
Abstract
Mitochondrial dysfunction and necroptosis are closely associated, and play vital roles in the medical strategy of multiple cardiovascular diseases. However, their implications in intracranial aneurysms (IAs) remain unclear. In this study, we aimed to explore whether mitochondrial dysfunction and necroptosis could be identified as valuable starting points for predictive, preventive, and personalized medicine for IAs. The transcriptional profiles of 75 IAs and 37 control samples were collected from the Gene Expression Omnibus (GEO) database. Differentially expressed genes (DEGs), weighted gene co-expression network analysis, and least absolute shrinkage and selection operator (LASSO) regression were used to screen key genes. The ssGSEA algorithm was performed to establish phenotype scores. The correlation between mitochondrial dysfunction and necroptosis was evaluated using functional enrichment crossover, phenotype score correlation, immune infiltration, and interaction network construction. The IA diagnostic values of key genes were identified using machine learning. Finally, we performed the single-cell sequencing (scRNA-seq) analysis to explore mitochondrial dysfunction and necroptosis at the cellular level. In total, 42 IA-mitochondrial DEGs and 15 IA-necroptosis DEGs were identified. Screening revealed seven key genes invovled in mitochondrial dysfunction (KMO, HADH, BAX, AADAT, SDSL, PYCR1, and MAOA) and five genes involved in necroptosis (IL1B, CAMK2G, STAT1, NLRP3, and BAX). Machine learning confirmed the high diagnostic value of these key genes for IA. The IA samples showed higher expression of mitochondrial dysfunction and necroptosis. Mitochondrial dysfunction and necroptosis exhibited a close association. Furthermore, scRNA-seq indicated that mitochondrial dysfunction and necroptosis were preferentially up-regulated in monocytes/macrophages and vascular smooth muscle cells (VSMCs) within IA lesions. In conclusion, mitochondria-induced necroptosis was involved in IA formation, and was mainly up-regulated in monocytes/macrophages and VSMCs within IA lesions. Mitochondria-induced necroptosis may be a novel potential target for diagnosis, prevention, and treatment of IA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Intracranial aneurysms (IA) are pathologically localized dilatations and thinning of the cerebral arterial wall, preferentially forming at the bifurcations of the circle of Willis [1]. Based on the shape, IAs can be divided into 3 types: saccular, fusiform, and dissecting. Saccular IAs constitute the majority, occurring in 1–2% of the population [2]. The number of annually detected IAs is continuously increasing with advances in imaging techniques [3]. Subarachnoid hemorrhage caused by IA rupture can be life-threatening with a mortality rate of approximately 35%, resulting in lasting disabilities and cognitive dysfunction [4]. Current treatments for IAs mainly include two approaches: microsurgical clipping and endovascular management, both of which are invasive, expensive, and exist complications. Therefore, early non-invasive prevention and intervention (e.g. drugs) are particularly vital for IA patients to decrease their health burden and promote health quality. Recently, the concept of predictive, preventive, and personalized medicine (PPPM/3PM) has been introduced for the management of vascular diseases [5] such as arterial stiffness [6], and stroke [7]. From the perspective of PPPM/3PM, we need to identify IA biomarkers for early detection and to understand the molecular mechanisms underlying drug development.
Mitochondria are essential regulators of apoptotic cell death, including necroptosis, ferroptosis, and pyroptosis [8]. Among these, necroptosis is the best-characterized form of regulated necrosis, and is mediated by RIPK3 and MLKL [9]. The mitochondrial-necroptosis axis involves in multiple disease occurrences and developments [10, 11]. It has been highlighted that vascular diseases have potentially benefited from the PPPM/3PM medical strategy targeting mitochondrial dysfunction and necroptosis. Previous research reported that mitochondrial dysfunction could predict the outcome of cardiovascular diseases linked to air pollution [12]. Thus, necroptosis may represent a novel therapeutic target for inhibiting the progression of cardiovascular diseases [13]. Mitochondrial dysfunction of vascular smooth muscle cells (VSMCs) drives the progression of aortic aneurysms [14]. RIPK3-mediated VSMC necroptosis promotes the pathogenesis of aortic aneurysms [15]. Both mitochondrial dysfunction and necroptosis are potential therapeutics for aortic aneurysms [13, 16]. However, studies on mitochondria and necroptosis in IAs are lacking. Identifying the roles of mitochondrial dysfunction and necroptosis in IAs may contribute to the development of better PPPM/3PM medical strategy for IAs.
Bioinformatics is a crucial component of the transition from traditional medicine to PPPM/3PM [5]. Through bioinformatics, this study aimed to investigate the molecular regulatory mechanism of the mitochondrial-necroptosis axis in IAs, which will contribute to the development of the predictive/diagnostic tools and targeted prevention and therapy from the perspective of PPPM/3PM. In our study, we collected mitochondria-related genes from the MitoCarta3.0 database [17] and necroptosis-related genes from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database. Aiming at mitochondrial dysfunction and necroptosis, the signature genes were selected using weighted gene co-expression network analysis (WGCNA), and phenotype scores were constructed using ssGSEA. We evaluated the association between these two phenotypes using functional enrichment crossover, correlation analysis, immune infiltration, and interaction networks. The key genes of these two phenotypes were validated by machine learning, including Random Forest (RF) and Sequential Minimal Optimization (SMO). Single-cell sequencing (scRNA-seq) was performed to investigate these two phenotypes at the cellular level. Collectively, with the framework of PPPM/3PM, our results elucidate the molecular mechanism of the mitochondrial-necroptosis axis in IAs, and provide a potential target for IA diagnosis, prevention, and treatment.
Methods
The detailed working flow chart is shown in Fig. 1.

The flow chart of this study
Data acquisition and processing
IA-related datasets were retrieved from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), including bulk-tissue mRNA sequence data (GSE75436, GSE15629, and GSE122897) and scRNA-seq data (GSE193533). A total of 75 IA samples and 37 control samples were included, and 4823 cells from the circle of Willis in the sham group and 9986 cells of that in the IA group were collected. The details of these datasets are listed in Table 1. In addition, 1136 human genes encoding proteins that strongly support mitochondrial localization were acquired from the MitoCarta3.0 (https://www.broadinstitute.org/mitocarta). One hundred and fifty-nine necroptosis-related genes were obtained from the KEGG Pathway database (https://www.genome.jp/dbget-bin/www_bget?pathway+hsa04217).
Identification of DEGs and IA-related genes
The training set GSE75436 was analyzed using the R package “limma” [18] (P < 0.05 and |log2(FoldChange)| > 1). Differentially expressed genes (DEGs) were identified using heatmaps and volcano plots. WGCNA is a crucial tool in bioinformatic analysis, which has been universally utilized in trait and gene association analysis [19]. In this study, we used the R package “WGCNA” to construct a co-expression network with the gene expression data of GSE75436 as input data and IA/control as trait data. First, the hclust function was used to perform sample clustering to remove outlier samples, and “method = average” was set as the parameter to calculate distance. Second, an appropriate soft threshold was identified to obtain a standard-scale free network. Third, a dynamic shear tree algorithm was employed to segment the modules, and Pearson’s correlation analysis was used to identify the modules related to IA.
Selection of IA-necroptosis-related DEGs (IA-Necroptosis DEGs) and IA-mitochondria-related DEGs (IA-Mito DEGs)
IA-Mito DEGs were acquired by intersecting the mitochondrial-localization protein-encoding genes, IA-related genes, and DEGs. IA-necroptosis DEGs were identified by intersecting necroptosis-related, and IA-related genes, and DEGs. To decrease the high false discovery rates reported in previous research [20], these genes were further screened using the Wilcox test between the IA and control samples (P < 0.05). Potential biological functions were identified by Gene Ontology (GO) and KEGG enrichment.
Phenotype scoring of mitochondrial dysfunction and necroptosis
IA-Mito DEGs and IA-Necroptosis DEGs were further screened to acquire key genes using the LASSO regression of the R package “glmnet” [21]. Response type was set as binomial and alpha was set as one. The phenotype scores of mitochondrial dysfunction and necroptosis were calculated using the ssGSEA algorithm of the R package “GSVA” [22]. The disparity of phenotype scores between the two groups and the correlation of phenotype scores were analyzed. Furthermore, according to the median values of phenotype scores, the IA and control samples were separately divided into high- and low-groups, and then GSEA analysis [23] was performed for all genes.
Immune infiltration analysis
Based on a gene set of 28 immune-related cells [24], the immune activity of each sample was evaluated by the ssGSEA algorithm of the R package “GSVA” [22]. The disparity of immune infiltration between the two groups and their correlations were analyzed. Furthermore, the association between the level of immune infiltration and the expression of mitochondrial dysfunction/necroptosis was investigated.
Construction of interaction networks
The protein-protein interaction (PPI) network between IA-Mito DEGs and IA-necroptosis DEGs was visualized using the STRING website (https://string-db.org) and Cytoscape software (version 3.9.1). The key genes screened from LASSO regression were set as the core of the PPI network, and the confidence level was set to 0.4. To explore the upstream regulation of key genes, we constructed the gene-microRNA-transcription factor (TF) interaction network through the online tool NetworkAnalyst [25]. The parameter of the minimum network was selected.
Validation of key genes
The expressions of key genes were extracted from the validating sets GSE15629 and GSE122897, and the Wilcoxon test was used to calculate and visualize the expression difference between IA and control samples. We also performed machine learning to evaluate the IA diagnosis value of key genes. To evaluate the classification performance, the 10-fold cross-validation of the RF algorithm was used in dataset GSE75436. The SMO algorithm was employed in the validating sets GSE15629 and GSE122897 with the training set GSE75436.
scRNA-seq analysis
For single-cell characteristics investigations, we analyzed the scRNA-seq dataset GSE193533, following the “Seurat” standard procedure [26]. Cells with less than 200 genes, more than 7000 counts in total, and more than 20% mitochondria genes were filtered out according to the previous reports [27]. The batch effect among samples was reduced using the R package “harmony” [28]. The top 2000 variably expressed genes were identified using the function “FindVariableFeatures”. The annotation of cell clusters was conducted according to the prior research [29]. Next, the level of mitochondrial dysfunction and necroptosis in each cell was quantified through the ssGSEA algorithm and key genes previously acquired. The expression disparity of these phenotypes was compared between IA and sham groups in each cell cluster.
Statistical analysis
All statistical analyses were performed using the R software (version 4.1.2). The difference of continuous variables was compared using the Wilcoxon test. The correlation of continuous variables was evaluated by the Pearson correlation. Data were visualized using the R package “ggplot2”. All tests were two-sided, and P < 0.05 was considered statistically significant. The significance level is denoted as follows: NS, not significant; *P < 0.05, **P < 0.01, and ***P < 0.001.
Result
Identification of DEGs and IA-Related genes
Based on the expression profile of GSE75436, IA and control groups were dramatically distinguished by PCA (Fig. 2A). Differential expression analysis showed that a total of 2508 DEGs between the IA and control groups, including 1310 genes upregulated and 1198 genes downregulated (Fig. 2B, C). Next, we performed the WGCNA analysis. The sample clustering diagram was shown in Fig. 2D. The correlation coefficient greater than 0.85 (the soft threshold β is 5) was highly correlated and suitable for constructing gene modules (Fig. 2E). The dynamic tree cut algorithm identified 13 gene modules (Fig. 2F). Among them, the darkorange, green, and darkseagreen modules were highly correlated with IAs (|R| > 0.6 and P < 0.01) (Fig. 2G). Therefore, the genes in these three modules were regarded as IA-related genes.

Identification of DEGs and IA-related genes in IA. A The PCA results of GSE75436. B Volcano plot showing DEGs in the IA and the normal samples. C Heat map of DEGs. D Sample clustering diagram of WGCNA. E Soft-thresholding filtering. F Clustering dendrogram of genes. G Correlation heatmap of gene modules and clinical features
Function crossover of gene between IA-Mito DEGs and IA-necroptosis DEGs
We first intersected the DEGs and IA-related genes, and then intersected with mitochondria-related genes and necroptosis-related genes respectively. In total, 44 IA-Mito DEGs and 15 IA-necroptosis DEGs were identified (Fig. 3A, and B). In addition, we screened these genes through the Wilcoxon test between IA and control groups. Two genes ALAS2 and CYP27B1 failed to show significant expression differences and therefore were eliminated, while the remaining 57 genes were used for subsequent analysis (Fig. 3C). Next, the gene functions were explored by GO and KEGG enrichment. In terms of IA-Mito DEGs, the most enriched GO terms were mitochondrial transport, apoptotic mitochondrial changes, T cells homeostasis, leukocyte homeostasis, and the like (Fig. 3D). The most overrepresented KEGG pathways were apoptosis, necroptosis, and so on (Fig. 3F). These results suggest that mitochondrial dysfunction may be related to apoptosis and immunity in IAs. As for IA-Necroptosis DEGs, GO analysis showed that programmed necrotic cell death, apoptotic mitochondrial changes, interleukin-1 beta production, and cytokine production involved in immune response were notable (Fig. 3E). KEGG analysis revealed that necroptosis, neutrophil extracellular trap formation, and inflammatory mediator regulation of TRP channels were enriched (Fig. 3G). These results suggest that necroptosis may be related to mitochondrial dysfunction and immunity in IAs. Overall, the enrichment results showed the gene function of IA-Mito DEGs and IA-necroptosis DEGs existed some level of crossover.

The function crossover of gene between IA Mito DEGs and IA-Necroptosis DEGs. A Venn diagram showing the overlap of DEGs, IA-related genes, and mitochondria-related genes. B Venn diagram showing the overlap of DEGs, IA-related genes, and Necroptosis-related genes. C Expression of 44 IA-Mito DEGs and 15 IA-Necroptosis DEGs in GSE75436. D and E GO enrichment results of 42 IA-Mito DEGs (D) and 15 IA-Necroptosis DEGs (E). F and G KEGG enrichment results of 42 IA-Mito DEGs (F) and 15 IA-Necroptosis DEGs (G)
Function crossover of phenotype scores between mitochondrial dysfunction and necroptosis
LASSO regression was used to identify key genes with the strongest capacity to predict IA occurrence (Fig. 4A, B). Seven key genes were obtained from 42 IA-Mito DEGs, including AADAT, BAX, HADH, KMO, MAOA, PYCR1, and SDSL. Five key genes were acquired from 15 IA-Necroptosis DEGs, including BAX, CAMK2G, IL1B, NLRP3, and STAT1. These key genes were applied to construct the phenotype scores by the ssGSEA algorithm. As shown in Fig. 4C, IA had a significantly higher level of phenotype scores of mitochondrial dysfunction and necroptosis (P < 0.001). The correlation between these two types of phenotype scores was highly close (R = 0.51) (Fig. 4D). Next, we divided the phenotype scores into high- and low-groups, performed the differential analysis, and conducted the GSEA enrichment analysis for the DEGs. Results showed that both the high mitochondrial dysfunction group and the high necroptosis group had increased levels of mitochondrial pathways (e.g. the release of cytochrome c from mitochondria, mitochondrial depolarization), necroptosis pathways, and immune pathways (e.g. T cell activation) (Fig. 4E, F). These crossover results further suggested the close correlation among mitochondrial dysfunction, necroptosis, and immunocyte infiltration in IAs.

The function crossover of phenotype scores between mitochondrial dysfunction and necroptosis. A LASSO regression screened out key genes of mitochondrial dysfunction from the 42 IA-Mito DEGs. B LASSO regression screened out key genes of necroptosis from the 15 IA-Mito DEGs. C Boxplot of phenotype scores of mitochondrial dysfunction and necroptosis between IA and normal samples. D The correlation matrix of phenotype scores of mitochondrial dysfunction and necroptosis. E GO enrichment analysis is based on the GSEA algorithm in key genes. F KEGG enrichment analysis is based on the GSEA algorithm in key genes
Correlation of immune infiltration to mitochondrial dysfunction and necroptosis
Considering the vital role of immune cells in IA development, we also estimated the level of immune infiltration. Among the 28 immune cells, there were 22 cell types with significant differences between IAs and the controls, and all of them showed higher expression in the IA group (Fig. 5A, B). The expression level of these types of immune cells showed a significantly strong association with each other (Fig. 5C). In addition, the correlation of immune infiltration to mitochondrial dysfunction and necroptosis was evaluated. The phenotype scores of mitochondrial dysfunction and necroptosis were positively correlated to the expression of the 22 types of immune cells. Among them, macrophage was top ranking, with its close relationship to mitochondrial dysfunction (R = 0.675, P < 0.001) and to necroptosis (R = 0.636, P < 0.001) (Fig. 5D).

The ssGSEA algorithm for analyzing immunocyte infiltration A Heatmap of 28 immune cell types. B The boxplot plot of 28 immune gene-sets content. C The correlation matrix of immune cells. D Correlation diagram for phenotype scores and immunocyte expression
Construction of interaction network and validation of key genes
After identifying the correlation between mitochondrial dysfunction and necroptosis, we constructed the PPI network between them. The PPI network included a total of 33 IA-Mito DEGs and 13 IA-Necroptosis DEGs, with the core of 7 key IA-Mito DEGs and 5 key IA-Necroptosis DEGs (Fig. 6A). In addition, the NetworkAnalyst tool was applied to predict the interaction network of key genes, microRNAs, and TFs. JUN, TP53, FOS, EGR1, MYC, MXI1, and USF1 were the common TFs targeting at least 3 key genes. Key genes AADAT and STAT1 had the most predicted microRNAs, including has-miR-129-5p, has-miR-203, and has-miR-199a-5p (Fig. 6B).

Construction of interaction network and validation of key genes. A The construction of PPI network among IA-Mito DEGs, IA-Necroptosis DEGs, and key genes. B Prediction of TFs and microRNAs for key genes of mitochondrial dysfunction and necroptosis. C Expression of 11 key genes in the validating datasets GSE15629 and GSE122897. D Evaluation of IA diagnosis value of key genes by the RF and SMO algorithms
Moreover, we used the datasets GSE15629 and GSE122897 to perform the validation of key genes. Most genes exhibited significant expression disparities between IA and control samples. The gene expression trends in the validating set were completely consistent with those in the training set GSE75436 (Fig. 6C). The machine learning showed the high IA diagnosis value of key genes in GSE75436 and GSE122897 (P < 0.05). However, their predictive capability in GSE15629 was unsatisfactory with P > 0.05, which may be influenced by the small samples (Fig. 6D). The predicting IA precisions of key genes were 0.774, 0.902, and 0.811 in GSE15629, GSE75436, and GSE122897. F-measures of the prediction were 0.770, 0.900, and 0.798 in GSE15629, GSE75436, and GSE122897.
Single-cell level expression of mitochondrial dysfunction and necroptosis
To better characterize mitochondrial dysfunction and necroptosis at the single-cell level, we performed scRNA-seq analysis in the circle of Willis of the mouse IA model. After data screening and integration as described in the Methods, we obtained the gene expression profiles of 4823 cells from the sham sample (GSM5813881), and 9986 cells from IA samples (GSM5813883, and GSM5813885) (Fig. 7A). Ten types of cell clusters were annotated and visualized, including VSMC, monocyte/macrophage (Mo/MΦ), and others (Fig. 7B). For ease of comparison, we divided the UMAP figure into 2 pieces: IA and sham (Fig. 7C). The IA samples had dramatically higher proportions of Mo/MΦ and lower percentages of VSMC than sham samples (Fig. 7D).

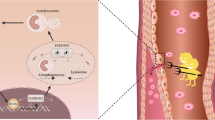
The expression of mitochondrial dysfunction and necroptosis at the cellular level. A Integration of multiple sample data using the R package harmony. B UMAP plot is colored by different cell types. C UMAP plot is colored by different cell types and divided by IA and sham groups. D Bar chart showing the percentage of different types of cells between IA and sham groups. E UMAP plot is colored by the expression of mitochondrial dysfunction. F Bar chart showing the expression disparity of mitochondrial dysfunction between IA and sham groups. G UMAP plot is colored by the expression of necroptosis. H Bar chart showing the expression disparity of necroptosis between IA and sham groups. I GO enrichment results of IA-Mito DEGs in the previously obtained PPI network. J Mechanism diagram of mitochondria-induced necroptosis in IAs
Next, we quantified the expression of mitochondrial dysfunction and necroptosis, through ssGSEA algorithms and key genes. These two phenotypes were differentially expressed in Mo/MΦ, VSMCs, and neutrophils between IA and sham samples. Compared with sham samples, IA had a significantly higher level of mitochondrial dysfunction and necroptosis in Mo/MΦ and VSMCs. However, the tendency of expression disparity of the two phenotypes was opposite in neutrophils (Fig. 7E–H). Since previous studies have reported a positive correlation between mitochondrial dysfunction and necroptosis [10, 11], we chose to focus on Mo/MΦ and VSMCs, rather than neutrophils. The GO enrichment of the previously obtained PPI network indicated that the reactive oxygen species (ROS) metabolic process was enriched in IA-Mito DEGs (Fig. 7I). Accordingly, combined with the literature review, we proposed the hypothesis that mitochondrial dysfunction (e.g. ROS) may induce the necroptosis of VSMCs and Mo/MΦ in IAs (Fig. 7J).
Discussion
Mitochondria are important organelles located in the cytoplasm of eukaryotic cells. Besides their roles in cellular metabolism and ATP generation, mitochondria also regulate multiple types of apoptotic cell death, such as necroptosis [8]. Necroptosis is not only caspase-independent but also inhibited by caspase activation, and its morphological features resemble necrosis, including cell swelling, organelle dysfunction, and plasma membrane rupture [30]. Although some studies have elucidated the promoting effects and therapeutic value of mitochondrial dysfunction and necroptosis in aortic aneurysms [14, 15], the relationship between these two phenotypes and IAs remains unclear.
This study focused on the roles of mitochondria dysfunction and necroptosis in IA formation and progression as diagnostic factors and therapeutic targets from the viewpoint of PPPM/3PM. We first used the WGCNA, differential gene analysis, and Wilcoxon test to identify signature genes, including 42 IA-Mito DEGs and 15 IA-necroptosis DEGs. Next, seven mitochondrial key genes and five necroptosis key genes were screened out using the LASSO algorithm. The phenotype scores were quantified through the ssGSEA algorithm. The close relationship between these two phenotypes was identified by function enrichment crossover, correlation analysis, immune infiltration, and interaction network. Subsequently, machine learning verified the high diagnostic value of key genes in IA. The scRNA-seq analysis revealed the concentrated expression of the mitochondrial-necroptosis axis in VSMCs and Mo/MΦ.
Accumulating evidence has demonstrated that mitochondria and necroptosis play crucial roles in aneurysm formation and progression. In our study, IA lesions were associated with significantly higher mitochondrial dysfunction and necroptosis rates. Key genes of these two phenotypes can well predict IA occurrence. Similarly, previous research found that mice with abnormal mitochondrial calcium uniporter channel complexes were prone to fatal abdominal aortic aneurysms [31]. Sustained increases in mitochondrial dysfunction and oxidative stress have been reported in VSMCs of abdominal aortic aneurysm [32]. RIP3-mediated VSMC necroptosis was actively involved in abdominal aortic aneurysm progression [33]. This preliminary evidence has layed the foundation for subsequent in-depth analysis.
Mitochondria are essential regulators of cellular necroptosis [9]. The mitochondrial-necroptosis axis is involved in multiple disease occurrences and developments [10, 11]. Chen et al. reported that RIPK1-mediated mitochondrial dysfunction contributed to compression-induced rat pulposus cell necroptosis [34]. Yang et al. found that MiR-7 mediated mitochondrial dysfunction triggered necroptosis in rhabdomyosarcoma [35]. The present study revealed that mitochondrial dysfunction and necroptosis existed the function crossover, high correlation, and network interaction, indicating that the mitochondrial-necroptosis axis may mediate IA pathogenesis. Among the factors inducing mitochondrial dysfunction, ROS (reactive oxygen species) accounts for a significant proportion [36, 37]. Mitochondrial-generated ROS can promote RIPK1 autophosphorylation to initiate necroptosis, ultimately leading to the formation of necrosomes [38, 39]. There has been a strong association between ROS and IA pathogenesis [40]. Cerebral macrophages and VSMCs utilized the main enzymatic sources of ROS to produce O2•− and H2O2 in response to hemodynamic stress, growth factors, and cytokines [41]. Our study indicated that key genes of mitochondrial dysfunction focus on ROS, implying a potentially induced role of ROS in the IA mitochondria-necroptosis axis.
The key genes of the IA mitochondrial-necroptosis axis were identified, including mitochondria genes of seven and necroptosis genes of five. These genes presented a strong interaction in the PPI network. Among them, IL1B[42], BAX [43], STAT1[44], and NLRP3[45] have been considered as potential biomarkers for IA formation and development. BAX, one of the BCL2 protein family members, is an apoptosis regulator. Under stress conditions, BAX would accumulate at the mitochondrial membrane, which results in the release of cytochrome c and triggers cell death [46]. However, the role of BAX in mitochondria-induced necroptosis has not been realved [47]. Moreover, NLRP3 acts as a sensor component of the NLRP3 inflammasome. Previous studies found that NLRP3 inflammasome could be activated by mitochondrial dysfunction [48], and contributes to the necroptosis occurence [49]. Notably, our machine learning analysis revealed that these key genes have a high diagnostic value for IA, which can assist clinicians in identifying early-stage patients.
TFs and microRNAs were involved in the regulation of mitochondrial-necroptosis axis [35]. We drew a microRNA-TF-gene regulatory network on mitochondrial dysfunction and necroptosis in IAs. Seven TFs (JUN, TP53, FOS, MYC, EGR1, MXI1, and USF1) regulated at least three key genes involved in mitochondrial dysfunction and necroptosis. Of them, c-JUN activation is linked to both mitochondrial dysfunction and necroptosis [50, 51]. MYC can increase mitochondrial oxidative phosphorylation and also impede mitophagy-dependent necroptosis [52, 53]. Additionally, JUN [54], TP53[55], FOS [56], SPI1[57], MYC [58], and HNF4A [59] have been proven to participate in IA pathogenesis. In terms of microRNA, we identified hsa-miR-610, hsa-miR-129-5p, hsa-miR-495, hsa-miR-203, and hsa-miR-199a-5p, all of which target the two or more key genes on the mitochondrial dysfunction and necroptosis in IAs. Among these, miR-199a-5p was significantly decreased in IA patients with poor prognosis and vasospasm [53]. MiR-199a-5p was also found to regulate mitochondrial function [60]. Altogether, these TFs and microRNA are promising targets for developing small molecular drugs and novel diagnostic tools in IAs.
To further elucidate the mechanism at the cellular level, we performed the scRNA-seq analysis. Results indicated that both mitochondrial dysfunction and necroptosis expression were significantly higher in Mo/MΦ and VSMC. Macrophages are known to be critical components of immune infiltration and can promote the expression of proteases that disrupt the internal elastic lamina and collagen matrix, leading to the initiation of IA [61]. The mitochondrial-necroptosis axis has been previously reported in macrophages. CIRP has induced mitochondrial DNA fragmentation and regulated macrophage necroptosis [62]. LRRK2 mutations have perturbed mitochondrial homeostasis and reprogrammed necroptosis pathways in macrophages [63]. The close correlation between the mitochondrial-necroptosis axis and immune infiltration in IAs has also been revealed through our functional enrichment and correlation analysis. Additionally, VSMC phenotypic switching from a contractile state to a synthetic state drove the IA formation and rupture [64]. Recent research has reported that mitochondrial DNA damage in VSMCs activated STING signaling and induced cellular necroptosis [65], suggesting the presence of the mitochondria-necroptosis axis in VSMCs. Collectively, mitochondria-induced necroptosis in macrophages and VSMCs may drive IA formation.
This study has some limitations. First, our data came from the GEO database, and the specific clinical data of each sample, such as gender, age, and complications, cannot be obtained, therefore it is not included in our research scope. Second, the sample size obtained from the database was limited. Therefore, studies with a large sample size are needed to thoroughly understand the specific roles of mitochondria and necroptosis in IA. Third, we did not perform in vitro experiments due to the difficulty of obtaining IA samples.
Conclusions and expert recommendations
In conclusion, our results strongly suggested that mitochondria-induced necroptosis was involved in IA formation. Its key genes had extremely high diagnostic values for IA occurrence. Furthermore, we demonstrated that this mechanism is mainly located in VSMCs and Mo/MΦ within IA tissues. The upregulation of mitochondria-induced necroptosis could be a novel potential target for predictive diagnosis, targeted prevention, and personalized treatment of IAs, which might promote the development of PPPM/3PM in IAs.
Predictive diagnosis and targeted prevention
From the perspective of PPPM/3PM, mitochondria-induced necroptosis may be a suitable genetic marker for the predictive diagnosis and targeted prevention of IA. The present study screened eleven key biomarker genes of mitochondria-induced necroptosis in IA through integrative bioinformatic approaches (mitochondrial dysfunction: KMO, HADH, BAX, AADAT, SDSL, PYCR1, MAOA; necroptosis: IL1B. CAMK2G, STAT1, NLRP3, BAX). Accordingly, phenotype scores were constructed. Individuals with higher phenotype scores of mitochondria dysfunction and necroptosis are more susceptible to IA. Machine learning further identified their high diagnosis value for IA occurrence. Actually, previous studies had reported that the genes on the mitochondria-induced necroptosis can be a precise method for the diagnosis and prevention of diseases [66]. This is the first time to correlate mitochondria-induced necroptosis with IA. Considering the genetic susceptibility of IAs, we recommend more genetic sequencing studies in terms of mitochondria-induced necroptosis, like exome and intron, to assess the risk of IA occurrence and rupture.
Personalized medicine
Although growing evidence suggested that both mitochondrial dysfunction and necroptosis are potential therapeutics for aortic aneurysm [13, 16], no study noted the application of these two phenotypes in IA treatment. Therefore, we recommend focusing on mitochondria-induced necroptosis in IAs. These eleven key biomarker genes can help researchers to design and develop novel small molecular drugs, which may inhibit IA formation and progression. The key gene-miRNA-TF regulatory network would also provide a unique benefit to developing new approaches in IA treatment. Of note, since the mitochondria-induced necroptosis is mainly located in Mo/MΦ, we strongly recommend developing customized immunotherapy in IAs from the perspective of PPPM/3PM.
Data availability
The datasets generated and/or analyzed during the current study are available in the GEO database, including the datasets GSE75436 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE75436), GSE15629 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE15629), GSE122897 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122897), and GSE193533 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE193533).
Code availability
All software applications used are included in this article.
Abbreviations
- DEG:
-
Differentially expressed gene
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene Ontology
- IA-Mito DEGs:
-
IA-mitochondria-related DEGs
- IA-Necroptosis DEGs:
-
IA-Necroptosis-related DEGs
- IA:
-
Intracranial aneurysm
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MMP:
-
Matrix metalloproteinases
- Mo/MΦ:
-
Monocyte/macrophage
- PPPM/3PM:
-
Predictive, preventive, and personalized medicine
- PPI:
-
Protein-protein interaction
- RF:
-
Random Forest
- ROS:
-
Reactive oxygen species
- SMO:
-
Sequential Minimal Optimization
- scRNA-seq:
-
Single-cell RNA sequencing
- TF:
-
Transcription factor
- VSMC:
-
Vascular smooth muscle cell
- WCGNA:
-
Weighted gene co-expression network analysis
References
Chen B, Tao W, Li S et al (2022) Medial gap: a structural factor at the arterial bifurcation aggravating hemodynamic insult. J Neuropathol Exp Neurol 29(4):282–290. https://doi.org/10.1093/jnen/nlac017
Brown RD Jr, Broderick JP (2014) Unruptured intracranial aneurysms: epidemiology, natural history, management options, and familial screening. Lancet Neurol 13(4):393–404. https://doi.org/10.1016/s1474-4422(14)70015-8
Etminan N, Rinkel GJ (2016) Unruptured intracranial aneurysms: development, rupture and preventive management. Nat Rev Neurol 12(12):699–713. https://doi.org/10.1038/nrneurol.2016.150
Neifert SN, Chapman EK, Martini ML et al (2021) Aneurysmal subarachnoid hemorrhage: the last decade. Transl Stroke Res 12(3):428–446. https://doi.org/10.1007/s12975-020-00867-0
Golubnitschaja O, Baban B, Boniolo G et al (2016) Medicine in the early twenty-first century: paradigm and anticipation-EPMA position paper 2016. Epma J 7(1):23. https://doi.org/10.1186/s13167-016-0072-4
Wu Z, Zhang H, Li Z et al (2022) Mutual effect of homocysteine and uric acid on arterial stiffness and cardiovascular risk in the context of predictive, preventive, and personalized medicine. Epma J 13(4):581–595. https://doi.org/10.1007/s13167-022-00298-x
Polivka J Jr, Polivka J, Pesta M et al (2019) Risks associated with the stroke predisposition at young age: facts and hypotheses in light of individualized predictive and preventive approach. Epma J 10(1):81–99. https://doi.org/10.1007/s13167-019-00162-5
Bock FJ, Tait SWG (2020) Mitochondria as multifaceted regulators of cell death. Nat Rev Mol cell Biol 21(2):85–100. https://doi.org/10.1038/s41580-019-0173-8
Pasparakis M, Vandenabeele P (2015) Necroptosis and its role in inflammation. Nature 15(7534):311–320. https://doi.org/10.1038/nature14191
Han Q, Zhang J, Sun Q, Xu Y, Teng X (2020) Oxidative stress and mitochondrial dysfunction involved in ammonia-induced nephrocyte necroptosis in chickens. Ecotoxicol Environ Saf 15:110974. https://doi.org/10.1016/j.ecoenv.2020.110974
Huang Z, Wang S, Yang Y et al (2023) Mitochondrial dysfunction promotes the necroptosis of Purkinje cells in the cerebellum of acrylamide-exposed rats. Food Chem Toxicol 171:113522. https://doi.org/10.1016/j.fct.2022.113522
Boovarahan SR, Kurian GA (2018) Mitochondrial dysfunction: a key player in the pathogenesis of cardiovascular diseases linked to air pollution. Rev Environ Health 27(2):111–122. https://doi.org/10.1515/reveh-2017-0025
Gupta K, Phan N, Wang Q, Liu B (2018) Necroptosis in cardiovascular disease-a new therapeutic target. J Mol Cell Cardiol 118:26–35. https://doi.org/10.1016/j.yjmcc.2018.03.003
Oller J, Gabandé-Rodríguez E, Ruiz-Rodríguez MJ et al (2021) Extracellular tuning of mitochondrial respiration leads to aortic aneurysm. Circulation 143(21):2091–2109. https://doi.org/10.1161/CIRCULATIONAHA.120.051171
Wang Q, Liu Z, Ren J, Morgan S, Assa C, Liu B (2015) Receptor-interacting protein kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circul Res 13(4):600–611. https://doi.org/10.1161/circresaha.116.304899
Cooper HA, Cicalese S, Preston KJ et al (2021) Targeting mitochondrial fission as a potential therapeutic for abdominal aortic aneurysm. Cardiovasc Res 22(3):971–982. https://doi.org/10.1093/cvr/cvaa133
Rath S, Sharma R, Gupta R et al (2021) MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res 8(D1):D1541-d1547. https://doi.org/10.1093/nar/gkaa1011
Ritchie ME, Phipson B, Wu D et al (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res Apr 20(7):e47. https://doi.org/10.1093/nar/gkv007
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinf 29:559. https://doi.org/10.1186/1471-2105-9-559
Li Y, Ge X, Peng F, Li W, Li JJ (2022) Exaggerated false positives by popular differential expression methods when analyzing human population samples. Genome Biol 15(1):79. https://doi.org/10.1186/s13059-022-02648-4
Engebretsen S, Bohlin J (2019) Statistical predictions with glmnet. Clin Epigenetics 23(1):123. https://doi.org/10.1186/s13148-019-0730-1
Hanzelmann S, Castelo R, Guinney J (2013) GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf 16:7. https://doi.org/10.1186/1471-2105-14-7
Subramanian A, Tamayo P, Mootha VK et al (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 25(43):15545–15550. https://doi.org/10.1073/pnas.0506580102
Charoentong P, Finotello F, Angelova M et al (2017) Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep 18(1):248–262. https://doi.org/10.1016/j.celrep.2016.12.019
Zhou G, Soufan O, Ewald J, Hancock REW, Basu N, Xia J (2019) NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res 2(W1):W234-w241. https://doi.org/10.1093/nar/gkz240
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36(5):411–420. https://doi.org/10.1038/nbt.4096
Li S, Zhang Q, Li J, Weng L (2022) Comprehensive analysis of autoimmune-related genes in amyotrophic lateral sclerosis from the perspective of 3P medicine. Epma J 13(4):699–723. https://doi.org/10.1007/s13167-022-00299-w
Korsunsky I, Millard N, Fan J et al (2019) Fast, sensitive and accurate integration of single-cell data with harmony. Nat Methods 16(12):1289–1296. https://doi.org/10.1038/s41592-019-0619-0
Martinez AN, Tortelote GG, Pascale CL et al (2022) Single-cell transcriptome analysis of the circle of willis in a mouse cerebral aneurysm model. Stroke 53(8):2647–2657. https://doi.org/10.1161/strokeaha.122.038776
Weinlich R, Oberst A, Beere HM, Green DR (2017) Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 18(2):127–136. https://doi.org/10.1038/nrm.2016.149
Bick AG, Wakimoto H, Kamer KJ et al (2017) Cardiovascular homeostasis dependence on MICU2, a regulatory subunit of the mitochondrial calcium uniporter. Proc Natl Acad Sci USA 114(43):E9096–E9104. https://doi.org/10.1073/pnas.1711303114
Tavris BS, Peters AS, Böckler D, Dihlmann S (2023) Mitochondrial dysfunction and increased DNA damage in vascular smooth muscle cells of abdominal aortic aneurysm (AAA-SMC). Oxid Med Cell Longev 2023:6237960. https://doi.org/10.1155/2023/6237960
Wang Q, Liu Z, Ren J, Morgan S, Assa C, Liu B (2015) Receptor-interacting protein kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circ Res 116(4):600–611. https://doi.org/10.1161/CIRCRESAHA.116.304899
Chen S, Lv X, Hu B et al (2018) Critical contribution of RIPK1 mediated mitochondrial dysfunction and oxidative stress to compression-induced rat nucleus pulposus cells necroptosis and apoptosis. Apoptosis 23(5–6):299–313. https://doi.org/10.1007/s10495-018-1455-x
Yang L, Kong D, He M et al (2020) MiR-7 mediates mitochondrial impairment to trigger apoptosis and necroptosis in Rhabdomyosarcoma. Biochim Biophys Acta Mol Cell Res 1867(12):118826. https://doi.org/10.1016/j.bbamcr.2020.118826
Raimondi M, Fontana F, Marzagalli M et al (2021) Ca(2+) overload- and ROS-associated mitochondrial dysfunction contributes to δ-tocotrienol-mediated paraptosis in melanoma cells. Apoptosis 26(5–6):277–292. https://doi.org/10.1007/s10495-021-01668-y
Zhao M, Wang Y, Li L et al (2021) Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 11(4):1845–1863. https://doi.org/10.7150/thno.50905
Schenk B, Fulda S (2015) Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 34(47):5796–5806. https://doi.org/10.1038/onc.2015.35
Zhang Y, Su SS, Zhao S et al (2017) RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun 8:14329. https://doi.org/10.1038/ncomms14329
Chalouhi N, Ali M, Jabbour P et al (2012) Biology of intracranial aneurysms: role of inflammation. J Cereb Blood Flow Metab: Official J Int Soc Cereb Blood Flow Metab 32(9):1659–1676. https://doi.org/10.1038/jcbfm.2012.84
Sheinberg DL, McCarthy DJ, Elwardany O et al (2019) Endothelial dysfunction in cerebral aneurysms. Neurosurg Focus 1(1):E3. https://doi.org/10.3171/2019.4.Focus19221
Gao Y, Zhao C, Wang J, Li H, Yang B (2020) The potential biomarkers for the formation and development of intracranial aneurysm. J Clin Neurosci 81:270–278. https://doi.org/10.1016/j.jocn.2020.09.072
Chen B, Zhou H, Zhou X, Yang L, Xiong Y, Zhang L (2022) Comprehensive analysis of endoplasmic reticulum stress in intracranial aneurysm. Front Cell Neurosci 16:865005. https://doi.org/10.3389/fncel.2022.865005
Tian T, Sun W, Du J, Sun Y (2022) Analysis of co-expression gene network associated with intracranial aneurysm and type 2 diabetes mellitus. Front Neurol 13:1032038. https://doi.org/10.3389/fneur.2022.1032038
Turhon M, Maimaiti A, Gheyret D et al (2022) An immunogenic cell death-related regulators classification patterns and immune microenvironment infiltration characterization in intracranial aneurysm based on machine learning. Front Immunol 13:1001320. https://doi.org/10.3389/fimmu.2022.1001320
Edlich F, Banerjee S, Suzuki M et al (2011) Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145(1):104–16. https://doi.org/10.1016/j.cell.2011.02.034
Jiang Y, Chen X, Yang G et al (2013) BaP-induced DNA damage initiated p53-independent necroptosis via the mitochondrial pathway involving bax and Bcl-2. Hum Exp Toxicol 32(12):1245–1257. https://doi.org/10.1177/0960327113488613
Kelley N, Jeltema D, Duan Y, He Y (2019) The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 20(13):3328. https://doi.org/10.3390/ijms20133328
Huang Y, Xu W, Zhou R (2021) NLRP3 inflammasome activation and cell death. Cell Mol Immunol 18(9):2114–2127. https://doi.org/10.1038/s41423-021-00740-6
Della-Flora Nunes G, Wilson ER, Hurley E et al (2021) Activation of mTORC1 and c-Jun by Prohibitin1 loss in Schwann cells may link mitochondrial dysfunction to demyelination. Elife. https://doi.org/10.7554/eLife.66278
Cao M, Chen F, Xie N et al (2018) c-Jun N-terminal kinases differentially regulate TNF- and TLRs-mediated necroptosis through their kinase-dependent and -independent activities. Cell Death Dis 15(12):1140. https://doi.org/10.1038/s41419-018-1189-2
Xiong J, Wang L, Fei XC et al (2017) MYC is a positive regulator of choline metabolism and impedes mitophagy-dependent necroptosis in diffuse large B-cell lymphoma. Blood Cancer J 7(7):e0. https://doi.org/10.1038/bcj.2017.61
Lee KM, Giltnane JM, Balko JM et al (2017) MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab 26(4):633–657. https://doi.org/10.1016/j.cmet.2017.09.009
Takagi Y, Ishikawa M, Nozaki K, Yoshimura S, Hashimoto N (2002) Increased expression of phosphorylated c-Jun amino-terminal kinase and phosphorylated c-Jun in human cerebral aneurysms: role of the c-Jun amino-terminal kinase/c-Jun pathway in apoptosis of vascular walls. Neurosurg 51(4):997–1002. https://doi.org/10.1097/00006123-200210000-00027
Jiang H, Ding Y, Wu L, Jiang C, Wang C (2022) The roles and diagnostic value of miRNA-1246 in the serum of patients with intracranial aneurysms. Transl Neurosci 1(1):172–180. https://doi.org/10.1515/tnsci-2022-0227
Zheng X, Xue C, Luo G, Hu Y, Luo W, Sun X (2015) Identification of crucial genes in intracranial aneurysm based on weighted gene coexpression network analysis. Cancer Gene Ther 22(5):238–245. https://doi.org/10.1038/cgt.2015.10
Zhao H, Li ST, Zhu J, Hua XM, Wan L (2019) Analysis of peripheral blood cells’ transcriptome in patients with subarachnoid hemorrhage from ruptured aneurysm reveals potential biomarkers. World Neurosurg 129:e16–e22. https://doi.org/10.1016/j.wneu.2019.04.125
Li X, Zhao H, Liu J, Tong J (2020) Long non-coding RNA MIAT Knockdown prevents the formation of intracranial aneurysm by downregulating ENC1 via MYC. Front Physiol 11:572605. https://doi.org/10.3389/fphys.2020.572605
Bo L, Wei B, Wang Z, Li C, Gao Z, Miao Z (2018) Bioinformatic analysis of gene expression profiling of intracranial aneurysm. Mol Med Rep 17(3):3473–3480. https://doi.org/10.3892/mmr.2017.8367
Li K, Zhao B, Wei D et al (2020) Long non-coding RNA ANRIL enhances mitochondrial function of hepatocellular carcinoma by regulating the MiR-199a-5p/ARL2 axis. Environ Toxicol 35(3):313–321. https://doi.org/10.1002/tox.22867
Frösen J, Cebral J, Robertson AM, Aoki T (2019) Flow-induced, inflammation-mediated arterial wall remodeling in the formation and progression of intracranial aneurysms. Neurosurg Focus 1(1):E21. https://doi.org/10.3171/2019.5.Focus19234
Li Z, Fan EK, Liu J et al (2017) Cold-inducible RNA-binding protein through TLR4 signaling induces mitochondrial DNA fragmentation and regulates macrophage cell death after trauma. Cell Death Dis 11(5):e2775. https://doi.org/10.1038/cddis.2017.187
Weindel CG, Martinez EL, Zhao X et al (2022) Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell 18(17):3214-3231e23. https://doi.org/10.1016/j.cell.2022.06.038
Starke RM, Chalouhi N, Ding D et al (2014) Vascular smooth muscle cells in cerebral aneurysm pathogenesis. Transl Stroke Res 5(3):338–346. https://doi.org/10.1007/s12975-013-0290-1
Luo W, Wang Y, Zhang L et al (2020) Critical role of cytosolic DNA and its sensing adaptor STING in aortic degeneration, dissection, and rupture. Circulation 7(1):42–66. https://doi.org/10.1161/circulationaha.119.041460
Cheng M, Lin N, Dong D, Ma J, Su J, Sun L (2021) PGAM5: a crucial role in mitochondrial dynamics and programmed cell death. Eur J Cell Biol 100(1):151144. https://doi.org/10.1016/j.ejcb.2020.151144
Acknowledgements
The authors thank the Fundamental Research Funds for the Central Universities of Central South University (Grant no: 2021zzts1036) for their support.
Funding
The Fundamental Research Funds for the Central Universities of Central South University (No. 2021zzts1036). National Natural Science Foundation of China (No. 81901268).
Author information
Authors and Affiliations
Contributions
BC, KX, RP, and LZ designed and drafted the manuscript; BC, KX, RP and LZ organized figures and edited legends. BC, KX, LY, HZ, RP and LZ revised the article; BC, KX, LY, HZ, RP and LZ conducted the data analysis; All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval and consent to participate
Data retrieved from the GEO and controlled-access databases were collected using IA and control tissues from patients who provided informed consent based on the guidelines laid out by the GEO Ethics, Law, and Policy Group.
Consent for publication
After reviewing the manuscript, all authors agreed to its publication in the current form.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, B., Xie, K., Zhang, J. et al. Comprehensive analysis of mitochondrial dysfunction and necroptosis in intracranial aneurysms from the perspective of predictive, preventative, and personalized medicine. Apoptosis 28, 1452–1468 (2023). https://doi.org/10.1007/s10495-023-01865-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-023-01865-x