0.25 and 3. They corresponded well with the amount of reagents used (Table 1). With Ac2O and IpeAc the acetylation reaction is almost quantitative. However, the reactions made with vinyl carboxylates (vinyl acetate and vinyl propionate) achieve a maximum DS value of ~ 1.5, e.g., the use of excess reagent, higher temperature or increased reaction time did not increase the maximum obtainable DS. In prior literature, the amount of the reagent required, typically acetic anhydride, has been 5–8 eq’s per AGU to reach almost complete esterification of hydroxyl groups (DS 0.64–2.92) (Heinze et al. 2005; Jogunola et al. 2016; Kosan et al. 2010; Wu et al. 2004). Acyl chlorides have also been used and full reactivity was achieved (DS 3) with 5 eq of reactant, as they are known to be more reactive (Barthel and Heinze 2006). However, the by-product from acid chlorides is hydrochloric acid, which may increase chances for depolymerization. For vinylic reagents, cellulose acylations have not been made in ILs, but in DMF and catalyzed with pyridine or potassium carbonate (K2CO3) (Çetin et al. 2009; Jebrane et al. 2011; Jebrane and Sebe 2007) or in DMSO, with sodium hydroxide (NaOH) addition (Chen et al. 2016). Unfortunately, only Chen et al. have reported the DS and DP values for the products. In their study the reaction was fast, but some degradation of the product occurred, since the DP’s decreased. The recovered DS values were high, more than 2.52. However, Jebrane and Sebe (2007) reported mass losses, probably due to the solubility of the product to the regeneration solvents.

Expansion of a 31P NMR spectrum of (Eucalyptus PHK pulp) the product from reaction of Ac2O (9 eq.) with cellulose at 70 °C in [DBNH][OAc]. The calculated DS of the product is 2.80
In general, in our reaction conditions the maximum DS was obtained under 2 h reaction time, for all of the reagents (Ac2O, VinAc, VinPr, and IpeAc). Even at moderate temperature (65 °C), the acetylation of cellulose hydroxyls is rapid and complete when the amount of Ac2O or IpeAc is sufficient. To our understanding, the reason for such rapid reaction must be the [DBNH][OAc] itself. It is a relatively effective IL for dissolution of cellulose, with potential for several equilibrating basic species in solution, which may catalyze the reaction. In prior literature, the yields of the acylated products have often not been reported. However, in the DMSO/TBAF solvent system, the conversion and yield of cellulose acetate is moderate with a prolonged reaction time (Heinze et al. 2003). The reason for such result is TBAF, which deacetylates the cellulose esters regioselectively from the positions of 2 and 3 (Zheng et al. 2013). With imidazolium-based e.g., [Emim][OAc], [Bmim][Cl], [Amim][Cl], ILs the dissolution times are longer, typically 3-24 h (Heinze et al. 2005), compared with our system (0.5–1 h). Reaching high DS often requires high temperature, 80–110 °C, long reaction times 3–24 h (Kakuchi et al. 2015), catalyst (Luan et al. 2013) or co-solvent (Jogunola et al. 2016; Kakuchi et al. 2017; Rinaldi 2011; Schenzel et al. 2014; Xu et al. 2013). Lower temperatures have also been reported but the reaction time has been 24 h and the product produced had a low DS of 0.64 (Wu et al. 2004). The role of the addition of the organic co-solvent is usually to keep the system homogeneous throughout the reaction, as the acylated cellulose (product) is not always soluble in pure IL. Further, organic co-solvents can be used to reduce the viscosity of the reaction media, and possibly to lower the capital and running costs, as less ionic liquid might be required. However, the use of co-solvents might complicate the recycling of the system.
High DS samples of cellulose acetate become soluble in CDCl3, which enables direct measurement of 1H NMR spectra in the solution state and thus provides another tool to assess DS values using NMR (see supporting information). The results from 1H and 31P NMR measurements agreed well with each other. Also the regioselectivity of the reaction was assessed with 1H NMR after perpropinylation (or peracylation) with propionic anhydride and 4-(dimethylamino)pyridine catalyst according to Tezuka and Tsuchiya (1995). In [DBNH][OAc], all the reagents showed typical reactivity with the –OH groups of cellulose (see supporting material), e.g., largest reactivity (preference) for the O-6 position (Liebert et al. 2005; Xu et al. 2011). The reactivity of the positions O-2 and O-3 was almost equal (6 > 2 ~ 3). Notably, this is not always the case, as with larger substituents O-3 position can be hindered (Xu et al. 2011) or the reactivity of the positions of O-2 and O-3 may vary, depending on the solvent and acylation reagent (Abe et al. 2016).
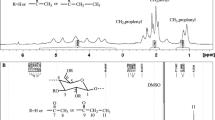
As cellulose is a linear homopolymer, cellulose triacetates obtained from reactions with excess of Ac2O and IpeAc, show only one 1H-13C HSQC correlation in the acetal region ~ δ 6/100 ppm (Fig. 4). However, in the case of the cellulose acylated with excess of vinyl carboxylates, several signals are detected around ~ 100 ppm in 13C. This is due to the fact that vinyl carboxylates react well to produce low DS value products but do not achieve full acetylation (max. DS ~ 1.5). Chemical shifts of ~ 100 ppm in 13C NMR are diagnostic for hemiacetal- and acetal structures. With lower DS values a wider range of substitution patterns are possible, as substitution of OH can be in various positions. Therefore, the HSQC spectra has multiple signals due to the different (acetylated) AGU combinations. For instance, a signal at 102 ppm in 13C NMR analysis is indicative for structures where the C-2 position is not acetylated (see supporting information).

Expansion of a 1H-13C HSQC spectra of acetylated products from the reactions of MCC with IpeAc (black) and Eucalyptus PHK pulp with Ac2O (red). Both products have DS of ~ 3. (Color figure online)
Previously, it has been shown that the reactivities of woody components and biopolymers are different when performing base catalyzed reactions with Ac2O and VinAc in DMF (Jebrane et al. 2011). However, in [DBNH][OAc], the reaction was fast with both Ac2O and VinAc, as after 30 min with 9 Eq (3 eq per hydroxyl group) of reagent at 70 °C, the Ac2O had already reacted fully, but VinAc reaction had stalled at its maximum. This can be seen from both the ATR-IR and the GPC analyses. From the ATR-IR spectra (Fig. 5) it is apparent that the ester carbonyl signal (1740 cm−1) has reached its largest intensity value after 30 min with Ac2O and after 10 min with VinAc. However, the signal intensity with VinAc is only half that of the Ac2O product. With prolonged reaction times (24 or 26 h) the carbonyl signal intensity remains the same with Ac2O reaction samples but slowly decreases with the VinAc samples. The GPC data (Fig. 6), from the same reactions show that compared to the unmodified cellulose sample, the molecular weights (MW) of the acetylated samples increase with low DS samples but cellulose undergoes slight hydrolytic degradation with prolonged reaction times (Table 1; Fig. 6). It is also evident that more hydrolysis occurs especially when reactant is used in high excess. With Ac2O, the released acetic acid could possibly cause the degradation of the cellulose backbone, prior being scavenged by the excess of DBN. However, there is an evident drop in DP with VinAc, VinPr and IpeAc with larger amounts of reagents. In the current system, the DP of some of the products decreased to almost 1/3 that of the original pulp (DPw = 2300), when the amount of reagent was 3 eq (Table 1). At the moment we have no proper explanation for this chain degradation. Somewhat speculatively we can argue that this effect is due to high reactivity of the vinylic reagents (local effects/energy).

Expansion of the normalized ATR-IR-spectra from cellulose acetylation products: Kinetic evaluation of samples from reactions made using 9 eq’s at 70 °C with Ac2O (a) and VinAc (b). The maximum signal is obtained in 10–30 min in both cases but the intensity of the carbonyl signal in reaction b is only half of that from reaction A. A scatter plot on the right shows how the intensity of the signal decreases in the vinyl acetate (b) reaction compared to reaction A

A bar chart demonstrating the variation in number average and weight average molecular weights DPn and DPw during kinetic evaluation of reactivity. The samples were from reactions made using 9 eq’s at 70 °C with Ac2O (a) and VinAc (b). The corresponding GPC molecular mass distribution (MMD) curves are presented on the right (see supporting information). (Color figure online)
In order to be able to calculate the yields from the esterification reactions, the real DS values for each sample were evaluated. The analysis of the DS values were performed using 31P NMR and the DS correlated average MW per AGU for each sample was calculated (3). In the formula (3) M(AGU) is the molecular weight of one anhydroglucose unit (AGU), DS(31P NMR) is the result obtained from the 31P NMR, M(Ac) is the molecular weight of the substituent, e.g., acetate and M(H) is the molecular weight of the substituted hydrogen. For instance, the calculated MW for cellulose acetate with a DS value of 0.89 is 199.6 g/mol for one AGU.
After calculating these two results the yield can be determined (Table 1). The DS correlated yields are from moderate to good, being more than 80% with all of the reagents. Even though a portion of the acetylated materials may be soluble in ethanol, water or acetone, the recovered yields were still quite high. Obviously, some material is lost in the washing and drying sequences but results are likely to be representative.
Recycling of solvent [DBNH][OAc]
[DBNH][OAc] can be purified by distillation, as it undergoes reverse reaction to DBN and acetic acid at elevated temperatures. (Parviainen et al. 2013) However, recycling of [DBNH][OAc] from aqueous solutions has some challenges as DBN can undergo hydrolysis, under certain conditions, when it is in contact with water. (Parviainen et al. 2015) However, alcoholysis under similar conditions does not proceed. Thus, n-butanol was selected to study the entrainment of water from the IL, to avoid hydrolysis. This approach is based on the fact that the boiling point of n-butanol (117.7 °C) is higher than for water. Yet, the strong interaction between n-butanol hydroxyl groups and IL might allow for selective distillation of the lower boiling water, even though water also strongly interacts with the ionic liquid. The hypothesis is that water can be completely entrained from the solvent system with successive additions of n-butanol. After the water was completely removed, the residual n-butanol was removed under high vacuum. It is not yet clear whether the evaporation of water could be azeotropic in this system, or whether the separation can be more related to the difference in boiling points between the pure liquids, as some ionic liquids are known to break azeotropes (Roughton et al. 2012). Quite likely, in the current case, the water removal is a combination of both of these. It is known that azeotropic distillation with n-butanol can remove a larger portion of water during distillation than for example ethanol or hexane (Haynes 2017). With other suitable water-azeotrope forming solvents it is difficult to remove all of the remaining water, since their boiling points are typically below 100 °C.
Finally, we analyzed the concentrated [DBNH][OAc] after all solvent evaporation. The 1H NMR spectrum showed (see supporting information) that some impurities remained but the major component was [DBNH][OAc]. By using a Kugelrohr evaporation apparatus, the IL was distilled into 4 fractions. The distillation was conducted at 0.7 mbar pressure at 140 and 160 °C. A 3-bulb setup was used, with two bulbs in the oven. The evaporated fractions were condensed at two different temperatures, with one sample for each temperature collected to the innermost bulb and one on the outermost bulb, using a dry ice-acetone cold trap (− 78 °C). The mass balances of the collected fractions (F1–F4) and distillation residue (R) are listed in the Table 2.
The corresponding 1H NMR spectra are shown in Fig. 7. In fraction 1, there are traces of n-butanol but DBN was the main product. In fraction 2, pure [DBNH][OAc] was obtained with good (80%) recovery. Fraction 3 contained only traces of the IL. Fraction 4 was the first sample that contained hydrolysis product. This was 5 wt% of 1-(3-aminopropyl)-2-pyrrolidonium [APPH] cation, which is obtained from hydrolysis of the [DBNH] cation. A distillation residue of 12 wt% was remaining but the procedure is unoptimised so we expect this to decrease.

1H NMR spectra of the collected distillation fractions (F1–F4) and distillation residue (R), after reaction, quenching and entraining of water using n-butanol. (Color figure online)
When we compare the HSQC spectra before and after the distillation we can see that some 1-(3-aminopropyl)-2-pyrrolidone acetate (APPAc), and hydrolyzed IL [APPH] are formed during the distillation (see supporting information). However, it is possible to convert the hydrolysis product back to DBN by distillation under certain conditions (unpublished). A possible circulation system for the entire process (i.e. cellulose acetylation and solvent recovery) is presented in the Fig. 8. Obviously, the use of water as the regeneration solvent in this process introduces the possibility of hydrolysis during recycling but this could be avoided either by using alcohol as regeneration solvent or as entrainer during the recycling stage.

A process flow scheme for acetylation and recovery of [DBNH][OAc]; the regeneration solvent is preferably water but can also be alcohols for the quenching and regeneration step
Conclusions
In the current work, we demonstrate the fast and efficient acylation of cellulose with isopropenyl acetate, vinyl acetate and vinyl propionate in [DBNH][OAc], in the absence of additional catalyst. Using these reagents, the byproducts are low boiling, such as acetaldehyde or acetone, allowing for separation from the reaction components by distillation. Acetylation was also achieved using Ac2O, where DBN was used to scavenge the released acetic acid. Under these conditions, no side products are formed but rather new IL is generated. The reactions with Ac2O and IpeAc were fast and essentially quantitative. No major differences in reactivity were found between these two reagents. IpeAc has the advantage that it needs no additional base during acetylation. However, it is a more expensive reagent and acetone must be separated after the reaction. The effect of the reaction temperature on the course of the reactions was examined, for Ac2O and VinAc. The minimum temperature used for the reactions in [DBNH][OAc] was close to the melting point of the pure IL (63 °C). At these temperatures the reactions were almost instantaneous.
We found that with Ac2O or IpeAc in [DBNH][OAc], it is possible to tune the DS values the products between 0.25 and 2.97. On the other hand, the vinylic reagents react well to produce low DS values but the maximum obtainable DS value is ~ 1.5. Comparison of the DS determination using 31P NMR and ATR-IR showed a strong linear correlation between these independent methods. Thus, a simple correlation can be used to determine DS, which does not require NMR.
In the current work, we have also demonstrated that the IL can be recovered and recycled by distillation. n-Butanol can be used to entrain water before distillation, thus, avoiding significant hydrolysis. The mass balances in these initial experiments are very promising. However, also other recycling systems for IL’s are to be considered to recognize the most cost-effective recycling pathways.
References
Abe M, Sugimura K, Nishio Y (2016) Regioselectivity in acetylation of cellulose in ionic liquids. ChemistrySelect 1:2474–2478. doi:10.1002/slct.201600520
Ahmad W, Ostonen A, Jakobsson K, Uusi-Kyyny P, Alopaeus V, Hyväkkö U, King AWT (2016) Feasibility of thermal separation in recycling of the distillable ionic liquid [DBNH][OAc] in cellulose fiber production. Chem Eng Res Des 114:287–298. doi:10.1016/j.cherd.2016.08.032
Barthel S, Heinze T (2006) Acylation and carbanilation of cellulose in ionic liquids. Green Chem 8:301–306. doi:10.1039/B513157J
Çetin NS, Tingaut P, Özmen N, Henry N, Harper D, Dadmun M, Sèbe G (2009) Acetylation of cellulose nanowhiskers with vinyl acetate under moderate conditions. Macromol Biosci 9:997–1003. doi:10.1002/mabi.200900073
Chen J, Xu J, Wang K, Cao X, Sun R (2016) Cellulose acetate fibers prepared from different raw materials with rapid synthesis method. Carbohyd Polym 137:685–692. doi:10.1016/j.carbpol.2015.11.034
Cross CF, Bevan EJ, Beadle C (1894) Plastic compound of cellulose. US Patent 520770A
Fukaya Y, Sugimoto A, Ohno H (2006) Superior solubility of polysaccharides in low viscosity, polar, and halogen-free 1,3-dialkylimidazolium formates. Biomacromol 7:3295–3297. doi:10.1021/bm060327d
Fukaya Y, Hayashi K, Wada M, Ohno H (2008) Cellulose dissolution with polar ionic liquids under mild conditions: required factors for anions. Green Chem 10(1):44–46. doi:10.1039/B713289A
Gericke M, Fardim P, Heinze T (2012) Ionic Liquids—promising but challenging solvents for homogeneous derivatization of cellulose. Molecules 17:7458–7502. doi:10.3390/molecules17067458
Hauru LKJ, Hummel M, King AWT, Kilpeläinen I, Sixta H (2012) Role of solvent parameters in the regeneration of cellulose from ionic liquid solutions. Biomacromol 13:2896–2905. doi:10.1021/bm300912y
Haynes WM (2017) Physical constants of organic compounds, interactive table: azeotropic temperature, pressure and composition. CRC Press/Taylor & Francis, Boca Raton
Heinze T, Liebert T, Pfeiffer KS, Hussain MA (2003) Unconventional cellulose esters: synthesis, characterization and structure-property relations. Cellulose 10(3):283–296
Heinze T, Schwikal K, Barthel S (2005) Ionic liquids as reaction medium in cellulose functionalization. Macromol Biosci 5:520–525. doi:10.1002/mabi.200500039
Heinze T, Dorn S, Schöbitz M, Liebert T, Köhler S, Meister F (2008) Interactions of ionic liquids with polysaccharides—2: cellulose. Macromol Symp 262:8–22. doi:10.1002/masy.200850202
Hinner LP, Wissner JL, Beurer A, Nebel BA, Hauer B (2016) Homogeneous vinyl ester-based synthesis of different cellulose derivatives in 1-ethyl-3-methyl-imidazolium acetate. Green Chem 18(22):6099–6107. doi:10.1039/C6GC02005D
Holding AJ, Heikkilä M, Kilpeläinen I, King AWT (2014) Amphiphilic and phase-separable ionic liquids for biomass processing. Chemsuschem 7:1422–1434. doi:10.1002/cssc.201301261
Isik M, Sardon H, Mecerreyes D (2014) Ionic liquids and cellulose: dissolution, chemical modification and preparation of new cellulosic materials. Int J Mol Sci 15:11922–11940. doi:10.3390/ijms150711922
Jebrane M, Sebe G (2007) A novel simple route to wood acetylation by transesterification with vinyl acetate. Holzforschung 61:143–147. doi:10.1515/HF.2007.026
Jebrane M, Pichavant F, Sèbe G (2011) A comparative study on the acetylation of wood by reaction with vinyl acetate and acetic anhydride. Carbohyd Polym 83:339–345. doi:10.1016/j.carbpol.2010.07.035
Jogunola O, Eta V, Hedenström M, Sundman O, Salmi T, Mikkola J-P (2016) Ionic liquid mediated technology for synthesis of cellulose acetates using different co-solvents. Carbohyd Polym 135:341–348. doi:10.1016/j.carbpol.2015.08.092
Kakuchi R et al (2015) Efficient and rapid direct transesterification reactions of cellulose with isopropenyl acetate in ionic liquids. RSC Adv 5:72071–72074. doi:10.1039/C5RA14408F
Kakuchi R et al (2017) A mechanistic insight into the organocatalytic properties of imidazolium-based ionic liquids and a positive co-solvent effect on cellulose modification reactions in an ionic liquid RSC. Advances 7:9423–9430. doi:10.1039/C6RA28659C
King AWT, Jalomaki J, Granstrom M, Argyropoulos DS, Heikkinen S, Kilpelainen I (2010) A new method for rapid degree of substitution and purity determination of chloroform-soluble cellulose esters, using 31P NMR. Anal Methods 2:1499–1505. doi:10.1039/C0AY00336K
King AWT, Asikkala J, Mutikainen I, Jaervi P, Kilpelaeinen I (2011) Distillable acid-base conjugate ionic liquids for cellulose dissolution and processing. Angew Chem Int Ed 50:6301–6305. doi:10.1002/anie.201100274
King AW, Parviainen A, Karhunen P, Matikainen J, Hauru LK, Sixta H, Kilpeläinen I (2012) Relative and inherent reactivities of imidazolium-based ionic liquids: the implications for lignocellulose processing applications. RSC Adv 2:8020–8026
Klemm D, Philipp B, Heinze T, Heinze U, Wagenknecht W (1998) Comprehensive cellulose chemistry functionalization of cellulose, vol 2. Wiley-VCH, Weinheim
Kosan B, Dorn S, Meister F, Heinze T (2010) Preparation and subsequent shaping of cellulose acetates using ionic liquids. Macromol Mater Eng 295:676–681. doi:10.1002/mame.201000022
Liebert T, Hussain MA, Heinze T (2005) Structure determination of cellulose esters via subsequent functionalization and NMR spectroscopy. Macromol Symp 223:79–92. doi:10.1002/masy.200550506
Luan Y, Zhang J, Zhan M, Wu J, Zhang J, He J (2013) Highly efficient propionylation and butyralation of cellulose in an ionic liquid catalyzed by 4-dimethylminopyridine. Carbohyd Polym 92:307–311. doi:10.1016/j.carbpol.2012.08.111
Michud A et al (2016) Ioncell-F: ionic liquid-based cellulosic textile fibers as an alternative to viscose and Lyocell. Text Res J 86:543–552. doi:10.1177/0040517515591774
Nawaz H, Pires PR, Bioni T, Arêas EG, El Seoud O (2014) Mixed solvents for cellulose derivatization under homogeneous conditions: kinetic, spectroscopic, and theoretical studies on the acetylation of the biopolymer in binary mixtures of an ionic liquid and molecular solvents. Cellulose 21:1193–1204. doi:10.1007/s10570-014-0184-8
Parviainen A et al (2013) Predicting cellulose solvating capabilities of acid-base conjugate ionic liquids. Chemsuschem 6:2161–2169. doi:10.1002/cssc.201300143
Parviainen A et al (2015) Sustainability of cellulose dissolution and regeneration in 1,5-diazabicyclo[4.3.0]non-5-enium acetate: a batch simulation of the IONCELL-F process. RSC Adv 5:69728–69737. doi:10.1039/C5RA12386K
Rinaldi R (2011) Instantaneous dissolution of cellulose in organic electrolyte solutions. Chem Commun 47:511–513. doi:10.1039/C0CC02421J
Rosenau T, Potthast A, Sixta H, Kosma P (2001) The chemistry of side reactions and byproduct formation in the system NMMO/cellulose (Lyocell process). Prog Polym Sci 26(9):1763–1837. doi:10.1016/S0079-6700(01)00023-5
Roughton BC, Christian B, White J, Camarda KV, Gani R (2012) Simultaneous design of ionic liquid entrainers and energy efficient azeotropic separation processes. Comput Chem Eng 42:248–262. doi:10.1016/j.compchemeng.2012.02.021
Ruokonen S-K et al (2016) Effect of ionic liquids on zebrafish (danio rerio) viability, behavior, and histology; correlation between toxicity and ionic liquid aggregation. Environ Sci Technol 50:7116–7125. doi:10.1021/acs.est.5b06107
Schenzel A, Hufendiek A, Barner-Kowollik C, Meier MAR (2014) Catalytic transesterification of cellulose in ionic liquids: sustainable access to cellulose esters. Green Chem 16:3266–3271. doi:10.1039/C4GC00312H
Sixta H et al (2015) Ioncell-F: a high-strength regenerated cellulose fibre. Nord Pulp Pap Res J 30:43–57
Stepan A, King AT, Kakko T, Toriz G, Kilpeläinen I, Gatenholm P (2013) Fast and highly efficient acetylation of xylans in ionic liquid systems. Cellulose 20:2813–2824. doi:10.1007/s10570-013-0028-y
Swatloski RP, Spear SK, Holbrey JD, Rogers RD (2002) Dissolution of cellulose with ionic liquids. J Am Chem Soc 124:4974–4975. doi:10.1021/ja025790m
Tezuka Y, Tsuchiya Y (1995) Determination of substituent distribution in cellulose acetate by means of a 13C NMR study on its propanoated derivative. Carbohydr Res 273(1):83–91
Tian D, Han Y, Lu C, Zhang X, Yuan G (2014) Acidic ionic liquid as “quasi-homogeneous” catalyst for controllable synthesis of cellulose acetate. Carbohyd Polym 113:83–90. doi:10.1016/j.carbpol.2014.07.005
Timpa JD (1991) Application of universal calibration in gel permeation chromatography for molecular weight determinations of plant cell wall polymers: cotton fiber. J Agric Food Chem 39:270–275. doi:10.1021/jf00002a010
Witos J, Russo G, Ruokonen S-K, Wiedmer SK (2017) Unraveling interactions between ionic liquids and phospholipid vesicles using nanoplasmonic sensing. Langmuir 33:1066–1076. doi:10.1021/acs.langmuir.6b04359
Wu J, Zhang J, Zhang H, He J, Ren Q, Guo M (2004) Homogeneous acetylation of cellulose in a new ionic liquid. Biomacromol 5:266–268. doi:10.1021/bm034398d
Xu D, Li B, Tate C, Edgar KJ (2011) Studies on regioselective acylation of cellulose with bulky acid chlorides. Cellulose 18:405–419. doi:10.1007/s10570-010-9476-9
Xu A, Zhang Y, Zhao Y, Wang J (2013) Cellulose dissolution at ambient temperature: role of preferential solvation of cations of ionic liquids by a cosolvent. Carbohyd Polym 92:540–544. doi:10.1016/j.carbpol.2012.09.028
Yang Y, Xie H, Liu E (2014) Acylation of cellulose in reversible ionic liquids. Green Chem 16:3018–3023. doi:10.1039/C4GC00199K
Zheng X, Gandour RD, Edgar KJ (2013) Probing the mechanism of TBAF-catalyzed deacylation of cellulose esters. Biomacromol 14:1388–1394. doi:10.1021/bm400041w
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kakko, T., King, A.W.T. & Kilpeläinen, I. Homogenous esterification of cellulose pulp in [DBNH][OAc]. Cellulose 24, 5341–5354 (2017). https://doi.org/10.1007/s10570-017-1521-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-017-1521-5