
Abstract
Pikeperch (Sander lucioperca) is a native fish species in the European and Asian basin, representing high natural and economical value. However, our information on the genetic background and diversity of European populations is still limited, despite of that the production and the number of bread stocks has increased significantly in the last decade. Our aim was to develop new useable species-specific microsatellites and compare genetic diversity of ten pikeperch population from the Danube drainage basin. Thirty-four novel species-specific DNA markers were isolated and seven polymorphic microsatellite markers were selected for the population genetic analysis. The results indicated strong anthropogenic effects among the populations. The FST estimated by the method of Weir and Cockerham (1984) was 0.214, showing moderate genetic difference among the populations. The Structure analysis and the neighbour-joining dendrogram are showing the same result: the ten examined populations aggregate six genetically distinct units. Significant lack of heterozygosity was detected in most of the populations. Presumably, it is indicating the effects of human activities and such as vigorous stocking of natural waters. In addition, the effect was proved that fish stocks involved in pond culture and RAS rearing do not necessarily originated from the geographically closest natural populations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pikeperch (Sander lucioperca) is a valuable and traditionally important carnivorous fish species in European and Asian Aquaculture. The species is becoming more and more popular among anglers and consumers. However, environmental pollution, loss of habitat, and overfishing reduced the number of pikeperch in natural waters, thus consumer demands cannot be fulfilled (Hedrick and Miller 1992). To overcome the problem, several European research projects were initiated to developing production technologies and techniques (Abdolmalaki and Psuty 2007; Björklund et al. 2007; Bódis and Bercsényi 2009; Menezes et al. 2013; Zakeś et al. 2013; Zarski et al. 2013; Blecha et al. 2016; Lappalainen et al. 2016). Classical intensive breeding and rearing methods can only be used with strong limitations due to the high stress sensitivity of the species and its need for a relatively large territory (especially in the breeding season) which is probably due to the predatory nature of the species (Molnár et al. 2004). If (when) the breeding space is too small, cannibalism begins to take place (Policar et al. 2013; Szkudlarek and Zakęś 2007). In addition pikeperch is highly sensitive to dissolved O2 levels and to mechanical injuries. There is significant progress in technology development of artificial propagation, incubation, pre-nursing, nursing, and formulated feed-based rearing of pikeperch (Lappalainen et al. 2003; Molnár et al. 2004; Blecha et al. 2016; Miroslav et al. 2016). Information is still scarce on the genetic structure and diversity of both natural water pikeperch populations and farmed stocks. However, it would be essential for the conservation of natural populations. Some of them are strongly influenced by anthropogenic affects, like uncontrolled stocking (Louati et al. 2016; Poulet et al. 2009; Salminen et al. 2012) and admixture (Eschbach et al. 2014) which are reduce the genetic differentiation among the natural populations.
On the other hand, genetic information and molecular genetic marker-based methods (e.g. marker-assisted selection, parentage analyses) are also important for efficient genetic improvement programmes, inbreeding, and selection programmes or for the maintenance of genetic variance of farmed stocks (Vandeputte et al. 2011; Kaczmarczyk and Fopp-Bayat 2013; Fopp-Bayat 2007). Until now, 27 polymorphic and species-specific microsatellite DNA markers were isolated from pikeperch (Kohlmann and Kersten 2008; Han et al. 2016) and some more microsatellites were described (Zardoya et al. 1996; Borer et al. 1999; Wirth et al. 1999; Leclerc et al. 2000; Li et al. 2007; Yang et al. 2009; Zhan et al. 2009; Grzybowski et al. 2010; Coykendall et al. 2014) from closely related species (walleye—Sander vitreus, yellow perch—Perca flavescens, perch—Perca fluviatilis). Although some of them (after adaptation) proved to be suitable for population genetic studies on the pikeperch too (Leclerc et al. 2000; Kusishchin et al. 2018), there is a significant need for new, well characterised, highly polymorphic genetic markers that are applicable for gaining new and more detailed information on the genetic variability and unique genetic characteristics of different populations and stocks of the species. In case of species where the genomic sequence is not available and therefore SNP markers are not applicable, microsatellite DNA markers are still widely used for population genetic studies. These codominant markers are usually located in the non-coding regions of the genome and therefore, the mutation rate is high and selection pressure against mutations is relatively low resulting in high variability (Sunnucks 2000). Due to their small size, easy amplification and fragment separation techniques and easy evaluation, microsatellites are ideal tools for conservation biology studies of less studied (fish) species (Chistiakov et al. 2006). With a sufficient, relatively large number of microsatellites, genetic mapping can be carried out (Botstein et al. 1980; Knapik et al. 1998) or breeding programmes can successfully be supported (Beuzen et al. 2000). Moreover, microsatellites can be applied in aquaculture during the selection of spawners based on genotype profiles (Kaczmarczyk and Fopp-Bayat 2013) or in genotyping of cryopreserved fish spermatozoa (Fopp-Bayat and Ciereszko 2012). In aquaculture genome engineering, microsatellites are generally used for identification of maternal or paternal inheritance (Fopp-Bayat 2007) or during the investigation of ploidy level of manipulated fish (Fopp-Bayat and Woznicki 2006).
The aim of the present study was to isolate and characterise novel microsatellite DNA markers from the pikeperch and to describe the genetic diversity, population genetic status, and the effects of breeding and stocking in a few natural populations and farmed stocks in the River Danube drainage system.
Materials and methods
Sampling
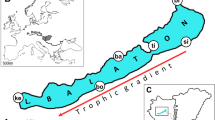
Fin samples were collected from 376 adult pikeperch individuals and stored in 96% ethanol. To reduce animal suffering cloves oil (Syzygium aromaticum) was used as an anaesthetic bath before sampling the fish (15 oil drops/10 l water). After the fish have become immobile, they were sampled (the outermost 0.5–1 cm2 of caudal fin were clipped) and transferred to clean water until they wake up. Samples were stored in ethanol on − 20 °C until utilisation. Samples (Table 1) were collected from fish farms located in Hungary (Dalmand [Da], N = 46; Attala [At], N = 21; Akasztó [Ak], N = 21; Nyíregyháza [Ny], N = 47), from two different RASs (Recirculating Aquaculture System) in Hungary (Győr [Gy], N = 21; Kisbajcs [Kb], N = 78) and one in Romania (Timișoara [Ti], N = 20) and from natural Hungary (Lake Balaton [Ba], N = 60) and Romania (estuary of Danube River [De], N = 48), and populations in Germany (Upper Danube [Ge], N = 14) (Fig. 1). However, the number of the available samples in case of some population (e.g. De or Ti) are too low to represent the full populations, but they are providing important data.

Geographical location of sampling sites. 1: Germany (Upper Danube, Ge); Hungary: 2: Győr (Gy), 3: Kisbajcs (Kb), 4: Lake Balaton (Ba), 5: Dalmand (Da), 6: Attala, (At), 7: Akasztó (Ak), 8: Nyíregyháza (Ny); Romania: 9: Timișoara (Ti), 10: Danube estuary (De)
Ethics
All procedures involving the handling and treatment of fish used during this study were approved by the Capital and Pest County Government Office for Food Chain Safety and Animal Health (permission number: XIV-I-001/2302-4/2012 and XIV-I-001/2304-4/2012).
DNA isolation
Standard phenol/chloroform (Rothi-Phenol, Carl Roth; Chloroform, Reanal) extraction method was used to isolate DNA (Blin and Stafford 1976). The quality of extracted DNA was tested by agarose gel electrophoresis and DNA concentration was measured by a spectrophotometer (IMPLEN, NanoPhotometer). Twenty-microgram DNA was separated for genomic library construction while the rest of the samples were diluted to 50 ng/μl concentration for microsatellite analysis.
Genomic library construction
The modified method of Glenn and Schable (2005) was used to construct a repeat-enriched genomic library. The DNA used for library construction was isolated from male individuals since in the examined species, the males are heterogametic and therefore, they carry both sex chromosomes (Rougeot et al. 2002). Twenty-microgram genomic DNA was digested by restriction endonucleases (Rsa I, Fermentas/HpyCH4 V, NewEngland BioLab) creating blunt ends. After separating the products on agarose gel, all fragments with the length of 300–1000 base pairs were re-isolated from the gel using a NucleoSpin Extract II kit (Macherey-Nagel). The DNA concentration was then measured by spectrophotometer (IMPLEN, NanoPhotometer), and following phosphatase treatment (Shrimp Alkaline Phosphatase, Fermentas) of 10-μg fragments, Box I linker (F: 5′-Phos-ATGTCTGAAGGTACCACTGCTGTCCGAAA-3′;R: 5′-CGGACAGCAGTGGTACCTTCAGACAT-3′) was ligated to them. Ligation was checked by a linker-specific PCR in a final volume of 25 μl: 1× Taq DNA polymerase buffer (Fermentas), 0.4 μM Box I reverse primer, 2 mM MgCl2 (Fermentas), 0.2 mM dNTP (Fermentas), 1 U Taq DNA polymerase (Fermentas), and 4 μl template (adapter connected DNA-fragment). The PCR profile of the reaction was the following: 2 min 94 °C, 35× 94 °C 20 s, 60 °C 30 s, 72 °C 3 min, and finally, 72 °C 5 min (Mastercycler 5341, Eppendorf). The PCR was followed by the collection of tandem repeats containing DNA fragments. The fragments were hybridised with a (CA)10 carrying oligonucleotide (3′ biotinylated) in a thermal cycler device using the following protocol: the initial denaturation (92 °C 5 min) was followed by the cooling of the reaction mixture from 70 to 50 °C with the decreasing of the temperature by 0.2 °C/s. The mixture was kept on 50 °C for 10 min and then it was cooled to 15 °C with the decreasing of the temperature by 0.1 °C/s. Hybridization complexes were bound to the surface of streptavidin covered magnetic beads (Dynabeads M-270 Streptavidin, Invitrogen) according to the method Glenn and Schable (2005). The resulting complexes were removed from the solution by a magnet. The repeat-containing DNA fragments were finally eluted by TLE solution (pH = 8.00) on 95 °C. The eluted single-stranded DNA was transformed to double-stranded DNA by the previously described PCR (Mastercycler 5341, Eppendorf) using adapter specific primers. Resulting double-stranded fragments were ligated to T-vector (pGEM-T Easy Vector System I, Promega) and transformed into competent Escherichia coli cells (XL10 GOLD, Stratagene) following the protocol described by the manufacturer. Colonies were screened by blue-white screening (Ullmann et al. 1967). Inserts were checked by agarose gel electrophoresis and colony PCR initiated at the T-vector coded M13 primer binding sites. The PCR was carried out in a final volume of 25 μl with the following ingredients: 1× Taq DNA Polymerase Buffer (Fermentas), 0.26 μM–0.26 μM M13 forward and reverse primers (F: 5′ TGTAAAACGACGGCCAGT 3′; R: 5′ CAGGAAACAGCTATGACC 3′), 2 mM MgCl2 (Fermentas), 0.2 mM dNTP (Fermentas), 1 U Taq DNA Polymerase (Fermentas), and a few cells of bacteria colonies as template. The PCR profile of the reaction was the following: 3× 95 °C 2 min, 55 °C 1 min, 72 °C 2 min, then 41× 95 °C 30 s, 55 °C 30 s, 72 °C 45 s, and finally, 72 °C 5 min. Fragments longer than 300 bp were cleaned up (PCR Advanced Clean Up System, Viogene) and the sequence of the inserts were determined (3130 Genetic Analyser, Applied Biosystems) using the SP6- (5′ CATACGATTTAGGTGACACTATAG 3′) and T7- (5′ TAATACGACTCACTATAGGG 3′) primers and 3.1 BigDye kit (Applied Biosystems). Sequences were aligned with MEGA5 software (Tamura et al. 2011) and the ones containing at least 5 dinucleotide repeats were selected and primers were designed to their flanking regions Primer3Plus software (Untergasser et al. 2007). Their optimal reaction parameters were determined.
Microsatellite analysis
Microsatellites were tested on a test panel of 8 individuals. After that, microsatellite analysis was carried out on the above mentioned populations by using PCR. At first, directly labelled (5′FAM) microsatellite specific forward primers were used (in case of MS 84 Sl, MS 146 Sl, MS 150 Sl, MS 192 Sl, MS 195 Sl, MS 198 Sl, MS 203 Sl, MS 260 Sl, and MS 268 Sl markers). Later, we chose a more simple method according to Shimizu et al. (2002) to reduce the costs of analysis and labour. The forward primers were elongated with a, 5′, 17 bp long tail sequence (tail; 5′ATTACCGCGGCTGCTGG- microsatellite specific sequence-3′) to provide attachment site for fluorescently labelled (dye: FAM or VIC or NED or PET) “universal” oligos (tail-specific oligo; 5′dye-ATTACCGCGGCTGCTGG-3′). One tail-specific oligo was added to the reaction mixtures for fluorescent labelling of amplicons. Table 2 summarises the ingredients of PCRs. The cycling conditions for the microsatellites amplification were 95 °C 2 min, 2× 95 °C 15 s, annealing temperature (see Table 3) 1 min, 72 °C 2 min, 35× or 45× (see Table 3) 95 °C 15 s, annealing temperature (see Table 3) 20 s, 72 °C 40 s, and finally, 72 °C 5 min by using Mastercycler 5341 (Eppendorf) thermal cycler. Table 3 contains primer sequences, annealing temperature values, and number of cycles during PCR, sequences of repeat regions, numbers and size ranges of detected alleles and GenBank access numbers. The length of amplified products was determined by 3130 Genetic Analyser (Applied Biosystems) using GeneScan 500 LIZ (Applied Biosystems) molecule weight marker in all cases except for MS 198 Sl marker where ILS 600 (Promega) molecule weight marker was used. Based on the preliminary results, seven markers (MS 192 Sl, MS 195 Sl, MS 198 Sl, MS 203 Sl, MS 260 Sl, MS 268 Sl, MS 397 Sl; Table 3) were chosen for population analysis.
Data analysis
Allele sizes were determined by GENEMAPPER VER. 4.0 (Applied Biosystems) These values served as a basis to calculate population genetic measures. EXCEL MICROSATELLITE TOOLKIT VER. 3.1.1 (Park 2001) was used to determine the mean number of alleles per locus and expected (HE) and observed heterozygosity (HO) levels in every single population. Allelic richness (Ar), FISFST, and pairwise FST values were calculated by using FSTAT VER. 2.9.3.2 (Goudet 1995). Bonferroni correction with 1000 permutation was used in FSTAT to analyses significance of FST values among sampling locations. Deviation from Hardy–Weinberg equilibrium values were determined by the GENPOP VER. 4.1.0 (Rousset 2008) software. Average gene diversity and the mean number of alleles per populations were calculated by the ARLEQUIN VER. 3.5 (Excoffier et al. 2005) software while private alleles and Nei’s genetic distances were determined and computed by GENALEX VER. 6.502 (Peakall and Smouse 2012; Smouse et al. 2015). For the construction of neighbour-Joining dendrogram, we used POPULATIONS VER. 1.2.32 (Langella 2002) and FIGTREE VER. 1.3.1 (Rambaut 2009) software. On the basis of results, the genetic structure of the examined stock (without the use of information on populations) was determined by STRUCTURE VER. 2.3.3 (Pritchard et al. 2000; Hubisz et al. 2009) software. This analysis was run with the following settings: length of burn-in period: 55,000; number of MCMC reps after burn-in: 555,000. Possible cluster distribution was analysed from K = 1 to K = 11 (Earl and von Holdt 2012). The number of most probable genetic clusters were determined on the basis of likelihood analysis of each K (L′(K), L′(K) and ΔK) values by STRUCTURE HARVESTER (Evanno et al. 2005; Earl and von Holdt 2012). MICRO-CHECKER VER. 2.2.3 (Van Oosterhout et al. 2004) was used to detect possible genotyping errors, allele dropout, and non-amplified alleles (null alleles). This software package can estimate the frequency of null alleles and can adjust the data set to account for the presence of null alleles.
Results
Two CA-dinucleotide enriched pikeperch genomic library were constructed with the use of two restriction enzymes (Rsa I, HpyCH4 V). Colony PCR confirmed insert DNA incorporation in case of 115 clones (out of 208 screened clones). Among these, 109 unique sequences were found, and 101 contained microsatellite repeat regions showing that the enrichment was highly effective (93%). New, unique sequences were deposited in GenBank (Table 3). Thirty-four sequences were suitable for the development of fully functional microsatellite DNA markers. These markers were tested on a test panel of eight pikeperch individuals to determine their functionality and characteristics.
All the 34 functional markers were proved to be polymorphic and the number of detected alleles ranged between 3 and 20 (Table 3). The highest number (20) of amplified alleles was found in case of the MS 260 Sl marker while MS 84 Sl, MS 192 Sl, MS 412 Sl, and MS 424 Sl were also found to be highly polymorphic. Seven of the newly developed microsatellite markers (MS 192 Sl, MS 195 Sl, MS 198 Sl, MS 203 Sl, MS 260 Sl, MS 268 Sl, MS 397 Sl) were applied to analyse ten populations (Ge, Kb, Gy, Ba, Ak, At, Da, Ny, Ti, and De) originating from the drainage system of the River Danube (Fig. 1). According to the Micro-Checker software, there is no large allele drop outs and stuttering errors. Possibility for null alleles was detected for MS 192 Sl in Ny and Da populations, MS 195 Sl in Ak and Da, MS 198 Sl in De, MS 203 Sl in Da, MS 260 Sl in Ny, and MS 397 Sl in At and Kb populations.
The highest number of alleles (17 and 20), the highest mean number of alleles per loci (7 and 6.7), and the highest allelic richness (10.2 and 8.6) values were detected in case of MS 192 Sl and MS 260 Sl markers respectively, while the total average allele number was also relatively high (9.14). The highest average number of alleles per locus was found surprisingly in the artificially established and bred Kisbajcs (Kb: 5.43) and Dalmand (Da: 4.71) stocks. The reason for this phenomenon can be explained with the establishment storey of these stocks. The farmers purchased broodfish from several different fish farms and natural waters at different times. If population size is also considered, the population from the Danube estuary (De) can be classified as a genetically rich population based on allelic richness values (Table 4).
The expected heterozygosity (HE) values ranged between 0.4518 and 0.5931 while the observed heterozygosity (HO) values were found between 0.4150 and 0.5665. The differences between HE and HO values were found to be non-significant in all but two case, meaning that only the Timișoara (Ti) and Győr (Gy) populations are in Hardy–Weinberg equilibrium. The highest average genetic diversity was found in the Danube estuary population (De) and in Attala (At) stock (Table 4). In case of the majority of examined populations and stocks, a low but significant lack of heterozygotes was detected while in case of Kisbajcs stock (Kb) the excess of heterozygotes was present (FIS = − 0.038).
During the examination of the different populations and stocks, it was recognised that although the genetic variability was not very high between the groups, the different populations and stocks possess individual characteristics such as private alleles. The highest number of individual alleles (4) was found in the Danube estuary (De) population while the Kisbajcs (Kb) and Győr (Gy) stocks had 3 and 3 private alleles, respectively. The MS 260 Sl marker showed nine private alleles while we found three private alleles in case of MS 397 Sl. The frequency of these private alleles is low, except two private alleles (171 and 173 bp) in the German (Ge) population on MS 260 Sl locus with a frequency of 0.179 and 0.143, respectively.
The FST value calculated for all populations and stocks was 0.214 showing a moderate genetic differentiation between these groups. The pairwise FST values between pairs of populations were also calculated and in some cases, the genetic differentiation was found high. The highest differentiation was found between two natural water populations. The lowest genetic difference was found between the artificial Dalmand and the natural Balaton stocks, confirming that the former originate from the later one. Furthermore, low genetic differentiation was found between the artificially propagated stocks (Table 5). When summarising the results, one can state that the most of population and stock pairs does not represent high genetic differentiation (FST < 0.25) which can be explained with the fact that broodstocks of intensive systems and fish farms were established with the use of individuals from different origins and fish were restocked to natural waters sometimes randomly from these stocks.
Nei’s genetic distances (Da) (Nei et al. 1983) were specified and based on the results a dendrogram (neighbour Joining) was constructed in order to represent the relationship between populations (Fig. 2). The highest genetic distance (0.807) was detected between the German (Ge) and the Győr (Gy) populations where the highest genetic differentiation was also described. Furthermore, high genetic distances were observed between the Timișoara (Ti) and German (Ge; Da = 0.758), the Kisbajcs (Kb) and German (Ge, Da = 0.778), and between the Danube estuary (De) and German (Ge; Da = 0.713) population pairs, respectively. The smallest genetic distance value was found between the Dalmand (Da) and Lake Balaton (Ba; Da = 0.040) stocks. Similarly, low levels were found between the Akasztó-Attala (Ak-At; Da = 0.073) and Kisbajcs-Nyíregyháza (Kb-Ny; Da = 0.085) stock pairs.

Dendrogram constructed from Nei’s genetic distances using neighbour-Joining method. The German (Ge), Timișoara (Ti), and Danube estuary (De) populations construct individual genetic units while Lake Balaton (Ba) and Dalmand (Da); Kisbajcs (Kb) and Nyíregyháza (Ny); Akasztó (Ak), Attala (At), and Győr (Gy) populations construct separate groups
The genetic structure of the examined populations was analysed assuming the presence of a different number of clusters based on the microsatellite results using Structure. The method of Evanno et al. (2005) implemented in Structure Harvester software indicated that the most probable number of clusters was K = 6 (Fig. 3) showing that the samples collected from ten different locations can be classified into six genetically different groups.

Results of the Structure analysis: the ten examined populations can be classified into six (K = 6) genetically different groups. De, Danube estuary; Ti, Timișoara; Ny, Nyíregyháza; Ak, Akasztó; Da, Dalmand; At, Attala; Ba, Lake Balaton; Kb, Kisbajcs; Gy, Győr; Ge, Upper Danube
Discussion
In the present study, natural water populations from the Danube catchment area and pikeperch stocks reared in extensive fish farms and in intensive systems in the basin of the river were examined. The newly developed species-specific microsatellite DNA markers could complement previously developed marker sets (some of them adapted from closely related species) and could provide a highly useful tool for gaining more information on the genetic background of the species.
Two microsatellite-enriched genomic libraries were constructed using the method (slightly modified) of Glenn and Schable (2005). Ninety-three percent of the specified sequences contained microsatellites and 33.6% of these were successfully developed into genetic markers. Thirty-four microsatellites were tested altogether for functionality and polymorphism. The method of enrichment proved to be very efficient in contrast to traditional isolation methods (Rassmann et al. 1991; Wu et al. 1994; Lench et al. 1996; Hunt et al. 1999) Compared to other enrichment-based microsatellite isolation methods, the efficiency results are still acceptable (Ostrander et al. 1992; Armour et al. 1994;Kandpal et al. 1994).
The newly developed markers were proved to be polymorphic; the use of the ones with higher polymorphism levels and allelic richness is recommended in further studies even in case of closely related species.
Seven out of the new markers (MS 192 Sl, MS 195 Sl, MS 198 Sl, MS 203 Sl, MS 260 Sl, MS 268 Sl, and MS 397 Sl) were used to describe the genetic structure, variability, and relatedness of ten populations and stocks originating from the Danube catchment area. We looked for the effects of increased stocking and intensive pikeperch farming on the natural populations and found that in case of the majority of markers and in all examined populations and stocks, the expected and observed heterozygosity differed significantly from each other. Some analyses reported populations with higher genetic variance. For example, Kusishchin et al. (2018) described that the pikeperch populations in Akhtuba and the Volga rivers have higher heterozygosity (0.792 and 0.750) than in some examined Baltic (0.59–0.71) (Khurshut and Kohlmann 2009; Säisä et al. 2010;) and Western European populations (< 0.67) (Eschbach et al. 2014) populations. However, “precise comparison is hardly possible,” because the experiments are used different marker sets. In a similar comparison, the heterozygosity of our examined populations are similar to other European populations (Table 4.), together with a small but significant lack of heterozygotes (FIS = 0.039). This phenomenon is probably due to overfishing and the low number of spawners (broodfish) used for artificial propagations. Heterozygosity could be maintained or increased with the implementation of long term, well-planned breeding programmes (based on parentage analyses, genotypes of the breeders, crossing rare and/or distant genotypes of the stock, or introducing new genotypes).
The analysis of the difference among the population did not show outstandingly high genetic difference (FST = 0.214); however, pairwise tests showed notable separation between the German (Ge) population and the Győr (Gy), Kisbajcs (Kb), Nyíregyháza (Ny), and Timișoara (Ti) stocks. The highest genetic differences were found between the population originating from the Upper Danube in Germany and all other populations. The highest number of private alleles was also described in the German (Ge) population (instead of the low number of analysed individuals). The segregation of this population is probably the consequence of geographic distance. The highest genetic diversity was found in Kisbajcs (Kb) intensively reared stock, which is most likely the result of recent broodfish import from different external resources or hybridization with pikeperch from unsteadied area.
This is also supported by the dendrogram based on the Nei’s genetic distances between population pairs, where the wild living populations can be found on the tree in relation to their geographic location (The Lake Balaton (Ba) population can be found between the Danube estuary (De) and Upper Danube (Ge) populations). The stocks reared in extensive fishponds and in intensive RAS cannot be fitted into the above mentioned system. The origin of these stocks can be clearly described and the (sometimes identical) genetic basis can be always identified. For example, the Kisbajcs (Kb) and Nyíregyháza (Ny) stocks (geographic distance ~ 310 km) are standing genetically closer to each other than the geographically close Kisbajc (Kb) and Győr (Gy) stocks (geographical distance ~ 7.2 km). The latter is an intensively selected and reared, artificial feed consuming stock that shows the highest genetic similarity to the Attala (At) stock (geographical distance ~ 145 km) and Akasztó (Ak) stock (geographical distance ~ 305 km), because Győr (Gy) stock was derived from these two (At, Ak) pond hatcheries (pers. comm.), while the Dalmand (Da) stock is standing genetically close to the Lake Balaton population, since the farm was established with broodfish from Lake Balaton. The study on the genetic structure of the populations showed the same results, the ten stocks can be clustered into six genetically different groups. The Dalmand stock originates from Lake Balaton, while Győr stock was established by brood fish from Akasztó and Attala and the Kisbajcs stock was selected from Nyíregyháza stock. The linkage between the detected alleles and the sex of the examined fish was also tested but no correlation was found. These results proved the presence of anthropogenic effects and draw attention to the increasing requirement of genetic conservation of natural pikeperch populations.
The results suggest that there is a continuous and direct connection between fishpond/intensive stocks and natural water populations. The broodstocks of intensive systems and fish farms are established by mixing individuals of different origin and fish are restocked to natural waters, sometimes randomly from these stocks.
The breeding programmes (Abdolmalaki and Psuty 2007; Björklund et al. 2007; Menezes et al. 2013; Lappalainen et al. 2016) and technological developments (Zakeś et al. 2013;Zarski et al. 2013; Blecha et al. 2016; Miroslav et al. 2016) have occurred in the last 10 years had fortunately only a minor effect on the natural populations of pikeperch in the Danube basin so far. But since there is a deviation from Hardy–Weinberg equilibrium due to overfishing and poorly planned restocking, the natural populations may face the loss of their original genetic variability. The intensification of breeding and selection programmes together with high levels of stocking can degrade the genetic basis of the species and can lead to the disappearance of varieties and to the uniformization of the different populations as it happened in case of the long time ago domesticated common carp (Hulak et al. 2010) and brown trout (Ward 2006). Recognising this phenomenon suggests that the conservation of pikeperch genetic resources became an actual issue.
Abbreviations
- Ak:
-
Akasztó fish farm
- Ar:
-
allelic richness
- At:
-
Attala fish farm
- Ba:
-
Lake Balaton
- Box I:
-
DNA linker
- Da:
-
Dalmand fish farm
- De:
-
estuary of Danube River
- FAM:
-
blue fluorescent dye
- Ge:
-
Germany Upper Danube population
- Gy:
-
Győr fish farm
- HE:
-
expected heterozygosity
- HO:
-
observed heterozygosity
- HpyCH4 V:
-
HpyCH4 V restriction endonucleases
- Kb:
-
Kisbajcs fish farm
- LIZ:
-
orange fluorescent dye
- MCMC:
-
Markov Chain Monte Carlo Bayesian approach)
- NED:
-
red fluorescent dye
- Ny:
-
Nyíregyháza fish farm
- PCR:
-
polymerase chain reaction
- PET:
-
yellow fluorescent dye
- RAS:
-
recirculating Aquaculture System
- Rsa I:
-
RsaI restriction endonucleases
- Ti:
-
Timișoara fish farm
- TLE:
-
Tris low EDTA solution
- VIC:
-
green fluorescent dye
References
Abdolmalaki S, Psuty I (2007) The effects of stock enhancement of pikeperch (Sander lucioperca) in Iranian coastal waters of the Caspian Sea. ICES J Mar Sci 64:973–980. https://doi.org/10.1093/icesjms/fsm062
Armour JAL, Neumann R, Gobert S, Jeffreys AJ (1994) Isolation of human simple repeat loci by hybridization selection. Hum Mol Genet 3:599–605. https://doi.org/10.1093/hmg/3.4.599
Beuzen ND, Stear MJ, Chang KC (2000) Molecular markers and their use in animal breeding. Vet J 160:42–52. https://doi.org/10.1053/tvjl.2000.0468
Björklund M, Aho T, Larsson LC (2007) Genetic differentiation in pikeperch (Sander lucioperca): the relative importance of gene flow, drift and common history. J Fish Biol 71:264–278. https://doi.org/10.1111/j.1095-8649.2007.01609.x
Blecha M, Flajshans M, Lebeda I, Kristan J, Svacina P, Policar T (2016) Triploidisation of pikeperch (Sander lucioperca), first success. Aquaculture 462:115–117. https://doi.org/10.1016/j.aquaculture.2016.05.016
Blin N, Stafford DW (1976) A general method for isolation of high molecular weight DNA from eukaryotes. Nucleic Acids Res 3:2303–2308. https://doi.org/10.1093/nar/3.9.2303
Bódis M, Bercsényi M (2009) The effect of different daily feed rations on the growth, condition, survival and feed conversion of juvenile pikeperch (Sander lucioperca) reared with dry feed in net cages. Aquac Int 17:1–6. https://doi.org/10.1007/s10499-008-9226-2
Borer SO, Miller LM, Kapuscinski AR (1999) Microsatellites in walleye Stizostedion vitreum. Mol Ecol 8(2):336–338. https://doi.org/10.1007/s12686-014-0275-8
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331. https://doi.org/10.1016/j.ajhg.2009.08.008
Chistiakov DA, Hellemans B, Volckaert FAM (2006) Microsatellites and their genomic distribution, evolution, function and applications: a review with special reference to fish genetics. Aquaculture 255:1–29. https://doi.org/10.1016/j.aquaculture.2005.11.031
Coykendall DK, Morrison CL, Stott W, Springmann MJ (2014) Development of eighteen microsatellite loci in walleye (Sander vitreus). Conserv Genet Resour 6(4):1019–1021. https://doi.org/10.1007/s12686-014-0275-8
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Eschbach E, Nolte AW, Kohlmann K, Kersten P, Kail J, Arlinghaus R (2014) Population differentiation of zander (Sander lucioperca) across native and newly colonized ranges suggests increasing admixture in the course of an invasion. Evol Appl 7:555–568. https://doi.org/10.1111/eva.12155
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals usingthe software STRUCTURE : a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinformatics Online 1:47–50. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Fopp-Bayat D (2007) Verification of meiotic gynogenesis in Siberian sturgeon (Acipenser baeri) using microsatellite DNA and cytogenetical markers. J Fish Biol 77:478–485. https://doi.org/10.1111/j.1095-8649.2007.01704.x
Fopp-Bayat D, Ciereszko A (2012) Microsatellite genotyping of cryopreserved spermatozoa for improvement of fish semen cryobanking. Cryobiology 65:196–201. https://doi.org/10.1016/j.cryobiol.2012.06.003
Fopp-Bayat D, Woznicki P (2006) Verification of ploidy level in sturgeon larvae. Aquac Res 37:1671–1675. https://doi.org/10.1111/j.1365-2109.2006.01614.x
Glenn TC, Schable NA (2005) Isolating microsatellite DNA loci. Methods Enzymol 395:202–222. https://doi.org/10.1016/S0076-6879(05)95013-1
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86(6):485–486. https://doi.org/10.1093/oxfordjournals.jhered.a111627
Grzybowski M, Sepulveda-Villet OJ, Stepien CA, Rosauer D, Binkowski F, Klaper R, Shepherd B, Goetz F (2010) Genetic variation of 17 wild yellow perch populations from the midwest and east coast analyzed via microsatellites. Trans Am Fish Soc 139(1):270–287. https://doi.org/10.1577/T07-276.1
Han X, Ling Q, Li C, Wang G, Xu Z, Lu G (2016) Characterization of pikeperch (Sander lucioperca) transcriptome and development of SSR markers. Biochem Syst Ecol 66:188–195. https://doi.org/10.1016/j.bse.2016.04.001
Hedrick PW, Miller PS (1992) Conservation genetics: techniques and fundamentals. Ecol Appl 2:30–46. https://doi.org/10.2307/1941887
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332. https://doi.org/10.1111/j.1755-0998.2009.02591.x
Hulak M, Kaspar V, Kohlmann K, Coward K, Tešitel J, Rodina M, Gela D, Kocour M, Linhart O (2010) Microsatellite-based genetic diversity and differentiation of foreign common carp (Cyprinus carpio) strains farmed in the Czech Republic. Aquaculture 298:194–201. https://doi.org/10.1016/j.aquaculture.2009.10.021
Hunt DH, Hutchinson WF, Carvalho GR (1999) An efficient method for PCR-based isolation of microsatellite arrays ( PIMA ). Mol Ecol 8:891–894. https://doi.org/10.1046/j.1365-294X.1999.00636.x
Kaczmarczyk D, Fopp-Bayat D (2013) Assemblage of spawning pairs based on their individual genetic profiles - as tool for maintaining genetic variation within sturgeon populations. Aquac Res 44:677–682. https://doi.org/10.1111/j.1365-2109.2011.03064.x
Kandpal RP, Kandpal G, Weissman SM (1994) Construction of libraries enriched for sequence repeats and jumping clones, and hybridization selection for region-specific markers. Proc Natl Acad Sci U S A 91:88–92. https://doi.org/10.1073/pnas.91.1.88
Khurshut E, Kohlmann K (2009) Application of nine species specific microsatellite loci to characterize three pikeperch (Sander lucioperca) populations from the Aral Sea basin in Uzbekistan. Environ Biotechnol 5(1):3–10. https://doi.org/10.1134/S0032945218030062
Knapik EW, Goodman A, Ekker M, Chevrette M, Delgado J, Neuhauss S, Shimoda N, Driever W, Fishman MC, Jacob HJ (1998) A microsatellite genetic linkage map for zebrafish (Danio rerio). Nat Genet 18:338–343. https://doi.org/10.1038/ng0498-338
Kohlmann K, Kersten P (2008) Isolation and characterization of nine microsatellite loci from the pike-perch, Sander lucioperca (Linnaeus, 1758). Mol Ecol Resour 8:1085–1087. https://doi.org/10.1111/j.1755-0998.2008.02166.x
Kusishchin KV, Ponomareva EV, Samoilov KY, Gruzdeva MA, Kholodova MV, Pavlov DS (2018) Morphological and genetic traits of pikeperch Sander lucioperca of the Volga-Akhtuba Aquatic System: on the spatial structure of a species in the Lower Volga Basin. J Ichthyol 58:318–332. https://doi.org/10.1134/S0032945218030062
Langella O (2002) POPULATIONS 1.2.28, a population genetics software. http://www.pge.cnrsgif.fr/bioinfo/populations. Accessed 2 April 2019
Lappalainen J, Dorner H, Wysujack K (2003) Reproduction biology of pikeperch (Sander lucioperca (L.)) - a review. Ecol Freshw Fish 12:95(2):95–95(2)106. https://doi.org/10.1034/j.1600-0633.2003.00005.x
Lappalainen A, Saks L, Šuštar M, Heikinheimo O, Jürgens K, Kokkonen E, Kurkilahti M, Verliin A, Vetemaa M (2016) Length at maturity as a potential indicator of fishing pressure effects on coastal pikeperch (Sander lucioperca) stocks in the northern Baltic Sea. Fish Res 174:47–57. https://doi.org/10.1016/j.fishres.2015.08.013
Leclerc D, Wirth T, Bernatchez L (2000) Isolation and characterization of microsatellite loci in the yellow perch (Perca flavescens), and cross-species amplification within the family Percidae. Mol Ecol 9:995–997. https://doi.org/10.1046/j.1365-294X.2000.00939-3.x
Lench NJ, Norris A, Bailey A, Booth A, Markham AF (1996) Vectorette PCR isolation of microsatellite repeat sequences using anchored dinucleotide repeat primers. Nucleic Acids Res 24(11):2190–2191. https://doi.org/10.1093/nar/24.11.2190
Li L, Wang HP, Givens C, Czesny S, Brown B (2007) Isolation and characterization of microsatellites in yellow perch (Perca flavescens). Mol Ecol Notes 7(4):600–603. https://doi.org/10.1111/j.1471-8286.2006.01645.x
Louati M, Kohlmann K, Ben Hassine OK, Kersten P, Poulet N, Bahri-Sfar L (2016) Genetic characterization of introduced Tunisian and French populations of pike-perch (Sander lucioperca) by species-specific microsatellites and mitochondrial haplotypes. Czech J Anim Sci 61(4):159–171. https://doi.org/10.17221/8847-CJAS
Menezes RF, Borchsenius F, Svenning JC, Søndergaard M, Lauridsen TL, Landkildehus F, Jeppesen E (2013) Variation in fish community structure, richness,and diversity in 56 Danish lakes with contrasting depth, size,and trophic state: does the method matter? Hydrobiologia 710:47–59. https://doi.org/10.1007/s10750-012-1025-0
Miroslav B, Jiri K, Tomas P, Blecha M, Kristan J, Policar T, Miroslav B, Jiri K, Tomas P (2016) Adaptation of intensively reared pikeperch (Sander Lucioperca) juveniles to pond culture and subsequent re-adaptation to a recirculation aquaculture system. Turk J Fish Aquat Sci 16(1):15–18. https://doi.org/10.4194/1303-2712-v16_1_02
Molnár T, Hancz C, Molnár M, Horn P (2004) The effects of diet and stocking density on the growth and behaviour of pond pre-reared pikeperch under intensive conditions. J Appl Ichthyol 20:105–109. https://doi.org/10.1046/j.1439-0426.2003.00529.x
Nei M, Tajima F, Tateno Y (1983) Accuracy of estimated phylogenetic trees from molecular dataii. gene frequency data. J Mol Evol 19:153–170. https://doi.org/10.1007/BF02300753
Ostrander EA, Jong PM, Rine J, Duyk G (1992) Construction of small-insert genomic DNA libraries highly enriched for microsatellite repeat sequences. Proc Natl Acad Sci U S A 89:3419–3423. https://doi.org/10.1073/pnas.89.8.3419
Park SDE (2001) Trypanotolerance in West African cattle and the population genetics effects of selection. Ph.D Thesis. University of Dublin, Dublin, p 2001
Peakall R, Smouse PE (2012) GenALEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Policar T, Stejskal V, Kristan J, Podhorec P, Svinger V, Blaha M (2013) The effect of fish size and stocking density on the weaning success of pond-cultured pikeperch Sander lucioperca L. juveniles. Aquac Int 21:869–882. https://doi.org/10.1007/s10499-012-9563-z
Poulet N, Balaresque P, Aho T, Björklund M (2009) Genetic structure and dynamics of a small introduced population: the pikeperch, Sander lucioperca, in the Rhône delta. Genetica 135(1):77–86. https://doi.org/10.1007/s10709-008-9260-z
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Rambaut A (2009) FigTree version 1.3.1, a graphical viewer of phylogenetic trees. http://tree.bio.ed.ac.uk. Accessed 2 April 2019
Rassmann K, Schlötterer C, Tautz D (1991) Isolation of simple-sequence loci for use in polymerase chain reaction-based DNA fingerprinting. Electrophoresis 12(2-3):113–118. https://doi.org/10.1002/elps.1150120205
Rougeot C, Jacobs B, Kestemont P, Melard C (2002) Sex control and sex determinism study in Eurasian perch, Perca fluviatilis, by use of hormonally sex-reversed male breeders. Aquaculture 211:81–89. https://doi.org/10.1016/S0044-8486(01)00893-6
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8(1):103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Säisä M, Salminen M, Koljonen ML, Ruuhijärvi J (2010) Coastal and freshwater pikeperch (Sander lucioperca) populations differ genetically in the Baltic Sea basin, Hereditas. Hereditas. 147:205–214. https://doi.org/10.1111/j.1601-5223.2010.02184.x
Salminen M, Koljonen ML, Säisä M, Ruuhijärvi J (2012) Genetic effects of supportive stockings on native pikeperch populations in boreal lakes - three cases, three different outcomes. Hereditas 149(1):1–15. https://doi.org/10.1111/j.1601-5223.2011.02230.x
Shimizu M, Kosaka N, Shimada T, Nagahata T, Iwasaki H, Nagai H, Shiba T, Emi M (2002) Universal fluorescent labeling (UFL) method for automated microsatellite analysis. DNA Res 9:173–178. https://doi.org/10.1093/dnares/9.5.173
Smouse PE, Whitehead MR, Peakall R (2015) An informational diversity framework, illustrated with sexually deceptive orchids in early stages of speciation. Mol Ecol Resour 15:1375–1384. https://doi.org/10.1111/1755-0998.12422
Sunnucks P (2000) Efficient genetic markers for population biology. Trends Ecol Evol 15(5):199–203. https://doi.org/10.1038/mt.2008.212
Szkudlarek M, Zakęś Z (2007) Effect of stocking density on survival and growth performance of pikeperch, Sander lucioperca (L.), larvae under controlled conditions. Aquac Int 15:67–81. https://doi.org/10.1007/s10499-006-9069-7
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. https://doi.org/10.1093/molbev/msr121
Ullmann A, Jacob F, Monod J (1967) Characterization by in vitro complementation of a peptide corresponding to an operator-proximal segment of the β-galactosidase structural gene of Escherichia coli. J Mol Biol 24(2):339–343. https://doi.org/10.1016/0022-2836(67)90341-5
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM (2007) Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35:71–74. https://doi.org/10.1093/nar/gkm306
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4(3):535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Vandeputte M, Rossignol MN, Pincent C (2011) From theory to practice: Empirical evaluation of the assignment power of marker sets for pedigree analysis in fish breeding. Aquaculture 314(1):80–86. https://doi.org/10.1016/j.aquaculture.2011.01.043
Ward RD (2006) The importance of identifying spatial population structure in restocking and stock enhancement programmes. Fish Res 80(1):9–18. https://doi.org/10.1016/j.fishres.2006.03.009
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution (N Y) 38:1358–1370. https://doi.org/10.2307/2408641
Wirth T, Saint-Laurent R, Bernatchez L (1999) Isolation and characterization of microsatellite loci in the walleye (Stizostedion vitreum), and cross-species amplification within the family Percidae. Mol Ecol 8(11):1960–1962. https://doi.org/10.1046/j.1365-294x.1999.00778-3.x
Wu KS, Jones R, Danneberger L, Scolnik PA (1994) Detection of microsatellite polymorphisms without cloning. Nucleic Acids Res 22:3257–3258. https://doi.org/10.1093/nar/22.15.3257
Yang X, Wang C, Wang J, Ma Y, Yin J, Wu H (2009) Isolation and characterization of 12 polymorphic microsatellite loci in Eurasian perch (Perca fluviatilus L.). Conserv Genet Resour 1:229–231. https://doi.org/10.1007/s12686-009-9056-1
Zakeś Z, Szczepkowski M, Partyka K, Wunderlich K (2013) Effect of gonadotropin hormonal stimulation on out-of-season propagation success of different year classes of indoor-reared pikeperch (Sander lucioperca (L.)). Aquac Int 21(4):801–810. https://doi.org/10.1007/s10499-012-9562-0
Zardoya R, Vollmer DM, Craddock C, Streelman JT, Karl S, Meyer A (1996) Evolutionary conservation of microsatellite flanking regions and their use in resolving the phylogeny of cichlid fishes (Pisces: Perciformes). Proc Biol Sci 263:1589–1598. https://doi.org/10.1098/rspb.1996.0233
Zarski D, Targońska K, Kaszubowski R, Kestemont P, Fontaine P, Krejszeff S, Kupren K, Kucharczyk D (2013) Effect of different commercial spawning agents and thermal regime on the effectiveness of pikeperch, Sander lucioperca (L.), reproduction under controlled conditions. Aquac Int 21(4):819–828. https://doi.org/10.1007/s10499-012-9597-2
Zhan A, Wang Y, Brown B, Wang H-P (2009) Isolation and characterization of novel microsatellite markers for yellow perch (Perca flavescens). Int J Mol Sci 10(1):18–27. https://doi.org/10.3390/ijms10010018
Acknowledgements
We wish to thank all colleagues who participated in sample collection.
Funding
Open access funding provided by Szent István University(SZIE). The research was supported by OTKA (Hungarian Scientific Research Fund) PD 79177 and the GINOP-2.3.2-15-2016-00025 projects.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed by the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kánainé Sipos, D., Kovács, G., Buza, E. et al. Comparative genetic analysis of natural and farmed populations of pike-perch (Sander lucioperca). Aquacult Int 27, 991–1007 (2019). https://doi.org/10.1007/s10499-019-00365-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10499-019-00365-7